

Summary
Nutrients are not only organic compounds fueling bioenergetics and biosynthesis, but also key chemical signals controlling growth and metabolism. Nutrients enormously impact the production of reactive oxygen species (ROS), which play essential roles in normal physiology and diseases. How nutrient signaling is integrated with redox regulation is an interesting but not fully understood question. Herein, we report that superoxide dismutase 1 (SOD1) is a conserved component of the mechanistic target of rapamycin complex 1 (mTORC1) nutrient signaling. mTORC1 regulates SOD1 activity through reversible phosphorylation at S39 in yeast and T40 in humans in response to nutrients, which moderates ROS level and prevents oxidative DNA damage. We further show that SOD1 activation enhances cancer cell survival and tumor formation in the ischemic tumor microenvironment, and protects against the chemotherapeutic agent cisplatin. Collectively, these findings identify a conserved mechanism by which eukaryotes dynamically regulate redox homeostasis in response to changing nutrient conditions.
eTOC Blurb
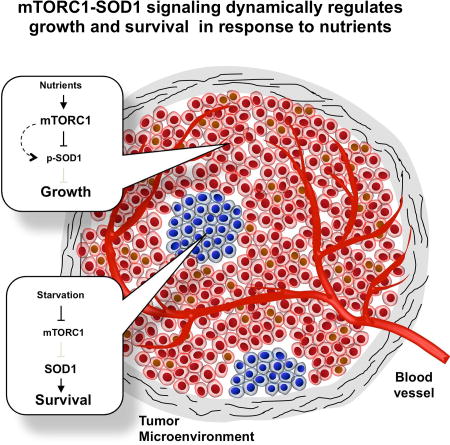
Tsang et al. show that SOD1 phosphorylation by mTOR provides a dynamic mechanism for eukaryotic cells to respond to changing nutrient conditions. It permits rapid growth in rich nutrients while confers resistance to oxidative stress during starvation. This mechanism contributes to cancer cell survival and chemoresistance in the ischemic microenvironment.
Introduction
Reactive oxygen species (ROS) are generated in eukaryotic cells in the form of superoxide cation (O2−) during respiration. Superoxide are subsequently converted to other reactive species such as H2O2 and hydroxyl radicals (Apel and Hirt, 2004). At modest levels, ROS serves as signaling molecules to promote growth and proliferation (D'Autreaux and Toledano, 2007; Finkel, 2011; Reczek and Chandel, 2015). However, under certain stress and pathological conditions such as hypoxia and tumorigenesis, excessive ROS is produced that can lead to cell and tissue damages through oxidization of DNA, lipids and proteins. The paradoxical role of ROS is nicely illustrated in human cancer. Aberrant metabolism leads to high ROS production, and uncontrolled growth and proliferation. High ROS also results in oxidative DNA mutagenesis that contributes to tumor progression. On the other hand, cancer cells often produce excessive amount of ROS, especially under the ischemic tumor microenvironment, causing severe cellular damage and death (Gorrini et al., 2013; Trachootham et al., 2009). Cancer cells must up-regulate anti-oxidative capacity to gain resistance to oxidative damage and enhance survival (Gorrini et al., 2013; Trachootham et al., 2009).
Superoxide dismutases (SODs) are antioxidant enzymes that catalyze the conversion of O2− to H2O2 and O2 (Miao and St. Clair, 2009). There are two conserved intracelluar SODs (Valentine et al., 2005): SOD1 (Cu/ZnSOD) and SOD2 (MnSOD). SOD1 is the major SOD that is widely distributed throughout the cytosol, mitochondrial intermembrane space and nucleus (Sturtz et al., 2001; Tsang et al., 2014). In contrast, SOD2 is localized exclusively in the mitochondrial matrix (Schieber and Chandel, 2014). SOD1 and SOD2 are critical to counter the superoxide production during mitochondrial respiration, providing a direct control of cellular ROS level as well as the first line of defense against oxidative damages. Increasing evidence also indicates that SOD1 acts as a regulatory protein for diverse cellular processes such as signaling and respiration (Che et al., 2016), and plays an important role in human diseases such as cancer (Glasauer et al., 2014; Papa et al., 2014).
Cells derive biochemical energy from nutrients in the form of ATP to fuel biosynthesis and growth, a process called energy metabolism. Eukaryotes have two main routes for energy production, glycolysis in the cytosol and oxidative phosphorylation (OXYPHOS) in the mitochondria. Environmental nutrients dictate which energetic pathway is used for ATP production. The budding yeast Saccharomyces cerevisiae predominantly uses glycolysis when glucose is available even in the presence of oxygen, a phenomenon called ‘aerobic glycolysis’ or the ‘Crabtree effect’. Yeast switch to OXYPHOS when only non-fermentable carbon source (e.g. glycerol) is available (Broach, 2012). Like yeast, cancer cells also predominantly use glycolysis for energy metabolism, a phenomenon called the ‘Warburg effect’ (Cairns et al., 2011; Hamanaka and Chandel, 2011; Koppenol et al., 2011; Warburg, 1956). When carbohydrates are limited or a high fat diet is provided, animal cells such as hepatocytes produce energy through β-oxidation and mitochondrial OXYPHOS by utilizing free fatty acids derived from food, intracellular lipid storage or the adipose tissue.
As the substrates for bioenergetic pathways, nutrients can directly impact ROS production. For instance, non-fermentable nutrients generate ATP through mitochondrial OXYPHOS, and as a result also superoxide. Nutrients are increasingly appreciated as mitogenic signals that control growth and metabolism (Dechant and Peter, 2008; Jorgensen et al., 2004; Zaman et al., 2008). Mechanistic target of rapamycin (mTOR) forms two distinct complexes, mTORC1 and mTORC2. mTORC1 is a nutrient sensor and a master regulator of cell growth and metabolism (Jewell and Guan; Kim et al., 2013; Laplante and Sabatini, 2012; Wullschleger et al., 2006). Here we show that mTORC1 regulates SOD1 activity in both yeast and human cells in response to nutrient availability. This regulation modulates cellular ROS levels to ensure adequate proliferation under nutrient-rich condition while minimize oxidative damages under nutrient stress. Our observations identify a conserved mechanism by which eukaryotic cells dynamically regulate redox homeostasis in response to changing nutritional environment.
Results
TORC1 interacts genetically with SOD1 and regulates SOD1 activity in yeast
Mutations in TORC1 pathway render differential sensitivity to a low, sub-inhibitory concentration of rapamycin in the presence of rich nutrient medium such as YPD or SC (Bertram et al., 2002; Chan et al., 2000). Deletion of a positive factor such as TOR1 exacerbates the reduction of TORC1 signaling, resulting a rapamycin hypersensitive phenotype compared with the wild type (WT) strain (Thomas et al., 2014). On the other hand, deletion of a negative factor such as GLN3, prevents activation of starvation response by rapamycin in the presence of rich nutrients, thereby conferring better growth or rapamycin resistance (Bertram et al., 2002; Chan et al., 2000). In a similar study, the sod1Δ mutation was shown to display the rapamycin resistant phenotype (Neklesa and Davis, 2008). Notably, the sod1Δ mutation confers a rapamycin resistant phenotype (Figure 1A). In fact, deletion of both SOD1 and its copper chaperone CCS1 (LYS7), an enzyme facilitate incorporate Cu2+ into the SOD1 catalytic site (Culotta et al., 1997; Wong et al., 2000), renders rapamycin resistance (Figure 1A). The rapamycin-resistant phenotype of sod1Δ is suppressed by a plasmid-borne SOD1 (Figure 1B). Consistently, deletion of SOD2 does not affect rapamycin sensitivity (Figure 1A). Thus, SOD1 genetically interacts with TORC1, suggesting that it plays a role in TORC1 signaling. Indeed, rapamycin rapidly activates SOD1’s enzymatic activity in a CCS1- and TORC1-dependent manner (Figure S1A, 1C and 1D). SOD1 activation is much less by rapamycin in yeast cells expressing a rapamycin-resistant TOR1 (TOR1-RR) allele (Figure 1E)(Zheng et al., 1995). Thus, the rapamycin effect on SOD1 activity is due to TORC1 inhibition. Furthermore, genetic inactivation of TORC1 in the tor1Δtor2-dg strain through TOR1 deletion and degron-mediated thermal inactivation of TOR2 (Dohmen et al., 1994), also enhances SOD1 activity (Figure 1F). The rapamycin resistant phenotype of sod1Δ was previously attributed to oxidative modification of TOR1’s FKBP12-rapamycin binding (Neklesa and Davis, 2008). However, our results revealed a functional relationship between TORC1 and SOD1.
Figure 1. TORC1 negatively regulates SOD1 activity in yeast.

(A) Deletion of SOD1 renders the rapamycin resistant phenotype in yeast. Wild type (WT), sod1Δ, sod2Δ and ccs1Δ cells were serially diluted (10-fold), spotted onto YPD plates without or with a sub-inhibitory concentration of rapamycin (1 nM), and incubated at 30°C for 2 (− rapamycin) to 5 (+ rapamycin) days.
(B) Plasmid-borne SOD1 suppresses the rapamycin resistant phenotype of sod1Δ strain. WT, sod1Δ and sod1Δ [SOD1-MYC9] yeast strains were assayed for rapamycin sensitivity as above.
(C) Rapamycin rapidly activates the activity of SOD1, but not SOD2. Yeast cells were treated with rapamycin for different times and the activity of SOD1 and SOD2 were examined by the in-gel SOD activity assay. Right panel shows quantification of the results (expressed as relative activity to time zero; mean ± S.D.; n = 3; * p < 0.05, Student’s t-test). sod1Δ strain was used as a negative control.
(D) Rapamycin activation of SOD1 is dependent on its copper chaperone CCS1. WT, sod1Δ and ccs1Δ cells were treated with rapamycin for different times. Right panel shows quantification of the results (mean ± S.D.; n = 3; * p < 0.05, Student’s t-test).
(E) SOD1 activation by rapamycin is a result of TORC1 inhibition. Yeast cells expressing TOR1 or TOR1-RR were treated with rapamycin and assayed for the SOD activity. Quantification of SOD1 activity is shown in the lower panel (mean ± S.D.; n = 3; * p < 0.05, Student’s t-test).
