

Abstract
A novel series of bis-indoles derived from naturally occurring marine alkaloid 4 were synthesized and evaluated as inhibitors of methicillin-resistant Staphylococcus aureus (MRSA) pyruvate kinase (PK). PK is not only critical for bacterial survival which would make it a target for development of novel antibiotics, but it is reported to be one of the most highly connected ‘hub proteins’ in MRSA, and thus should be very sensitive to mutations and making it difficult for the bacteria to develop resistance. From the co-crystal structure of cis-3–4-dihydrohamacanthin B (4) bound to S. aureus PK we were able to identify the pharmacophore needed for activity. Consequently, we prepared simple direct linked bis-indoles such as 10b that have similar anti-MRSA activity as compound 4. Structure–activity relationship (SAR) studies were carried out on 10b and led us to discover more potent compounds such as 10c, 10d, 10k and 10m with enzyme inhibiting activities in the low nanomolar range that effectively inhibited the bacteria growth in culture with minimum inhibitory concentrations (MIC) for MRSA as low as 0.5 μg/ml. Some potent PK inhibitors, such as 10b, exhibited attenuated antibacterial activity and were found to be substrates for an efflux mechanism in S. aureus. Studies comparing a wild type S. aureus with a construct (S. aureus LAC Δpyk::ErmR) that lacks PK activity confirmed that bactericidal activity of 10d was PK-dependant.
Keywords: Antibacterial, MRSA, Pyruvate kinase, Bis-indole
1. Introduction
Multidrug-resistant bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA), have developed numerous resistance mechanisms in response to antibiotic pressure.1 There is an increasing incidence of MRSA infections in hospitals worldwide and they have begun to penetrate into the general community. The vast majority of current antibiotics in use are directed to critical proteins unique to the bacteria and without human homologs to avoid mechanism based toxicity. This has severely limited the available targets for drug design.
Recently, pyruvate kinase (PK) was identified as a highly interconnected essential hub protein in MRSA, with structural features distinct from the human homologs, as a novel drug target.2–4 This was based on the supposition that hub proteins are not only critical for bacterial survival but should be very sensitive to mutations5 and targeting them should reduce the potential for development of resistance strains and species. In silico library screening, initially directed to putative binding sites unique to MRSA PK, combined with enzyme assays identified several active MRSA PK inhibitors including compound 16 (Fig. 1). Compound 1 was very selective for the bacterial enzyme compared to four human PK isoforms (M1, M2, R and L) and it did not inhibit growth of HeLa cells indicating a lack of overt toxicity to mammalian cells. Structure–activity relationship (SAR) studies were initiated which led to the identification of more potent enzyme inhibitors (such as 2) and which showed effective inhibition of a wide panel of gram positive bacterial growth, with potencies comparable to standard antibiotics such as vancomycin.3,7 In addition, the MIC was not significantly increased even after 25 bacterial passages in culture with compound 2 at the highest sub-lethal concentration,4 which confirmed that MRSA PK is an essential target less prone to developing resistance. X-ray crystal structures for 1 and 3 bound to MRSA282 PK were obtained which revealed that both compounds bind to a flat lipophilic pocket at the minor interfaces in the homo-tetrameric enzyme structure. This pocket was found to be modified and not accessible in the human PK enzymes.
Figure 1.
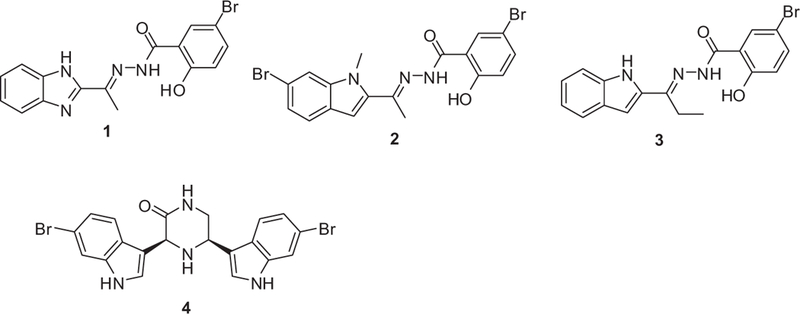
MRSA PK inhibitors 1–4.
More recently, Zoraghi et al.8 screened the inhibitory potential of a natural marine product library of 968 crude benthic invertebrate extracts and identified cis-3–4-dihydrohamacanthin B (4) as a potent inhibitor of MRSA PK with an IC50 of 16 nM. Compound 4 also exhibited anti-bacterial activity with MIC of 12.5 μg/ml (tested against S. aureus stains RN4229 and MRSA252). They were able to derive an X-ray crystal structure of 4 bound to MRSA PK and found that it binds to the same site as the hydrazone compounds 1 and 3.
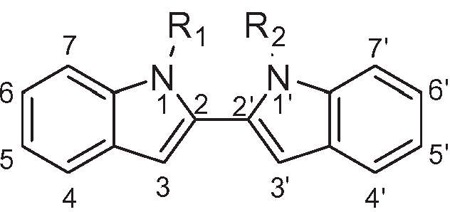
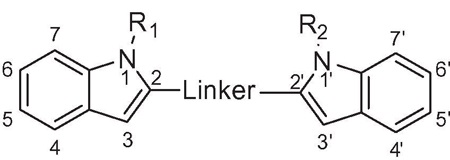
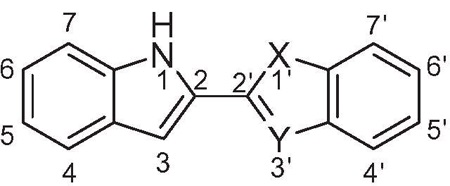
From examination of the crystal structure of cis-3–4-dihydro-hamacanthin B (4) bound to S. aureus PK, it is apparent that the two indole moieties lie in a linear relationship to each other and are essentially in the same plane. The compound is anchored by symmetric hydrogen bonds between Ser362 and Ser365 from chains A and B, respectively, and the indole nitrogens (Fig. 2a). The indole phenyl rings have prominent hydrophobic interactions with Ile361 and His365. The two bromine atoms are oriented towards the interior of the binding site in the deep hydrophobic pocket formed by Thr353, Ser354, Ala358, and Leu370. The symmetrical nature of the binding pocket was mirrored by the pseudo-symmetrical properties of the ligand. Hence it appears that both the indoles in the scaffold are critical for binding and the structure suggested that by linking the two indole at the C-2 position and removing the lactam ring of 4, one might derive compounds such as compound 10b (Fig. 2b) having all the necessary elements to bind tightly to MRSA PK.
Figure 2.
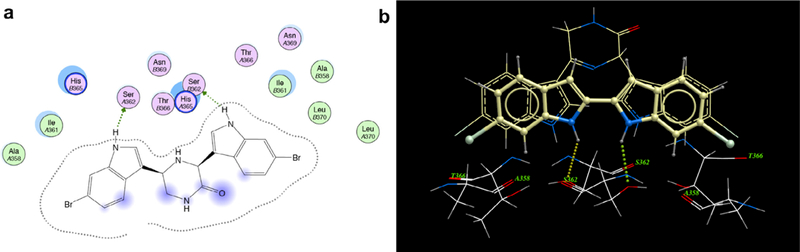
Binding mode of compound 4 at the MRSA PK tetramer interface binding site; (a) a two-dimensional map (MOE software) of the binding interactions between 1 and the interface site based on its co-crystallization with MRSA PK. Green arrows depict hydrogen-accepting interactions between 4 and MRSA PK residues from the interface (left). (b) Modeled overlay of compound 10b with 4 in the MRSA binding site (right) (ICM software from PDB data file 3t07).
In this paper, we present a detailed account of SAR for enzyme inhibitory and optimization of antibacterial activity for such an extensive series of bis-indoles.
2. Results
2.1. Chemistry
The syntheses of all target compounds were carried out as described in Schemes 1–6. The indole NH was first protected with a phenylsulfonyl group to give intermediate 6 which was subsequently iodinated at the 2-position to give 2-iodoindole 7 by treating 6 with LDA followed by the addition of diiodoethane (Scheme 1). We did attempt to couple 7 with the boronic acid 9 under standard Suzuki–Miyura conditions but no desired product was isolated. Fortunately, the coupling reaction of boronic acid 9 with the unprotected indole 8, (obtained by hydrolysis of compound 7), proceeded smoothly to give the desired adduct. Finally, removing the Boc protecting group with TFA gave the desired compound 10. In order to prepare the alkylated bis-indole 12, 8 was first alkylated with alkyl halide to give intermediate 11, which was subsequently coupled with boronic acid 9. Compound 22 was prepared in a similar manner where an appropriate 2-iodo-hetrocycle 21 was coupled with boronic acid 9 under standard Suzuki–Miyura conditions and finally the Boc protecting group was removed with TFA (Scheme 2).
Scheme 1.
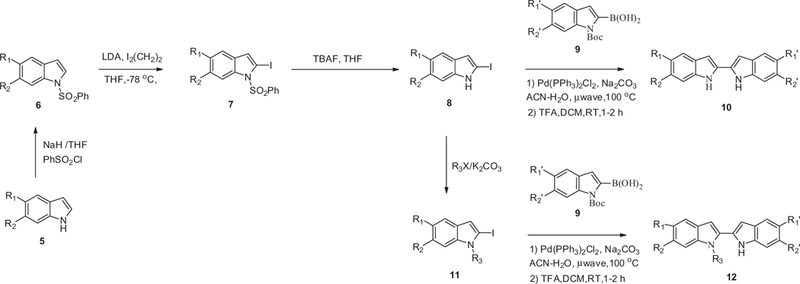
General synthesis of bis-indoles 10 and 12.
Scheme 6.
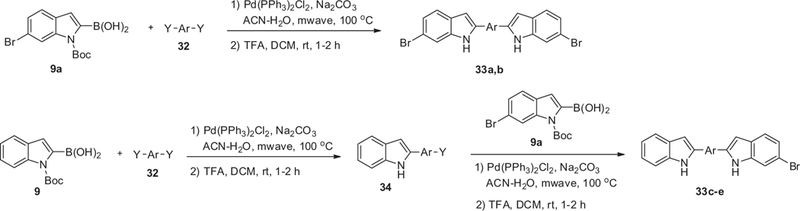
General synthesis of bis-indoles with an aryl linker.
Scheme 2.
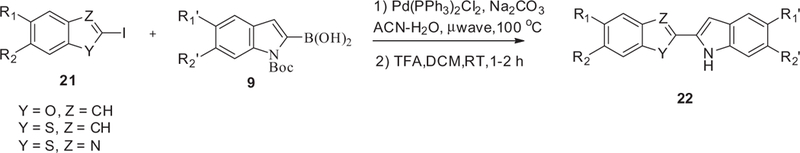
General synthesis of mono-indole coupled with heterocycles.
Compound 14 was prepared from 8b where it was first treated with alkyl bromide and then the ester was hydrolyzed with LiOH to give the corresponding carboxylic acid derivative 13 which was then coupled with 9a (Scheme 3). The carboxylic acid on 14 was then reacted with morpholine and HBTU to give compound 15. Treating intermediate 8b with 2-bromoethanol gave alcohol 12 that was then coupled with boronic acid 9a and subsequent removal of Boc protecting group with TFA gave compound 17. Compound 20 was prepared from alcohol 12 which was first converted to the mesylate and then displaced by an amine to give intermediate 19 which was subsequently coupled with 9a followed by the removal of the Boc protecting group.
Scheme 3.
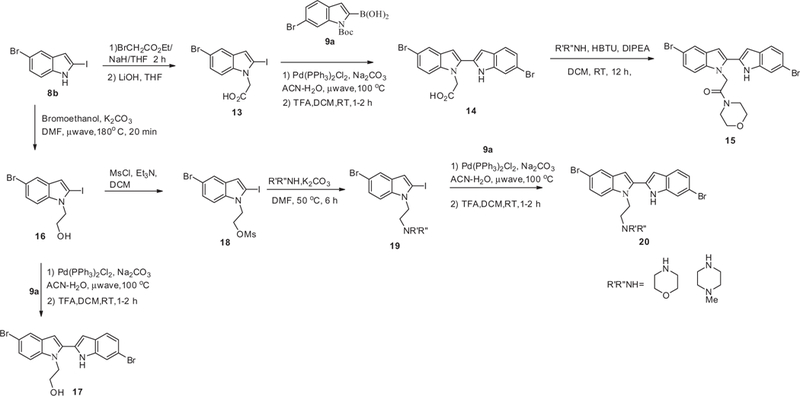
General synthesis of bis-indoles 14, 15, 17 and 20.
2-Acetylene-indole 24 was prepared by coupling 2-iodoindole 7 with TIPS-acetylene using Sonogashira coupling condition with PdCl2(PPh3)2 and CuI, and then the phenylsulfonyl protecting group was removed with TBAF (Scheme 4). A second Sonogashira coupling of intermediate 24 with 7 followed by removal of the phenylsulfonyl group gave compound 25. Treating 25 with MeI gave a mixture of mono-methylated compound 26a and dimethylated compound 26b. Attempts were made to reduce the acetylene linker of compound 25 as a route to synthesize 27b; however, we were only able to isolate the mono-brominated compound 27a, which was nonetheless useful for our SAR study (Scheme 5). Compound 27b was finally synthesized by cross-coupling two molecules of 6-bromo-1H-indole-2-carbaldehyde using titanium tetrachloride and zinc dust, which also gave 28a as a bi-product. Compound 27c was prepared from alcohol 29 where it was first converted to Wittig salt 30 (Scheme 5). Compound 30 was then coupled with the corresponding aldehyde to give 31 and finally the phenylsulfonyl protecting groups were removed with Cs2CO3 to give the desired adduct 27c. The double bond of compound 27c was reduced by hydrogenation over Pt/C to give compound 28b.
Scheme 4.
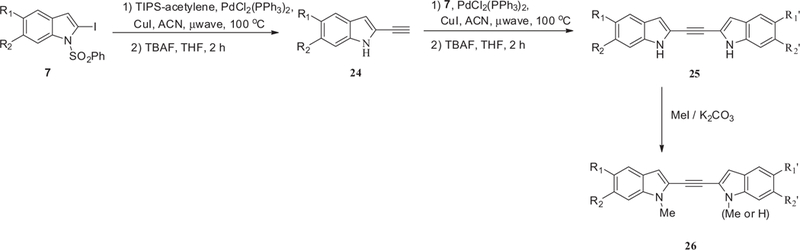
General synthesis of bis-indoles with acetylene linker.
Scheme 5.
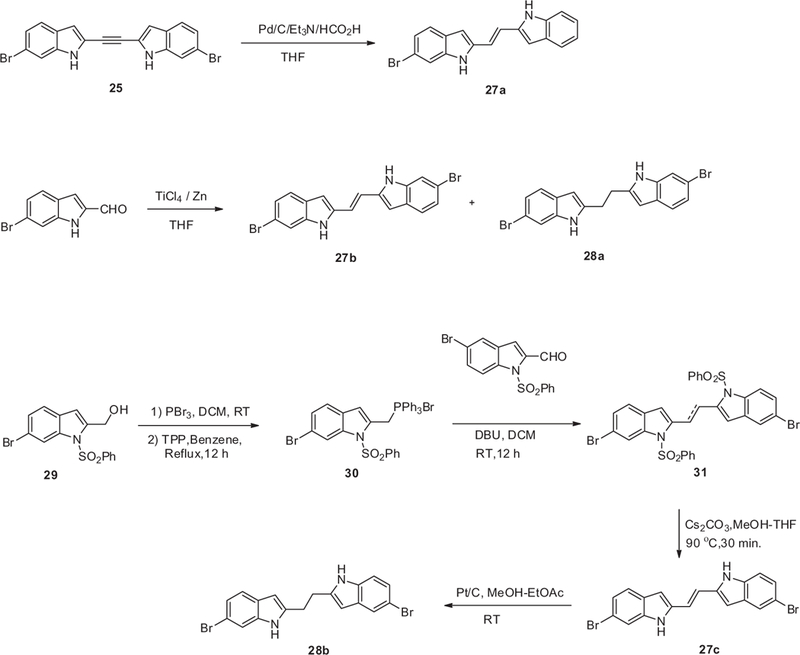
General synthesis of bis-indoles 27 and 28.
Symmetrical bis-indoles 33a and 33b were prepared by double Suzuki–Miyura reaction of boronic acid 9 with aryl di-halide 32 followed by the removal of the Boc protecting group with TFA (Scheme 6). In order to prepare unsymmetrical bis-indoles 33c–e, aryl di-halide was first coupled with 1 equiv of boronic acid 9 to give intermediate 34 which was consequently coupled with a different boronic acid 9a and finally the Boc group was cleaved with TFA to give the desired compounds.
