

Abstract
Craniosynostosis is defined as congenital premature fusion of one or more cranial sutures. While the genetic basis for about 30% of cases is known, the causative genes for the diverse presentations of the remainder of cases are unknown. The recently discovered cranial suture stem cell population affords an opportunity to identify early signaling pathways that contribute to craniosynostosis. We previously demonstrated that enhanced BMP signaling in neural crest cells (caA3 mutants) leads to premature cranial suture fusion resulting in midline craniosynostosis. Since enhanced mTOR signaling in neural crest cells leads to craniofacial bone lesions, we investigated the extent to which mTOR signaling is involved in the pathogenesis of BMP-mediated craniosynostosis by affecting the suture stem cell population. Our results demonstrate a loss of suture stem cells in the caA3 mutant mice by the newborn stage. We have found increased activation of mTOR signaling in caA3 mutant mice during embryonic stages, but not at the newborn stage. Our study demonstrated that inhibition of mTOR signaling via rapamycin in a time specific manner partially rescued the loss of the suture stem cell population. This study provides insight into how enhanced BMP signaling regulates suture stem cells via mTOR activation.
Keywords: BMP Smad signaling, mTOR, neural crest cells, craniosynostosis, suture
Introduction
Craniosynostosis is defined as a congenital premature fusion of one or more cranial sutures. The cranial sutures are actively growing sites that support the growth of the calvaria and facial bones. Premature fusion occurs in about 1 in 2500 live births, which leads to facial deformity and restricted brain growth (Mishina & Snider, 2014; Morriss-Kay & Wilkie, 2005; Wilkie et al., 2010). Approximately 30% of cases of craniosynostosis have a known genetic cause, with more than 20 genetic mutations attributed to craniosynostosis. The majority of observed cases have no known genetic etiology emphasizing the need for continued study into this field (Lajeunie, Le Merrer, Bonaiti-Pellie, Marchac, & Renier, 1995; Mishina & Snider, 2014; Twigg & Wilkie, 2015; Wilkie et al., 2010; Wilkie & Morriss-Kay, 2001).
Sutures are fibrous connective tissue found between the bones in the cranial vault and base. They are composed of two osteogenic fronts separated interproximal by suture mesenchyme. In mice, the suture remains patent for the animal’s lifetime. In humans, the cranial sutures start to fuse by 3–4 months of age while other sutures fuse between 20 to 30 years of age (Badve, K, Iyer, Ishak, & Khanna, 2013; Senarath-Yapa et al., 2012). Recent studies have identified Gli1 positive cells as the stem cell of the craniofacial suture (Zhao et al., 2015). Gli1 positive cells are restricted to the suture mesenchyme of craniofacial bones and undifferentiated cells; ablation leads to craniosynostosis, skull growth arrest and compromised injury repair (Zhao et al., 2015). GLI1 is a robust activator and potentiates the transcriptional output of Hedgehog signaling (Hui & Angers, 2011). Hedgehog signaling regulates the expression of many transcriptional regulators during neural tube patterning (Briscoe, Pierani, Jessell, & Ericson, 2000; Dessaud et al., 2010). Gli1 expression is highest during the early stages of facial development (Du et al., 2012). Taken together, these results suggest that Hedgehog-GLI axis plays an important role during craniofacial development for the formation of sutures and Gli1 expression can serve as a marker to trace stem cell/progenitor cells within the suture mesenchyme.
Neural crest cells represent a transient cell population capable of self-renewal and multipotency (Crane & Trainor, 2006; Kalcheim, 2018). Cranial neural crest cells arise from the anterior portion of the neural tube and migrate superficially to give rise to the majority of anterior craniofacial structures and differentiate into several different cell types including osteoblasts and chondrocytes (G. Chen et al., 2017; Mishina & Snider, 2014; Noden & Trainor, 2005). Thus, imbalance of cranial neural crest cells between their proliferation, cell fate specification and cell death can result in abnormal craniofacial development including craniosynostosis.
Bone morphogenetic proteins (BMPs) are members of the transforming growth factor β (TGF-β) superfamily. These proteins were initially discovered for their role in inducing bone formation in soft tissue (Grafe et al., 2017; Urist, 1965). BMPs play an important role in the development and formation of a number of craniofacial structures (Nie, Luukko, & Kettunen, 2006). Binding of BMP ligands to their receptors results in the phosphorylation of downstream Smad proteins affecting a number of pathways associated with neurogenesis, gastrulation, and apoptosis (Grafe et al., 2017; Miyazono, Kamiya, & Morikawa, 2010; Nie et al., 2006). BMP signaling has been recognized as an important patterning signal for neural crest formation (Liem, Tremml, Roelink, & Jessell, 1995; Trainor, Melton, & Manzanares, 2003; Tribulo, Aybar, Nguyen, Mullins, & Mayor, 2003). Gain of function mutations in Msx2, a transcription factor regulated by BMP-SMAD signaling, leads to craniosynostosis (Liu et al., 1995; Pelegrino Kde et al., 2012). Noggin, an antagonist of BMP, is expressed in patent sutures and ectopic expression prevents normal fusion of posterior frontal suture (Greenwald et al., 2001; Warren et al., 2001). Our previous work demonstrated that a small increase in BMP-SMAD signaling in mouse neural crest cells leads to premature fusion of the anterior frontal suture, which can be rescued by Bmpr1a heterozygosity (Komatsu et al., 2013; Pan et al., 2017).
Mammalian/mechanistic target of rapamycin (mTOR) was discovered in the early 1990s (Brown et al., 1994; Sabatini, Erdjument-Bromage, Lui, Tempst, & Snyder, 1994; Sabers et al., 1995), and despite the elucidation of the mTOR complexes, a number of interactions and downstream targets of mTOR are still unknown. mTOR has been shown to regulate essential cell processes and integrates signals related to nutrient availability, energy status, cell stresses, growth factors and homeostasis (Laplante & Sabatini, 2012). Deregulation of mTOR signaling contributes to the pathogenesis of human diseases ranging from cancer to neurodegenerative diseases (Maiese, Chong, Shang, & Wang, 2013; Sabatini, 2006). mTOR has been shown to play a role in hematopoietic stem cell self-renewal and differentiation (C. Chen, Liu, Liu, & Zheng, 2009). mTOR activation induces stem cell exhaustion which reduces tissue repair and promotes tissue dysfunction (Castilho, Squarize, Chodosh, Williams, & Gutkind, 2009). mTOR determines proliferation versus quiescence of stem cells in the adult forebrain (Paliouras et al., 2012). PI3K/mTOR/AKT have been shown to maintain pluripotency in stem cells (Singh et al., 2012; Yu & Cui, 2016). TSC1/TSC2 complexes negatively regulate mTOR complex (mTORC) by converting Rheb into its inactive GDP-bound state (Inoki, Li, Xu, & Guan, 2003; Inoki, Zhu, & Guan, 2003; Tee et al., 2002).