(F) Genetic inactivation of TORC1 stimulates SOD1 activity. WT and tor1Δtor2-dg strains were shifted to the restrictive temperature (37°C) for different times, and SOD1 activity was determined by the in-gel assay. Lower panel shows quantification of SOD1 activity (mean ± S.D.; n = 3; * p < 0.05, Student’s t-test).
See also Figures S1.
SOD1 is important for nutrients to regulate ROS in yeast
Because TORC1 is a major mediator of nutrient signaling, we asked if nutrients regulate SOD1 activity. Indeed, starvation results in rapid activation of SOD1 (Figure 2A). Changing from glucose to a non-fermentable carbon source (e.g. glycerol), a condition known to turn on mitochondrial OXYPHOS, significantly enhances SOD1 activity (Figure 2B). As expected, these conditions inhibit TORC1 signaling as judged by decreased phosphorylation of MAF1 (Figures S1B and S1C), a known TORC1 substrate (Wei et al., 2009; Wei and Zheng, 2010). We next investigated how different nutrient conditions affect cellular ROS and the role of SOD1. In wild type (WT) yeast cells in glucose culture, ROS level is virtually undetectable, which increases moderately following change from glucose to glycerol medium (Figures 2C and 2D). In sod1Δ cells in glucose medium, ROS level is also relatively low though the superoxide level is noticeable (Figures 2C and 2D). After the glucose-to-glycerol switch, however, both superoxide and peroxide (DHR staining) are drastically elevated in sod1Δ cells (Figures 2C and 2D). Consistently, changing from glucose to glycerol causes an increase in γ-H2AX level in WT cells and this increase is exacerbated by the sod1Δ mutation (Figure 2E). Similar results were obtained with glucose starvation (Figures S1D, S1E and S1F). These results show that SOD1 is important for moderating ROS level and preventing oxidative DNA damage in response to changing nutrient conditions.
Figure 2. SOD1-dependent regulation of cellular ROS by nutrients in yeast.

(A) Glucose starvation activates SOD1. WT yeast cells were starved from glucose (SC without glucose) for different times. SOD1 activity was determined by the in-gel assay. Right panel shows quantification of SOD1 activity (mean ± S.D.; n = 3; * p < 0.05, Student’s t-test).
(B) Changing nutrients from glucose to non-fermentable carbon source activates SOD1. WT yeast cell culture was changed from glucose to glycerol medium for different times. SOD1 activity was determined by the in-gel assay. Right panel shows quantification of SOD1 activity (mean ± S.D.; n = 3; * p < 0.05, Student’s t-test).
(C) SOD1 is important for preventing excessive superoxide under different nutrient conditions. WT and sod1Δ cell cultures were changed from glucose to glycerol medium for 3 hr. Cellular superoxide was analyzed by staining with dihydroethidium (DHE). Images were captured by fluorescence microscopy. Scale bar, 10 µm. Right panel, quantification of cells with different staining intensity (N > 100).
(D) SOD1 is important for preventing excessive ROS under different nutrient conditions. WT and sod1Δ cells cultured in glucose medium were changed to glycerol medium for 3 hr and analyzed for general ROS by staining with dihydrorhodamine (DHR). Images were captured by fluorescence microscopy. Scale bar, 10 µm. Right panel shows quantification of cells with different staining intensity (N > 100).
(E) SOD1 is important for preventing oxidative DNA damage under different nutrient conditions. Oxidative DNA damage was performed by immunofluorescence (IF) with γ-H2AX antibody under the same conditions as in Figure 2C. Scale bar, 10 µm. Right panels show quantification of positively stained cells (mean ± S.D., n = 3, * p < 0.05, Student’s t-test). See also Figure S1.
TORC1 phosphorylates SOD1 at S39 in a nutrient-dependent manner in yeast
Rapid activation of SOD1 by rapamycin suggests that TORC1 regulates SOD1 through a posttranslational mechanism. SOD1 protein from cells cultured in glucose can be resolved into two distinct electrophoretic forms (Figure 3A). The slow migrating form disappears and the fast migrating form becomes predominant following rapamycin treatment or change from glucose to glycerol medium (Figures 3A, 3C and 3D). Mass spectrometry analysis reveals that SOD1 is phosphorylated at multiple serine/threonine sites (Table S1). In particular, S39 is phosphorylated in a rapamycin-sensitive manner. SOD1 has a highly negatively charged surface but contains a positively charged tunnel, which helps guide the flow of negatively charged O2− substrates into the active site (Getzoff et al., 1983)(Figure 3B). Remarkably, S39 is located at the entry site of this tunnel. Phosphorylation generates negative charge at this position, which may impede the substrate flow into the catalytic site and hence reduce the catalytic activity. To determine the effect of S39 phosphorylation, we made the S39A mutation to mimic the dephosphorylated state, or the S39D/E mutation to mimic the phosphorylated state. Interestingly, on the SDS polyacrylamide gel, SOD1S39A co-migrates with the fast electrophoretic form, while SOD1S39D and SOD1S39E co-migrate with the slow electrophoretic form (Figures 3C and 3D). Moreover, the mobility of these mutants is no longer altered by rapamycin or nutrient change (Figures 3C and 3D). Thus, TORC1 regulates SOD1 phosphorylation in response to different nutrient conditions.
Figure 3. TORC1 interacts with SOD1 and phosphorylates SOD1 at S39 in yeast.

(A) Rapamycin induces electrophoretic shift of SOD1 protein. Yeast cells expressing SOD1-Myc9 were treated with 100 nM rapamycin for different times. The electrophoretic mobility of SOD1 was analyzed by immunoblot with a Myc-specific antibody.
(B) S39 is located at the entry of a positively charged tunnel (circled by a dotted line) that guides the inflow of negatively charged O2− substrates to the active site. Shown are 3D crystal structures of yeast SOD1 with electrostatic surface with S39 in unphosphorylated (top) or modeled phosphorylated state (bottom). Blue, electropositive charge; White, neural; Red, electronegative charge; Electrostatic potential, kcal mol−1 e−1.
(C) S39 is phosphorylated in a rapamycin-sensitive manner. Yeast cells expressing WT or mutant SOD1-Myc9 were treated with 100 nM rapamycin for 30 min. The electrophoretic mobility of SOD1-Myc9 was analyzed by immunoblot with a Myc-specific antibody.
(D) S39 phosphorylation is regulated by nutrients. Yeast cells expressing WT or S39 mutant SOD1-Myc9 proteins were shifted from glucose (D, dextrose) to glycerol medium (G) for 3 hrs. Electrophoretic mobility of SOD1-Myc9 was immunoblot with a Myc-specific antibody.
(E) TORC1 interacts with SOD1 in a nutrient-dependent manner. Yeast cells expressing SOD1-Myc9 were shifted from glucose (D, dextrose) to glucose or glycerol (G) medium for 3 hr. TORC1 was immunoprecipitated with a TOR1 specific antibody. TORC1 interaction with SOD1-Myc9 was analyzed by immunoblot.
(F) TORC1 phosphorylates SOD1 at S39. Immunoprecipitated TORC1 from cells cultured in glucose medium was incubated with bacterially produced recombinant GST-SOD1WT or GST-Sod1S39A in the presence of γ-[32P]-ATP. Phosphorylation of GST-SOD1 proteins was detected by autoradiography.
See also Figures S1 and S2; Table S1.
To further understand the relationship between TORC1 and SOD1, we investigated a possible interaction between TORC1 and SOD1. Indeed, TOR1 forms a complex with SOD1 as detected by co-immunoprecipitation (Figure 3E). Moreover, this TORC1-SOD1 interaction only occurs when yeast cells are cultured in glucose but not in glycerol (Figure 3E). To explore the possibility of SOD1 as a substrate for TORC1, we immunoprecipitated TORC1 using a TOR1-specific antibody and incubated it with recombinant GST-SOD1 fusion proteins in the presence of [32P]-ATP. TORC1 phosphorylates GST-SOD1 but not GST-SOD1S39A (Figure 3F), indicating that TORC1 specifically phosphorylates SOD1 at S39. Together, these data demonstrate that TORC1 interacts with SOD1 and phosphorylates S39 in a nutrient-dependent manner.
Reversible phosphorylation of SOD1 at S39 is important to control cellular ROS in response to changing nutrient conditions in yeast
To investigate the role of S39 phosphorylation, we measured the enzymatic activity of bacterially expressed GST-SOD1WT and GST-SOD1S39E proteins. The activity of GST-SOD1S39E is significantly lower than that of GST-SOD1WT (Figure S1G). In contrast, S39 mutations do not affect the interaction between SOD1 and CCS1 in the absence or presence of rapamycin (Figure S1H). These observations suggest that S39 phosphorylation directly regulates SOD1 activity. As seen earlier, SOD1WT is activated when cells are cultured in glycerol. In contrast, the activity of SOD1S39A and SOD1S39D/E remains constant regardless of the nutrient status (Figures 4A and 4B). Moreover, SOD1S39A activity is constitutively high, which is similar to that of activated SOD1WT in glycerol medium. On the other hand, SOD1S39D/E activity is constitutively low that is comparable to that of SOD1wt in glucose medium. Similar results were seen with rapamycin treatment (Figure S1I). These data indicate that SOD1 activity is modulated by nutrients through TOR-dependent S39 phosphorylation.
Figure 4. Nutrient regulation of SOD1 and cellular ROS is mediated by S39 phosphorylation in yeast.

(A) Nutrients regulate SOD1 activity through S39 phosphorylation. Yeast cells expressing WT and S39 mutant SOD1 were changed from glucose to glycerol medium for 3 hr. SOD1 activity and protein were measured by in gel SOD assay and immunoblot, respectively.
(B) Quantification of SOD1 activity in Figure 4A. Data represent mean ± S.D. from three independent experiments. * p < 0.05, Student’s t-test.
(C) S39 phosphorylation is important to control cellular superoxide in response to different nutrient conditions. Yeast cells expressing WT and mutant SOD1 cultured in glucose were changed to glycerol medium for 3 hrs. Cellular superoxide was analyzed by DHE staining. Scale bar, 10 µm.