2.2. Biological results and SAR studies
All compounds prepared were screened against in an in vitro enzyme assay to assess MRSA PK inhibitory activity. Cellular antimicrobial activity was evaluated for ability to fully inhibit the growth of S. aureus ATCC 29213 in culture (determining minimum fully inhibitory concentration (MIC) values) (Tables 1–3). Selectivity for the MRSA PK over the four human isoforms was confirmed for a selected series of compounds (Table 4). Cytotoxicity was evaluated for selected compounds with HEK 293 cells and they were found not to be significantly cytotoxic at concentration up to 100 µg/ml (Table 5).
Table 1.
PK inhibitory activity and antimicrobial activity of substituted bis-indoles
| |||||
|---|---|---|---|---|---|
| Analogs | IC50a (nM) | MICb (μg/ml) | R1 | R2 | Substitutions |
| 10a | 21.4 | 2.0 | H | H | 6-Bromo |
| 10b | 7.0 | 16.0 | H | H | 6,6′-Dibromo |
| 10c | 2.2 | 0.3 (3) | H | H | 6,5′-Dibromo |
| 10d | 2.0 | 0.3 | H | H | 6-Bromo-5′-chloro |
| 10e | 2.8 | 0.3 | H | H | 6-Bromo-5′-fluoro |
| 10f | 2.5 | >64 | H | H | 6-Bromo-5′-methoxy |
| 10g | 2.2 | >64 | H | H | 6-Bromo-5′-phenyl |
| 10h | 36% @ 10 μM | ND | H | H | 5-Bromo |
| 10i | 3.0 | >64 (2) | H | H | 5,5′-Dibromo |
| 10j | 39% @ 10 μM | ND | H | H | 5,6-Dibromo |
| 10k | 1.5 | 0.5 | H | H | 5,6,6′-Tribromo |
| 10l | 2.0 | >64 | H | H | 5,5′,6,6′-Tetrabromo |
| 10m | 1.6 | 1.0 | H | H | 5,6,5′-Tribromo |
| 12a | 1.0 | >64 (2) | H | CH3 | 6,5′-Dibromo |
| 12b | 25% @ 10 μM | ND | CH3 | H | 6,5′-Dibromo |
| 12c | 1.9 | >64 | H | CH2OCH3 | 6,5′-Dibromo |
| 14 | 11.1 | >64 | H | CH2COOH | 6,5′-Dibromo |
| 15 | 6.0 | >64 | H | 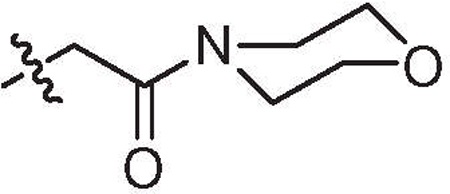 |
6,5′-Dibromo |
| 17 | 1.3 | 2.0 | H | CH2CH2OH | 6,5′-Dibromo |
| 20a | 2.0 | 4.0 | H | 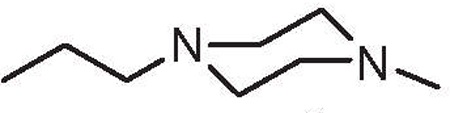 |
6,5′-Dibromo |
| 20b | 1.2 | >64 | H | 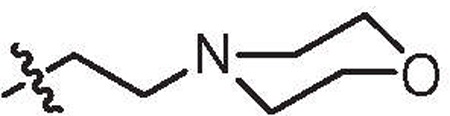 |
6,5′-Dibromo |
IC50 values are calculated from a triplicate 15-point titration. Alternatively the % inhibition at the highest concentration tested is presented
Minimum concentration to give >98% inhibition of growth of S. aureus ATCC 29213 (single determination or average of (n) determinations). Control MIC (vancomycin) is 1 μg/ml.
Table 3.
PK inhibitory activity and antimicrobial activity of compounds with modification on the linker
| ||||||
|---|---|---|---|---|---|---|
| Analogs | IC50a (nM) | MICb (μg/ml) | Linker | R1 | R2 | Substitutions |
| 10a | 21.4 | 2.0 | ||||
| 25a | 7.1 | >64 |  |
H | H | 6-Bromo |
| 25b | 2.4 | >64 |  |
H | H | 6,6′-Dibromo |
| 25c | 4.7 | 16 |  |
H | H | 6,5′-Dibromo |
| 26a | 13.7 | >64 | 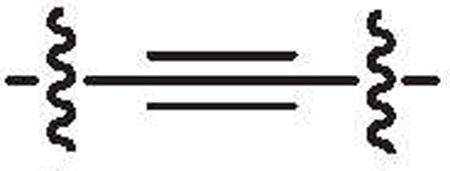 |
CH3 | H | 6,6′-Dibromo |
| 26b | 27% @ 10 μM | ND | 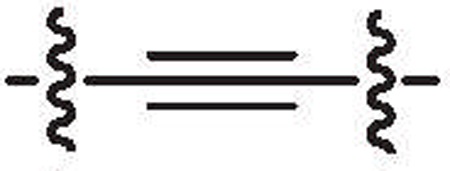 |
H | CH3 | 6,6′-Dibromo |
| 27a | 4.5 | >64 | 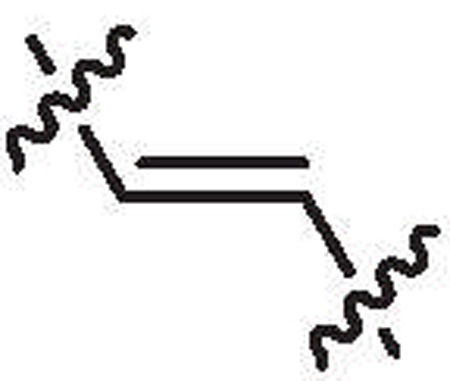 |
H | H | 6-Bromo |
| 27b | 3.9 | >64 | 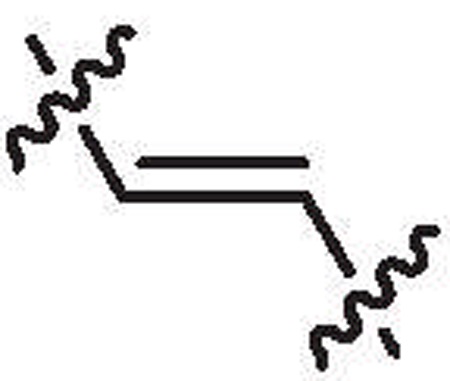 |
H | H | 6,6′-Dibromo |
| 27c | 4.1 | >64 | 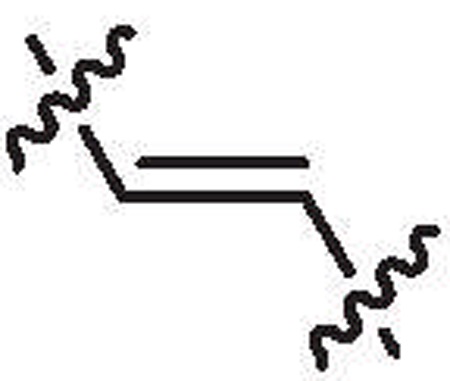 |
H | H | 6,5′-Dibromo |
| 28a | 6.5 | >64 | 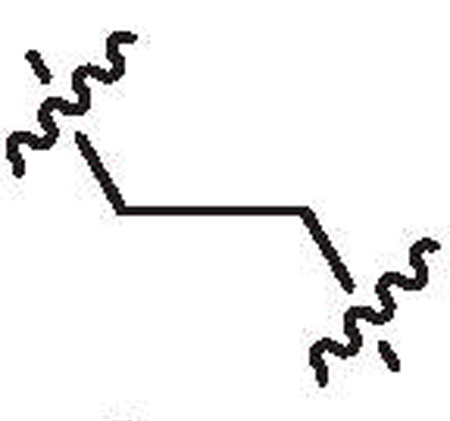 |
H | H | 6,6′-Dibromo |
| 28b | 6.3 | >64 | 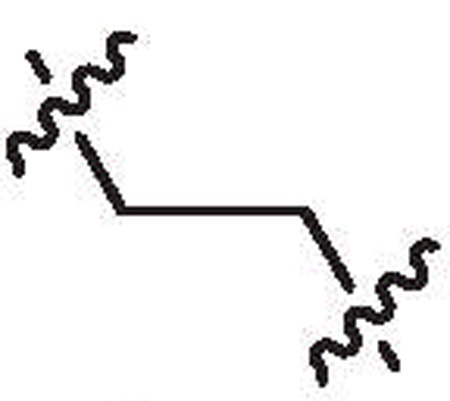 |
H | H | 6,5′-Dibromo |
| 33a | 54% @ 1 μM | >64 | 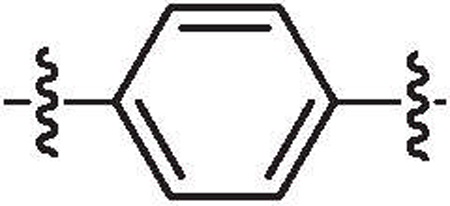 |
H | H | 6,6′-Dibromo |
| 33c | 5.6 | >64 | 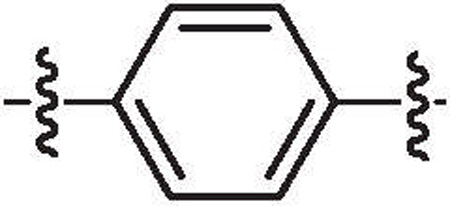 |
H | H | 6-Bromo |
| 33d | 1.8 | 2.0 | 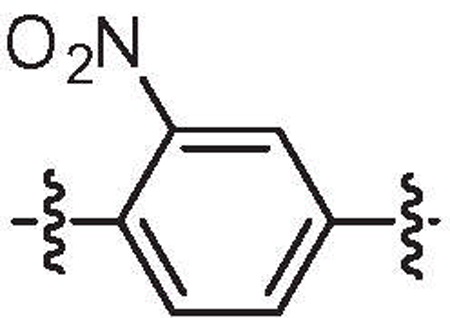 |
H | H | 6-Bromo |
| 33e | 2.1 | >64 | 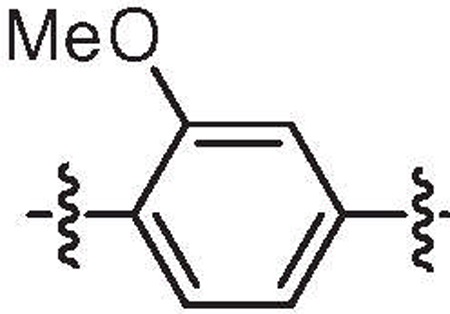 |
H | H | 6-Bromo |
| 33f | 36% @ 10 μM | ND | 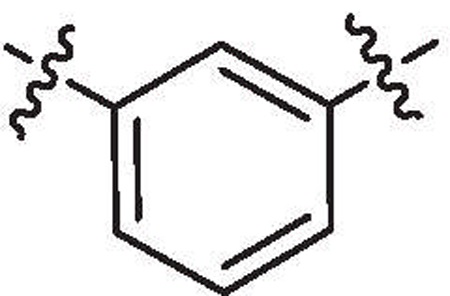 |
H | H | 6-Bromo |
| 33b | 28% @ 1 μM | >64 | 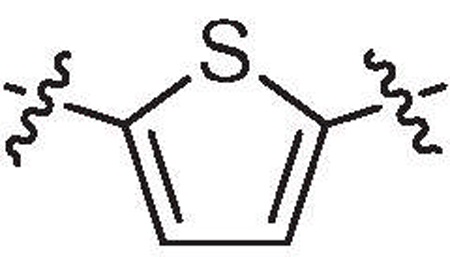 |
H | H | 6,6′-Dibromo |
IC50 values are calculated from a triplicate 15-point titration. Alternatively the % inhibition at the highest concentration tested is presented.
Minimum concentration to give >98% inhibition of growth of S. aureus ATCC 29213 (single determination). Control MIC (vancomycin) is 1 μg/ml.
Table 4.
Inhibition of enzymatic activities of MRSA PK and human PK isoforms at 10 μM
| Analogs | % Inhibition of MRSA PK | % Inhibition of human M1 | % Inhibition of human M2 | % Inhibition of human R | % Inhibition of human L |
|---|---|---|---|---|---|
| 10b | 84.9 (2) | 10.3 (2) | 0.1 (2) | 18.3 (2) | 7.9 (2) |
| 10c | 97.8 | 6.9 | 8.7 | 13.6 | 6.1 |
| 10i | 71 | 12.3 | 0.5 | 24.4 | 9.8 |
| 10k | 94.1 | 8.6 | 15.8 | 33.4 | 23.7 |
| 10l | 97.6 | 11.1 | 2.3 | 20.7 | 8.2 |
| 12a | 99.0 | 13.9 | 9.1 | 2.7 | 2.8 |
| 17 | 100.9 | 14.6 | 12.1 | 25.2 | 6.8 |
| 20a | 100.1 | 3.2 | 1.7 | 26.1 | 16.8 |
| 20b | 100.6 | 13.1 | 3.5 | 50.7 | 32.1 |
| 22a | 89.8 | 12.4 | 3.0 | 21.6 | 7.8 |
| 22b | 95.9 | 5.3 | 0.4 | −5.5 | −6.0 |
| 25c | 86.3 | 19.5 | 8.7 | 27.8 | 14.0 |
| 27a | 93.4 | 16.1 | −0.9 | 23.8 | 12.9 |
| 28a | 93.4 | 9.9 | 1.3 | 24.1 | 9.7 |
| 33d | 86.2 | 9.5 | 6.8 | 0.0 | −8.9 |
| 33e | 62.2 | 7.3 | 14.6 | 13.5 | 4.3 |
Table 5.
Mammalian cell cytotoxicity of selected compounds
| Analogues | % Cell viability found on incubation with HEK 293 cell line |
||
|---|---|---|---|
| 1 μg/ml | 10 μg/ml | 100 μg/ml | |
| 10b | 103 | 106 | 118 |
| 10f | 107 | 88 | 72 |
| 10k | 102 | 71 | 63 |
| 12c | 123 | 102 | 73 |
| 27a | 107 | 114 | 115 |
| 28a | 104 (2) | 92 (2) | 85 (2) |
2.2.1. SAR for in vitro inhibition of MRSA PK
To further improve and optimize biological activity we set out to develop SAR for both MRSA PK enzyme and the cellular antibacterial activity in parallel. For the purpose of clarity we will discuss the SAR derived for inhibition of the MRSA PK enzyme separately from the cellular antibacterial activity.
We systematically evaluated the effect of substitution on bis-indole scaffold (Table 1), replacing one indole ring with a number of heterocycles (Table 2) and modifying the central linking moiety (Table 3).
Table 2.
PK inhibitory activity and antimicrobial activity of compounds 22a–g
| |||||
|---|---|---|---|---|---|
| Analogs | IC50a (nM) | MICb (μg/ml) | X | Y | Substitutions |
| 10a | 21.4 | 2.0 | |||
| 22a | 151.7 | >64 | S | N | 6-Bromo |
| 22b | 107.6 (2) | >64 | O | CH | 6-Bromo |
| 22c | 14.0 | >64 | S | CH | 6-Bromo |
| 22d | 358.1 | >64 | S | N | 5-Bromo |
| 22e | 45% @ 10 μM | ND | O | CH | 5-Bromo |
| 22f | 42% @ 1 μM | >64 | S | CH | 5-Bromo |
| 22g | 4.1 | >64 | S | N | 5,6′-Dibromo |
IC50 values are calculated from a triplicate 15-point titration or are an average of (n) such determinations as indicated. Alternatively the % inhibition at the highest concentration tested is presented.
Minimum concentration to give >98% inhibition of growth of S. aureus ATCC 29213 (single determination). Control MIC (vancomycin) is 1 μg/ml.
The directly linked 6,6ʹ-dibromo-1H,1ʹH-2,2ʹ-biindole (10b) was prepared and we were pleased to note that it was a potent inhibitor of the MRSA PK enzyme with an IC50 of 7.0 nM and that it also gave an MIC against S. aureus of 16 µg/ml (Table 1). To evaluate the role of the two bromines, the mono-brominated compound 10a was prepared and was found to be about threefold less active. However, the asymmetrically substituted 6,50-dibrominated com-pound 10c was more potent with IC50 of 2.2 nM. It was found that the 5ʹ-bromine could be substituted with chloro (10d), fluoro (10e), methoxy (10f) or even with a relatively bulky group such as phenyl (10g) without significant loss of potency suggesting that there are still some room in the binding pocket which might be further exploited to improve activity.