Previous studies have identified crosstalk between the BMP and the mTOR signaling pathway. BMP2 induces the PI3K-mTOR signaling cascade in lung cancer cells, (Langenfeld, Kong, & Langenfeld, 2005) whereas BMP7 regulates food intake in an mTOR-p70S6K dependent manner in the central nervous system (Townsend et al., 2012). BMP7 stimulates osteogenesis and adipogenesis in primary cultures of fetal rat calvaria mediated by mTOR signaling (Yeh, Ma, Ford, Adamo, & Lee, 2013). BMP and concomitant activation of the mTOR pathway leads to fibrosis and ectopic bone formation in fibrodysplasia ossificans progressiva (Agarwal et al., 2015). Rapamycin, an inhibitor of mTOR signaling, increases osteoblastic differentiation by modulating mTOR and BMP/Smad signaling in human embryonic stem cells (Lee et al., 2010).
The goal of this study was to evaluate how mTOR signaling is involved in the pathogenesis of BMP-mediated craniosynostosis by disrupting the balance between differentiation and proliferation in the suture stem cell population. Cross-talk between mTOR and BMP signaling pathways is critical for stem cell proliferation and quiescence. The contribution of mTOR signaling is a key factor in the pathogenesis of BMP-mediated craniosynostosis by disrupting the normal balance of cell proliferation and death in the suture stem cell population.
Results
Identification and localization of a Gli1-positive stem cell population in caA3 mutant mice
We used our previously generated transgenic mice that conditionally express a constitutively active form of Bmpr1a (caBmpr1a, aka caAlk3) that develop craniofacial abnormalities including craniosynostosis when crossed with P0-cre mice that express a Cre-recombinase in a neural crest-specific promoter (caA3 mutant hereafter) (Hayano, Komatsu, Pan, & Mishina, 2015; Kamiya et al., 2008; Komatsu et al., 2013; Pan et al., 2017; Yamauchi et al., 1999). These mice developed premature fusion of cranial sutures including the nasal and the anterior frontal sutures before weaning stage along with a large osseous defect at the level of the posterior frontal suture (Supplementary Fig. 1) (Komatsu et al., 2013; Pan et al., 2017). To identify the effect of enhanced BMP signaling on the mesenchymal stem cell population that gives rise to the cranial suture (Zhao et al., 2015) these mice were crossed with a Gli1-LacZ mouse line, which expresses LacZ in Gli1 expressing cells (Bai, Auerbach, Lee, Stephen, & Joyner, 2002). We observed expression of Gli1-positive cells throughout the calvaria in newborn control mice with localization of the Gli1-positive population to the sutures by postnatal day 6 (PN6) (Fig. 1Aa, b). By postnatal day 20 (PN20), the localization of the Gli1-positive cells to the sutures was evident with the decrease in the length of anterior posterior growth of the skull (Fig. 1Ac). Interestingly, the caA3 mutants displayed changes to the calvaria by newborn stage with loss of Gli1 expression in the frontal suture and nasal suture when compared to control (Fig. 1Ad, arrow and arrowhead, respectively). This decrease in Gli1 expression continued through to PN6 with loss of Gli1 expression within the anterior frontal and nasal sutures (Fig. 1Ae, arrow and arrowhead, respectively). By PN20, premature fusion of the anterior frontal suture was observed in the caA3 mutant skulls while the anterior frontal suture in the control mouse remained patent and were positive for Gli1 expression (Fig. 1Ac, f). At PN20 the mutant mice displayed a shortened snout and a decrease in anterior posterior length of anterior frontal and nasal bones.
Figure 1. Reduction of suture stem cell population in caA3 mutant mice (caA3 mut).

A. Whole heads of caA3 mut mice carrying Gli1-lacZ were stained with X-gal at newborn stage (a, d), postnatal (PN) day 6 (b, e) and day 20 (c, f). Approximate positions of sutures are shown in (a). af, anterior frontal; cr, coronal; fm, frontomaxillary; fms, frontal-maxillary-sphenoid; lm, lambdoid; n, nasal; pf, posterior frontal. Arrowheads in d, e, loss of Gli1 expression in the nasal suture; arrows in d, e, f, loss of Gli1 expression in the frontal suture.
B. Histological observations at different anterior-posterior levels. Arrows in (a) and (d) indicate the nasal suture. Areas pointed by arrows in (b) and (e) are enlarged at (c) and (f). Areas pointed by arrows in (g) and (j) are enlarged at (h) and (k). Boxes in (g) and (j) are enlarged at (i) and (l). fr, frontal bone; mx, maxilla; ns, nasal bone; sp; sphenoid. Bar = 100 μm.
We compared histological observation of various sutures in frontal sections at different anterior-posterior levels at newborn stage. At the level of the nasal suture, the anterior end of the nasal bone and the frontomaxillary suture, Gli1-positive cells were depleted within the caA3 mutants compared to controls (Fig. 1Ba–f). At the anterior end of the sphenoid, the frontal maxillary sphenoid suture and the anterior frontal suture, decreases in the Gli1-positive population in the caA3 mutants were observed (Fig. 1Bg–m, p). In non-neural crest derived tissue such as the coronal suture and lambdoid suture, levels of Gli1-positive cells were comparable between controls and caA3 mutants (Fig. 1Bn, o, q, r). These results further confirm loss of the Gli1-positive population in neural crest derived sutures of the caA3 mutant mice compared to the control mice. These changes to the stem cell population begin by newborn stage and contribute to the morphological changes observed by PN20.
mTOR signaling is upregulated in caA3 mutant mice
Previous studies have identified crosstalk between the BMP signaling and mTOR signaling pathways (Langenfeld et al., 2005; Townsend et al., 2012; Yeh et al., 2013). The Tsc1/Tsc2 complex inhibits Rheb and blocks the mTOR signaling pathway. Activated mTOR initiates a phosphorylation cascade targeting p70S6K and activated p70S6K (phosoho-p70S6K, pp70S6K) further phosphorylates ribosomal protein S6 (Fig. 2A). Given the role mTOR plays in stem cell maintenance and proliferation, we investigated a potential role of mTOR in our BMP mediated craniosynostosis mouse model. At embryonic stage 10.5 (E10.5) head regions were isolated from control and caA3 mutant mice and levels of mTOR signaling were measured by Western blot. caA3 mutants showed higher levels of phospho-SMAD1/5/9 (pSMAD1/5/9), and increased mTOR signaling as demonstrated by increased levels of phospho-mTOR (pmTOR), pp70SK6 and phospho-S6 (pS6) (Fig. 2B, C). At this stage, mTOR signaling detected by phospho-S6 immunofluorescence was observed within the mesenchymal tissues throughout the cranial region and more intense signals were detected in caA3 mutant embryos, which is in concordant with the western measurements (Fig. 2D).
Figure 2. mTOR signaling is increased in the caA3 mut embryos.

A. Schematic representation of mTOR signaling pathway.
B. Protein lysate prepared from the head region of control and caA3 mut embryos (E10.5) were subjected for Western blot analyses to quantify levels of mTOR signaling components.
C. Levels of phosphorylation of each signaling component normalized by total proteins were quantified. Fold increases of caA3 mutants compared with controls are shown. n=3. *, p<0.05; **, p<0.01.