(D) Quantification of results from Figure 4C (N > 100).
(E) Cells expressing WT and mutant SOD1 in glucose medium were changed from glucose to glycerol medium and incubated for 3 hr. Oxidative DNA damage was analyzed by IF with γ-H2AX antibody (green). Nuclei were visualized by DAPI (blue). D, dextrose; G, glycerol.
(F) Quantification of results from Figure 4E. Data represent mean ± S.D. from three independent experiments. * p < 0.05, Student’s t-test. Scale bar, 10 µm.
See also Figure S2.
We next investigated the role of SOD1 phosphorylation in redox regulation by measuring ROS in cells expressing WT or mutant SOD1. In glucose medium, O2− is undetectable as judged by DHE staining in SOD1WT and SOD1S39A cells, while at a low level in SOD1S39D/E cells (Figures 4C and 4D). The total ROS level as judged by DHR staining is undetectable in all four type of cells (Figure S2A). This small change in O2− in SOD1S39D and SOD1S39E cells does not appear to significantly impact the overall ROS level. In glycerol medium, however, there is an elevated level of both O2− and overall ROS in the SOD1wt cells, which is tempered in SOD1S39A cells but exacerbated in SOD1S39D/E cells (Figures 4C, 4D and S2A). In glucose medium, DNA oxidation as measured by 8-OxoG staining and DNA damage as measured by γ-H2AX staining are low in SOD1WT and SOD1S39D/E cells, and are even lower in SOD1S39A cells (Figures S2B, 4E and 4F). Under glycerol culture condition, there is a significant increase in DNA oxidation and damage in SOD1wt cells, which is tempered in SOD1S39A cells but exacerbated in SOD1S39D/E cells (Figures S2B, 4E and 4F). This increased DNA damage is attenuated by treatment with Tempol, a cell-permeable SOD mimetic (Figure S2C). Lipid oxidation as judged by malondialdehyde (MDA) is comparably low in glucose in both SOD1wt and SOD1S39A cells, and is similarly increased in glycerol medium (Figure S2D). In contrast, there is already a high level of lipid oxidation in SOD1S39E cells in glucose culture, which is further increased in glycerol, indicating that lipid oxidation appears to be much more sensitive to differential SOD1 regulation. Together, these observations demonstrate that reversible S39 phosphorylation is important for maintenance of redox homeostasis and prevention of oxidative damage under different nutrient conditions in yeast.
mTORC1 regulates SOD1 through T40 phosphorylation in human cells
Rapamycin treatment of HEK293, Hep3B, A549 and MCF7 cells, representing immortalized or cancer cells of human kidney, liver, lung and breast tissue origins, leads to rapid activation of SOD1 (Figure 5A), indicating that mTORC1 also negatively regulates SOD1 in mammalian cells. Unlike yeast SOD1, the endogenous SOD1 protein in Hep3B cells can only be resolved by 2D gel electrophoresis into distinct electrophoretic forms in a rapamycin- and phosphatase-sensitive manner (Figure 5B). Overlaying the 3D structures of yeast and human SOD1 reveals that T40 of human SOD1 is located at the same position as S39 of yeast SOD1 (Figure 5C). When expressed in Hep3B cells, human SOD1-GFP and SOD1-Flag also exhibit distinct electrophoretic forms in a rapamycin- and phosphatase-sensitive manner (Figure 5D and 5E). Similarly, glucose deprivation by itself also leads to increased SOD1 phosphorylation (Figure S3A and S3B). In vitro kinase assay shows that mTORC1 phosphorylates recombinant His6-SOD1, but not His6-SOD1T40A (Figure S3C). Substitution of T40 in human SOD1 to A or E leads to electrophoretic mobility mimicking the dephosphorylated and phosphorylated forms, respectively (Figure 5D and 5E). Moreover, the T40A mutation activates SOD1 activity while the T40E mutation inhibits SOD1 activity (Figures 5F and 5G). Moreover, the activity of SOD1T40A and SOD1T40E no longer changes in response to rapamycin treatment (Figure 5G). These results indicate that mTORC1 regulates SOD1 through T40 phosphorylation. Of note, S39/T40 is located on a flexible loop exposed to the surface (Figure 5C). The sequence downstream of the phosphorylation site, but not the one immediately surrounding the site is highly conserved. This suggests that this is a distinct substrate recognizing mechanism for mTORC1, which is potentially interesting to pursue in the near future.
Figure 5. mTORC1 regulates human SOD1 activity through T40 phosphorylation.

(A) Rapamycin activates endogenous SOD1 activity in human cells. HEK293T, Hep3B, A549 and MCF7 cells were treated with 10 nM rapamycin for different times. SOD1 activity and protein expression were measured by the in-gel assay and immunoblot, respectively. Lower panels show quantification of SOD1 activity relative to time 0. (Mean ± S.D., n = 3, *p < 0.05, Student’s t-test).
(B) mTORC1 regulates phosphorylation of the endogenous SOD1. Hep3B cells were treated with 10 nM rapamycin and the endogenous SOD1 protein was analyzed by 2D gel electrophoresis and immunoblot. The bottom panel shows SOD1 sample (0 min) treated with calf intestine phosphatase (CIP).
(C) 3D protein structure of yeast SOD1 (top), human SOD1 (middle) and merged image (bottom). Arrows indicate the overlapping position of S39 in SOD1 and T40 in SOD1.
(D, E) mTORC1 regulates phosphorylation of SOD1-GFP (D) and SOD1-Flag (E). WT and mutant SOD1-GFP or SOD1-Flag expressed in Hep3B cells treated with or without rapamycin were analyzed by 2D gel electrophoresis and immunoblot.
(F) Activity of SOD1WT-GFP, SOD1T40A-GFP and SOD1T40E -GFP expressed in Hep3B cells as determined by the in-gel SOD assay. Right panel shows quantification of the results (mean ± S.D.; n = 3, * p < 0.05, Student’s t-test).
(G) mTORC1 regulates SOD1 activity through T40 phosphorylation. Hep3B cells transiently expressing SOD1WT-GFP, SOD1 T40A-GFP or SOD1T40E -GFP were treated with or without 10 nM rapamycin for 1 hour. SOD1 activity was measured by in-gel hSOD assay. SOD1-GFP expression was determined by immunoblot. Right panel shows quantification of the results (mean ± S.D.; n = 3, * p < 0.05, Student’s t-test).
See also Figure S3.
Regulation of SOD1 is important for moderating oxidative stress and sustaining cancer cell survival under ischemic tumor microenvironment
Unicellular organisms such as yeast must obtain nutrients from the environment that tend to drastically fluctuate. In contrast, mammalian cells have steady access to nutrients through the circulation. However, ischemia is one condition that results in severe nutrient and oxygen deprivation due to restricted blood supply. It is a major cause for human diseases such as heart disease and stroke. Cancer cells can be severely starved from nutrients and oxygen in an ischemic tumor microenvironment due to poor tumor vascularization (Gorrini et al., 2013; Trachootham et al., 2009), making cancer an excellent model to study nutrient regulation of redox homeostasis in mammals. As reported previously (Qiao et al., 2016; Semenza, 2000), deprivation of nutrients and oxygen, an ischemic condition, leads to rapid inhibition of mTORC1 signaling as shown by the decreased phosphorylation of mTORC1 effectors S6K1 and S6, but activation of AMPK and HIF1α, two hypoxic markers (Figure 6A). Similarly to the yeast results, human SOD1 but not SOD2 becomes rapidly activated (Figure 6A). However, the activity of SOD1T40A and SOD1T40E remain unchanged (Figure 6B), indicating that SOD1 regulation by ischemia through reversible T40 phosphorylation.
Figure 6. mTORC1-dependent regulation of SOD1 is important for mitigating oxidative DNA damage in human cells in an ischemic environment.

(A) SOD1 is activated by an ischemic environment. Hep3B cells were subjected to an ischemic condition (1% oxygen and minus glucose) for different times. The activity of SOD1, SOD2 and mTORC1 signaling was followed by in-gel assay and immunoblot. Right panel shows quantification of SOD1 and SOD2 activity, and P-S6K1(T389)(mean ± S.D., N = 3, * p < 0.05, Student’s t-test). HIF1a and p-AMPK were used as ischemic markers.
(B) Ischemic condition activates SOD1 in a T40-dependent manner. Hep3B cells transiently expressing SOD1WT-GFP, SOD1T40A-GFP and SOD1T40E-GFP proteins were subjected to ischemia for 3 hours. SOD1-GFP activity and expression were determined by the in-gel assay and immunoblot, respectively. Bottom, quantification of SOD1 activity (mean ± S.D., N = 3, * p < 0.05, Student’s t-test).
(C) Regulation of SOD1 by mTORC1 is important for cancer cell survival in an ischemic environment. Hep3B cells stably expressing WT and mutant SOD1 under normal or ischemic conditions (−/+ N-acetyl cysteine, NAC) were assayed for cell death by Trypan blue staining after 24 hrs. Data represent mean ± S.D. (N = 3, * p < 0.05, Student’s t-test).
(D) Protective effects of WT and mutant SOD1 against oxidative DNA damage in an ischemic environment. Hep3B cells stably expressing SOD1WT-GFP, SOD1T40A-GFP and SOD1T40E-GFP proteins were subjected to ischemia for 3 hours and analyzed for oxidative DNA damage by immunofluorescence staining with an 8-OxoG antibody. Scale bar, 20 µm.
(E) Quantification of results from Figure 6D. Data represent mean ± S.D. (N = 3, * p < 0.05, Student’s t-test).
(F) Protective effects of WT and mutant SOD1 against DNA damage in an ischemic environment. Hep3B cells stably expressing SOD1WT-GFP, SOD1T40A-GFP and SOD1T40E-GFP proteins were subjected to ischemia for 3 hours and analyzed for DNA damage by immunofluorescence staining with a p-H2A.X(S139)-specific antibody. As a positive control for DNA damage, Hep3B cells were treated with different concentrations of H2O2 for 20 min. Scale bar, 20 µm.
(G) Quantification of results from Figure 6F. Data represent mean ± S.D. (N = 3, * p < 0.05, Student’s t-test).