We next prepared the 5ʹ-monobrominated bis-indole analog (10h) and noted a significant drop in activity. This suggested that at least one bromine atom in the 6-position of one of the indole moieties was critical for activity. On the other hand, however, the 5,5ʹ dibromo bis-indole (10i) was found to be very potent with an IC50 of 3 nM. It is not clear why 10h was not active whereas 10i is potent but one possible explanation could be that one of the bro-mines of 10i is oriented towards the interior of the binding pocket and the other bromine is facing outwards, thus placing the indole NH towards the interior to provide a necessary hydrogen bonding with Ser362. Mysteriously, the 5,6 dibromo compound 10j was not potent whereas the 5,6,6ʹ- and 5,6,5ʹ-tribromo compounds 10k and 10m and tetrabromo bis-indole 10l were found to be very potent. The activity of these relatively large molecules was in keeping with there being still more space in the binding pocket to be exploited (discussed later, Table 3).
Next we investigated the effect of substitution on the NH of in-dole. The mono-N-methyl bis-indole 12a was very active with an IC50 of 1.0 nM whereas the isomeric mono-N-methyl bis-indole 12b was almost inactive. This further confirmed that the presence of a bromine atom at the 6-position relative to the indole NH is important for activity. It appears that the bromine and NH of 6-bromo-indole fragment binds very tightly to the pocket and there is no room for further substitution in that region of the molecule. The methyl group in 12a is most likely oriented towards the outside (water side) of the binding site and that explains why compounds 12c, 14, 15, 17, 20a and 20b with bulky groups attached to the nitrogen atom of the 5-bromo indole are still very potent. There was no further improvement in activity by either introducing polar (12c, 15, 17 and 20b), acidic (14) or basic (20a) groups at NH of the second indole.
Our next step was to investigate whether both indoles are critical for binding or if one can be replaced with other heterocycles. Keeping the 6-bromoindole element constant and replacing the second indole with benzothiazole (22a) or benzofuran (22b) led to a 5–8-fold decrease in activity (compared to 10a). However, the benzothiophene derivative (22c) is slightly more potent than 10a suggesting that one indole can indeed be replaced with benzo-thiophene. In the cases where we kept one 5-bromoindole constant and replaced the other indole with benzothiazole (22d), benzofuran (22e) or benzothiophene (22f), there was a significant drop in activity as expected from earlier observation. Nevertheless, 6-bromothiazole analog (22g) was about fivefold more potent than 10a and comparable in potency to 5,5ʹ-dibromo-bis-indole 10i, and hence might have a similar mode of binding.
Next we investigated whether a spacer moiety could be placed between the two indoles. Compound 25a, with an acetylene linker, was found to be threefold more potent than direct linked compound (10a) indicating that the binding pocket has more linear space. Next we made the 6,6ʹ-dibromo analog (25b) and 6,5ʹ-dibromo analog (25c) with an acetylene linker, but there was no significant improvement in potency. One indole NH could be methylated (26a) without significant loss in activity, but as expected, methylation of both NH (26b) led to a compound that was essentially inactive. Surprisingly 27a and 27b with an ethylene linker was almost equipotent as the corresponding acetylene linker. We had anticipated a loss in potency as the two indoles should adopt a linear staggered conformation with ethylene linker that should displace one of the indoles within the binding pocket but evidently this is not detrimental. More interestingly, compounds (28a and 28b) with the fully saturated ethane linker moiety were still very potent, both with IC50 of 6 nM. Compounds 28a and 28b presumably are able to orient in a staggered linear and planer conformation similar to 27a and 27b to bind in the flat lipophilic pocket similar to the natural product 4. In order to make the spacer slightly longer we synthesized 1,4-phenyl linked compound 33a. There was a significant drop in activity observed for 33a (as com-pared to 25a) and modelling suggests that it might be now too long to fit in the binding pocket. Subsequently, we made a slightly shorter analog 33c (by removing one bromine atom) and this compound was found to be very potent (IC50 of 5.6 nM). Hence it appears that 33c is occupying most of the binding pocket and it defines the breadth of the site.
It was noted that if 33 was modelled into the binding site, the central ring was flanked above and below by the His-365 residues. Considering that these interactions might lead to some potential for charge transfer interaction, we investigated the effect of placing an electron-withdrawing group (33d) or an electron-releasing group (33e) on the phenyl linker. However both 33d and 33e were about 2–3 fold more potent than 33c so it is not clear if such an interaction exists. 1,3-Phenyl-linked (33f) and 2,5-thiophene linked (33b) were essentially inactive which demonstrates that both indoles have to be able to adopt an essentially linear attitude for significant binding.
2.2.2. SAR for anti-S. aureus activity
For those tested, compounds with low potency against MRSA PK (IC50 >100 nM) were also poorly effective (MIC >64 µg/ml) against S. aureus ATCC95923. However while many potent inhibitors of MRSA PK gave low MICs (e.g., compound 10d), other potent ana-logs had high MICs. This suggested that issues such as solubility, cellular penetration or active export from the bacterial cells might impair full translation of activity. The MIC of the initial lead compound 10b was similar to that reported for cis-3–4-dihydroham-acanthin B (4). However, compound 10a with one less bromine was about eightfold more potent than 10b despite it being three-fold less potent in MRSA PK enzyme assay. Direct linked bis-indole compounds 6,5ʹ-dibromo (10c), 6-bromo-5-chloro (10d) and 6-bromo-5-fluoro (10e) were most potent with MICs of 0.3 µg/ml. However, although 5,5ʹ-dibromo derivative 10i and tetrabromo 10l were equipotent in vitro compared to 10b–10e, they had much higher MICs (>64 µg/ml). Both tribromo substituted compounds 10k and 10m showed excellent MICs.
Many of these compounds had limited solubility that might limit ability to access and to penetrate cells and therefore a number of potentially solubilizing groups were installed on one of the indole NHs (see compounds 12c, 14, 15, 17, 20a and 20b). However, although all these compounds were potent inhibitors of MRSA PK, only the hydroxyethyl analog 17 and piperazinylethyl derivative 20a showed reasonable MICs. Hence, it appears that there was no clear correlation between the MIC and polarity. In the cases where one indole was replaced with a heterocycle (22a–g), none gave significant MIC despite being potent in the enzyme assay (e.g., compound 22g). Of all the compounds made with a spacer between the two indoles only 25c and 33d gave significant MIC values further exemplifying the lack of correlation of enzymatic activity and anti-bacterial activity. Of all the phenyl-linked compounds made, only 33d bearing an electron-with drawing group was MIC active.
2.2.3. Evaluation of the effect of verapamil on the MIC of selected PK inhibitors
Considering the divergent anti-S. aureus activity observed among very close structural analogs such as 10b and 10l compared to 10c, 10d or 10e, in spite of similar IC50s for inhibition of MRSA PK, it seemed unlikely that cell penetrability or solubility could explain these discrepancy. One possibility is that compounds such as 10b may be substrates for putative bacterial efflux systems, such as has been proposed to impart resistance of many bacteria to antibiotics.9 The antihypertensive drug, verapamil has been shown to be an inhibitor the ABC class of pumps in Gram-positive bacteria such as Staphylococci and verapamil has been demonstrated to inhibit efflux pumps in S. aureus.10,11 We therefore evaluated the effect of verapamil on the observed MIC for several key compounds. MICs were determined for S. aureus ATCC92953 in the absence and presence of verapamil (200 mg/L, a concentration previously reported as an optimal sub-inhibitory concentration11). Vancomycin was used as control and gave the same MIC (2 μg/ml) in presence and absence of verapamil as did compound 10d (MIC = 0.5 μg/ml). However, the MIC of 10l was shifted at least fourfold (from >64 to 16 μg/ml) by verapamil while the MIC of 10b was dramatically shifted, 32-fold (from 16 to 0.5 μg/ml) (Fig. 3).
Figure 3.
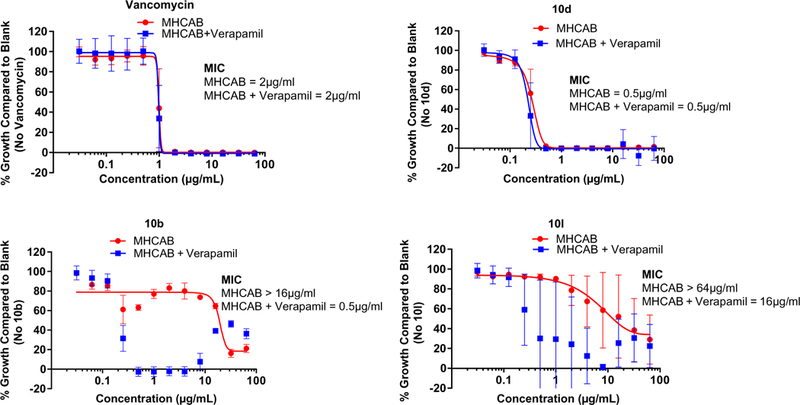
The MIC determinations of 10d, 10b and 10l in the presence and absence of efflux inhibitor Verapamil with S. aureus (ATCC29213). Assay of vancomycin, 10d and 10b was performed in triplicate. Compound 10l was performed in duplicate.
2.2.4. Evaluation of efficacy of selected MRSA PK inhibitors against MRSA
To further investigate the antibacterial properties of this class of PK inhibitors, the three of the most potent compounds (10c, 10d and 10e) from the SAR study were tested in vitro for their antibacterial activities against MRSA strain MW2 (USA400) with vancomycin as positive control. In each case an MIC of 0.5 µg/ml was observed while vancomycin gave an MIC of 2 µg/ml (Fig. 4).
Figure 4.
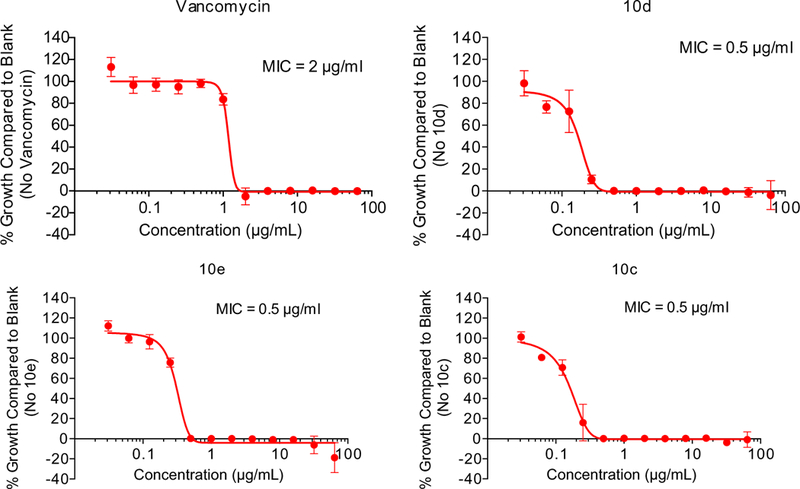
MIC determinations of 10c, 10d and 10e with MRSA strain MW2 (USA 400).
2.2.5. Evaluation of MRSA PK inhibitors in bacteria pyruvate kinase deficient S. aureus cells
To implicate 10d-mediated inhibition of S. aureus PK as the direct cause of bacterial killing, we constructed a S. aureus LAC Δpyk::ErmR mutant. Essentially, we replaced the pyk gene on the chromosome of S. aureus with an ErmR cassette using a standard allelic exchange procedure as previously described.12 Previous work has described PK as an essential enzyme required for growth.2,3 In each of these previous studies however, the growth medium used contained primarily glycolytic carbon sources such as glucose (G). By designing media lacking carbohydrates but replete with pyruvate (P), PK becomes dispensable and a mutant can therefore be made that has relatively no growth defect under these specific conditions (Fig. 5 shows that in TSB-G+P, the Δpyk and WT grow identically in the absence of 10d). Initial characterization of the mutant revealed that it exhibits an aerobic growth defect when grown on Brain–Heart Infusion Broth (BHI) but not on Tryptic Soy Broth (TSB) without glucose, provided the medium is supplemented with 1% sodium pyruvate (TSB-G+P) (Fig. 5). The growth defect exhibited by S. aureus Δpyk::ErmR on BHI was reversible via complementation using a plasmid expressing pyk from a constitutive promoter (data not shown). These data indicate that the metabolic activity of PK is required for aerobic growth of S. aureus on glycolytic carbon sources (e.g., BHI) but not certain combinations of gluconeogenic carbon sources (e.g., TSB-G+P).
Figure 5.
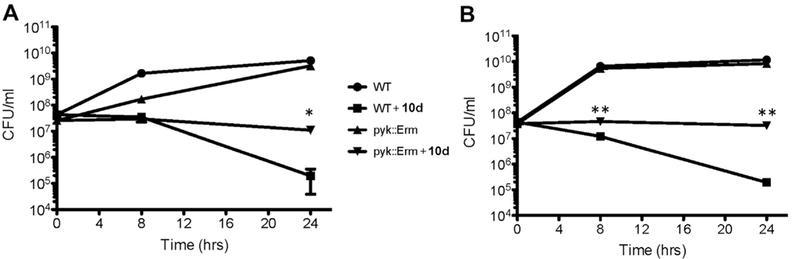
Resistance of S. aureus LAC ∆pyk::ErmR to killing by compound 10d. (A) Resistance of S. aureus LAC ∆pyk::ErmR to killing by 1.6 μM 10d during aerobic growth in BHI. (B) Resistance of S. aureus LAC ∆pyk::ErmR to killing by 1.6 μM 10d during aerobic growth in TSB without glucose supplemented with 1% sodium pyruvate. Significance was assessed between WT LAC and ∆pyk::ErmR LAC treated with 10d at 8 and 24 h using a Students two-sided t-test (*p-value ⩽0.05, **p-value ⩽0.0001). Error bars = ±SD. (n = 3).
We next examined the bacteriostatic and bactericidal effects of 10d on wild type (WT) S. aureus as well as the Δpyk::ErmR. We found that, while 10d was highly bactericidal to WT S. aureus, the drug merely acted statically on the Δpyk::ErmR mutant in BHI medium (Fig. 5A). The Δpyk::ErmR mutant grows slower than WT in BHI medium and to test whether this had any effect on 10d toxicity, we tested the compound’s effect in media in which the mutant has no growth defect. As in BHI, 10d was unable to kill the Δpyk::ErmR mutant in TSB-G+P as it did WT S. aureus (Fig. 5B). Thus, while 10d efficiently eliminated growth of both WT and Δpyk::ErmR S. aureus (no significant difference in 10d MIC for either strain), the bactericidal activity of this compound is completely dependent on PK activity (Fig. 5).
2.2.6. Selectivity and cytotoxicity
For selected compounds selectivity for MRSA PK relative to the mammalian PK isoforms M1, M2, R and L (Table 4) was determined using enzymes and methods as previously described.4,8 Those compounds tested for selectivity generally showed no significant inhibition at the highest concentration tested (10 μM) and only 20b showed inhibition of a mammalian PK isoform of greater than 50% (51% for the R isoform). Cytotoxicity was evaluated for selected compounds with HEK 293 cells as previously described4,8 and they were found not to be significantly cytotoxic at concentration up to 100 μg/ml (Table 5).
3. Conclusions
Based on observations from the published crystal structure of a naturally occurring marine alkaloid 4 bound to the tetrameric MRSA PK enzyme, a novel series of bis-indoles were [stat] designed and prepared and shown to be very potent inhibitors of MRSA pyruvate kinase. Compounds such as 10c, 10d, 10e, 10k, 10m and 12a are among the most potent inhibitors of this enzyme identified to this date with IC50s of 1–3 nM. Numerous analogs were synthesized in order to define structure–activity relationships and to further optimize the physiochemical properties for effective inhibition of bacterial growth. Lead compounds were very selective for the bacterial PK over the human isoforms. Although there was, overall, a poor correlation between the in vitro enzyme inhibitory activity and anti-bacterial activity against S. aureus ATCC 29213, some compounds showed excellent MIC values of as low as 0.3 μg/ml while lacking significant mammalian cytotoxicity.
Considering that this lack of correlation might be due (at least in part) to selective active efflux from the bacteria, studies were carried out on the effect of the known efflux inhibitor, verapamil, on the MIC values of selected close structural analogs, 10a, 10d and 10l. These studies revealed a pronounced enhancement of MIC values for 10a and 10l but not for 10d (or vancomycin control) indicating that at least for these key compounds the attenuated antibacterial activity is due to active efflux mechanisms.
Compounds 10c, 10d and 10e were further evaluated for efficacy against MRSA strain MW2 where MICs (0.5 μg/ml) similar to those obtained for S. aureus ATCC 29213 were observed. In previous studies on MRSA pyruvate kinase inhibitors4,8 potency against S. aureus ATCC 29213 was found to be representative of potency against MRSA strains and thus we infer that this will also be the case for this class of compounds.