D. mTOR signaling levels were examined using a phospho-S6 antibody on frontal sections of cranial regions of E10.5 embryos. Nuclear DNA was stained with DAPI (blue). LNP, lateral nasal process, MNP, medial nasal process, NT, neural tube, *, presumptive choroid plexus. Approximate positions of each section are also shown (a, b, nasal level, c, d, before eye level). Bar = 100 μm.
mTOR signaling levels remain elevated in caA3 mutant from embryonic stage E10.5 until newborn stage
Since mTOR signaling levels were elevated at E10.5 in caA3 mutant embryos, we sought to determine the extent to which these levels remained high during embryonic development. We observed phospho-S6 levels, an important downstream signaling target of mTOR, during embryogenesis. At E13.5 more mTOR signaling was observed as demonstrated by more intensive phospho-S6 staining in caA3 mutant embryos (Fig. 3A, arrows). At E17.5 while mTOR signaling was downregulated in the presumptive area for the frontal suture (a mesenchymal derived region located between two osteogenic fronts) in the controls, mTOR signaling persisted in the corresponding area of the caA3 mutants (Fig. 3B, boxes). mTOR activity in the mesenchymal regions of the caA3 mutants were significantly higher than that in controls (Fig, 3E). At newborn stage, levels of mTOR signaling in the mesenchymal regions of the most posterior end of the nasal suture did not show statistic significance (Fig. 3C, E). While those in the mesenchymal regions between the two osteogenic fronts of the anterior frontal suture were not statistically significant between two genotypes (Fig. 3D, E), the frontal sutures in the caA3 mutant mice did not show the same organization as the controls and showed distinctive immunofluorescent patterns suggesting that the phenotypic changes we found in the caA3 mutants were already evident by newborn stage (Fig. 3C, D, E). These results confirm that mTOR signaling remains elevated in the presumptive suture areas in caA3 mutants from E10.5 until late gestation. Given the changes that we found in the Gli1-lacZ staining pattern at newborn stage, we hypothesized that an increase in mTOR signaling during embryonic development contributes to the changes in the organization of the calvarial bones and sutures contributing to the craniosynostosis phenotype.
Figure 3. mTOR signaling is transiently increased during embryogenesis.
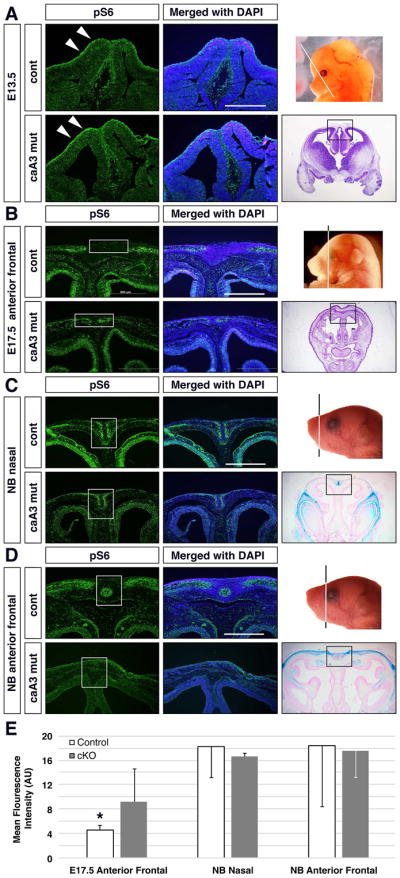
mTOR signaling levels were examined using a phospho-S6 antibody on frontal sections of cranial regions from E13.5 (A, eye level), E17.5 (B, anterior frontal suture level) and newborn (NB) (C, D, nasal and anterior suture levels, respectively) mice. Nuclear DNA was stained with DAPI (blue). White arrows in A indicate different signaling levels at the surface of embryos. White boxes in B indicate presumptive suture area negative for mTOR signaling in control embryos. White boxes in C and D indicate suture areas where control mice show more intense immunosignals. Quantification of mean fluorescence of the suture area in caA3 mutant and control images for E17.5, NB nasal and NB Anterior Frontal shown (E) *, p<0.05; n>3 per group. Bar = 500 μm.
Inhibition of mTOR signaling via rapamycin has a stage sensitive impact on the craniosynostosis phenotype
To investigate a potential role of elevated mTOR signaling during embryogenesis in BMP mediated craniosynostosis, we injected pregnant female mice with rapamycin, a pharmacological inhibitor of mTOR, to decrease mTOR signaling in utero (Lai, Lilley, Sanes, & McMahon, 2013). We previously demonstrated that administration of LDN193189, an inhibitor for BMP-Smad signaling from E14.5 significantly restores suture patency and craniofacial morphology (Komatsu et al., 2013). Since P0-Cre expression starts at E8.5 (G. Chen et al., 2017) and because we demonstrated that mTOR signaling is increased at E10.5, we compared two time windows, i.e. 1 mg/kg of daily injection from E14.5 (treatment 1, Tx 1) and from E8.5 (treatment 2, Tx 2) until pups were born (Fig. 4A). We also set up 5 mg/kg of one-time injection at E8.5 (treatment 3, Tx 3). Immunofluorescent detection demonstrated that rapamycin injections successfully decreased mTOR signaling as evidenced by lowered phospho-S6 staining at newborn stage for Tx 1 and Tx 2 (Fig. 4B). Pups treated with Tx 3 showed slightly more intensive mTOR signaling than other treatments, probably due to the short half life of rapamycin (Arriola Apelo et al., 2016).
Figure 4. Rapamycin treatment reduces facial abnormalities caused by increased BMP signaling.

A. Three injection regimens for rapamycin treatment. Tx 1, 1 mg/kg daily injection from E14.5 to E18.5. Tx 2, 1 mg/kg daily injection from E8.5 to E18.5. Tx 3, 5 mg/kg at E8.5 only.
B. Immunohistochemistry for pS6 at newborn stage showed reduction of mTOR signaling by rapamycin treatment in caA3 mutants (Tx 1).
C. Lateral view of newborn stage pups. Typical images of vehicle treated (Vehicle Tx) and rapamycin treated (Tx 3, Rapamycin Tx) pups are shown.
D. Measurement of the naso-frontal angle. Rapamycin treated caA3 mut embryos using Tx 1 did not show significant improvement while treated using Tx 2 and Tx 3 showed significant improvement on this measurement. n> 6 for each group. n.s., no significance; *, p<0.05; **p<0.01; ***, p<0.001.
E. Measurement of the nasal and frontal lengths. Actual lengths are normalized by ear widths of each mice. Rapamycin treated caA3 mutant embryos using Tx 1 did not show significant improvement while treated using Tx 2 and Tx 3 showed tendency of improvement on the relative nasal length. No significant changes were found in the relative frontal length between genotypes and treatments. n> 6 for each group. n.s., no significance; *, p<0.05; **p<0.01; ***, p<0.001.
One distinct morphological change we noted was the alteration of the naso-frontal angle measured from nasion to nasale (nasion defined as the most caudal point of the nasal bone and nasale as the most rostral point of the nasal bone) in the mutant mice. Figure 4C and 4D demonstrate the change in this angle comparing the control and caA3 mutant with and without rapamycin treatment. The results demonstrate a significant decrease in the naso-frontal angle of the vehicle-treated caA3 mutants compared to the vehicle-treated controls. This decrease in the naso-frontal angle was rescued to nearly control levels via rapamycin injections starting at E8.5 for both Tx 2 and Tx 3. Interestingly, injections that began later in mutant mice E14.5 did not have the same rescue effect, and the naso-frontal angle remained decreased, similar to what is observed in the vehicle treated mutants.