See also Figures S4 and S5.
Ischemia, or deprivation of glucose or oxygen leads to elevated ROS in Hep3B cells expressing SOD1T40E-GFP, but to a much less extent in SOD1WT-GFP and SOD1T40A-GFP expressing cells (Figure S4A). Consistently, Hep3B cells expressing SOD1T40E-GFP, but not SOD1WT-GFP and SOD1T40A-GFP, are considerably more susceptible to ischemia-induced cell death, and DNA oxidation and DNA damage (Figures 6C, 6D, 6E, 6F and 6G). The ischemia-induced cell death in SOD1T40E-GFP expressing Hep3B cells is largely reversed by the ROS scavenger N-acetyl cysteine (NAC) (Figure 6C) or the SOD mimetic Tempol (Figure S4B, S4C and S4D). Tempol similarly tempers the elevated DNA oxidation and damage in SOD1T40E-GFP expressing cells under the ischemic condition (Figures S4D and S4E). Rapamycin also partially reverses ischemia-induced cell death and oxidative DNA damage (Figure S5A, S5B and S5C). Some chemotherapeutic agents such as cisplatin elicit superoxide production, which is recognized as an important anticancer mechanism (Miyajima et al., 1997). Interestingly, rapamycin or expression of SOD1T40A, but not SOD1T40E in Hep3B cells, attenuates the increase of superoxide caused by cisplatin (Figure S5D). These results indicate that the oxidative DNA damage and cell death under ischemia is caused by elevated ROS.
Tumor spheroids cultured in vitro exhibit characteristics of solid tumors in vivo, including elevated ROS and oxidative stress due to limited nutrients and oxygen inside tumor spheroids. To assess the role of SOD1 in tumor development, we performed tumor spheroid formation assay using Hep3B cells expressing WT or mutant SOD1. Compared with SOD1WT-GFP expressing cells, SOD1T40A-GFP expressing cells form tumor spheroid more efficiently (Figure 7A). In contrast, SOD1T40E-GFP expressing cells form tumor spheroid much less efficiently. Consistently, SOD1T40A-GFP-expressing tumor spheroids have lower ROS and cell death, while SOD1T40E-GFP tumor spheroids have much higher ROS and cell death (Figures 7B, 7C, 7D and 7E). Essentially the same results were obtained with tumor spheroids derived from the A549 non-small cell lung cancer (NSCLC) cells (Figure S6A, S6B, S6C, S6D and S6E). Consistently, rapamycin treatment reduces ROS level in A549 tumor spheroids and apoptosis (Figure S6F and S6G). Xenograft tumors derived from Hep3B cells expressing SOD1T40A-GFP grow faster than the SOD1WT and SOD1T40E-GFP tumors, and continue to grow after the SOD1WT-GFP and SOD1T40E-GFP tumors have plateaued in growth (Figures 7F, 7G and 7H). In contrast, the growth of SOD1T40E-GFP tumors is attenuated. Consistently, the SOD1T40E-GFP tumors exhibit elevated oxidative DNA damage as shown by positive γH2AX staining (Figure 7I). Together, these results indicate that regulation of T40 phosphorylation is important for SOD1 to moderate cellular ROS and maintain cancer survival in an ischemic environment.
Figure 7. mTORC1-dependent regulation of SOD1 is important for cancer cell survival and tumor growth in an ischemic environment.

(A) Activation of SOD1 enhances tumor spheroid formation. Hep3B cells stably expressing WT and mutant SOD1-GFP proteins were assayed for spheroid formation in suspension culture. Three typical tumor spheroid morphologies were quantified: A, complete tumor spheroid; B, partially mature tumor spheroid; C, immature tumor spheroid. Data represent mean ± S.D. of three independent experiments (N = 18 in each experiment, * p < 0.05).
(B) Activation of SOD1 reduces ROS levels in tumor spheroids. Tumor spheroids derived from Hep3B cells stably expressing different SOD1-GFP proteins (green) were stained for DHE (red). Shown are representative confocal microscopic images. Boxed area is enlarged to show more detailed DHE staining. Scale bars, 100 µm.
(C) Quantification of the DHE staining results. Data represent mean ± S.D. of three independent experiments (two-tailed t-test).
(D) Activation of SOD1 reduces cancer cell death in tumor spheroids. Tumor spheroids derived from Hep3B cells stably expressing different SOD1-GFP proteins (green) were analyzed for cell death by propidium iodine (PI) staining (red). Shown are representative confocal microscopic images. Scale bars, 100 µm.
(E) Quantification of the PI staining results. Data represent mean ± S.D. of three independent experiments (two-tailed t-test).
(F) Activation of SOD1 promotes xenograft tumor growth. Hep3B cells stably expressing different SOD1-GFP proteins were assayed for xenograft tumor growth in athymic nude mice.
(G, H) Shown are tumor images and weights at the end of the study (mean ± S.D, N = 6, *** p < 0.05, Student’s t-test).
(I) Shown are representative γH2AX immunofluorescence staining (red) images in Hep3B xenograft tumor sections. Tumor cell nuclei were stained by DAPI (blue). Scale bars, 20 µm. See also Figure S6.
Discussion
Nutrients supply building blocks for biomass generation and for producing energy to fuel biochemical reactions. They also serve as chemical signals that dictate cell growth and metabolism. Cells have elaborate mechanisms sensing different types of nutrients, allowing them to adjust and reprogram biochemical pathways to utilize them accordingly. When glucose is available, yeast cells prefer the use of glycolysis to generate energy. Mitochondrial OXYPHOS is up-regulated when non-fermentable carbon source is available instead of glucose. Such a nutrient-dependent energetic switch is preserved in mammals. For example, glucose is preferentially utilized for rapid energy production in muscle and cancer cells, while fatty acid is used through mitochondrial respiration. Because mitochondrial respiration is a major source of cellular ROS, ROS production can change significantly with nutrients. Indeed, ROS is rapidly elevated when cultured mammalian cells, such as hepatocytes, are exposed to starvation or free fatty acids (Scherz-Shouval and Elazar, 2009; Soardo et al., 2011). Likewise, when yeast cells are subjected to starvation or non-fermentable carbon source, ROS production is increased (Figure 2).
ROS is a double-edged sword: it promotes growth and metabolism yet excessive ROS causes cellular damage and even cell death (D'Autreaux and Toledano, 2007; Finkel, 2011; Reczek and Chandel, 2015). Because different nutrient conditions can have enormous impact on ROS production, ROS must be tightly regulated accordingly to support optimal proliferation yet provide adequate protection against oxidation. Reversible phosphorylation of SOD1 by mTORC1 enables nutrient signaling to directly control the level of superoxide, the primary source of cellular ROS, providing a highly effective and dynamic response to changing nutrient conditions. It is interesting to note that SOD1 has also been shown to repress respiration and promote glycolysis in the presence of glucose and oxygen (Reddi and Culotta, 2013). Previous studies in yeast also revealed that TORC1 controls chronological lifespan, which is dependent on mitochondrial respiration and ROS production (Pan et al., 2011). Thus, SOD1 appears to be a key component of nutrient signaling that modulates energy metabolism, redox homeostasis and cellular aging.
ROS has been linked to a plethora of human diseases including aging, cancer, chronic inflammation, diabetes, ischemia/reperfusion injury, neurological disorders (e.g. amyotrophic lateral sclerosis, Parkinson’s disease) and traumatic brain injury (Brieger et al., 2012). mTORC1 regulation of SOD1 has implications in these human diseases. For example, cancer cells in the interior of solid tumors are often deprived of nutrients and oxygen due to poor vascularization. Our results show that activation of SOD1 promotes cancer cell survival under this ischemic tumor environment. While SOD1T40A, the more active mutant promotes the formation of tumor spheroids and growth of xenograft tumors, SOD1T40E, the less active mutant has the opposite effect. Consistently, dietary restriction in Sod1−/− mice was shown to significantly reduce the burden and severity of neoplastic lesions (Zhang et al., 2013), and targeting SOD1 has potent anticancer activity against KRAS transformed non-small cell lung cancer (NSCLC) in an experimental mouse model (Glasauer et al., 2014). These observations provide insight into the role of SOD1 in tumorigenesis, and into the mechanism of therapeutic targeting SOD1. Human cancer clinical trials have been carried out with combination of cisplatin and everolimus, a rapamycin analog, but met with little success (Jovanović et al., 2017). The opposing role of cisplatin and everolimus on cellular ROS may be antagonistic to their therapeutic benefit. Further investigation of SOD1 in nutrient signaling in the context of ROS-related diseases should shed light on the underlying pathobiology and therapeutic outcomes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for reagents and resources as well as further information of this study should be directed to and will be fulfilled by the Lead Contact, X.F. Steven Zheng (zhengst@cinj.rutgers.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Xenograft tumors were generated as described previously (Li et al., 2016; Wu et al., 2015; Zhang et al., 2017). Briefly, four-week old BALB/c female nude mice were ordered from the Beijing Vital River Laboratory Animal Technology (11400700232586) and were used to generate xenograft tumors. Animal care and handling were performed according to the Sun Yat-Sen University Cancer Center guidelines. The animal study was approved by the Animal Ethics Committee of Sun Yat-Sen University Cancer Center.
Human Cell Lines
HEK293T, Hep3B and MCF7 cells were cultured at 37°C in DMEM containing 10% fetal bovine serum (FBS) and antibiotics in a humidified incubator with 5% CO2. A549 cells were cultured in RPMI containing 10% fetal bovine serum (FBS) and antibiotics. HEK293FT cells were cultured in DMEM medium containing 10% FBS supplemented with 0.1 mM MEM Non-Essential Amino Acids, 1 mM sodium pyruvate and 2 mM L-glutamine as described in the manufacturer’s user guide (Thermo Fisher Scientific).