In order to gain further insight into the mechanism of action of these compounds on S. aureus we evaluated the effects of the compound 10d on a S. aureus mutant where the gene for pyruvate kinase (Pyk) is knocked out. These cells are viable when cultured in a medium lacking usable carbohydrate and additionally supplemented with pyruvate. These studies showed that the bacteriostatic and bactericidal effects of 10d on S. aureus occur by different mechanisms. The bacteriostatic effects of 10d are pyk-independent while the bactericidal effects of 10d are pyk-independant. The bactericidal mechanism of 10d does not require PK metabolic activity (10d is bactericidal to S. aureus on TSB-G+P, a growth media where pyk::ErmR exhibits no growth defects). This supports the model of pyruvate kinase as a highly interactive protein and the expectation that pyruvate kinase inhibitors of this class may yield effective anti-MRSA compounds with low potential for development of resistance.
The lead compounds (10c, 10d, 10e, 10k and 10m) with best MIC values are currently being evaluated to determine in vivo absorption and tissue exposure in preparation for evaluation in in vivo animal infection models. These studies will be reported elsewhere.
4. Experimental section
4.1 Chemistry
4.1.1. General synthesis methods
1H and 13C NMR spectra were recorded with either Bruker Avance II™ 600 MHz, Bruker Avance III™ 500 MHz or Bruker Avance III™ 400 MHz. Processing of the spectra was performed with MestRec™ software. The high-resolution mass spectra were recorded either in positive or negative ion-mode with an ESI or multimode ESI/APCI ion source on an Agilent™ 6210 Time-of-Flight LC/MS mass spectrometer. Analytical thin-layer chromatography (TLC) was performed on aluminum plates pre-coated with silica gel 60F-254 as the absorbent. The developed plates were air-dried, exposed to UV light and/or dipped in KMnO4 solution and heated. Column chromatography was performed with silica gel 60 (230–400 mesh). Automated flash chromatography was carried out on Biotage Isolera Flash Purification Systems using commercial 50 μm silica gel cartridges. Purity (>95%) for all final compounds was confirmed by analytical reverse-phase HPLC utilizing a Dikma Technologies™ Inspire® C18 reverse-phase analytical column (4.6 × 150 mm) using ACN and H2O with 0.1% formic acid as eluent. All HPLC purifications were carried out using an Agilent™ C18 reverse-phase preparatory column (21.2 × 250 mm) using ACN and H2O with 0.1% formic acid as eluent.
4.1.1.1. General procedure for the synthesis of 1-(phenylsulfonyl)-1H-indole (6).
To a stirred solution of an appropriate indole (1 mmol) in THF (25 ml) at 0 °C was added NaH (60% in oil, 1.2 mmol) gradually. After stirring at room temperature for 10 min benzenesulphonyl chloride (1.2 mmol) was added and the mixture was further stirred for 2 h. The reaction was quenched with saturated ammonium chloride solution and extracted with EtOAc (2 × 50 ml). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was purified by automated flash chromatography using EtOAc and hexanes as eluents to give the desired product.
4.1.1.2. General procedure for the synthesis of 2-iodo-(phenylsulfonyl)-1H-indole (7).
To a stirred solution of 6 (1 mmol) in anhydrous THF (10 ml) at −78 °C was added a solution of LDA (1.5 mmol) in THF (5 ml). The mixture was stirred for at −78 °C for 100 min and then warmed to 0 °C for 30 min. The solution was re-cooled to −78 °C and then either a solution of 1,2-diiodo ethane or molecular iodine (1.5 mmol) in THF (10 ml) was added. The reaction mixture was stirred at 0 °C for 15 min and then allowed for warm to room temperature for 1 h. The reaction was quenched with saturated NH4Cl solution and extracted with EtOAc (2 × 50 ml). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was purified by automated flash chromatography using EtOAc and hexanes as eluents to give the desired product.
4.1.1.3. 6-Bromo-2-iodo-1-(phenylsulfonyl)-1H-indole (7a).
Yield = 53%, white solid. 1H NMR (CDCl3, 400 MHz): δ 8.16 (d, J = 9.0 Hz, 1H), 7.87 (d, J = 7.6 Hz, 2H), 7.58 (t, J = 7.5 Hz, 1H), 7.5 (d, J = 1.6 Hz, 1H), 7.46 (t, J = 7.8 Hz, 2H), 7.37 (dd, J = 9.0, 1.9 Hz, 1H), 6.93 (s, 1H). HRMS calcd for (C14H9BrINO2S–H)− 460.8582; found 460.85998.
4.1.1.4. 5-Bromo-2-iodo-1-(phenylsulfonyl)-1H-indole (7b).
Yield = 54%, white solid. 1H NMR (CDCl3, 400 MHz): δ 8.15 (d, J = 9.0 Hz, 1H), 7.87 (d, J = 8.5 Hz, 2H), 7.56 (t, J = 8.1 Hz, 1H), 7.53 (d, J = 1.7 Hz, 1H), 7.44 (t, J = 7.5 Hz, 2H), 7.37 (dd, J = 2.0, 9.0 Hz, 1H), 6.91 (s, 1H).
4.1.1.5. 5-Chloro-2-iodo-1-(phenylsulfonyl)-1H-indole (7c).
Yield = 75%, white Solid. 1H NMR (CDCl3, 400 MHz): δ 8.21 (d, J = 9.0 Hz, 1H), 7.84–7.90 (m, 2H), 7.55–7.61 (m, 1H), 7.46 (dt, J = 7.4, 1.8 Hz, 2H), 7.38 (d, J = 1.9 Hz, 1H), 7.24 (dd, J = 9.0, 2.2 Hz, 1H), 6.93 (d, J = 0.7 Hz, 1H). 13C NMR (CDCl3, 125 MHz): 138.12, 136.95, 134.44, 132.87, 129.82, 129.42, 127.31, 126.80, 125.21, 123.43, 119.36, 116.55 ppm. HRMS calcd for (C14H9ClINO2S–H)− 416.9087; found 416.9094.
4.1.1.6. 5-Fluoro-2-iodo-1-(phenylsulfonyl)-1H-indole (7d).
Yield = 94%, white solid. 1H NMR (CDCl3, 400 MHz): δ 8.23 (dd, J = 9.2, 4.4 Hz, 1H), 7.84–7.89 (m, 2H), 7.54–7.61 (m, 1H), 7.42–7.48 (m, 2H), 6.97–7.08 (m, 2H), 6.95 (d, J = 0.5 Hz, 1H). HRMS calcd for (C14H9FINO2S–H)− 400.9383; found 400.9392.
4.1.1.7. General procedure for the synthesis of substituted 2-iodo-1H-indole (8).
To a stirred solution of compound 7 (1 mmol) in THF (20 ml) at room temperature was added a solution of TBAF (1 ml, 1 M in THF, 1 mmol). The mixture was stirred at ambient temperature for 5 h and then partitioned between EtOAc (100 ml) and H2O (50 ml). The organic phase was washed with brine (50 ml), dried over anhydrous Na2SO4 and concentrated. The residue was purified by automated flash chromatography using EtOAc and hexanes as eluents to give compound 8.
4.1.1.8. 6-Bromo-2-iodo-1H-indole (8a).
Yield = 48%, white solid. 1H NMR (CDCl3, 400 MHz): δ 8.07 (br s, 1H), 7.46–7.50 (m, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.19 (dd, J = 8.4, 1.7 Hz, 1H), 6.69 (dd, J = 2.0, 0.9 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz): 139.38, 128.33, 122.17, 120.23, 114.01, 112.90, 110.84, 79.73. HRMS calcd for (C8H5BrIN–H)− 320.8650; found 320.8658.
4.1.1.9. 5-Bromo-2-iodo-1H-indole (8b).
Yield = 92%, white solid. 1H NMR (DMSO-d6, 400 MHz): δ 11.9 (br s, 1H), 7.65 (d, J = 1.6 Hz, 1H), 7.28 (d, J = 8.6 Hz, 1H), 7.15 (dd, J = 1.9, 8.6 Hz, 1H), 6.6 (s, 1H).
4.1.1.10. 5-Chloro-2-iodo-1H-indole (8c).
Yield = 96%, pale brown solid. 1H NMR (CDCl3, 400 MHz): δ 8.12 (br s, 1H), 7.51 (d, J = 2.0 Hz, 1H), 7.24 (d, J = 8.7 Hz, 1H), 7.09 (dd, J = 8.6, 2.0, 1H), 6.67 (d, J = 1.2 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz): δ 137.16, 130.31, 123.94, 121.09, 117.68, 111.88, 110.33, 80.64. HRMS calcd for (C8H5ClIN–H)− 276.9155; found 276.9158.
4.1.1.11. 5-Fluoro-2-iodo-1H-indole (8d).
Yield = 85%, white solid. 1H NMR (CDCl3, 400 MHz): δ 8.08 (br s, 1H), 7.22–7.25 (m, 1H), 7.19 (dd, J = 9.4, 2.5 Hz, 1H), 6.89 (td, J = 9.1, 2.5 Hz, 1H), 6.68 (dd, J = 2.0, 0.8 Hz, 1H).
4.1.1.12. 5,6-Dibromo-2-iodo-1H-indole.
Yield = 89%, white solid. 1H NMR (CDCl3, 400 MHz): δ 8.09 (br s, 1H), 7.80 (s, 1H), 7.69 (d, J = 0.7 Hz, 1H), 6.64 (dd, J = 0.8, 2.3 Hz, 1H).
4.1.1.13. General procedure for the synthesis of substituted 2-iodo-1-methyl-1H-indole (11).
A mixture of an appropriate 2-iodo-1H-indole 8 (1 mmol), K2CO3 (2 mmol) and methyl iodide (1.5 mmol) in DMF was stirred at room temperature for 3 d. The reaction mixture was partitioned between EtOAc (100 ml) and H2O (50 ml). The organic phase was washed with brine (50 ml), dried over anhydrous Na2SO4 and concentrated. The residue was purified by automated flash chromatography using EtOAc and hexanes as eluents to afford compound 11.
4.1.1.14. 6-Bromo-2-iodo-1-methyl-1H-indole (11a).
Yield = 97%, white solid. 1H NMR (CDCl3, 400 MHz): δ 7.46–7.47 (m, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.17 (dd, J = 8.4, 1.7 Hz, 1H), 6.76 (d, J = 0.8 Hz, 1H), 3.72 (s, 3H).
4.1.1.15. 5-Bromo-2-iodo-1-methyl-1H-indole (11b).
Yield = 99%, white solid. 1HNMR(CDCl3, 500 MHz): δ 7.63 (d, J = 1.6 Hz, 1H), 7.22 (dd, J = 8.7, 1.8 Hz, 1H), 7.16 (d, J = 8.7 Hz, 1H), 6.72 (s, 1H), 3.73 (s, 3H). HRMS calcd for (C9H7BrIN) 334.8807; found 334.8780.
4.1.1.16. Synthesis of 5-bromo-2-iodo-1-(methoxymethyl)-1H-indole (11c).
To a stirred slurry of NaH (22 mg, 60% in oil, 0.94 mmol) in THF (5 ml) and DMF (1 ml) at 0 °C was added a solution of 8b (200 mg, 0.63 mmol) in THF (5 ml). After stirring for 1 h methoxymethyliodide (64 μL, 0.75 mmol) was added and the mixture was further stirred for 2 h. The reaction mixture was partitioned between Et2O (100 ml) and H2O (50 ml). The organic phase was washed with brine (50 ml), dried over anhydrous Na2-SO4 and concentrated. The residue was purified by automated flash chromatography using EtOAc and hexanes as eluents to afford 11c (178 mg, 77%) as yellow solid. 1H NMR (DMSO-d6, 500 MHz): δ 7.70 (d, J = 1.8 Hz, 1H), 7.60 (d, J = 8.7 Hz, 1H), 7.26 (dd, J = 8.7, 1.9 Hz, 1H), 6.85 (s, 1H), 5.52 (s, 2H), 3.19 (s, 3H). 13C NMR (DMSO-d6, 125 MHz): 136.66, 131.25, 124.32, 121.35, 112.96, 112.51, 112.31, 88.25, 76.62, 55.46. HRMS calcd for (C10H9BrINO) 364.8192; found 364.8923.
4.1.1.17. Synthesis of 2-(5-bromo-2-iodo-1H-indol-1yl)acetic acid (13).
To a stirred slurry of NaH (144 mg, 60% in oil, 6.0 mmol) in DMF (5 ml) at 0 °C was added a solution of 8b (960 mg, 3.0 mmol) in DMF (15 ml). The mixture was stirred at 0 °C for 30 min and then at rt for an additional 30 min. Ethyl 2-iodoacetate (225 μL, 3.6 mmol) was added and the mixture was stirred at rt for 16 h. The reaction mixture was diluted with EtOAc and then washed with 1 M HCl followed by brine, dried over anhydrous Na2SO4 and concentrated. The residue was dissolved in a mixture of THF and LiOH solution at 0 °C and stirred at rt until the completion of the reaction as indicated by TLC. The reaction mixture was acidified and extracted with EtOAc. The organic phase was washed with brine, dried over anhydrous Na2SO4 and concentrated. The crude product was purified by automated flash chromatography to give white solid (1.01 g, 89%). 1H NMR (DMSO-d6, 500 MHz): δ 13.17 (br s, 1H), 7.69 (d, J = 1.8 Hz, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.21 (dd, J = 8.7, 2.0 Hz, 1H), 6.81 (s, 1H), 5.0 (s, 2H). 13C NMR (DMSO-d6, 125 MHz): 169.46, 136.74, 130.82, 123.92, 121.11, 112.36, 112.26, 111.22, 89.24, 48.26. HRMS calcd for (C10H7BrINO2–H)− 378.8705; found 378.8691.
4.1.1.18. Synthesis of 2-(5-bromo-2-iodo-1H-indol-1-yl)ethanol (16).
To a solution of 8b (38 mg, 0.11 mmol) in DMF (1 ml) was added bromoethanol (14 mg, 0.12 mmol) and K2CO3 (49 mg, 0.36 mmol). The reaction heated at 180 °C with lwave for 30 min and then partitioned between EtOAc and H2O. The organic phase was washed with brine, dried over anhydrous Na2SO4 and concentrated. The residue was purified by automated flash chromatography using EtOAc and hexanes as eluents to afford 16 (24 mg, 59%) as white solid. 1H NMR (CDCl3, 500 MHz): δ 7.65 (s, 1H), 7.28 (d, J = 7.7 Hz, 1H), 7.23 (dd, J = 8.7, 1.8 Hz, 1H), 6.75 (s, 1H), 4.32 (t, J = 5.6 Hz, 2H), 3.9–3.97 (m, 2H).
4.1.1.19. Synthesis of 2-(5-bromo-2-iodo-1H-indol-1-yl)ethyl-methanesulfonate (18).
To a stirred solution of 16 (3.0 g, 8.2 mmol) in DCM (20 ml) at 0 °C was added Et3N (1.8 ml, 12.4 mmol) and methanesulfonyl chloride (0.64 ml, 8.2 mmol). The reaction mixture was stirred at rt for 1 h and then quenched by the addition of ice. The reaction mixture was extracted into DCM (3 × 10 ml) and the combined extract was washed with brine, dried over anhydrous Na2SO4 and concentrated to give essentially pure compound 14 as brown solid (2.98 g, 82%). 1H NMR (CDCl3, 500 MHz): δ 7.66 (m, 1H), 7.24–7.28 (m, 2H), 6.78 (s, 1H), 4.45–4.55 (m, 4H), 2.71 (s, 3H). HRMS calcd for (C11H11BrINO3S) 442.8688; found 442.8686.
4.1.1.20. General procedure for the synthesis of 2-iodoindole derivative (19).
To a stirred solution of the intermediate 18 (1 mmol) in DMF (3 ml) was added K2CO3 (1.5 mmol) and the corresponding amine (3 ml). The mixture was heated at 50 °C for 12–20 h and then partitioned between EtOAc and H2O. The organic phase was washed with saturated NH4Cl, brine, dried over anhydrous Na2SO4 and concentrated. The residue was purified by automated flash chromatography using EtOAc and hexanes as eluents to afford the desired product 19.
4.1.1.21. 5-Bromo-2-iodo-1-(2-(4-methylpiperazin-1-yl)ethyl)-1H-indole (19a).