We also took linear measurements of the lengths of the nasal portions and the frontal portions, respectively, and normalized by the ear width as shown in Fig. 4E. Similar to the naso-frontal angle, slight improvements were observed in the relative nasal lengths by rapamycin treatment Tx 2 and Tx 3, while no changes were found in the relative frontal lengths neither between genotypes nor treatments (Fig. 4E). Absolute lengths of the nasal and frontal portions showed similar patterns with normalized lengths (data not shown).
Treatment with rapamycin impacts the Gli1-expressing cranial suture cell population in a stage dependent manner
To identify how inhibition of mTOR impacted the Gli1 positive cell population, we stained whole skulls with X-gal to look at changes in the Gli1-lacZ pattern. The control mice treated with rapamycin demonstrated the same X-gal staining pattern as vehicle treated control mice (Fig. 5A). Following rapamycin treatment 3 (Tx 3), caA3 mutant mice demonstrated a partial increase in the Gli1 positive population within areas that were negative in vehicle treated mutants. This change was most evident between the nasal and frontal bones in the location of the nasal and anterior frontal suture, respectively (Fig. 5A, white arrowheads and double-headed arrows, C, D). In the vehicle treated caA3 mutants, Gli1 positive cells were found only in the posterior half of the nasal suture, whereas Gli1 positive cells were found in the entire portion of the nasal suture in rapamycin treated caA3 mutants (Fig. 5A, white arrowheads). Interestingly, when comparing treatment groups at E8.5 (Tx 2) and E14.5 (Tx 1) we observed an exacerbation of the loss of Gli1 positive cells in this area from treatment group 1, i.e. total loss of Gli1 positive cells in the nasal suture, wider distances between frontal bones (Fig. 5B, double-headed arrows) and parietal bones (Fig. 5B, opposed arrowheads), respectively, as detailed below. Thus, it appears that early inhibition of mTOR signaling mitigates loss of the Gli1 positive population when compared to later inhibition of mTOR (Fig. 5B, C, D).
Figure 5. Rapamycin treatment reduces distance between skull bones.

A. control and caA3 mut newborn skulls were stained with X-gal after vehicle treatment or rapamycin treatment (Tx 3, 5 mg/kg at E8.5).
B. caA3 mut newborn skulls treated with rapamycin (Tx 1, 1mg/kg daily injection from E14.5 or Tx 2, 1 mg/kg daily injection from E8.5)
C. Distance between the frontal bones (a) and between the parietal bones (b) were measured. n> 3 for each group. #, p=0.1, *, p<0.05; **p<0.01; ***, p<0.001.
D. Lengths of the nasal (c) and the frontal (d) bones and the distance between eyes were measured. n> 3 for each group. *, p<0.05; **p<0.01; ***, p<0.001.
Several measurements were used to quantify the loss of Gli1 positive cells within these specific locations of the calvaria. First, we measured the distances between bones (Fig. 5C), including the distance between the X-gal stained regions of the frontal bones (a), and the distance between the X-gal stained regions of the parietal bones (b). Our results revealed significant differences in distances between the frontal bones or the parietal bones in Tx 2 and Tx 3 groups compared with vehicle treated mutants (Fig. 5B, double-headed arrows, Fig. 5C). Interestingly, the distance between parietal bones in Tx 1 was larger than that of the vehicle treated, which further reinforces the idea that timing of rapamycin treatment is important for morphological rescue (Fig. 5B, opposed arrowheads, Fig. 5C).
Next, we also measured the sagittal length of the two most anterior bones: the nasal and the frontal bones. We previously reported that both bones are shorter in the caA3 mutant mice in association with a wider distance between the eyes (hypertelorism) (Komatsu et al., 2013; Pan et al., 2017). Similar to the distances between bones, nasal bone length and distance between eyes were significantly rescued in the mutant mice in Tx 2 and Tx 3 groups but not in the Tx 1 treatment group (Fig. 5D).
Rapamycin treatment prevents cell death of the Gli1-expressing cranial suture cell
We previously reported an increase of cell death in the suture mesenchyme of the affected sutures in the caAlk3 mutant mice and suppression of cell death by inhibiting p53 activity can restore the craniofacial deformity (Hayano et al., 2015; Komatsu et al., 2013). To gain insight into how rapamycin treatment results in partial rescue of the cranial deformity, we investigated how rapamycin treatments alter cell death in the Gli1-lacZ expressing cells. At the newborn stage, very limited TUNEL-positive cells were found in close proximity to the suture mesenchyme of the nasal and the anterior frontal sutures in the vehicle-treated and rapamycin treated control mice (Fig. 6, top row). In the vehicle treated caA3 mutant mice, massive cell death was observed in the same area (Fig. 6, second row). While rapamycin treatment from E14.5 (Tx 1) did not improve the levels of cell death, rapamycin treatment at or beginning from E8.5 (Tx 2 or Tx 3) significantly reduced cell death in the nasal suture comparable to the control mice (Fig. 6, bottom row). At the anterior frontal suture level, some levels of cell death were still observed in Tx 2 and Tx 3 treated caA3 mutant mice demonstrating an interesting association with different degrees of rescues in different sutures, i.e. more robust rescue in the nasal sutures compared to the anterior frontal sutures (Fig. 5A, B).
Figure 6. Suppression of cell death in the suture mesenchyme by rapamycin treatment.

Frontal sections of lacZ (blue) stained heads from new born stage mice were made and levels of cell death were measured by TUNEL (orange). The nasal suture (upper panels) and the anterior frontal suture (lower panels) from control and caA3 mutant mice received different treatments are shown. cont, caBmpr1a(+);P0-Cre(−), caA3 mut, caBmpr1a(+);P0-Cre(+). Bar = 100 μm.
Increase in mTOR signaling from birth does not rescue BMP-mediated craniosynostosis phenotype
Since rapamycin injections and inhibition of mTOR signaling beginning at E8.5 demonstrated therapeutic potential, we superimposed heterozygous null mutation of Tsc1 to the caA3 mutant mice (caA3mut; Tsc1+/−) which have increased mTOR signaling from birth (Kobayashi et al., 2001). Tsc1 complexes negatively regulate mTOR complex 1 (mTORC1) by converting Rheb into its inactive GDP-bound state (Inoki, Li, et al., 2003; Tee et al., 2002). We hypothesized that upregulation of mTOR signaling would exacerbate the loss of the Gli1 positive population and the BMP-mediated craniosynostosis phenotype. Tsc1 heterozygous mutant embryos showed an increase of mTOR signaling at E9.5, although this increase was less than what was observed in the Tsc1 homozygous mutant (Tsc1−/−) embryos (Supplementary Fig. 2). Interestingly, Tsc1+/− and Tsc1−/− mice showed increases of BMP-Smad signaling, and thus we expected the phenotypes to worsen in caA3mut; Tsc1+/− mice. However, the caA3 mut;Tsc1+/− mice did not significantly impact the change in the naso-frontal angle or the Gli1 positive cell population measured by distances between the frontal bones or parietal bones, lengths of the nasal or the frontal bones, or distances between eyes (Fig. 7A–C). Western blot analyses using embryonic heads at E10.5 revealed that BMP signaling levels measured by pSmad1/5/9 was higher in caA3 mutant embryos and Tsc1 heterozygous mutant embryos as shown in other figures (Fig. 2 and Supplementary Fig. 2). The compound mutant embryos (caA3mut; Tsc1+/−) showed lower levels of pSmad1/5/9 than Tsc1 heterozygous mutant embryos with similar increased levels of mTOR signaling found in Tsc1 heterozygous mutant embryos. Although the mechanism to explain this unexpected interaction needs to be explored further, this can explain why the double mutant mice did not develop more severe craniofacial phenotypes.