Yeast Strains
Yeast strains used in this study are derived from FM391/S288C (MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0) or W303 (MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100) as listed in the Key Resources Table. Yeast strains were cultured in yeast extracts peptone-dextrose (YPD) or synthetic complete medium (SC) at 30 °C. For metabolic switch experiments, yeast strains were grown in SC or YP medium containing 2% dextrose or 2% glycerol. For glucose starvation, cells were incubated in YP or SC medium without the addition of dextrose.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-histone H2A | Abcam | Cat#ab13923; RRID: AB_300750 |
| Rabbit anti-Phospho-p70 S6 Kinase (Thr389) (108D2) | Cell Signaling Technology | Cat#9234S; RRID: AB_2269803 |
| Rabbit anti-Phospho-S6 Ribosomal Protein (Ser235/236) (91B2) | Cell Signaling Technology | Cat#4857S; RRID: AB_2181035 |
| Rabbit anti-4E-BP1 | Cell Signaling Technology | Cat#9452; RRID: AB_331692 |
| Rabbit anti-HIF-1α (D2U3T) | Cell Signaling Technology | Cat#14179S; RRID: AB_2622225 |
| Rabbit anti-Phospho-H2AX (Ser139) (20E3) | Cell Signaling Technology | Cat#9718; RRID: AB_2118009 |
| Rabbit anti-Phospho-AMPKα (Thr172) (40H9) | Cell Signaling Technology | Cat#2535; RRID: AB_331250 |
| Rabbit anti-DYKDDDDK Tag Antibody | Cell Signaling Technology | Cat#2368; RRID: AB_2217020 |
| Rabbit anti-β-Tubulin (9F3) | Cell Signaling Technology | Cat#2128S; RRID: AB_823664 |
| Rabbit anti-β-Actin (13E5) | Cell Signaling Technology | Cat#4970S; RRID: AB_2223172 |
| Horse anti-mouse horseradish peroxidase-conjugated | Cell Signaling Technology | Cat#7076; RRID: AB_330924 |
| Goat anti-rabbit horseradish peroxidase-conjugated | Cell Signaling Technology | Cat#7074; RRID: AB_2099233 |
| Mouse anti-GST | Cell Signaling Technology | Cat#2624; RRID: AB_2189875 |
| Mouse anti-Myc (9E10) | Harlan Laboratories | Cat#200613 |
| Mouse anti-8-Hydroxydeoxyguanosine | Millipore | Cat#MAB3560; RRID: AB_94925 |
| Rabbit anti-phospho histone H2A (Ser129) | Millipore | Cat#07-745-I; RRID: AB_492642 |
| Rabbit anti-SOD2 | Millipore | Cat#06-984; RRID: AB_310325 |
| Mouse anti-Penta-His | Qiagen | Cat#34660; RRID: AB_2619735 |
| Rabbit anti-SOD1 | Santa Cruz | Cat#sc-17767; RRID: AB_628301 |
| Rabbit anti-goat horseradish peroxidase-conjugated | Thermo-Fisher | Cat#A16142; RRID: AB_2534813 |
| Goat anti-mouse Alexa Fluor 594-conjugated | Thermo-Fisher | Cat#A11005; RRID: AB_141372 |
| Goat anti-mouse Alexa Fluor 488-conjugated | Thermo-Fisher | Cat#A11001; RRID: AB_2534069 |
| Goat anti-rabbit Alexa Fluor 594-conjugated | Thermo-Fisher | Cat#R37117; RRID: AB_2556545 |
| Goat anti-rabbit Alexa Fluor 546-conjugated | Thermo-Fisher | Cat#A11010; RRID: AB_2534077 |
| Rabbit anti-TAP tag | Thermo-Fisher | Cat#CAB1001; RRID: AB_10709700 |
| Bacterial and Virus Strains | ||
| E. coli DH5α | Transgen | Cat#CD201 |
| E. coli BL21 (DE3) | Transgen | Cat#CD601 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rapamycin | LC Laboratories | Cat#NC9362949 |
| Dihydrorhodamine 123 | Thermo-Fisher | Cat#D632 |
| Dihydroethidium | Thermo-Fisher | Cat#D11347 |
| TEMPOL | Sigma-Aldrich | Cat#176141 |
| Nitroblue tetrazolium | Sigma-Aldrich | Cat#N6876 |
| Anti-FLAG M2 affinity gel | Sigma-Aldrich | Cat#F2426 |
| Glucose-free DMEM | Thermo-Fisher | Cat#11966025 |
| Lipofectamine™ 3000 | Thermo-Fisher | Cat#L3000015 |
| Glutathione Agarose | Thermo-Fisher | Cat#16100 |
| Critical Commercial Assays | ||
| Lipid Peroxidation (MDA) Assay Kit | Abcam | Cat#ab118970 |
| Universal Kinase Assay Kit | Abcam | Cat#ab138879 |
| QIAexpress Ni-NTA Fast start Kit | Qiagen | Cat#30600 |
| Deposited Data | ||
| Original data | Mendeley data | http://dx.doi.org/10.17632/rvbvczzd5d.1 |
| Experimental Models: Cell Lines | ||
| Human: Hep3B | ATCC | Cat#HB-8064 |
| Human: Hep3B (SOD1-GFPWT) | This study | N/A |
| Human: Hep3B (SOD1-GFPT40A) | This study | N/A |
| Human: Hep3B (SOD1-GFPT40E) | This study | N/A |
| Human: A549 | ATCC | Cat#CCL-185 |
| Human: A549 (SOD1-GFPWT) | This study | N/A |
| Human: A549 (SOD1-GFPT40A) | This study | N/A |
| Human: A549 (SOD1-GFPT40E) | This study | N/A |
| Human: HEK293FT | Thermo-Fisher | Cat#R70007 |
| Human: HEK293T | ATCC | Cat#CRL-3216 |
| Human: MCF7 | ATCC | Cat#HTB-22 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BALB/c female nude mice | Beijing Vital River Laboratory Animal Technology | Stock#: 11400700232586 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 | Tsang et al., 2014 | FM391/S288C |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 tor1 Δ::KanMX pYDF80-TOR1S1792I | Li et al., 2006 | SZy 997 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX pRS415 | This study | SZy 1050 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX pRS415(SOD1WT-MYC9) | This study | SZy 1051 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX pRS415(SOD1S39A-MYC9) | This study | SZy 2542 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX pRS415(SOD1S39D-MYC9) | This study | SZy 2543 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX pRS415(SOD1S39E-MYC9) | This study | SZy 2544 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX | Dharmacon | 16913 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod2Δ::KanMX | Dharmacon | 16605 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 ccs1Δ::KanMX | Dharmacon | 10614 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 pRS415 | This study | SZy 2550 |
| S. cerevisiae MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100 tor1 Δ::KanMX tor2-dg | This study | SZy 2551 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 SOD1-TAP::HIS3 | This study | SZy 2552 |
| S. cerevisiae MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100 | Tsang et al., 2014 | W303 |
| S. cerevisiae MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100 MAF1-MYC9::TRP1 | This study | SZy 1701 |
| S. cerevisiae MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100 MAF1-MYC9::TRP1 pRS426(SOD1-MYC9) | This study | SZy 3000 |
| S. cerevisiae MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100 MAF1-MYC9::TRP1 pRS426(SOD1S39A-MYC9) | This study | SZy 3001 |
| S. cerevisiae MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100 MAF1-MYC9::TRP1 pRS426(SOD1S39E-MYC9) | This study | SZy 3002 |
| S. cerevisiae MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100 MAF1-MYC9::TRP1 pRS426(SOD1S39D-MYC9) | This study | SZy 3003 |
| S. cerevisiae MATa ura3-1 leu2-3,-112 his3-11,-15 trp1-1 ade2-1 can1-100 MAF1-MYC9::TRP1 pRS426 | This study | SZy 3004 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX CCS1-TAP::His1 pRS415(SOD1WT-MYC9) | This study | SZy3005 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX CCS1-TAP::His1 pRS415(SOD1S39A-MYC9) | This study | SZy3006 |
| S. cerevisiae MATa hisΔ1 leu2Δ0 met15Δ0 ura3Δ0 sod1Δ::KanMX CCS1-TAP::His1 pRS415(SOD1S39E-MYC9) | This study | SZy3007 |
| Oligonucleotides | ||
| Primers for S39A mutagenesis: Forward: CGAGATCGCTGGTAACGCTCCTAACGCAGAACGTG Reverse: CACGTTCTGCGTTAGGAGCGTTACCAGCGATCTGC | This study | N/A |
| Primers for S39D mutagenesis: Forward: TTACGAGATCGCTGGTAACGATCCTAACGCAGAACGTGG Reverse: CCACGTTCTGCGTTAGGATCGTTACCAGCGATCTCGTAA | This study | N/A |
| Primers for S39E mutagenesis: Forward: CTTACGAGATCGCTGGTAACGAGCCTAACGCAGAACGTGGGTT Reverse: AACCCACGTTCTGCGTTAGGCTCGTTACCAGCGATCTCGTAAG | This study | N/A |
| Primers for GST-SOD1 construct: Forward: CCGGAATTCATGGTTCAAGCAGTCGCAGT Reverse: CCGCTCGAGGTTGGTTAGACCAATGACACCAC | This study | N/A |
| Primers for T40A mutagenesis: Forward: TCCATGCAGGCCTTCAGCCAGTCCTTTAATGCTTC-3 Reverse: GAAGCATTAAAGGACTGGCTGAAGGCCTGCATGGA | This study | N/A |
| Primers for T40E mutagenesis: Forward: TGGAATCCATGCAGGCCTTCCTCCAGTCCTTTAATGCTTCCC C Reverse: GGGGAAGCATTAAAGGACTGGAGGAAGGCCTGCATGGATTCCA | This study | N/A |
| Primers for gap repair: Forward: CTCTTCGCTATTACGCCAGC Reverse: ATTAATGCAGCTGGCACGAC | This study | N/A |
| Primers for construction of CCS1-TAP strains Forward: TGA ACC ACC CAG AAA ACG AG Reverse: AGG ATT GGA AAC CGG CTT TG | This study | N/A |
| Recombinant DNA | ||
| FLAG-mTORWT | This study | pSZ92 |
| FLAG-mTORD2338E (Kinase Dead) | This study | pSZ96 |
| HIS6-hSOD1WT | This study | pSZ1366 |
| HIS6-hSOD1T40A | This study | pSZ1366-b |
| GST-4EBP1 | This study | pSZ1130 |
| hSOD1WT-GFP | Stevens et al., 2010 | Addgene plasmid, Cat#26407 |
| hSOD1T40A-GFP | This study | pSZ1458 |
| hSOD1T40E-GFP | This study | pSZ1459 |
| hSOD1WT-FLAG | Somwar et al., 2011 | N/A |
| hSOD1T40A-FLAG | This study | pSZ1460 |
| hSOD1T40E-FLAG | This study | pSZ1461 |
| SOD1WT-MYC9 | This study | pSZ374 |
| SOD1S39A-MYC9 | This study | pSZ1398a |
| SOD1S39D-MYC9 | This study | pSZ1398b |
| SOD1S39E-MYC9 | This study | pSZ1398c |
| GST-SOD1WT | This study | pSZ1410 |
| GST-SOD1S39A | This study | pSZ1410b |
| Software and Algorithms | ||
| Protein Data Bank | Hart et al., 1999 | 2JCW |
| Protein Data Bank | Banci et al., 1999 | 1DSW |
| Other | ||
METHOD DETAILS
Plasmids, Strains and Cell Lines
The yeast SOD1-Myc9 plasmids were described previously (Tsang et al., 2014). The yeast SOD1S39A/D/E and human SOD1T40A/E mutants were generated by PCR-directed mutagenesis using the QuikChange II Site-Directed Mutagenesis Kit from Agilent Technologies. GST-SOD1WT, GST-SOD1S39A and GST-SOD1S39E plasmids were constructed by PCR cloning of SOD1WT, SOD1S39A or SOD1S39E ORF into pGEX-4T-1 plasmid using the EcoRI and XhoI restriction sites. TOR1-RR and tor1Δtor2dg expressing plasmids were described before (Li et al., 2006). The human SOD1-GFP expressing plasmid was obtained from Addgene (Plasmid #26407). Overexpression of yeast SOD1WT and SOD1S39 mutants was generated by gap repair in SZy1701 strain. Briefly, pRS415-SOD1-MYC9 was used as the template for PCR to synthesize a fragment containing SOD1-MYC9 with the primers. Then, the plasmid pRS426 was digested with BamHI, HindIII and SalI. The PCR fragment and the cut pRS426 were co-transformed into the yeast strains SZy1701 in Ura− and Trp− SC selection medium. To construct yeast strains endogenously expressing both CCS1-TAP and SOD1WT, SOD1S39A or SOD1S39E, a PCR-fragment containing the 3’end of the CCS1 ORF and TAP tagged sequence amplified from the TAP-tag strain collection (Thermo Fisher Scientific) was transformed into SZy3000, SZy3001 and SZy3002, respectively, using SC-Leu-His as selection medium. The human SOD1-Flag expressing plasmid was a generous gift from Dr. Harold E. Varmus and constructed in pCMV6-Entry vector as described (Somwar et al., 2011). All plasmid constructs were verified by sequencing and showed proper expression.