Yield = 89%, white solid. 1H NMR (CDCl3, 500 MHz): δ 7.61–7.64 (m, 1H), 7.19–7.21 (m, 2H), 6.69 (s, 1H), 4.20–4.30 (m, 2H), 2.35–2.65 (m, 10H), 2.29 (s, 3H). 13C NMR (125 MHz): 136.02, 131.22, 124.66, 122.06, 113.30, 111.53, 111.01, 103.91, 57.18, 55.01, 53.46, 46.14, 45.21. HRMS calcd for (C15H19BrIN3) 446.9807; found 446.9814.
4.1.1.22. 4-(2-(5-Bromo-2-iodo-1H-indol-1-yl)ethyl)morpholine (19b).
Yield = 55%, brown solid. 1H NMR (CDCl3, 400 MHz): δ 7.65–7.64 (m, 1H), 7.26–7.22 (m, 2H), 6.72 (s, 1H), 4.27 (t, J = 7.2 Hz, 2H), 3.70 (br s, 4H), 2.63 (t, J = 7.0 Hz, 2H), 2.53 (br s, 4H).
4.1.1.23. General procedure for the synthesis of compounds 10, 12, 14, 17, 20, 22, 33 and 24.
A solution of boronic acid 9 (1 mmol), iodo-heterocycle (8, 11, 21, 32 or 34) (1 mmol), Na2CO3 (1 M aqueous solution, 3.5 mmol) in ACN (5 ml) was purged with argon for 10 min followed by the addition of Pd(PPh3)2Cl2 catalyst (10 mol %). The mixture was heated in a sealed tube with μwave at 110 °C until all the staring material was consumed as indicated by TLC (typically in about 40–60 min). The reaction mixture was partitioned between EtOAc (100 ml) and H2O (50 ml). The organic phase was washed with brine (50 ml), dried over anhydrous Na2SO4 and concentrated. The residue was taken up in DCM (10 ml) and then TFA (1 ml) was added. After stirring at room temperature for 2 h, solvent was removed and the crude product was purified by automated flash chromatography using either EtOAc and hex-anes or MeOH and DCM as eluents to give the desired adduct.
4.1.1.24. 6-Bromo-1H,1ʹH-2,2ʹ-biindole (10a).
Compound 10a was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and tert-butyl 2-iodo-1H-indole-1-carboxylate.
Yield = 35%, pink solid. Mp = 199–201 °C. 1H NMR (DMSO-d6, 400 MHz): δ 11.71 (s, 1H), 11.59 (s, 1H), 7.51–7.59 (m, 3H), 7.41 (d, J = 8.1 Hz, 1H), 7.09–7.16 (m, 2H), 7.02 (t, J = 7.4 Hz, 1H), 6.92–6.95 (m, 2H). 13C NMR (DMSO-d6, 150 MHz): 137.04, 136.85, 132.34, 130.69, 128.20, 127.42, 122.21, 121.92, 121.65, 120.15, 119.48, 114.14, 113.35, 111.13, 98.79, 98.37. HRMS calcd for (C16H11BrN2 H) 310.0106; found 310.0107.
4.1.1.25. 6,6′-Dibromo-1H,1′H-2,2′-biindole (10b).
Compound 10b was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 6-bromo-2-iodo-1H-indole (8a).
Yield = 63%, white solid. Mp = 266–267 °C. 1H NMR (500 MHz, DMSO-d6): δ 11.77 (s, 2H), 7.56 (s, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.15 (dd, J = 1.6 Hz, zH8.4 Hz, 2H), 6.95 (d, J = 1.1 Hz, 2H). 13C NMR (125 MHz, DMSO-d6): δ 137.8 (2C), 131.8 (2C), 127.4 (2C), 122.4 (2C), 121.9 (2C), 114.4 (2C), 113.5 (2C), 99.0 (2C). HRMS calcd for (C16H10Br2N2 H) 388.9118, found 388.9123.
4.1.1.26. 5,6-Bibromo-1H,1′H-2,2′-biiindole (10c).
Compound 10c was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 5-bromo-2-iodo-1H-indole (8b).
Yield = 45%, pale brown solid. Mp = 270–272 °C. 1H NMR (DMSO-d6, 400 MHz): δ 11.82 (s, 1H), 11.78 (s, 1H), 7.79 (s, 1H), 7.53–7.57 (m, 2H), 7.37 (d, J = 8.5 Hz, 1H), 7.23 (d, J = 8.6 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 6.97 (s, 1H), 6.93 (s, 1H). 13C NMR (DMSO-d6, 100 MHz): 137.86, 135.68, 132.28, 131.74, 130.26, 127.38, 124.30, 122.43, 122.28, 121.88, 114.47, 113.58, 113.01, 111.98, 99.11, 98.43. HRMS calcd for (C16H10Br2N2–H)− 387.9211; found 387.9227.
4.1.1.27. 6′-Bromo-5-chloro-1H,1′H-2,2′-biindole (10d).
Compound 10d was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 5-chloro-2-iodo-1H-indole (8c).
Yield = 60%, white solid. Mp = 257–259 °C. 1H NMR (DMSO-d6, 500 MHz): δ 11.80 (s, 1H), 11.77 (s, 1H), 7.64 (d, J = 1.8 Hz, 1H), 7.52–7.55 (m, 2H), 7.40 (d, J = 8.6 Hz, 1H), 7.14 (dd, J = 8.4, 1.7 Hz, 1H), 7.11 (dd, J = 8.6, 2.0 Hz, 1H), 6.95 (d, J = 1.5 Hz, 1H), 6.92 (d, J = 1.5 Hz, 1H). 13C NMR (DMSO-d6, 100 MHz): 137.83, 135.44, 132.44, 131.77, 129.51, 127.36, 123.99, 122.41, 121.89, 121.77, 119.23, 114.43, 113.55, 112.54, 99.04, 98.53. HRMS calcd for (C16H10BrClN2–H)− 343.9716; found 343.9727.
4.1.1.28. 6′-Bromo-5-fluoro-1H,1′H-2,2′-biindole (10e).
Compound 10e was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 5-fluoro-2-iodo-1H-indole (8d).
Yield = 61%, white solid. Mp = 246–248 °C. 1H NMR (DMSO-d6, 600 MHz): δ 11.73 (s, 1H), 11.68 (s, 1H), 7.51–7.56 (m, 2H), 7.38 J = 8.4, 1.8 Hz, 1H), 6.90–6.97 (m, 3H). 13C NMR (DMSO-d6, 151 MHz): δ 157.68 (d, J = 231.7 Hz), 138.26, 134.13, 133.10, 132.42, 129.06 (d, J = 10.6 Hz), 127.86, 122.87, 122.31, 112.48 (d, J = 9.8 Hz), 114.82, 114.02, 110.46 (d, J = 26.1 Hz), 105.13 (d, J = 23.4 Hz), 99.48 (d, J = 4.7 Hz), 99.27. HRMS calcd for (C16H10BrFN2–H)− 328.0011; found 328.0019.
4.1.1.29. 6′-Bromo-5-methoxy-1H,1′H-2,2′-biindole (10f).
Compound 10f was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and tert-butyl 2-iodo-5-methoxy-1H-indole-1-carboxylate.
Yield = 16%, pale brown solid. Mp = 240–241 °C. 1H NMR (DMSO-d6, 500 MHz): δ 11.67 (s, 1H), 11.43 (s, 1H), 7.50–7.54 (m, 2H), 7.28 (d, J = 8.7 Hz, 1H), 7.12 (dd, J = 8.4, 1.7 Hz, 1H), 7.07 (d, J = 2.4 Hz, 1H), 6.89 (s, 1H), 6.84 (s, 1H), 6.76 (dd, J = 8.7, 2.4 Hz, 1H), 3.77 (s, 3H). 13C NMR (DMSO-d6, 150 MHz): 153.68, 137.69, 132.51, 132.10, 131.19, 128.73, 127.50, 122.24, 121.61, 113.99, 113.40, 112.20, 111.75, 101.67, 98.80, 98.17, 55.30. HRMS calcd for (C17H13BrN2O–H) 340.0211; found 340.0217.
4.1.1.30. 6′-Bromo-5-phenyl-1H,1′H-2,2′-biindole (10g).
Compound 10g was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and tert-butyl 2-iodo-5-phenyl-1H-indole-1-carboxylate.
Yield = 17%, pale yellow solid. Mp = 237–239 °C. 1H NMR (DMSO-d6, 600 MHz): δ 11.78 (s, 1H), 11.69 (s, 1H), 7.85–7.86 (m, 1H), 7.68–7.70 (m, 2H), 7.51–7.56 (m, 2H), 7.42–7.50 (m, 3H), 7.31 (t, J = 7.4 Hz, 1H), 7.14 (dd, J = 8.4, 1.7 Hz, 1H), 7.0 (s, 1H), 6.96 (s, 1H). 13C NMR (DMSO-d6, 151 MHz): 141.62, 137.79, 136.60, 132.25, 132.00, 131.52, 128.95, 128.76 (2C), 127.46, 126.63 (2C), 126.27, 122.31, 121.74, 121.36, 118.17, 114.19, 113.45, 111.48, 99.30, 98.60. HRMS calcd for (C22H15BrN2 –H)− 386.0419; found 386.0429.
4.1.1.31. 5-Bromo-1H,1′H-2,2′-biindole (10h).
Compound 10h was prepared from (5-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9b) and tert-butyl 2-iodo-1H-indole-1-carboxylate.
Yield = 50%, white solid. 1H NMR (DMSO-d6, 400 MHz): δ 11.77 (s, 1H), 11.61 (s, 1H), 7.77 (d, J = 1.7 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.37 (d, J = 8.6 Hz, 1H), 7.22 (dd, J = 8.6, 1.9 Hz, 1H), 7.10–7.15 (m, 1H), 7.0–7.04 (m, 1H), 6.95 (d, J = 1.3 Hz, 1H), 6.92 (d, J = 1.4 Hz, 1H). 13C NMR (DMSO-d6, 100 MHz): 137.0, 135.60, 132.91, 130.73, 130.37, 128.30, 124.03, 122.12, 121.96, 120.18, 119.50, 112.92, 111.87, 111.14, 99.02, 97.94. HRMS calcd for (C16H11BrN2 H) 310.0106; found 310.0107.
4.1.1.32. 5,5′-Bibromo-1H,1′H-2,2′-biindole (10i).
Compound 10i was prepared from (5-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9b) and 5-bromo-2-iodo-1H-indole (8b).
Yield = 65%, brown solid. Mp = 304–306 °C. 1H NMR (DMSO-d6, 400 MHz): δ 11.83 (s, 2H), 7.79 (d, J = 1.7 Hz, 2H), 7.36 (d, J = 8.6 Hz, 2H), 7.23 (dd, J = 8.6, 1.9 Hz, 2H), 6.94 (d, J = 1.4 Hz, 2H). 13C NMR (DMSO-d6, 100 MHz): 135.68, 132.22, 130.22, 124.33, 122.30, 113.03, 111.96, 98.52. HRMS calcd for (C16H11BrN2–H)− 387.9211; found 387.9221.
4.1.1.33. 5,6-Dibromo-1H,1′H-2,2′-biindole (10j).
Compound 10j was prepared from (1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9c) and 5,6-dibromo-2-iodo-1H-indole (8e).
Yield = 53%, white solid. Mp = 233 °C –(decomposition). 1H NMR (400 MHz, DMSO-d6): δ 11.86 (s, 1H), 11.66 (s, 1H), 8.00 (s, 1H), 7.74 (s, 1H), 7.59 (d, J = 7.7 Hz, 1H), 7.42 (d, J = 8.0 Hz, 1H), 7.14 (t, J = 7.2 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 6.97 (s, 1H), 6.94 (s,1H). 13C NMR (100 MHz, DMSO-d6): δ 137.0, 136.7, 133.8, 130.2, 129.6, 128.2, 124.1, 122.1, 120.3, 119.5, 115.4, 115.3, 113.5, 111.2, 99.4, 97.8. HRMS calcd for (C16H10Br2N2–H)− 388.9118; found 388.9108.
4.1.1.34. 5,6,6′-Tribromo-1H,1′H-2,2′-biindole (10k).
Compound 10k was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 5,6-dibromo-2-iodo-1H-indole (8e).
Yield = 24%, pale yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 11.95 (s, 1H), 11.87 (s, 1H), 8.02 (s, 1H), 7.75 (s, 1H), 7.75 (s, 1H), 7.56 (s, 1H), 7.55 (d, J = 8.6 Hz, 1H), 7.15 (dd, J = 1.7 Hz, 8.4 Hz, 1H), 6.98 (br s, 1H), 6.94 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 137.9, 136.7, 133.2, 131.3, 129.5, 127.3, 124.3, 122.5, 122.0, 115.6, 115.5, 114.6, 113.64, 113.60, 99.5, 98.3. HRMS calcd for (C16-H9Br3N2–H)− 466.8223; found 466.8223.
4.1.1.35. 5,5′,6,6′-Tetrabromo-1H,1′H-2,2′-biindole (10l).
Compound 10l was isolated as a minor bi-product during the synthesis of 10k from 9a and 8e.
Yield = 7%, pale yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 11.98 (bd, J = 0.8 Hz, 2H), 8.04 (s, 2H), 7.75 (d, J = 0.6 Hz, 2H), 6.97 (d, J = 1.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 136.8 (2C), 132.6 (2C), 129.3 (2C), 124.5 (2C), 115.9 (2C), 115.6 (2C), 113.8 (2C), 98.9 (2C). HRMS calcd for (C16H8Br4N2–H)− 546.7308; found 546.7292.
4.1.1.36. 5,5′,6-Tribromo-1H,1′H-2,2′-biindole (10m).
Compound 10m was prepared from (5-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9b) and 5,6-dibromo-2-iodo-1H-indole (8e).
Yield = 49%, white solid. 1H NMR (400 MHz, DMSO-d6): δ 12.04 (s, 1H), 11.97 (s, 1H), 8.02 (s, 1H), 7.79 (d, J = 1.8 Hz, 1H), 7.75 (s,1H), 7.37 (d, J = 8.6 Hz, 1H), 7.23 (dd, J = 1.9 Hz, 8.6 Hz, 1H), 6.95 (br s, 2H). 13C NMR (100 MHz, DMSO-d6): δ 136.8, 135.7, 133.2, 131.8, 130.1, 129.5, 124.5, 124.3, 122.4, 115.58, 115.55, 113.6, 113.1, 112.0, 98.9, 98.4. HRMS calcd for (C16H9Br3N2–H)− 466.8223; found 466.8211.
4.1.1.37. 5,6′-Dibromo-1-methyl-1H,1′H-2,2′-biindole (12a).
Compound 12a was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 5-bromo-2-iodo-1-methyl-1H-indole (11b).
Yield = 53%, white solid. Mp = 180–182 °C. 1H NMR (DMSO-d6, 400 MHz): δ 11.78 (s, 1H), 7.82 (d, J = 1.9 Hz, 1H), 7.51–7.61 (m, 3H), 7.31 (dd, J = 8.7, 2.0 Hz, 1H), 7.18 (dd, J = 8.4, 1.8 Hz, 1H), 6.91 (s, 1H), 6.89 (s, 1H), 3.96 (s, 3H). 13C NMR (DMSO-d6, 125 MHz): 137.58, 137.0, 133.90, 130.03, 128.85, 127.41, 124.25, 122.45, 122.31, 122.06, 114.75, 113.68, 112.35, 112.14, 101.76, 100.61, 31.78. HRMS calcd for (C17H12Br2N2–H)− 401.9367; found 401.9384.
4.1.1.38. 5′,6-Dibromo-1-methyl-1H,2′H-2,2′-biindole (12b).
Compound 12b was prepared from (5-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9b) and 6-bromo-2-iodo-1-methyl-1H-indole (8a).
Yield = 48%, pale yellow solid. Mp = 192–194 °C. 1H NMR (DMSO-d6, 400 MHz): δ 11.76 (s, 1H), 7.84 (s, 1H), 7.78 (d, J = 1.9 Hz, 1H), 7.57 (d, J = 8.4, 1H), 7.39 (d, J = 8.6 Hz, 1H), 7.26 (dd, J = 8.6, 1.9 Hz, 1H), 7.21 (dd, J = 8.4, 1.7 Hz, 1H), 6.92 (d, J = 0.6 Hz, 1H), 6.86 (d, J = 1.5 Hz, 1H). 13C NMR (DMSO-d6, 100 MHz): 139.13, 135.39, 133.41, 130.59, 130.25, 126.04, 124.57, 122.71, 122.37, 121.92, 114.76, 113.16, 112.97, 111.92, 101.48, 101.06, 31.78. HRMS calcd for (C17H12Br2N2--H)− 401.9367; found 401.9376.