Figure 7. An increase of mTOR signaling did not change morphologies.

A. Lateral view of newborn control (caBmpr1a(+);P0-Cre(−)) and caA3 mut (caBmpr1a(+);P0-Cre(+)) mice with or without heterozygous null mutation of Tsc1 (Tsc1+/− or Tsc1+/+).
B. Top view of skull after X-gal staining.
C. Morphometric measurements. The naso-frontal angle (described in Fig. 4), distance between the frontal bones, ratios of distances between the frontal bones to those between the parietal bones, lengths of the nasal and the frontal bones and the distance between eyes (described in Fig. 5) were measured. n> 11 for each group. *, p<0.05; **p<0.01; n.s., no significance.
D. Western blot analyses of E10.5 embryonic heads for mTOR signaling activity. Each band intensity was normalized by GAPDH. Typical examples of each genotype were shown.
Discussion
Our study demonstrates that the BMP-mediated craniosynostosis phenotype can be rescued in part through inhibition of mTOR signaling by rapamycin. Using the Gli1 positive cell population as a marker for putative calvarial suture stem cell, we were able to demonstrate an association of loss of this population in our caAlk3 (caA3) mutant mice with early fusion of the anterior frontal and nasal bones. The depletion of the Gli1 positive population is evident at newborn stage in the caA3 mutant mice. We observed loss of Gli1 positive stem cells within the caA3 mutants specific to sutures of the calvaria that are neural crest derived, while mesenchymal derived sutures had Gli1 positive stem cell numbers similar to that observed in the controls.
Taking into account the role mTOR plays in stem cell differentiation and proliferation in combination with the previously identified crosstalk between the mTOR and BMP signaling pathways (Agarwal et al., 2015; Langenfeld et al., 2005; Townsend et al., 2012) (Lee et al., 2010; Yeh et al., 2013), we explored a potential role of mTOR signaling in the development of BMP mediated craniosynostosis. We demonstrated that as early as E10.5, the caA3 mutant embryos exhibited elevated levels of mTOR signaling. Immunofluorescence images at E13.5 and 17.5 demonstrate that this increase in mTOR signaling is localized to the location of the future suture site for the anterior frontal suture, which is affected by enhanced BMP signaling that fuse prematurely contributing to the craniosynostosis phenotype. In contrast, at newborn stage, cells actively receiving mTOR signaling were reduced in presumptive areas for both the anterior and posterior frontal sutures in the caA3 mutants. There is accumulating evidence that mTOR signaling is essential for normal skeletal growth (J. Chen & Long, 2018; Linder et al., 2018). In contrast, it has been suggested that mTOR complex 1 (mTORC1) inhibition promotes stem cell maintenance by preventing stem cell hyper-proliferation, terminal differentiation, and exhaustion of the stem cell pool. Deletion of negative regulators of mTORC1 including TSC1 (C. Chen et al., 2008; Gan et al., 2008) and PTEN (Yilmaz et al., 2006; Zhang et al., 2006) results in defects in quiescence and long-term repopulating abilities leading to depletion of hematopoietic stem cells. Our previous studies also demonstrated that inhibition of mTOR signaling through mTORC1 prevents differentiation of dental stem cells (J. K. Kim, Baker, Nor, & Hill, 2011) and bone marrow mesenchymal stem cells (J. Kim et al., 2012; Sun et al., 2013). Taken together, it is possible that increased mTOR signaling in the caA3 mutants causes depletion of stem cell/progenitor population during mid to late gestation resulting in a decrease of osteogenic cells at newborn stage that are positive for mTOR signaling. We believe that this is illustrated by the results of our mTOR inhibition where we observe that the earlier inhibition of mTOR, as in treatment 2 (Tx 2) and treatment 3 (Tx 3), contributes to a rescue cell death of the stem cell population while later inhibition does not (Figure 5, 6).
Because the impact on the Gli1 positive cell population was evident by newborn stage and increased mTOR signaling was observed as early as E10.5, we inhibited mTOR signaling during embryonic stages in an attempt to rescue the mutant phenotypes. We observed an increase in the Gli1 positive population following treatment with rapamycin at E8.5, but not at E14.5. While not significant, there is a trend towards a decrease in distances between the nasal or anterior frontal bones indicative of an increase in the Gli1 positive population. We believe that the change observed in the Gli1 positive cell population in the caA3 mutant mice is a result of the depletion of the stem cell population that is necessary to maintain the neural crest derived cranial sutures. A likely explanation for this depletion is an increase in apoptosis of the stem cell population. We previously reported that BMP signaling induces p53-mediated apoptosis by preventing p53 degradation in the developing nasal cartilage in the caA3 mutant mice (Hayano et al., 2015). It is reported that rapamycin treatment can suppress p53 function to prevent apoptosis (Ding et al., 2015; Krzesniak, Zajkowicz, Matuszczyk, & Rusin, 2014), thus one potential explanation is that rapamycin treatment partially prevents cell death of Gli1-expressing cells. As shown in Fig. 6, massive cell death signals were found in vehicle treated caA3 mutant mice or rapamycin treated caA3 mutant mice from E14.5 (Tx 1) while minimal cell death was observed in the mutant mice treated by rapamycin at or from E8.5 (Tx 2 and Tx 3). These results support our hypothesis well.
It has been reported that rapamycin treatment activates the BMP-Smad pathway (van der Poel, Hanrahan, Zhong, & Simons, 2003; Wahdan-Alaswad et al., 2012) due to the dissociation of FKBP12 from the BMP type 1 receptor (Wang et al., 1996). Previous studies have alluded to the differential impact of rapamycin on mTORC1 and Akt signaling and rapamycin’s contributions to apoptosis and cell survival. Studies have shown that rapamycin treatment may have varying effects on the treatment of cancer cells due to changes in mTORC2 and Akt activity (Sabatini, 2006). These studies provide some insight into how rapamycin treatment may in fact be inhibiting mTOR while differentially affecting BMP signaling. This may explain why we are unable to fully capture the effect of loss of mTOR in the Gli1 positive population that contributes to the change in suture width as opposed to our previous studies that rescue craniofacial abnormalities by a BMP-Smad signaling inhibitor and p53 inhibitor (Hayano et al., 2015; Komatsu et al., 2013). There have been other cranial suture stem cell markers identified besides Gli1, such as Axin2 (Maruyama, Jeong, Sheu, & Hsu, 2016). Future studies looking at a combination of markers to help identify the cranial suture stem cell markers may provide insight in the effects of elevated BMP and mTOR signaling on various cell populations contributing to formation and maintenance of the sutures.
Because Tsc1 heterozygous mutant embryos with increased mTOR signaling showed increased BMP-Smad signaling, we expected that superimposition of Tsc1 heterozygosity to the caAlk3 mutants might show more robust abnormalities. However, we found no difference between the Tsc1 heterozygous and the Tsc1 control group in the caAlk3 mutant mice. A possible explanation for this is that levels of increases of both signaling activities in Tsc1 heterozygous mice are not high enough to change the outcomes that we measured here. Since Tsc1 homozygous embryos die around E10.5 (Kobayashi et al., 2001), future studies investigating the impact on homozygous Tsc1 mutation using its conditional allele may provide additional insight into elevated levels of mTOR signaling on the craniosynostosis phenotype.