All mutant yeast strains were generated by standard yeast transformation (Tsang et al., 2014). The CCS1-TAP strains were constructed using the following primers: CCS1-inΔF(587): 5’-TGA ACC ACC CAG AAA ACG AG-3’; CCS1ΔR: 5’-AGG ATT GGA AAC CGG CTT TG-3’. Transfection of Hep3B, A549 and HEK293FT cells were performed using Lipofectamine® 3000 Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. For establishment of stable cell lines, Hep3B and A549 cells were transfected with WT and mutant SOD1-GFP plasmids or the empty vector using Lipofectamine® 3000 Transfection Reagent (Thermo Fisher Scientific). After 48 h of transfection, Geneticin (G418, Invitrogen) was added to the culture medium at a concentration of 1 mg/ml. After 2 weeks of selection, clones were pooled and sorted by fluorescence-activated cell sorting in order to control for ectopic protein expression variations during stable cell selection.
SOD Activity Assay
Superoxide dismutase activity was performed as described previously (Tsang et al., 2014) with minor modification. Briefly, yeast cells were washed with phosphate buffer (PB) (0.05 M KH2PO4 and K2HPO4, pH 7.8), and lysed with glass beads by vortexing in PB supplemented with 0.1% Triton X-100, and protease and phosphatase inhibitor cocktails (Roche). 1–10 µg proteins were separated in 12% native PAGE gel for SOD1 and SOD2 activities, respectively. Mammalian cells were washed three times in phosphate-buffered saline (PBS) (KCl 2.7 mM, KH2PO4, 1.5 mM, NaHPO4 8 mM and NaCl 136.9 mM, pH 7.0), lysed in 50 mM phosphatase buffer (pH 7.8) containing protease and phosphatase inhibitor cocktail (Roche) with a Bioruptor (Diagenode) (30 seconds on and 30 seconds off at midpower) for 10 min. Forty to eighty µg proteins were separated in 12% native PAGE gel. Native PAGE gels were stained with 2.43 mM nitro blue tetrazolium chloride (Sigma), 0.14 M riboflavin-5’-phosphate (Sigma) and 28 mM TEMED (Bio-Rad) for 20 min at room temperature in darkness. To visualize SOD1 and SOD2 activities, gels were rinsed with water twice and placed on a light box for 60 min. SOD1 and SOD2 bands were identified either by sodium cyanide treatment in the gel staining step (Weydert and Cullen, 2010), by genetic deletion (yeast) or siRNA mediated knockdown (mammalian cells).
Indirect Immunofluorescence Microscopy
Yeast indirect immunofluorescence studies were performed as described (Tsang and Zheng, 2009). Briefly, after treatment, cells were resuspended in phosphate buffer (40 mM KPO4, 0.5 mM MgCl2, pH 6.5) and fixed with 3.7% formaldehyde for 1 h. Cell wall was then digested with 0.25 mg/ml zymolyase in sobitol buffer (40 mM KPO4, 0.5 mM MgCl2, 1.2 M sorbitol, pH 6.5) until spheroplasts were formed. After dehydration with methanol and acetone treatment, spheroplasts were blocked with 1% bovine serum albumin (BSA) in phosphate buffered saline (PBS) for 15 min, followed by incubation with primary antibodies overnight at 4 °C. Anti-γH2AX was used at a dilution of 1:500. The antibody-antigen complexes were detected with Alexa Fluor 488- or Alexa Fluor 594-congulated secondary antibody (1:200 dilution). DNA was stained for 15 min with 50 ng/ml 4’,6-diamidino-2-phenylindole (DAPI) in antifade mounting medium (Vector Laboratories). Fluorescent images were captured with an Olympus fluorescence microscope equipped with a digital camera. Texas Red filter (Olympus U-N41004), FITC/EGFP/BODIPY filter (Olympus U-N41001) and DAPI/Hoechst/AMCA filter (Olympus U-N31000) were used for detection of the red, green fluorescence signal of SOD1 and the blue fluorescent signal of nucleus, 28 respectively. DIC was used to capture the cell morphology. For 8-OxoG staining, cells were treated with 1 mg/ml RNase A for 30 min at 37 °C after fixation. Cells were then incubated in 3 N HCl at room temperature for 30 min, and in blocking buffer (4% BSA, 0.5% Triton X-100 in PBS) for 1 hour at room temperature. Cells were incubated with primary antibody against 8-Oxoguanine (8-OxoG) (mouse monoclonal IgG, MAB 3560, clone 483.15, 1:200 dilution, Millipore) at 4 °C overnight, followed by Alexa Fluor 594-conjugated anti mouse IgG. DNA was counterstained with 50 ng/ml DAPI. Quantitative analysis was determined by counting the percentage of positive staining cells. A minimum of 100 cells were counted per sample.
Yeast Cell Extracts and Western Blot Analysis
Disruption of cells was carried out by glass beads with vortexing in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, plus protease and phosphatase inhibitor cocktails, Roche). Protein samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and then transferred to 0.45 µm PVDF membranes. SOD1-Myc9- and TAP-tagged proteins were detected by anti-Myc (9E10, 1:10,000 dilution) and anti-TAP (1:1,000 dilution) antibodies, respectively. After washing with TBST, the membrane was developed using the enhanced chemiluminescent (ECL) detection system.
TORC1 Immunoprecipitation
For Tor1 immunoprecipitation (IP), yeast cells with/without SOD1-Myc9 expression were cultured to early log phase, and then incubated in glucose- or glycerol-containing medium for 3 hours. Cells were washed twice with water and resuspended in the IP buffer (50 mM Tris-HCl, pH 7.5, 0.5% Triton X-100, 150 mM NaCl, 5% glycerol, 2 mM PMSF, protease and phosphatase inhibitor cocktails). After cell lysis with glass beads, lysates were collected by centrifugation at 13,000 rpm and 3000 µg total protein in 0.5 ml was incubated with anti-Tor1 antibody overnight at 4°C with rotation. Protein G sepharose beads were then added to the antibody conjugated complex and incubated at 4 °C with rotation. After 2 hours, beads were washed three times with washing buffer A (50 mM Tris-HCl, pH 7.5, 0.5% Triton X-100, 150 mM NaCl, 5% glycerol, 2 mM PMSF), once with washing buffer B (50 mM Tris-HCl, pH 7.5, 0.5% Triton X-100, 500 mM NaCl, 5% glycerol, 2 mM PMSF), and twice with washing buffer C (50 mM Tris-HCl, pH 7.5, 150 mM NaCl). The bound materials were eluted from the beads by boiling in SDS protein sample buffer. Tor1 and SOD1-Myc9 were analyzed by Western blot with anti-Tor1 (1:2,000) and 9E10 29 (1:10,000) antibodies, respectively. For CCS1 IP, protein G sepharose beads and normal mouse IgG were used for pull-down of CCS1-TAP. CCS1-TAP and SOD1-Myc9 were analyzed by Western blot with anti-TAP (1:1,000) and 9E10 (1:5,000) antibodies, respectively.
Structural analysis of yeast and human SOD1 proteins
Crystal structure of yeast SOD1 (Protein Data Bank entry 2JCW (Hart et al., 1999)) and human SOD1 (Protein Data Bank entry 1DSW (Banci et al., 1999)) were analyzed using images generated by Jmol software.