4.1.1.39. 5,6′-Dibromo-1-(methoxymethyl)-1H,1′H-2,2′-biindole (12c).
Compound 12c was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 5-bromo-2-iodo-1-(methoxymethyl)-1H-indole (11c).
Yield = 65%, white solid. Mp = 199–201 °C. 1H NMR (DMSO-d6, 500 MHz): δ 11.74 (s, 1H), 7.87 (d, J = 1.9 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.55–7.61 (m, 2H), 7.36 (dd, J = 8.3, 2.4 Hz, 1H), 7.17 (dd, J = 8.4, 1.8 Hz, 1H), 6.98 (s, 1H), 6.95 (s, 1H), 5.71(s, 2H) 3.30 (s, 3H). 13C NMR (DMSO-d6, 125 MHz): 137.69, 137.46, 133.97, 129.50, 129.18, 127.40, 124.95, 122.55, 122.45, 122.26, 114.87, 113.67, 113.14, 112.51, 102.23, 101.93, 74.19, 55.55. HRMS calcd for (C18H14Br2N2O–H)− 431.9473; found 431.9485.
4.1.1.40. 2-(5,6′-Dibromo-1H,1′H-[2,2′-biindol]-1-yl)acetic acid (14).
Compound 14 was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 2-(5-bromo-2-iodo-1H-indol-1yl)acetic acid (13).
Yield = 33%, white solid. Mp = 321–323 °C. 1H NMR (DMSO-d6, 400 MHz): δ 11.77 (s, 1H), 7.84 (d, J = 1.9 Hz, 1H), 7.52–7.60 (m, 3H), 7.31 (dd, J = 8.7, 2.0 Hz, 1H), 7.17 (dd, J = 8.5, 1.7 Hz, 1H), 6.91 (s, 1H), 6.69 (d, J = 1.5 Hz, 1H), 5.24 (s, 2H). 13C NMR (DMSO-d6, 150 MHz): 170.07, 137.48, 137.29, 133.85, 129.69, 129.03, 127.34, 124.57, 122.51, 122.39, 122.13, 114.84, 113.67, 112.71, 112.34, 101.64, 100.79, 46.05. HRMS calcd for (C18H12Br2N2O2–H)− 445.9266; found 445.9272.
4.1.1.41. 2-(5,6′-Dibromo-1H,1′H-[2,2′-biindol]-1-yl)ethanol (17).
Compound 17 was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 2-(5-bromo-2-iodo-1H-indol-1-yl)ethanol (16).
Yield = 8%, pale white solid. 1H NMR (DMSO-d6, 500 MHz): δ 11.71 (s, 1H), 7.81 (d, J = 1.9 Hz, 1H), 7.54–7.59 (m, 3H), 7.30 (dd, J = 8.7, 2.0 Hz, 1H), 7.18 (dd, J = 8.4, 1.8 Hz, 1H), 6.91 (s, 1H), 6.86 (s, 1H), 5.17 (t, J = 5.1 Hz, 1H), 4.47 (t, J = 5.9 Hz, 2H), 3.78 (q, J = 5.6 Hz, 2H). 13C NMR (DMSO-d6, 100 MHz): 137.48, 136.82, 133.84, 130, 129.06, 127.36, 124.22, 122.42, 122.26, 122.07, 114.66, 113.70, 112.74, 112.39, 101.74, 101.32, 59.97, 46.44. HRMS calcd for (C18H14Br2N2O–H)− 431.9473; found 431.9483.
4.1.1.42. 5,6′-Dibromo-1-(2-(4-methylpiperazin-1-yl)ethyl)-1H,1′H-2,2′-biindole (20a).
Compound 20a was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 5-bromo-2-iodo-1-(2-(4-methylpiperazin-1-yl)ethyl)-1H-indole (19a).
Yield = 57%, pale white solid. Mp: 140 °C. 1H NMR (CDCl3, 500 MHz): δ 11.94 (s, 1H), 7.81 (s, 1H), 7.52–7.61 (m, 3H), 7.31 (d, J = 8.7 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 6.89 (s, 1H), 6.84 (s, 1H), 4.51 (t, J = 6.4 Hz, 2H), 2.62 (t, J = 6.4 Hz, 2H), 2.15–2.45 (m, 8H), 2.11 (s, 3H). 13C NMR (150 MHz): 137.47, 136.46, 133.66, 130.04, 129.12, 127.38, 124.34, 122.45, 122.41, 122.10, 114.65, 113.72, 112.54, 112.49, 101.71, 101.50, 56.77, 54.39, 52.79, 45.38, 42.41. HRM Scalcd for (C23H24Br2N4 H) 514.0368; found 514.0372.
4.1.1.43. 4-(2-(5,6′-Dibromo-1H,1′H-[2,2′-biindol]-1-yl)ethyl)morpholine (20b).
Compound 20b was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 4-(2-(5-bromo-2-iodo-1H-indol-1-yl)ethyl)morpholine (19b).
Yield = 22%, white form. 1H NMR (400 MHz, DMSO-d6): δ 11.89 (s, 1H), 7.81 (d, J = 1.3 Hz, 1H), 7.59–7.56 (m, 3H), 7.31 (dd, J = 1.4 Hz, 8.7 Hz, 1H), 7.18 (dd, J = 1.2 Hz, 8.4 Hz, 1H), 6.89 (s, 1H), 6.85 (s, 1H), 4.54 (t, J = 6.4 Hz, 2H), 3.43 (br s, 4H), 2.60 (t, J = 6.3 Hz, 2H), 2.30 (br s, 4H). 13C NMR (100 MHz, DMSO-d6): δ 137.5, 136.5, 133.7, 130.0, 129.1, 127.3, 124.3, 122.4, 122.3, 122.1, 114.6, 113.7, 122.5, 122.4, 101.7, 101.4, 66.0, 57.3, 53.5, 1720 N. S. Kumar et al. / Bioorg. Med. Chem. 22 (2014) 1708–1725 42.0. HRMS calcd for (C22H21Br2ON3–H)− 504.0105, found 504.0116.
4.1.1.44. 2-(6-Bromo-1H-indol-2-yl)benzo[d]thiazole (22a).
Compound 22a was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 2-iodobenzo[d]thiazole (21a).
Yield = 43%, pink solid. 1H NMR (DMSO-d6, 600 MHz): δ 12.35 (s, 1H), 8.16 (ddd, J = 8.0, 1.1, 0.6 Hz, 1H), 8.04 (ddd, J = 8.2, 1.1, 0.6 Hz, 1H), 7.63–7.64 (m, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.57 (ddd, J = 8.2, 7.2, 1.2 Hz, 1H), 7.48 (ddd, J = 8.2, 7.2, 1.2 Hz, 1H), 7.29 (dd, J = 2.2, 0.8 Hz, 1H), 7.21 (dd, J = 8.4, 1.8 Hz, 1H). 13C NMR (DMSO-d6, 150 MHz): 159.40, 153.21, 150.64, 138.43, 134.23, 131.92, 126.85, 126.78, 125.60, 123.21, 123.10, 122.50, 122.41, 116.61, 114.63, 105.08. HRMS calcd for (C15H9BrN2S–H)− 327.9670; found 327.9674.
4.1.1.45. 2-(Benzofuran-2-yl)-6-bromo-1H-indole (22b).
Compound 22b was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 2-iodobenzofuran (21b).
Yield = 40%, pale white solid. Mp = 196–198 °C. 1H NMR (DMSO-d6, 400 MHz): δ 12.02 (s, 1H), 7.71 (d, J = 7.1 Hz, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.60 (s, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.25–7.37 (m, 3H), 7.18 (dd, J = 8.4, 1.7 Hz, 1H), 7.0 (s, 1H). 13C NMR (DMSO-d6, 100 MHz): 154.0, 149.12, 137.97, 129.34, 128.55, 127.11, 124.75, 123.44, 122.75, 122.26, 121.55, 115.11, 113.92, 111.0, 102.28, 100.45. HRMS calcd for (C16H10BrNO–H)− is 310.9946; found 310.9948.
4.1.1.46. 2-(Benzo[b]thiophen-2-yl)-6-bromo-1H-indole (22c).
Compound 22c was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and 2-iodobenzo[b]thiophene (21c).
Yield = 57%, white solid. Mp = 250–252 °C. 1H NMR (DMSO-d6, 400 MHz): δ 11.91 (s, 1H), 7.94–7.98 (m, 1H), 7.86 (dd, J = 7.1, 1.5 Hz, 1H), 7.82 (s, 1H), 7.54–7.57 (m, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.33–7.43 (m, 2H), 7.14 (dd, J = 8.4, 1.8 Hz, 1H), 6.83 (d, J = 1.4 Hz). 13C NMR (DMSO-d6, 150 MHz): 139.98, 138.35, 138.03, 135.14, 132.93, 127.38, 124.99, 124.86, 123.75, 122.67, 122.47, 122.01, 119.87, 114.87, 113.67, 100.86. HRMS calcd for (C16H10BrNS–H)− 326.9717, found 326.9708.
4.1.1.47. 2-(5-Bromo-1H-indol-2-yl)benzo[d]thiazole (22d).
Compound 22d was prepared from (5-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9b) and 2-iodobenzo[d]thiazole (21a).
Yield = 35%, colorless solid. Mp = 208–210 °C. 1H NMR (DMSO-d6, 400 MHz): δ 12.43 (s, 1H), 8.15 (d, J = 7.9 Hz, 1H), 8.05 (d, J = 8.1 Hz, 1H), 7.84 (d, J = 1.5 Hz, 1H), 7.57 (t, J = 7.7 Hz, 1H), 7.42–7.50 (m, 2H), 7.34 (dd, J = 8.7, 1.8 Hz, 1H), 7.22 (s, 1H). 13CNMR (DMSO-d6, 100 MHz): 159.35, 153.22, 136.32, 134.22, 132.31, 129.50, 126.80, 126.44, 125.58, 123.30, 122.44 (2C), 114.22, 112.59, 104.24. HRMS calcd for (C15H9BrN2S–H)− 327.9670; found 327.9676.
4.1.1.48. 2-(Benzofuran-2-yl)-5-bromo-1H-indole (22e).
Compound 22e was prepared from (5-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9b) and 2-iodobenzofuran (21b).
Yield = 38%, white solid. Mp = 236–238 °C. 1H NMR (DMSO-d6, 400 MHz): δ 12.06 (s, 1H), 7.80 (d, J = 1.9 Hz, 1H), 7.70–7.73 (m, 1H), 7.61–7.66 (m, 1H), 7.40 (d, J = 8.6 Hz, 1H), 7.25–7.38 (m,4H), 6.97 (d, J = 1.5 Hz, 1H). 13C NMR (DMSO-d6, 150 MHz): 154.09, 154.08, 149.06, 149.04, 135.82, 129.91, 129.81, 128.50, 124.98, 124.80, 123.44, 122.64, 121.29, 113.42, 112.20, 111.01, 102.41, 99.81. HRMS calcd for C16H10BrNO is 310.9946; found 310.9954.
4.1.1.49. 2-(Benzo[b]thiophen-2-yl)-5-bromo-1H-indole (22f).
Compound 22f was prepared from (5-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9b) and 2-iodobenzo[b]thiophene (21c).
Yield = 57%, white solid. Mp = 269–271 °C. 1H NMR (DMSO-d6, 400 MHz): δ 12.0 (s, 1H), 7.98–8.01 (m, 1H), 7.86–7.90 (m, 1H), 7.85 (s, 1H), 7.76 (d, J = 1.8 Hz, 1H), 7.35–7.44 (m, 3H), 7.25 (dd, J = 8.6, 1.9 Hz, 1H), 6.82 (d, J = 1.6 Hz, 1H). 13C NMR (DMSO-d6, 100 MHz): 139.97, 138.37, 135.90, 134.64, 133.38, 130.18, 124.97, 124.89, 124.83, 123.75, 122.48, 122.38, 120.03, 113.24, 112.15, 100.26. HRMS calcd for (C16H10BrNS–H)− 326.9717; found 326.9722.
4.1.1.50. 6-Bromo-2-(5-bromo-1H-indol-2-yl)benzo[d]thiazole (22g).
Compound 22g was prepared from (5-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9b) and 6-bromo-2-iodobenzo[d]thiazole (21c).
Yield = 48%, yellow solid. Mp = 246–248 °C. 1H NMR (DMSO-d6, 400 MHz): δ 12.45 (s, 1H), 8.48 (d, J = 2.0 Hz, 1H), 7.97 (d, J = 8.7 Hz, 1H), 7.87 (d, J = 1.8 Hz, 1H), 7.72 (dd, J = 8.7, 2.0 Hz, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.36 (dd, J = 8.7, 1.9 Hz, 1H), 7.28 (d, J = 1.5 Hz, 1H). 13C NMR (DMSO-d6, 150 MHz): 160.26, 152.30, 136.42, 136.23, 131.87, 129.93, 129.45, 126.64, 125.01, 123.85, 123.38, 118.00, 114.26, 112.66, 104.67. HRMS calcd for (C15H8Br2N2S–H)− 405.8775; found 405.8765.
4.1.1.51. 2-(4-Bromo-3-nitrophenyl)-1H-indole (34a).
Compound 34a was prepared from (1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid and 1,4-dibromo-2-nitrobenzene.
Yield = 38%, brown form. 1H NMR (400 MHz, CDCl3): δ 8.45 (br s,1H), 7.96 (d, J = 2.0 Hz, 1H), 7.76 (dd, J = 2.0 Hz, 8.4 Hz, 1H), 7.65 (dd, J = 0.9 Hz, 7.9 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H) 7.42 (dd, J = 0.8 Hz, 8.2 Hz, 1H), 7.28–7.24 (m, 1H), 7.17–7.13 (m, 1H), 6.73–6.72 (m, 1H).
4.1.1.52. 2-(4-Bromo-3-methoxyphenyl)-1H-indole (34b).
Compound 34b was prepared from (1-(tert-butox-ycarbonyl)-1H-indol-2-yl)boronic acid and 1,4-dibromo-2-methoxybenzene.
Yield = 57%, white solid. 1H NMR (DMSO-d6, 600 MHz): δ 11.59 (s, 1H), 7.63 (d, J = 8.2 Hz, 1H), 7.57 (d, J = 2.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.42–7.39 (m, 2H), 7.14–7.11 (m, 1H), 7.02–7.00 (m, 2H). 13C NMR (DMSO-d6, 150 MHz): 156.2, 137.6, 137.2, 133.7, 133.6, 129.0, 122.4, 120.7, 120.0, 119.0, 111.8, 109.8, 109.5, 100.1, 56.9.
4.1.1.53. 1,4-Bis-(6-bromo-1H-indol-2-yl)benzene (33a).
Compound 33a was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) (2 equiv) and 1,4-diiodobenzene.
Yield = 67%, white solid. 1H NMR (600 MHz, DMSO-d6): δ 11.75 (s, 2H), 7.96 (s, 4H), 7.55 (s, 2H), 7.51 (d, J = 8.4 Hz, 2H), 7.14 (dd, J = 1.7 Hz, 8.4 Hz, 2H), 7.02 (d, J = 1.5 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): δ 138.2 (2C), 138.0 (2C), 130.8 (2C), 127.7 (2C), 125.5 (4C), 122.3 (2C), 121.8 (2C), 114.2 (2C), 113.6 (2C), 99.2 (2C). HRMS calcd for (C22H14Br2N2–H)− 464.9432; found 464.9441.
4.1.1.54. 2,5-Bis-(6-bromo-1H-indol-2-yl)thiophene (33b).
Compound 33b was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) (2 equiv) and 2,5-diiodothiophene.
Yield = 88%, yellow solid. Decomposes >228 °C. 1H NMR (600 MHz, DMSO-d6): δ 11.81 (s, 2H), 7.55 (s, 2H), 7.53 (br s, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.15 (dd, J = 1.7 Hz, 8.4 Hz, 2H), 6.76 (d, J = 1.2 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): δ 137.8 (2C), 133.8 (2C), 132.8 (2C), 127.5 (2C), 124.9 (2C), 122.6 (2C), 121.7 (2C), 114.4 (2C), 113.5 (2C), 99.3 (2C). HRMS calcd for (C20H12Br2N2-S–H)− 470.8995, found 470.9004.
4.1.1.55. 2-(4-(1H-Indol-2-yl)phenyl)-6-bromo-1H-indole (33c).
Compound 33c was prepared from (6-bromo-1-(tert-butoxycarbonyl)-1H-indol-2-yl)boronic acid (9a) and tert-butyl 2-(4-iodophenyl)-1H-indole-1-carboxylate.