We were able to demonstrate that treatment with rapamycin was able to rescue the BMP mediated craniosynostosis phenotype by inhibiting mTOR starting at embryonic stage 8.5. These results provide important novel therapeutic possibilities for rapamycin treatment and the role of mTOR signaling in the development of BMP mediated craniosynostosis.
Materials and Methods
Generation of mutant mice and embryos
Generations of the mutant mouse lines, P0-Cre, caBmpr1a, Gli1-LacZ, Tsc1 mutant mouse lines, were reported previously (Bai et al., 2002; Kamiya et al., 2008; Kobayashi et al., 2001; Yamauchi et al., 1999). We first crossed ca-Bmpr1a (Kamiya et al., 2008) with P0-Cre mice (C57BL/6J-Tg(P0-Cre)94Imeg (ID 148) provided by CARD, Kumamoto University, Japan) (Yamauchi et al., 1999) to generate caBmpr1a+/−;P0-Cre+/− mice. Subsequently, these mice were bred with Gli1-LacZ (Bai et al., 2002) mice to obtain ca-Bmpr1a+/−;P0-Cre+/−;Gli1-LacZ+/− (caAlk3 mutant or caA3 mut). caBmpr1a+/−;P0-Cre+/− mice; Gli1-LacZ+/− mice were also crossed with Tsc1+/− mice (Kobayashi et al., 2001) to obtain ca-Bmpr1a+/−;P0-Cre+/−; Gli1-LacZ+/−;Tsc1+/− (caA3 mut; Tsc1+/−) mice. Littermates that did not carry either ca-Bmpr1a or P0-Cre or were wild type for Tsc1 were used as controls. All mice were maintained on a mixed 129S6 and C57BL6/J background. Timed mating was determined by noting the presence of a vaginal plug as day 0.5 for staging embryo collections and estimating date of birth. The tail of each embryo or newborn mouse was subjected to DNA extraction for genotyping. They were housed in cages in a 20°C room with a 12-hour light/dark cycle. All mouse experiments were performed in accordance with University of Michigan guidelines covering the humane care and use of animals in research. All animal procedures used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan (Protocol #PRO00005716 and #PRO00007715).
Rapamycin Injection
Rapamycin (LC Laboratories) was reconstituted in absolute ethanol at 10 mg/ml and diluted in 5% Tween-80 (Sigma-Aldrich) and 5% PEG-400 (Hampton Research). Mice received 1 mg/kg rapamycin by intraperitoneal (I.P.) injection daily either from E14.5 (Tx 1) or E8.5 (Tx 2) as described by previous experiments (Castilho et al., 2009; C. Chen et al., 2008; Lai et al., 2013). Some mice received 5 mg/kg rapamycin one time at E8.5 (Tx 3).
LacZ Staining and histologic observation
For LacZ staining, embryos were fixed in 4% PFA at 4°C for 10 minutes, rinsed with LacZ rinse (0.1% Na Deoxycholate, 0.2% NP40, 2 mM MgCl2 in 0.1M sodium phosphate buffer, pH 7.3) and stained overnight with LacZ staining solution. The LacZ staining solution was made using 1 mg/ml X-gal, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide in LacZ rinse. Embryos are then washed, re-fixed with 4% PFA to take photographs. The newborn heads were decalcified in 14% EDTA overnight at 4°C before frozen embedding. Sections were counter-stained with Eosin.
Immunohistochemistry
Embryos were fixed in 4% PFA at 4°C for 2 hours, incubated in 30% sucrose/PBS at 4°C overnight, embedded in O.C.T. compound (Sakura Finetek, Tokyo, Japan), and serially sectioned at 10 μm. The samples were incubated with a rabbit anti-phospho S6 (Ser240/244) (1:200, #5364, Cell Signaling, Billerica, MA, USA) antibody at 4°C overnight. Alexa Fluor 488 donkey anti-rabbit IgG (1:100, A21206, Invitrogen, Carlsbad, CA, USA) was used as a secondary antibody. Sections were mounted with ProLong Gold antifade reagent with DAPI (P36935, Invitrogen).
Western blot analysis
Whole cell lysates were prepared from E10.5 embryo head or E9.5 embryo using NP40 lysis buffer (50 mM Tris-HCl, pH7.4, 200 mM NaCl, 2 mM MgCl2, 1% Nonidet P-40 (NP-40), 1 mM PMSF, 1 μg/ml leupeptin, 2 μg/ml aprotinin, and 1 μg/ml pepstatin). Proteins were separated on Novex 4–20 % Tris-Glycine Gel (Invitrogen) and transferred to PVDF membrane. The membranes were incubated with 5% milk for 1 h and incubated with primary antibodies overnight at 4°C. Primary antibodies were used as follows: phospho-Smad1/5/9 antibody (1:1000, #13820, Cell Signaling), phospho-mTOR (Ser2448) (1:1000, #2971, Cell Signaling), mTOR (1:1000, #2972, Cell Signaling), phospho-S6K1 (Thr389) (1:500, #9205, Cell Signaling), S6K1 (1:1000, #9202, Cell Signaling), phospho-S6 (Ser235/236) (1:2000, #2211, Cell Signaling), phospho-S6 (Ser240/244) (1:2000, #5364, Cell Signaling), S6 (1:2000, #2217, Cell Signaling), TSC1 (1:1000, #6935, Cell Signaling), and β-actin (1:1000, #4970, Cell Signaling). Blots were incubated with peroxidase-coupled secondary antibodies (Promega) for 1 h, and protein levels were detected with SuperSignal West Pico or Femto Chemiluminescent Substrate (Thermo Scientific). Images were quantified by ImageJ software (NIH).
Statistical analysis
Morphometric measurements were made using NIH ImageJ 1.50i at defined anatomical landmarks. A two-variable linear regression model was calculated by SPSS 23.0 (IBM) to model the impacts of two independent variables on the phenotype between control and mutant mice. Models include an interaction term to account for the confounding effects of two independent variables acting in concert. The mean fluorescence intensity was calculated using NIH ImageJ 1.51j8. An outline was drawn around the area of interest along with background readings. The background mean intensity was subtracted from sample mean intensity and multiplied by the sample area to calculate the mean fluorescent intensity. Statistical analysis was completed with unpaired Student t-test.
Supplementary Material
microCT images and alizarin red-alcian blue staining images followed by X-gal staining to visualize Gli1-expressing cells at PN17 manifest premature fusion of cranial sutures including the nasal and the anterior frontal sutures (white arrows), and a large osseous defect at the level of the posterior frontal suture (white boxes).
A. Tsc1+/− mice were bred with Tsc1+/− mice to isolate embryos at E9.5.
B. PCR to confirm genotypes.
C. Signaling levels of mTOR and BMP were quantified using protein from whole embryos.
Tsc1 homozygous mutants show a robust increase in mTOR signaling while heterozygous mutants show a small increase. Interestingly, BMP-Smad signaling is also increased in these mutant embryos.
Acknowledgments
YM is supported by NIH (R01DE020843), and PHK is supported by NIH (R01DE016530). KK is supported by TEAM NIH (T32DE007057).