Measurement of yeast TORC1 signaling activity
Yeast TORC1 signaling activity was determined as described (Wei et al., 2009; Wei and Zheng, 2009). Briefly, cells were cultured in SC medium to early log phase. Cells were collected and lysed in lysis buffer (50mM Tris–HCl pH 7.5, 150mM NaCl, 0.5mM EDTA, 0.5% NP-40, 2mM PMSF, Roche protease complete inhibitor cocktail and phosSTOP tablet) with glass beads with vortexing at 4 °C. Crude lysates were cleared by centrifugation at 14,000 rpm and protein samples were separated by SDS-PAGE. Maf1-Myc9 mobility shifts were detected by anti-Myc (9E10, 1:3000 dilution) antibody and used to monitor TORC1 activity (Wei et al., 2009).
Lipid peroxidation assay
Lipid peroxidation was determined using the Lipid Peroxidation (MDA) Assay Kit (Abcam) according to the manufacturer’s instructions. After treatments, cells were harvested and homogenized on ice by glass beads with vortexing in MDA lysis buffer supplemented with butylated hydroxytoluene, centrifuged for 10 min at 13,000 × g to remove insoluble material. Sample or standard (200 µl) was mixed with 600 µl of thiobarbituric acid solution, incubated at 95°C for 60 min and cooled to room temperature in an ice bath for 10 min. Each sample and standard (200 µl) was pipetted into a 96-well plate (clear bottom black plate) and fluorometric measurement was taken on a microplate reader at Ex540 nm/Em590 nm. Absorbance value of the blank (water) was used for background subtraction.
In vitro Kinase Assay
The in vitro TORC1 kinase assays were performed as described (Wei et al., 2009) with minor modifications. Briefly, cells were lysed in IP buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 30 mM EDTA, 0.003% Triton X-100, 2 mM PMSF, Roche complete protease inhibitor cocktail and phosSTOP tablets). TORC1 was affinity-purified onto Protein G sepharose beads by a Tor1 antibody, and incubated with 20 µCi [γ-32P]-ATP in 30 µl kinase buffer (10 mM PBS pH 7.5, 4 mM MgCl2, 100 µM ATP, 10 mM DTT, 0.003% Triton X-100, 20% glycerol, protease inhibitor cocktail) and 0.6 µg purified recombinant bacterial GST-SOD1 proteins for 30 min at 30°C. Kinase assay was stopped by heating at 100°C for 5 min in SDS protein sample buffer. The samples were separated on SDS polyacrylamide gels. Protein phosphorylation was detected by autoradiography.
Measurement of Cellular ROS
Fluorescence microscopy was used to determine the cellular reactive oxygen species (ROS) and superoxide levels as described (Tsang et al., 2014). Briefly, intracellular ROS was monitored by staining the cells with 5 µg/ml dihydrorhodamine (DHR) with shaking at 30 °C. Cells were then washed with PBS twice and viewed through a fluorescence microscope with a FITC filter (Olympus U-N41001). Intracellular superoxide levels were monitored by dihydroethidium (DHE) staining. Cells were incubated with 2.5 µg/ml DHE for 10 min with shaking at 30 °C. Cells were then washed with PBS twice and viewed through a fluorescence microscope with a Texas Red filter (Olympus U-N41004). For acquisition of bright-field images, an Olympus microscope equipped with a differential interference contrast system was used. Numbers of DHE- and DHR-stained cells exhibiting weak, moderate and strong signals were counted for quantification analysis.
Two-dimensional (2D) Gel Electrophoresis
Procedure for 2D gel electrophoresis was performed using the ReadyPrep 2-D Starter Kit (Bio-Rad). Briefly, cells were lysed in 2D-sample solubilization solution (8M urea, 2 mM tributylphosphine (TBP), 4% CHAPS, 0.2% Bio-Lyte Ampholyte (range 4/6), and protease and phosphatase inhibitor cocktails, Roche) with a Bioruptor (Diagenode) (30 seconds on and 30 seconds off at mid-power) for 10 min. Cell extracts were treated with 200U DNase I (Boehringer Mannheim) for 20 min. Protein samples were diluted in rehydration buffer (sample solubilization solution plus 0.0002% bromophenol blue), applied to 7-cm immobilized pH gradient (IPG) strips (pH 4–7), and incubated overnight for sample loading and rehydration. Strips were then isoelectrically focused on a Protein IEF Cell (Bio-Rad) for 14,000 V-hr. Following isoelectric 31 focusing, the strips were incubated in equilibration buffer I (6 M urea, 2% SDS, 0.05 M Tris-HCl [pH 8.8], 20% glycerol, 2% dithiothreitol) for 10 min. The strips were then incubated with equilibration buffer II (6 M urea, 2% SDS, 0.05 M Tris-HCl [pH 8.8], 20% glycerol, 2.5% iodoacetamide) for 10 min. Second-dimensional separation was performed on 12% (for endogenous SOD1) or 10% (SOD1-GFP and SOD1-Flag) SDS-PAGE. SOD1 proteins were then transferred to PVDF membranes, and detected by Western blot with human anti-SOD1 antibody (1:2,000). For the phosphatase treatment, the cell extract was incubated with calf intestinal phosphatase (CIP, 20 units, Roche) for 1 hour at 37 °C.
Ischemic Treatment
Cells were seeded and incubated overnight in a 5% CO2-air atmosphere at 37°C. Cells were then washed three times with PBS and the culture plates were replenished with glucose-free DMEM medium (Gibco, 11966-025). The culture plates were incubated in hypoxia glove box (Coy Laboratory Products Inc) with 1% O2, 5% CO2 and 94% N2 at 37°C to achieve the ischemic conditions.
Cancer Cell Growth and Death Assays
Relative cell growth was determined by seeding cells at 5000 cells per well in complete medium in normoxic conditions in 96-well plates. Cell growth was measured by Sulforhodamine B (SRB) assay (Sigma) as described in manufacturer’s protocol. Cell proliferation rate was also measured by total cell counts by cell viability analyzer (Vi-Cell, Beckman Coulter). For cell death assays, 0.1–0.3×106 cells were seeded as triplicate sets on 12-well culture plates and incubated in corresponding conditions. For cell harvest, total culture medium was collected. Adhered cells were washed with PBS and collected by trypsinization. The culture medium, rinsed PBS and trypsinized cells were then pooled and resuspended. Percentage of cell death was determined by trypan blue staining using a cell viability analyzer (Vi-Cell, Beckman Coulter).
In vitro Kinase Assay for mTOR
The Universal fluorometric kinase assay kit (Abcam) was used to detect the in vitro kinase activity of the mTOR proteins. Briefly, HEK293FT cells that had been transfected with pBJF-Flag-mTORWT or pBJF-mTORD2338E (kinase dead) were extracted in ice-cold mTOR IP buffer (20 mM Tris [pH 7.5], 20 mM NaCl, 1 mM EDTA, Roche protease complete inhibitor 32 cocktail and phosSTOP tablet) by sonication, and the extracts were centrifuged at 10,000 × g for 20 min at 4°C. mTOR proteins were immunoprecipitated from the supernatant with the anti-Flag M2 affinity beads (Sigma), and the immunoprecipitate was washed 4 times with mTOR IP buffer. As substrates for the kinase assays, GST fusion protein of 4EBP1 (GST-4EBP1) was purified from DH5α transformed with pGEX4T1-4EBP1 plasmid using glutathione agarose beads (Thermo Fisher Scientific) following the manufacturer’s protocol. His6 fusion proteins of SOD1WT and SOD1T40A were expressed from BL21 cells that had been transfected with pGBHT-SOD1WT and pGBHT-SOD1T40A, respectively. The His6-fusion proteins were then purified using QIAexpress Ni-NTA Fast start Kit (Qiagen) following manufacturer’s instructions. Kinase reaction was started by adding the kinase reaction mixture (ADP assay buffer, 100 µM ATP, 10 mM MgCl2, 4 mM MnCl2, Roche protease inhibitor cocktail) containing 3 µg of GST-4EBP1, His6-SOD1WT or His6-SOD1T40A as substrate, to the purified immunoprecipitated mTOR proteins and incubated for 30 min at 30°C. To monitor ADP formation, 20 µl of the kinase reaction mixture was combined with 20 µl of ADP sensor buffer and 10 µl of ADP sensor. The mixture was incubated in dark for 1 hour. Fluorescent readings were taken with a microplate reader at Ex540 nm/Em590 nm.
3D spheroid culture
The Hep3B spheroids were cultured as described (Grimes et al., 2014) with minor modification. Briefly, Hep3B cells were seeded in regular culture medium at a density of 20,000 cells per well on a 2% agar-coated 48-well plates. After the indicated time of culture in 5% CO2 at 37 °C, 3D Hep3B spheroid structures were imaged using phase-contrast microscopy and the size of spheroids was measured (n=7~33) with NIS-Elements BR software. For analysis of ROS in Hep3B spheroids, day 1 spheroids were incubated in normal Hep3B culture medium containing 10 µM Dihydroethidium (DHE) and 5 µg/ml Hoechst 33342 at 37 °C for 10 min, and then immediately imaged using confocal microscope. Lack of Hoechst 33342 signal in the inner cells was used as the criteria for focusing the center of spheroids. Fluorescence intensity and area of DHE positive cells and the total area of each spheroid (as determined by GFP-positive area) are analyzed using imageJ. For analysis of cell death in Hep3B spheroids, day 5 spheroids were incubated in normal culture medium containing 5 µM propidium iodide (PI) and 5 µg/ml Hoechst 33342 at 37 °C for 10 min, washed once with PBS and then the center of the spheroids (kept in normal culture medium) were immediately imaged using confocal microscope. Hoechst 33342 staining was used to mark the spheroid boundary and determine the center of spheroids while imaging. Fluorescence 33 intensity and area of PI positive cells and the total area of each spheroid (determined by GFP-positive area) were analyzed using imageJ. The A549 spheroids were cultured essentially the same as Hep3B spheroids described above. For analysis of ROS in A549 spheroids, spheroid in each culture well was incubated in normal culture medium containing 10 µM DHE at 37 °C for 10 min. Spheroids were then trypsinized and the average cellular DHE intensity was monitored by flow cytometry. For analysis of cell death in A549 spheroids, spheroids were incubated in normal culture medium containing 5 µM PI at 37 °C for 10 min. Spheroids were then trypsinized and the average cellular PI intensity was monitored by flow cytometry.