Yield = 89%, Yellow solid. Mp P300 °C. 1H NMR (400 MHz, DMSO-d6): δ 11.74 (s, 1H), 11.57 (s, 1H), 7.97 (d, J = 8.7 Hz, 1H), 7.94 (d, J = 9.1 Hz, 1H), 7.55 (s, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.42 (d, J = 8.1 Hz, 1H), 7.15–7.09 (m, 2H), 7.03–6.98 (m, 3H). 13C NMR (150 MHz, DMSO-d6): δ 138.3, 138.0, 137.2, 137.1, 131.3, 130.4, 128.6, 127.7, 125.5, 125.3 (2C), 125.31 (2C), 122.3, 121.7, 120.0, 119.4, 114.1, 113.6, 111.2, 99.1, 99.0. HRMS calcd for (C22H15BrN2–H)− 387.0328; found 387.0333.
4.1.1.56. 2-(4-(1H-Indol-2-yl)-3-nitrophenyl)-6-bromo-1H-indole (33d).
Compound 33d was prepared from 9a and 34a.
Yield = 57%, brown solid. (Mixture of isomers ~5:1) 1H NMR (400 MHz, DMSO-d6): δ 11.97 (d, J = 1.3 Hz, 1H), 11.60 (d, J = 1.3 Hz, 1H), 8.45 (d, J = 1.8 Hz, 1H), 8.25 (dd, J = 1.8 Hz, 8.2 Hz, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.60–7.55 (m, 3H), 7.43 (d, J = 8.1 Hz, 1H), 7.15–7.06 (m, 3H), 7.03 (dd, J = 7.0 Hz, 7.9 Hz, 1H), 6.59 (d,J = 1.4 Hz, 1H). 13C NMR (150 MHz, DMSO-d6): δ 148.9, 138.3, 137.1, 135.9, 132.1, 131.9, 131.4, 128.3, 128.1, 127.4, 124.4, 122.8, 122.4, 122.3, 120.5, 119.9, 119.6, 115.2, 113.9, 111.5, 101.7, 101.2. HRMS calcd for (C22H14BrN3O2–H)− 432.0179; found 432.0195.
4.1.1.57. 2-(4-(1H-Indol-2-yl)-2-methoxyphenyl)-6-bromo-1H-indole (33e).
Compound 33e was prepared from 9a and 34b.
Yield = 11%, yellow solid. 1H NMR (600 MHz, DMSO-d6): δ 11.62 (d, J = 1.0 Hz, 1H), 11.37 (d, J = 1.0 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.65 (d, J = 1.3 Hz, 1H), 7.63 (s, 1H), 7.59 (dd, J = 1.6 Hz, 8.1 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.44 (d, J = 8.1 Hz, 1H), 7.14–7.11 (m, 2H), 7.06–7.01 (m, 3H), 4.09 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 156.4, 137.3, 137.2, 137.2, 135.4, 132.6, 128.6, 128.0, 127.2, 122.0, 121.8, 121.5, 120.1, 119.5, 118.9, 117.6, 113.8, 113.7, 111.2, 108.4, 101.7, 99.5, 55.9. HRMS calcd for (C23H17BrN2O–H)− 417.0434; found 417.0442.
4.1.1.58. 2-(3-(1H-Indol-2-yl)phenyl)-6-bromo-1H-indole (33f).
Compound 33f was prepared from 9a and tert-butyl 2-(3-iodo-phenyl)-1H-indole-1-carboxylate.
Yield = 86%, white solid. Mp = 224–226 °C. 1H NMR (600 MHz, DMSO-d6): δ 11.78 (s, 1H), 11.60 (s, 1H), 8.38 (s, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.58–7.54 (m, 4H), 7.44 (d, J = 7.4 Hz, 1H), 7.17–7.01 (m, 5H). 13C NMR (150 MHz, DMSO-d6): δ 138.5, 138.0, 137.3, 137.1, 132.9, 132.3, 129.5, 128.6, 127.6, 124.2, 123.9, 122.3, 121.9, 121.8, 121.7, 120.1, 119.4, 114.2, 113.7, 111.3, 99.3, 99.2. HRMS calcd for (C22H15BrN2–H)− 387.0328; found 387.0335.
4.1.1.59. Specific procedure for the synthesis of 2-(5,6′-dibromo-1H,1′H-[2,2′-biindol]-1-yl)-1-morpholinoethanone (15).
To a stirred solution of compound 14 (50 mg, 0.11 mmol), morpholine (10 µL, 0.12 mmol) and HBTU (44 mg, 0.12 mmol) in DMF (1 ml) at 0 °C was added DIPEA (61 µL, 0.35 mmol) and the resulting solution stirred at rt for 12 h. The mixture was diluted with DCM (10 ml) and washed with 1 N HCl, saturated aqueous NaHCO3, brine and concen-trated. The crude product was purified by automated flash chroma-tography to give compound 15 as white solid (38 mg, 68%).
Mp = 320–322 °C. 1H NMR (DMSO-d6, 400 MHz): δ 11.17 (s, 1H), 7.65 (s, 1H), 7.50 (s, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.19 (d, J = 8.7 Hz, 1H), 7.11 (d, J = 8.5 Hz, 1H), 7.07 (d, J = 8.5 Hz, 1H), 6.71 (s, 1H), 6.37 (s, 1H), 5.10 (s, 2H), 3.62 (br s, 2H), 3.54 (br s, 2H). HRMS calcd for (C22H19Br2N3O2–H)− 514.9844; found 514.9867.
4.1.1.60. General procedure for the synthesis of substituted 2-ethynyl-1H-indole (24) and substituted bis-indole (25).
A solution of 2-iodoindole 7 (1 mmol) and acetylene derivative (1 mmol) in THF (5 ml) was purged with argon for 10 min followed by triethylamine (3.5 mmol), CuI (0.2 mmol) and Pd(Ph3P)2Cl2 as catalyst (10 mol %). The mixture was heated in lwave at 100 °C for 30 min. After completion of the reaction as monitored by TLC, water was added and the mixture extracted with EtOAc (2 20 ml). Combined organic layer was washed with brine, dried over anhydrous Na2SO4 and evaporated in vacuo. The residue was purified by automated flash chromatography and then taken up in THF (5 ml) followed by the addition if TBAF (1 M in THF, 1 mmol). The mixture was stirred at rt for 2 h and then partitioned between EtOAc (50 ml) and H2O (50 ml). The organic phase was washed with brine (50 ml), dried over anhydrous Na2SO4 and concentrated. The crude product was purified by automated flash chromatogra-phy using EtOAc and hexanes as eluents to give the desired product.
4.1.1.61. 6-Bromo-2-ethynyl-1H-indole (24a).
Compound 24a was prepared from 7a and TIPS-acetylene.
Yield = 52%, dark brown solid. 1H NMR (CDCl3, 400 MHz): δ 8.20 (s, 1H), 7.48 (s, 1H), 7.44 (d, J = 8.3, 1.0 Hz, 2H), 7.23 (dd, J = 8.5, 1.6 MHz, 1H), 6.68 (s, 1H), 3.33 (s, 1H). 13C NMR (CDCl3, 100 MHz): 136.59, 126.23, 123.98, 122.17, 118.29, 117.47, 113.54, 109.61, 81.31, 75.55. HRMS calcd for (C10H6Br2N–H)− 218.9684; found 218.9687.
4.1.1.62. 5-Bromo-2-ethynyl-1H-indole (24b).
Compound 24b was prepared from 7b and TIPS-acetylene.
Yield = 63%, brown solid. 1H NMR (CDCl3, 400 MHz): δ 8.23 (s, 1H), 7.72 (d, J = 1.32 Hz, 1H), 7.32 (dd, J = 8.7, 1.8 Hz, 1H), 7.19 (d, J = 8.7 Hz, 1H), 6.75 (d, J = 1.5 Hz, 1H), 3.33 (s, 1H). 13C NMR (126 MHz, CDCl3): 134.59, 129.20, 126.82, 123.52, 118.93, 113.96, 112.38, 109.15, 81.54, 75.77. HRMS calcd for (C10H6BrN–H)− 218.9684; found 218.9685.
4.1.1.63. 2-((1H-Indol-2-yl)ethynyl)-6-bromo-1H-indole (25a).
Compound 25a was prepared from 24a and 2-iodo-1-(phenylsulfo-nyl)-1H-indole.
Yield = 64%, pale brown solid; Mp = 240–242 °C; 1H NMR (DMSO-d6, 600 MHz): δ 11.94 (s, 1H), 11.78 (s, 1H), 7.57 (dd, J = 7.9, 0.6 Hz, 1H), 7.52–7.54 (m, 2H), 7.36 (dd, J = 8.2, 0.9 Hz, 1H), 7.17–7.22 (m, 2H), 7.06 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.89 (dd, J = 2.0, 0.7 Hz, 1H), 6.88 (dd, J = 2.0, 0.9 Hz, 1H). 13C NMR (DMSO-d6, 151 MHz): δ 150.63, 137.31, 136.61, 127.15, 126.22, 123.23, 122.91, 122.22, 120.50, 119.97, 118.80, 117.50, 115.92, 113.78, 113.39, 108.33, 108.16, 85.62, 84.50. HRMS calcd for (C18-H11BrN2–H)− 334.0106; found 334.0113.
4.1.1.64. 1,2-Bis-(6-bromo-1H-indol-2-yl)ethyne (25b).
Compound 25b was prepared from coupling 7a with 24a.
Yield = 50%, yellow solid. 1H NMR (600 MHz, CDCl3): δ 8.24 (br s, 2H), 7.52 (s, 2H), 7.47 (d, J = 8.2 Hz, 2H), 7.26 (m, 2H), 6.84 (s, 2H). 13C NMR (150 MHz, CDCl3): δ 137.0 (2C), 126.5 (2C), 124.3 (2C), 122.2 (2C), 118.6 (2C), 117.7 (2C), 113.8 (2C), 109.7 (2C), 85.1 (2C). HRMS calcd for (C18H10Br2N2–H)− 412.9118; found 412.9126.
4.1.1.65. 6-Bromo-2-((5-bromo-1H-indol-2-yl)ethynyl)-1H-indole (25c).
Compound 25c was prepared from 7b and 24a. Yield = 32%, pale yellow solid; Mp = 236–237 °C; 1H NMR (CDCl3, 600 MHz): δ 12.03 (s, 1H), 11.96 (s, 1H), 7.77 (d, J = 1.8 Hz, 1H), 7.52–7.55 (m, 2H), 7.33 (d, J = 8.7 Hz, 1H), 7.30 (dd, J = 8.6, 1.9 Hz, 1H), 7.19 (dd, J = 8.5, 1.6 Hz, 1H), 6.93 (m, 1H), 6.86 (m, 1H). 13C NMR (CDCl3, 150 MHz): 137.78, 135.60, 129.35, 126.61, 126.12, 123.37, 122.97, 122.72, 119.45, 118.88, 116.50, 114.26, 113.75, 112.90, 108.93, 108.14, 85.50, 85.38. HRMS calcd for (C18H10Br2N2–H)− 412.9118; found 412.9102.
4.1.1.66. Synthesis of compounds 26a and 26b.
A mixture of 25b (70 mg, 0.17 mmol), K2CO3 (94 mg, 0.65 mmol) and methyl io-dide (19 µL, 0.30 mmol) in DMF (1.5 ml) was stirred at 30 °C for 3δ and then partitioned between EtOAc (20 ml) and H2O (10 ml). The organic phase was washed with brine (10 ml), dried over anhy-drous Na2SO4 and concentrated. The crude product was purified by automated flash chromatography using EtOAc and hexanes as eluents to give compounds 26a and 26b.
4.1.1.67. 6-Bromo-2-((6-bromo-1H-indol-2-yl)ethynyl)-1-methyl-1H-indole (26a).
Yield = 40%, yellow solid. 1H NMR (600 MHz, DMSO-d6): δ 11.99 (s, 1H), 7.79 (s, 2H), 7.56 (s, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.23 (dd, J = 1.6 Hz, 8.4 Hz, 1H), 7.19 (dd, J = 1.7 Hz, 8.5 Hz, 1H), 6.96 (s, 2H), 3.89 (s, 6H). 13C NMR (150 MHz, DMSO-d6): δ 138.0, 137.4, 126.2, 125.6, 123.2, 123.0, 122.4, 122.3, 121.7, 118.4, 116.4, 116.1, 113.8, 113.1, 108.5, 107.6, 88.4, 83.6, 30.9. HRMS calcd for (C19H12Br2N2–H)− 426.9275; found 426.9279.
4.1.1.68. 1,2-Bis-(6-bromo-1-methyl-1H-indol-2-yl) ethyne (26b).
Yield = 47%, pale brown solid. 1H NMR (600 MHz, DMSO-d6): δ 7.82 (s, 2H), 7.55 (d, J = 8.3 Hz, 2H), 7.24 (d, J = 8.3 Hz, 2H), 7.03 (s, 2H), 3.90 (s, 6H). 13C NMR (150 MHz, DMSO-d6): δ 138.0 (2C), 125.6 (2C), 123.2 (2C), 122.5 (2C), 121.5 (2C), 116.5 (2C), 113.1 (2C), 108.1 (2C), 86.9 (2C), 31.0 (2C). HRMS calcd for (C20H14Br2N2+H)+ 442.9577; found 442.9562.
4.1.1.69. Synthesis of (E)-2-(2-(1H-indol-2-yl)vinyl)-6-bromo-1H-indole (27a).
To a stirred solution of 25b (72 mg, 0.17 mmol) and Et3N (0.34 ml, 2.4 mmol) in THF (2 ml) at 50 °C under Ar was added formic acid (88%, 77 µL, 1.8 mmol) followed by addition of 10% Pd/C (8 mg). More 10% Pd/C (8 mg) was added every 15 min to a total of 40 mg and the mixture was heated for an additional 2 h, filtered through a pad of silica which was thoroughly washed with EtOAc (2×10 ml). The filtrate was washed with H2O (10 ml), brine (10 ml), dried over anhydrous Na2SO4 and concentrated. The crude product was purified by automated flash chromatography using EtOAc and hexanes as eluents to give compounds 27a as yellow solid (4 mg, 7%). 1H NMR (600 MHz, DMSO-d6): δ 11.57 (s, 1H), 11.42 (s, 1H), 7.50 (d, J = 7.9 Hz, 1H), 7.49 (br s, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.20 (d, J = 16.5 Hz, 1H), 7.18 (d, J = 16.5 Hz, 1H), 7.11–7.09 (m, 2H), 6.96 (dd, J = 7.1 Hz, 7.8 Hz), 6.57 (s, 2H). 13C NMR (150 MHz, DMSO-d6): δ 138.2, 137.7, 137.5, 136.5, 128.4, 127.6, 122.18, 122.17, 121.6, 120.0, 119.3, 119.1, 117.7, 114.5, 113.3, 110.9, 103.2, 102.6. HRMS calcd for (C18H13BrN2–H)− 335.0189, found 335.0195.
4.1.1.70. Synthesis of compounds 27b and 28a.
To a stirred solution of 6-bromo-1H-indole-2-carbaldehyde (80 mg, 0.36 mmol) and TiCl4 (58 ll, 0.53 mmol) in THF (5 ml) under Ar was added Zn dust (70 mg, 1.1 mmol) gradually over 15 min and the resulting mixture was refluxed for 3 h. After cooling to rt 10% aqueous solution of K2CO3 (1 ml) was added and the reaction mix-ture was stirred at rt for 16 h. The mixture was partitioned between EtOAc (50 ml) and H2O (20 ml). The organic phase was washed with brine (20 ml), dried over anhydrous Na2SO4 and con-centrated. The crude product was purified by automated flash chromatography using EtOAc and hexanes as eluents to give com-pounds 27b and 28a.
4.1.1.71. (E)-1,2-Bis-(6-bromo-1H-indol-2-yl)ethane (27b).
Yield = 20%, yellow solid. Decomposes >300 LC. 1H NMR (600 MHz, DMSO-d6): δ 11.59 (s, 2H), 7.49 (s, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.20 (s, 2H), 7.11 (dd, J = 1.7 Hz, 8.4 Hz, 2H), 6.60 (d, J = 0.9 Hz, 2H). 13C NMR (150 MHz, DMSO-d6): δ 138.2 (2C), 137.4 (2C), 127.5 (2C), 122.2 (2C), 121.7 (2C), 118.5 (2C), 114.6 (2C), 113.3 (2C), 103.0 (2C). HRMS calcd for (C18H12Br2N2 H) 414.9275; found 414.9280.
4.1.1.72. 1,2-Bis-(6-bromo-1H-indol-2-yl)ethane (28a).