We thank Dr. Kenichi Yamamura for providing P0-Cre mice. We would like to express our appreciation to Drs. Fei Liu, Vesa Kaartinen, Yoshihiro Komatsu, Maiko Omi, Xiaoxi Wei and Honghao Zhang for helpful discussions. YM is supported by NIH (R01DE020843), and PHK is supported by NIH (R01DE016530). KK is supported by TEAM NIH (T32DE007057). JY is supported in part by the grant-in-aid from the National Natural Science Foundation of China (31500788) and the Fundamental Research Fund for the Central Universities of China (410500114). KK thanks Jason Kramer. SH thanks Natsuko, Toru and Kaoru Hayano.
Authors’ roles: Study design: KK, WBS, JY, JKK, PHK, YM. Study conduct, data collection, and data analysis: KK, WBS, JY, SH, MT, HP, JKK. Provide critical materials: SH and YM: Drafting manuscript: KK, WBS, JY, YM. Approving final version of manuscript: KK, WBS, JY, SH, MT, HP, JKK, PHK, and YM. KK and YM take responsibility for the integrity of the data analysis.
Footnotes
All authors state that they have no conflicts of interest.
See Supplemental Figure 1 and 2.
References
- Agarwal S, Loder SJ, Brownley C, Eboda O, Peterson JR, Hayano S, … Levi B. BMP signaling mediated by constitutively active Activin type 1 receptor (ACVR1) results in ectopic bone formation localized to distal extremity joints. Dev Biol. 2015;400(2):202–209. doi: 10.1016/j.ydbio.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, … Lamming DW. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell. 2016;15(1):28–38. doi: 10.1111/acel.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badve CA, KMM, Iyer RS, Ishak GE, Khanna PC. Craniosynostosis: imaging review and primer on computed tomography. Pediatr Radiol. 2013;43(6):728–742. doi: 10.1007/s00247-013-2673-6. quiz 725–727. [DOI] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129(20):4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Pierani A, Jessell TM, Ericson J. A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell. 2000;101(4):435–445. doi: 10.1016/s0092-8674(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, Castro A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A. 2010;107(28):12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell. 2009;5(3):279–289. doi: 10.1016/j.stem.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205(10):2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009;2(98):ra75. doi: 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ishan M, Yang J, Kishigami S, Fukuda T, Scott G, … Liu HX. Specific and spatial labeling of P0-Cre versus Wnt1-Cre in cranial neural crest in early mouse embryos. Genesis. 2017;55(6) doi: 10.1002/dvg.23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Long F. mTOR signaling in skeletal development and disease. Bone Res. 2018;6:1. doi: 10.1038/s41413-017-0004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JF, Trainor PA. Neural crest stem and progenitor cells. Annu Rev Cell Dev Biol. 2006;22:267–286. doi: 10.1146/annurev.cellbio.22.010305.103814. [DOI] [PubMed] [Google Scholar]
- Dessaud E, Ribes V, Balaskas N, Yang LL, Pierani A, Kicheva A, … Sasai N. Dynamic assignment and maintenance of positional identity in the ventral neural tube by the morphogen sonic hedgehog. PLoS Biol. 2010;8(6):e1000382. doi: 10.1371/journal.pbio.1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K, Wang H, Wu Y, Zhang L, Xu J, Li T, … He J. Rapamycin protects against apoptotic neuronal death and improves neurologic function after traumatic brain injury in mice via modulation of the mTOR-p53-Bax axis. J Surg Res. 2015;194(1):239–247. doi: 10.1016/j.jss.2014.09.026. [DOI] [PubMed] [Google Scholar]
- Du J, Fan Z, Ma X, Wu Y, Liu S, Gao Y, … Wang S. Different expression patterns of Gli1-3 in mouse embryonic maxillofacial development. Acta Histochem. 2012;114(6):620–625. doi: 10.1016/j.acthis.2011.11.007. [DOI] [PubMed] [Google Scholar]
- Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, … DePinho RA. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008;105(49):19384–19389. doi: 10.1073/pnas.0810584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafe I, Alexander S, Peterson JR, Snider TN, Levi B, Lee B, Mishina Y. TGF-beta Family Signaling in Mesenchymal Differentiation. Cold Spring Harb Perspect Biol. 2017 doi: 10.1101/cshperspect.a022202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald JA, Mehrara BJ, Spector JA, Warren SM, Fagenholz PJ, Smith LE, … Longaker MT. In vivo modulation of FGF biological activity alters cranial suture fate. Am J Pathol. 2001;158(2):441–452. doi: 10.1016/s0002-9440(10)63987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayano S, Komatsu Y, Pan H, Mishina Y. Augmented BMP signaling in the neural crest inhibits nasal cartilage morphogenesis by inducing p53-mediated apoptosis. Development. 2015;142(7):1357–1367. doi: 10.1242/dev.118802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–537. doi: 10.1146/annurev-cellbio-092910-154048. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Kalcheim C. Neural crest emigration: From start to stop. Genesis. 2018 doi: 10.1002/dvg.23090. [DOI] [PubMed] [Google Scholar]
- Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronenberg HM, … Mishina Y. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development. 2008;135(22):3801–3811. doi: 10.1242/dev.025825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung Y, Sun H, Joseph J, Mishra A, Shiozawa Y, … Taichman RS. Erythropoietin mediated bone formation is regulated by mTOR signaling. J Cell Biochem. 2012;113(1):220–228. doi: 10.1002/jcb.23347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Baker J, Nor JE, Hill EE. mTor plays an important role in odontoblast differentiation. J Endod. 2011;37(8):1081–1085. doi: 10.1016/j.joen.2011.03.034. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, … Hino O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci U S A. 2001;98(15):8762–8767. doi: 10.1073/pnas.151033798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu Y, Yu PB, Kamiya N, Pan H, Fukuda T, Scott GJ, … Mishina Y. Augmentation of Smad-dependent BMP signaling in neural crest cells causes craniosynostosis in mice. J Bone Miner Res. 2013;28(6):1422–1433. doi: 10.1002/jbmr.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzesniak M, Zajkowicz A, Matuszczyk I, Rusin M. Rapamycin prevents strong phosphorylation of p53 on serine 46 and attenuates activation of the p53 pathway in A549 lung cancer cells exposed to actinomycin D. Mech Ageing Dev. 2014;139:11–21. doi: 10.1016/j.mad.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Lai LP, Lilley BN, Sanes JR, McMahon AP. Lkb1/Stk11 regulation of mTOR signaling controls the transition of chondrocyte fates and suppresses skeletal tumor formation. Proc Natl Acad Sci U S A. 2013;110(48):19450–19455. doi: 10.1073/pnas.1309001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajeunie E, Le Merrer M, Bonaiti-Pellie C, Marchac D, Renier D. Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet. 1995;55(4):500–504. doi: 10.1002/ajmg.1320550422. [DOI] [PubMed] [Google Scholar]
- Langenfeld EM, Kong Y, Langenfeld J. Bone morphogenetic protein-2-induced transformation involves the activation of mammalian target of rapamycin. Mol Cancer Res. 2005;3(12):679–684. doi: 10.1158/1541-7786.MCR-05-0124. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Yook JY, Son MY, Kim MJ, Koo DB, Han YM, Cho YS. Rapamycin promotes the osteoblastic differentiation of human embryonic stem cells by blocking the mTOR pathway and stimulating the BMP/Smad pathway. Stem Cells Dev. 2010;19(4):557–568. doi: 10.1089/scd.2009.0147. [DOI] [PubMed] [Google Scholar]