Yield = 53%, yellow solid. 1H NMR (600 MHz, DMSO-d6): δ 11.18 (s, 2H), 7.46 (s, 2H), 7.46 (d, J = 8.3 Hz, 2H), 7.04 (d, J = 8.3 Hz, 2H), 6.21 (s, 2H), 3.11 (s, 4H). 13C NMR (150 MHz, DMSO-d6): δ 140.4 (2C), 136.8 (2C), 127.3 (2C), 121.4 (2C), 120.8 (2C), 113.1 (2C), 112.7 (2C), 98.7 (2C), 27.0 (2C). HRMS calcd for (C18H14Br2N2–H)−416.9431; found 416.9435.
4.1.1.73. Synthesis of (6-bromo-1-(phenylsulfonyl)-1H-indol-2-yl)methanol (29).
6-Bromo-N-benzenesulfonate (1.79 g, 5.3 mmol) in THF (20 ml) was cooled to –78 °C and LDA 1.8 M in THF (4.46 ml, 8.0 mmol) was added. After 30 min at –78 °C and 30 min at rt the mixture was cooled to –78 °C and paraformalde-hyde (206 mg, 6.89 mmol) was added all at once. The reaction was warmed to rt and stirred for 12 h and then quenched by addition of saturated NH4Cl. The mixture was extracted with EtOAc, the organics were washed with brine, dried over Na2SO4, concentrated and purified by automated flash chromatography using EtOAc and hexanes as eluents to give compound 29.
Yield = 72%, white solid; 1H NMR (CDCl3, 500 MHz): δ 8.25 (d, J = 0.6 Hz, 1H), 7.83 (dd, J = 1.1, 8.5 Hz, 1H), 7.61–7.56 (m, 1H), 7.47 (dd, J = 5.0, 10.8 Hz, 1H), 7.38–7.33 (m, 1H), 6.62 (s, 1H), 4.88 (s, 1H). 13C NMR (CDCl3, 126 MHz): 140.72, 138.27, 137.70, 134.47, 129.72, 128.01, 127.39, 126.51, 122.44, 118.88, 117.48, 111.12, 58.53.
4.1.1.74. Synthesis of (E)-6-bromo-2-(2-(5-bromo-1-(phenylsul-fonyl)-1H-indol-2-yl)vinyl-1-(phenulsulfonyl)-1H-indole (31).
Compound 29 (1.0 g, 2.54 mmol) was treated in dry ether (15 ml) at 0 °C with PBr3 (240 μl, 2.4 mmol) added slowly and then the reaction was stirred at rt for 30 min. After addition of aq KBr, the mixture was extracted with ether and the organics were washed with brine, dried over Na2SO4, concentrated to provide the crude bromide as a brown solid which was dissolved in DCM (20 ml) and triphenylphosphine (744 mg, 2.84 mmol) was added and the mixture was stirred at rt overnight. The solvent was removed in vacuo and the residue was suspended in EtOAc (15 ml), sonicated and the solid collected by filtration. The crude Wittig reagent (1.0 g, 1.49 mmol) was dissolved in 1:1, THF/MeOH (20 ml) and DBU (285 μl, 2.02 mmol) was added followed by 5-bromo-N-benzenesulfonylindole (490 mg, 1.35 mmol). The mixture was stirred at rt for 3 h and then the solvent was removed under reduced pressure and the residue was partitioned between water and EtOAc, and the aqueous layers were washed with EtOAc, the organic extracts were combined, washed with brine dried over Na2SO4, concentrated and purified by automated flash chromatog-raphy using EtOAc and hexanes as eluents to give compound 31 as a yellow solid.
Yield 36%; 1H NMR (DMSO-d6, 500 MHz): δ 8.23 (s, 1H), 8.04 (d, J = 8.9 Hz, 1H), 7.85 (d, J = 1.9 Hz, 1H), 7.83–7.48 (m, 15H), 7.35 (s, 1H), 7.27 (s, 1H). 13C NMR (DMSO-d6, 151 MHz): 139.40, 138.69, 137.45, 137.14, 136.81, 136.72, 135.60, 135.10, 135.02, 131.62, 130.20, 130.09, 130.03, 129.92, 128.77, 128.05, 127.65, 126.45, 126.24, 126.21, 123.81, 123.25, 121.68, 121.58, 118.16, 117.13, 117.02, 116.50, 110.27, 109.61. HRMS calcd for (C30H20Br2N2O4S2-–H)− 692.9153; found 692.9158.
4.1.1.75. Synthesis of (E)-6-bromo-2-(2-(5-bromo-1H-indol-2-yl)vinyl)-1H-indole (27c).
Compound 31 (20 mg, 0.29 mmol) was dissolved in 1 ml of THF/MeOH, (2:1) and Cs2CO3 (28 mg, 0.86 mmol) was added and the mixture has heated in a lwave reactor at 90 °C for 30 min. The reaction was cooled, solvents were removed and the residue was stirred with H2O (1 ml) for 10 min and then the mixture was extracted with DCM. The extracts were dried (Na2SO4), concentrated to dryness and the residue was purified by automated flash chromatography using EtOAc and hexanes as eluents to give compound 27c as a yellow solid.
Yield = 88%, yellow solid; 1H NMR (DMSO-d6, 500 MHz): δ 11.65 (s, 1H), 11.60 (s, 1H), 7.68 (d, J = 1.6 Hz, 1H), 7.50 (s, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 8.5 Hz, 1H), 7.22–7.17 (m, 3H), 7.11 (dd, J = 1.7, 8.4 Hz, 1H), 6.60 (s, 1H), 6.56 (s, 1H). 13C NMR (DMSO-d6, 126 MHz): 138.27, 137.96, 137.38, 136.11, 130.33, 127.52, 124.44, 122.23, 122.05, 121.72, 118.86, 118.47, 114.69, 113.29, 112.84, 111.74, 103.12, 102.3. HRMS calcd for (C18H12Br2-N2–H)− 412.9289; found 412.9251.
4.1.1.76. Synthesis of 6-bromo-2-(2-(5-bromo-1H-indol-2-yl)ethyl)-1H-indole (28b).
Compound 27c (48 mg) was dissolved in 1:1 EtOAc/MeOH (2 ml) and 5 mg 10% Pt-C was added and the mixture was stirred under a H2 atmosphere at rt following the reaction by TLC. When complete, the mixture was filtered, concentrated to dryness and the residue was purified by automated flash chromatography using EtOAc and hexanes as eluents to give compound 28b as a white solid.
Yield = 81%. Mp = 208–210 °C; 1H NMR (DMSO-d6, 500 MHz): δ 11.23 (s, 1H), 11.17 (s, 1H), 7.58 (d, J = 1.7 Hz, 1H), 7.45 (s, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 8.6 Hz, 1H), 7.10 (dd, J = 1.9, 8.5 Hz, 1H), 7.04 (dd, J = 1.8, 8.3 Hz, 1H), 6.20 (s, 1H), 6.18 (s, 1H), 3.14 (s, 4H). 13C NMR (DMSO-d6, 126 MHz): 141.00, 140.39, 136.83, 134.61, 130.18, 127.27, 122.51, 121.41, 121.32, 120.86, 113.14, 112.69, 112.53, 111.15, 98.68, 98.22, 27.04 (2C). HRMS calcd for (C18H14Br2N2–H)− 414.9445; found 414.9470.
4.2. PK enzymatic assays, cell-based assays and cytotoxicity
4.2.1. Pyruvate kinase enzymes and inhibitory activity testing
The pyruvate kinase enzymes MRSA PK and human PK isoforms were identical to and were sourced as His-tagged constructs as described in Ref. 4. Synthetic candidate MRSA PK inhibitors were diluted in 100% DMSO and assayed for their abilities to inhibit enzymatic activities of MRSA PK and (for selected compounds) human PKs as described in Ref. 4.
4.2.2. In vitro susceptibility testing Staphylococcus aureus (ATCC29213)
Determinations of in vitro susceptibility and MIC determinations in S. aureus ATCC29213 were carried out as described in Ref. 4 except that serial (twofold) dilutions of test compounds were done in 100% DMSO and added to give a final concentration of DMSO of 5% in brain heart infusion (BHI) broth (Difco).
4.2.3. In vitro susceptibility testing (MRSA)
MIC values for selected antimicrobial actives were determined with MRSA strain MW2 (USA400) using the 96-well microtiter CLSI recommended standard for twofold serial broth microdilution method13 as described previously.4,7 Each compound was prepared in DMSO with twofold serial dilutions to give a final concentration of 64–0.031 μg/ml. 10 μl of the compound solution was then added, in duplicate, to either, 190 μl of cation adjusted Mueller Hinton broth (CAMHB) or 190 μl CAMHB containing ~2.5 × 105 CFU/ml of bacteria (final compound concentration 64–0.031 μg/ml). Culture plates were then incubated for 18–24 h at 37 °C and the optical density at 590 nm (OD590) was measured using a Tecan M200 (Tecan). The absorbance control values for the series containing CAMHB and inhibitor were subtracted as background from the corresponding infected wells. The MIC was defined as the lowest concentration of test compound leading to complete inhibition of cell growth in relationship to compound-free control wells as determined by optical density. Vancomycin was used as a reference compound. Experiments were replicated at least three times to verify reproducibility using the above conditions.
4.2.4. The effect of efflux inhibitor verapamil on in vitro susceptibility testing
MIC values for selected antimicrobial actives were determined in the presence of efflux inhibitor verapamil with S. aureus (ATCC29213) using the 96-well microtiter procedure described above. A sub-inhibitory concentration of verapamil (200 mg/L)11 was added to CAMHB and all assays were preformed as described above with either CAMHB or CAMHB containing verapamil (vCAMHB). Culture plates were then incubated for 18–24 h at 37 LC and the optical density at 600 nm (OD600) was measured using the PowerWave™ XS (BioTek). The absorbance control values for the series containing CAMHB or vCAMHB and inhibitor were subtracted as background from the corresponding infected wells. The MIC was defined as the lowest concentration of test compound leading to complete inhibition of cell growth in relationship to compound-free control wells as determined by optical density. Vancomycin was used as a reference compound. All com-pounds with the exception of 10l, were replicated at least three times to verify reproducibility using the above conditions. Compound 10l was replicated twice due to a shortage of the compound.
4.2.5. Construction of Pyk-deficient bacterial strains and growth conditions
The S. aureus ∆pyk::ErmR LAC mutant (AR0891) was constructed by amplifying ~1 kb 5′ and 3′ homology fragments sur-rounding the pyk gene (SACOL1745) with the following primers: pyk-5.1A (tggtagaattcGCAGTTTTAACTAGTGGTGG) pyk-5.1B (tggta-gaattcGCTGGTCCAA-TTGTACATAC), pyk-3.1A (gtaggatccACGATTG ATGCTGCTCAAGG), pyk-3.1B (gtagg-atccAGAAGGTGTTTGATCTTG AGC). The resulting fragments were then cloned into the EcoRI and BamHI sites flanking an ErmR cassette, which was cloned into the SmaI site of the Escherichia coli/S. aureus shuttle vector pBT214 to yield pNV026. Following transformation of pNV026 into S. aureus SF8300, allelic exchange was performed as previously described12 using TSB without glucose supplemented with 1% sodium pyruvate (P). The pyk::ErmR mutation was then transduced into S. aureus LAC using bacteriophage Φ80.
4.2.6. Bacterial killing assay
Wild-type S. aureus LAC and S. aureus LAC ∆pyk::ErmR (AR0891) were grown overnight in TSB without glucose supplemented with 1% sodium pyruvate with shaking at 37 °C. Overnight cultures were washed 2× with sterile PBS and then diluted to an OD600 = 0.1 (~2.5 × 107 cfu/ml) in pre-warmed (37 °C) media (either BHI or TSB without glucose supplemented with 1% sodium pyruvate) ±1.6 μM 10d. Cultures were then grown at 37 °C with shaking at 250 rpm. Viable bacterial were enumerated by dilution plating at 0, 8, and 24 h post-inoculation using Tryptic Soy Agar (TSA) without glucose supplemented with 1% sodium pyruvate.
4.2.7. Mammalian cytotoxicity
Mammalian cytotoxicity was performed using procedures and statistical analysis previously described.4
Acknowledgments
The authors thank Dr. Jianghong An of the Genome Sciences Centre of Vancouver for valuable assistance in preparing Figure 2. The authors acknowledge Genome British Columbia, the Natural Sciences and Engineering Council of Canada (NSERC) and the Genome BC-CDRD Innovation Fund for financial support. R.N.Y. acknowledges the support of Simon Fraser University, the British Columbia Government Leading Edge Endowment Fund and Merck Canada Inc.
Abbreviations:
- BHI
brain–heart infusion broth
- MRSA
methicillin-resistant Staphylococcus aureus
- PK
pyruvate kinase
- MIC
minimal inhibitory concentration
- TSA
tryptic soy agar
- TSB
tryptic soy broth
References and notes
- 1.Wright GD Curr. Opin. Chem. Biol 2003, 7, 563. [DOI] [PubMed] [Google Scholar]
- 2.Cherkasov A; Hsing M; Zoraghi R; Foster LJ; See RH; Stoynov N; Jiang J; Kaur S; Lian T; Jackson L; Gong H; Swayze R; Amandoron E; Hormozdiari F; Dao P; Sahinalp C; Santos-Filho O; Axerio-Cilies P; Byler K; McMaster WR; Brunham RC; Finlay BB; Reiner NE J. Proteome Res 2011, 10, 1139. [DOI] [PubMed] [Google Scholar]
- 3.Zoraghi R; See RH; Gong H; Lian T; Swayze R; Finlay BB; Brunham RC; McMaster WR; Reiner NE Biochemistry 2010, 49, 7733. [DOI] [PubMed] [Google Scholar]
- 4.Zoraghi R; See RH; Axerio-Cilies P; Kumar NS; Gong H; Moreau A; Hsing M; Kaur S; Swayze RD; Worrall L; Amandoron E; Lian T; Jackson L; Jiang J; Thorson L; Labriere C; Foster L; Brunham RC; McMaster WR; Finlay BB; Strynadka NC; Cherkasov A; Young RN; Reiner NE Antimicrob. Agents Chemother 2011, 55, 2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fraser HB; Hirsh AE; Steinmetz LM; Scharfe C; Feldman MW Science 2002, 296, 750. [DOI] [PubMed] [Google Scholar]
- 6.Axerio-Cilies P; See RH; Zoraghi R; Worral L; Lian T; Stoynov N; Jiang J; Kaur S; Jackson L; Gong H; Swayze R; Amandoron E; Kumar NS; Moreau A; Hsing M; Strynadka NC; McMaster WR; Finlay BB; Foster LJ; Young RN; Reiner NE; Cherkasov A ACS Chem. Biol 2012, 7, 350. [DOI] [PubMed] [Google Scholar]
- 7.Kumar NS; Amandoron EA; Cherkasov A; Finlay BB; Gong H; Jackson L; Kaur S; Lian T; Moreau A; Labrière C; Reiner NE; See RH; Strynadka NC; Thorson L; Wong EWY; Zoraghi R; Young RN Bioorg. Med. Chem 2012, 20, 7069. [DOI] [PubMed] [Google Scholar]
- 8.Zoraghi R; Worrall L; See RH; Strangman W; Popplewell WL; Gong H; Samaai T; Swayze RD; Kaur S; Vuckovic M; Finlay BB; Brunham RC; McMaster WR; Davies-Coleman MT; Strynadka NC; Andersen RJ; Reiner NE J. Biol. Chem 2011, 286, 44716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zechini B; Versace L Recent Patents Anti-Infect. Drug Discovery 2009, 4, 37. [DOI] [PubMed] [Google Scholar]
- 10.Costa SS; Falcao C; Viveiros M; Machado D; Martins M; Melo-Cristino J; Amaral L; Couto I BMC Microbiol 2011, 11, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa SS; Junquiueira E; Palma C; Viveiros M; Melo-Cristino J; Amaral L; Couto I Antibiotics 2013, 2, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuller JR; Vitko NP; Perkowski EF; Scott E; Khatri D; Spontak JS; Thurlow LR; Richardson AR Front. Cell. Infect. Microbiol 2011, 1, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watts JL; Shryock TR; Apley M; Bade DJ; Brown SD; Gray JT; Heine H; Hunter RP; Mevius DJ; Papich MG; Silley P; Zurenk GE Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals. In Approved Standard, 3rd ed. In ; Clinical and Laboratory Standards Institute, 2008; 28(8),. M31–A3. [Google Scholar]
- 14.Brückner R FEMS Microbiol. Lett 1997, 151, 1. [DOI] [PubMed] [Google Scholar]