- Liem KF, Jr, Tremml G, Roelink H, Jessell TM. Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell. 1995;82(6):969–979. doi: 10.1016/0092-8674(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Linder M, Hecking M, Glitzner E, Zwerina K, Holcmann M, Bakiri L, … Sibilia M. EGFR controls bone development by negatively regulating mTOR-signaling during osteoblast differentiation. Cell Death Differ. 2018 doi: 10.1038/s41418-017-0054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YH, Kundu R, Wu L, Luo W, Ignelzi MA, Jr, Snead ML, Maxson RE., Jr Premature suture closure and ectopic cranial bone in mice expressing Msx2 transgenes in the developing skull. Proc Natl Acad Sci U S A. 1995;92(13):6137–6141. doi: 10.1073/pnas.92.13.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Shang YC, Wang S. mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. 2013;19(1):51–60. doi: 10.1016/j.molmed.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Jeong J, Sheu TJ, Hsu W. Stem cells of the suture mesenchyme in craniofacial bone development, repair and regeneration. Nat Commun. 2016;7:10526. doi: 10.1038/ncomms10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle. 2014;13(9):1400–1412. doi: 10.4161/cc.28401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y, Snider TN. Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp Cell Res. 2014;325(2):138–147. doi: 10.1016/j.yexcr.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147(1):35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207(5):637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X, Luukko K, Kettunen P. BMP signalling in craniofacial development. Int J Dev Biol. 2006;50(6):511–521. doi: 10.1387/ijdb.052101xn. [DOI] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207(5):575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliouras GN, Hamilton LK, Aumont A, Joppe SE, Barnabe-Heider F, Fernandes KJ. Mammalian target of rapamycin signaling is a key regulator of the transit-amplifying progenitor pool in the adult and aging forebrain. J Neurosci. 2012;32(43):15012–15026. doi: 10.1523/JNEUROSCI.2248-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Zhang H, Abraham P, Komatsu Y, Lyons K, Kaartinen V, Mishina Y. BmpR1A is a major type 1 BMP receptor for BMP-Smad signaling during skull development. Dev Biol. 2017;429(1):260–270. doi: 10.1016/j.ydbio.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pelegrino KO, Sugayama S, Lezirovitz K, Catelani AL, Kok F, de Chauffaille ML. MSX2 copy number increase and craniosynostosis: copy number variation detected by array comparative genomic hybridization. Clinics (Sao Paulo) 2012;67(8):981–985. doi: 10.6061/clinics/2012(08)23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78(1):35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270(2):815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- Senarath-Yapa K, Chung MT, McArdle A, Wong VW, Quarto N, Longaker MT, Wan DC. Craniosynostosis: molecular pathways and future pharmacologic therapy. Organogenesis. 2012;8(4):103–113. doi: 10.4161/org.23307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AM, Reynolds D, Cliff T, Ohtsuka S, Mattheyses AL, Sun Y, … Dalton S. Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell. 2012;10(3):312–326. doi: 10.1016/j.stem.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Kim JK, Mortensen R, Mutyaba LP, Hankenson KD, Krebsbach PH. Osteoblast-targeted suppression of PPARgamma increases osteogenesis through activation of mTOR signaling. Stem Cells. 2013;31(10):2183–2192. doi: 10.1002/stem.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and −2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99(21):13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend KL, Suzuki R, Huang TL, Jing E, Schulz TJ, Lee K, … Tseng YH. Bone morphogenetic protein 7 (BMP7) reverses obesity and regulates appetite through a central mTOR pathway. FASEB J. 2012;26(5):2187–2196. doi: 10.1096/fj.11-199067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor PA, Melton KR, Manzanares M. Origins and plasticity of neural crest cells and their roles in jaw and craniofacial evolution. Int J Dev Biol. 2003;47(7–8):541–553. [PubMed] [Google Scholar]
- Tribulo C, Aybar MJ, Nguyen VH, Mullins MC, Mayor R. Regulation of Msx genes by a Bmp gradient is essential for neural crest specification. Development. 2003;130(26):6441–6452. doi: 10.1242/dev.00878. [DOI] [PubMed] [Google Scholar]
- Twigg SR, Wilkie AO. A Genetic-Pathophysiological Framework for Craniosynostosis. Am J Hum Genet. 2015;97(3):359–377. doi: 10.1016/j.ajhg.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urist MR. Bone: formation by autoinduction. Science. 1965;150(3698):893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- van der Poel HG, Hanrahan C, Zhong H, Simons JW. Rapamycin induces Smad activity in prostate cancer cell lines. Urol Res. 2003;30(6):380–386. doi: 10.1007/s00240-002-0282-1. [DOI] [PubMed] [Google Scholar]
- Wahdan-Alaswad RS, Bane KL, Song K, Shola DT, Garcia JA, Danielpour D. Inhibition of mTORC1 kinase activates Smads 1 and 5 but not Smad8 in human prostate cancer cells, mediating cytostatic response to rapamycin. Mol Cancer Res. 2012;10(6):821–833. doi: 10.1158/1541-7786.MCR-11-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Li BY, Danielson PD, Shah PC, Rockwell S, Lechleider RJ, … Donahoe PK. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell. 1996;86(3):435–444. doi: 10.1016/s0092-8674(00)80116-6. [DOI] [PubMed] [Google Scholar]
- Warren SM, Greenwald JA, Spector JA, Bouletreau P, Mehrara BJ, Longaker MT. New developments in cranial suture research. Plast Reconstr Surg. 2001;107(2):523–540. doi: 10.1097/00006534-200102000-00034. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, … Wall SA. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126(2):e391–400. doi: 10.1542/peds.2009-3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001;2(6):458–468. doi: 10.1038/3507660135076601. [pii] [DOI] [PubMed] [Google Scholar]
- Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, … Yamamura K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol. 1999;212(1):191–203. doi: 10.1006/dbio.1999.9323. [DOI] [PubMed] [Google Scholar]
- Yeh LC, Ma X, Ford JJ, Adamo ML, Lee JC. Rapamycin inhibits BMP-7-induced osteogenic and lipogenic marker expressions in fetal rat calvarial cells. J Cell Biochem. 2013;114(8):1760–1771. doi: 10.1002/jcb.24519. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143(17):3050–3060. doi: 10.1242/dev.137075. [DOI] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, … Li L. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441(7092):518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zhao H, Feng J, Ho TV, Grimes W, Urata M, Chai Y. The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat Cell Biol. 2015;17(4):386–396. doi: 10.1038/ncb3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
microCT images and alizarin red-alcian blue staining images followed by X-gal staining to visualize Gli1-expressing cells at PN17 manifest premature fusion of cranial sutures including the nasal and the anterior frontal sutures (white arrows), and a large osseous defect at the level of the posterior frontal suture (white boxes).
A. Tsc1+/− mice were bred with Tsc1+/− mice to isolate embryos at E9.5.
B. PCR to confirm genotypes.
C. Signaling levels of mTOR and BMP were quantified using protein from whole embryos.
Tsc1 homozygous mutants show a robust increase in mTOR signaling while heterozygous mutants show a small increase. Interestingly, BMP-Smad signaling is also increased in these mutant embryos.