Flow Cytometry Analysis
Fluorescence signals of DHE- and PI-stained cells were analyzed by the flow cytometer (BD Accuri C6). A minimum of 5,000 cells were examined for each assay. Polygonal gating was used for excluding debris and aggregates. DHE and PI red fluorescence were analyzed in the FL-2 channel. The mean fluorescence intensity was monitored using the BD Accuri C6 software.
Mammalian DNA damage assay
For oxidative DNA damage analysis, cells were fixed with 4% formaldehyde solution (in PBS) for 15 min, rinsed with PBS for three times, permeabilized in PBS containing 0.2% Triton X-100 for 10 min at room temperature, and treated with 1 mg/ml RNase A for 30 min at 37 °C. Cells were then incubated in 3 N HCl at room temperature for 30 min to denature DNA, and in blocking buffer (4% BSA, 0.5% Triton X-100 in PBS) for 1 hour at room temperature. To assess the oxidative DNA damage, cells were incubated with primary antibody against 8-Oxoguanine (8-OxoG) (mouse monoclonal IgG, MAB 3560, clone 483.15, 1:200 dilution, Millipore) at 4 °C overnight, followed by Alexa Fluor 594- or Alexa Fluor 546-conjugated anti mouse IgG. DNA was counterstained with 50 ng/ml DAPI. Microscopic analysis was performed with an Olympus fluorescence microscope equipped with a digital camera. Quantitative analysis of oxidative DNA damage was determined by counting the percentage of positive 8-OxoG staining cells. DNA damage was also analyzed by fluorescence analysis using phospho-H2AX antibody, a specific indicator of DNA double-strand break response. Essentially same procedure was carried out as described above except that the RNase A and HCl treatments were omitted and anti-phospho-H2AX antibody (Ser139, 20E3) was used as the primary antibody (1:200 dilution, Cell Signaling Technology #9718). Nuclei with prominent phospho-H2AX staining (> 10 punta 34 per nucleus) were considered as phosphor-H2AX positive cells. At least 200 nuclei were counted for quantification for each sample.
Xenograft tumor assay and immunohistochemistry
Hep3B cells were resuspended in PBS and 5 × 106 cells were then injected subcutaneously into the right flank of each mouse. Tumor volumes were calculated as follows: volume (mm3) = length× (width)2× 0.5. Harvested tumor tissues were fixed in 4% formalin for HE staining or immunohistochemistry (IHC) assay. IHC staining of Ki-67 was used to evaluate cell proliferation as described previously.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are expressed as mean ± standard deviation (SD) from three or more independent experiments, and the level of significance between two groups was determined by Student’s t test. Statistical significance was considered when p < 0.05. All of the statistical details of experiments can be found in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The raw data files for images are available at Mendeley (http://dx.doi.org/10.17632/rvbvczzd5d.1).
Supplementary Material
Highlights.
SOD1 is a conserved effector of mTORC1 signaling in eukaryotic cells
Nutrients restrain SOD1 through mTORC1-dependent phosphorylation to promote growth
Starvation stimulate SOD1 activity to prevent oxidative damage and enhance survival
SOD1 enhances cancer cell survival and chemoresistance in ischemic microenvironment
Acknowledgments
We thank Dr. Bing Xia, Ren Wang, Xiaowen Wang and Tzung-Ju Wu for technical assistance. The work is supported by the Rutgers Cancer Institute of New Jersey Shared Resources for Advanced Microscopy, Biomedical Informatics, Biometrics, Biospecimen Repository and Histopathology Service, and Flow Cytometry/Cell Sorting. This project was supported by NIH (R01 grants CA123391, CA166575, CA163591, CA188096, CA130893), NSF/NIH/DoE-DBI1338415, the National Natural Science Foundation of China (No: 81372564, 81372600, 81572440, 81671148, 81730081), the Natural Science Foundation of Guangdong Province for Distinguished Young Scholar (No: 2015A030306047) and the Research Fund of State Key Laboratory of Oncology in South China.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
X.F.Z. conceptualized and directed the overall project; C.K.T., M.C., X.C., Y.M.Q., I.D., C.Y. and X.X.L. designed and/or performed experiments, C.K.T., X.C., Y.M.Q. and C.Y. prepared figures, C.K.T., B.V. and S.K.B. performed 3D-modeling of the effect of SOD1 phosphorylation; E.W. assisted with hypoxic studies; C.K.T., X.X.L., C.N.Q., L.W.F., H.Y.W., E.W., S.K.B. and X.F.Z. provided funding and/or research space; C.K.T. and X.F.Z. wrote the manuscript; C.K.T., E.W., S.K.B. and X.F.Z. edited and proof read the manuscript.
Declaration of Interests
The authors declare no known conflict of interest.
References
- Apel K, Hirt H. REACTIVE OXYGEN SPECIES: Metabolism, Oxidative Stress, and Signal Transduction. Annual Review of Plant Biology. 2004;55:373–399. doi: 10.1146/annurev.arplant.55.031903.141701. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Del Conte R, Fadin R, Mangani S, Silvia Viezzoli M. The solution structure of a monomeric, reduced form of human copper, zinc superoxide dismutase bearing the same charge as the native protein. JBIC Journal of Biological Inorganic Chemistry. 1999;4:795–803. doi: 10.1007/s007750050353. [DOI] [PubMed] [Google Scholar]
- Bertram PG, Choi JH, Carvalho J, Chan T-F, Ai W, Zheng XFS. Convergence of TOR-Nitrogen and Snf1-Glucose Signaling Pathways onto Gln3. Mol Cell Biol. 2002;22:1246–1252. doi: 10.1128/MCB.22.4.1246-1252.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieger K, Schiavone S, Miller FJ, Jr, Krause KH. Reactive oxygen species: from health to disease. Swiss medical weekly. 2012;142:w13659. doi: 10.4414/smw.2012.13659. [DOI] [PubMed] [Google Scholar]
- Broach JR. Nutritional Control of Growth and Development in Yeast. Genetics. 2012;192:73–105. doi: 10.1534/genetics.111.135731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Chan T-F, Carvalho J, Riles L, Zheng XFS. A chemical genomics approach toward understanding the global functions of the target of rapamycin protein (TOR) PNAS. 2000;97:13227–13232. doi: 10.1073/pnas.240444197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che M, Wang R, Li X, Wang HY, Zheng XF. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug discovery today. 2016;21:143–149. doi: 10.1016/j.drudis.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culotta VC, Klomp LWJ, Strain J, Casareno RLB, Krems B, Gitlin JD. The Copper Chaperone for Superoxide Dismutase. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- D'Autreaux B, Toledano M. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- Dechant R, Peter M. Nutrient signals driving cell growth. Curr Opin Cell Biol. 2008;20:678–687. doi: 10.1016/j.ceb.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Dohmen RJ, Wu P, Varshavsky A. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. The Journal of Cell Biology. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- Glasauer A, Sena LA, Diebold LP, Mazar AP, Chandel NS. Targeting SOD1 reduces experimental non-small-cell lung cancer. J Clin Invest. 2014;124:117–128. doi: 10.1172/JCI71714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Grimes DR, Kelly C, Bloch K, Partridge M. A method for estimating the oxygen consumption rate in multicellular tumour spheroids. Journal of the Royal Society, Interface. 2014;11:20131124. doi: 10.1098/rsif.2013.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Cell biology. Warburg effect and redox balance. Science. 2011;334:1219–1220. doi: 10.1126/science.1215637. [DOI] [PubMed] [Google Scholar]
- Hart PJ, Balbirnie MM, Ogihara NL, Nersissian AM, Weiss MS, Valentine JS, Eisenberg D. A Structure-Based Mechanism for Copper–Zinc Superoxide Dismutase. Biochemistry. 1999;38:2167–2178. doi: 10.1021/bi982284u. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Guan K-L. Nutrient signaling to mTOR and cell growth. Trends in Biochemical Sciences. 38:233–242. doi: 10.1016/j.tibs.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004;18:2491–2505. doi: 10.1101/gad.1228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanović B, Mayer IA, Mayer EL, Abramson VG, Bardia A, Sanders ME, Kuba MG, Estrada MV, Beeler JS, Shaver TM, et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel With or Without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clinical Cancer Research. 2017 doi: 10.1158/1078-0432.CCR-16-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Buel GR, Blenis J. Nutrient regulation of the mTOR complex 1 signaling pathway. Molecules and cells. 2013;35:463–473. doi: 10.1007/s10059-013-0138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini D. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tsang C, Watkins M, Bertram P, Zheng X. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature. 2006;442:1058–1061. doi: 10.1038/nature05020. [DOI] [PubMed] [Google Scholar]
- Li Y, Tsang CK, Wang S, Li XX, Yang Y, Fu L, Huang W, Li M, Wang HY, Zheng XF. MAF1 suppresses AKT-mTOR signaling and liver cancer through activation of PTEN transcription. Hepatology (Baltimore, Md) 2016;63:1928–1942. doi: 10.1002/hep.28507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao L, St Clair DK. Regulation of superoxide dismutase genes: Implications in disease. Free Radical Biology and Medicine. 2009;47:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima A, Nakashima J, Yoshioka K, Tachibana M, Tazaki H, Murai M. Role of reactive oxygen species in cis-dichlorodiammineplatinum-induced cytotoxicity on bladder cancer cells. Br J Cancer. 1997;76:206–210. doi: 10.1038/bjc.1997.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa TK, Davis RW. Superoxide anions regulate TORC1 and its ability to bind Fpr1:rapamycin complex. Proc Natl Acad Sci U S A. 2008;105:15166–15171. doi: 10.1073/pnas.0807712105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Schroeder Elizabeth A, Ocampo A, Barrientos A, Shadel Gerald S. Regulation of Yeast Chronological Life Span by TORC1 via Adaptive Mitochondrial ROS Signaling. Cell Metabolism. 2011;13:668–678. doi: 10.1016/j.cmet.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa L, Hahn M, Marsh EL, Evans BS, Germain D. SOD2 to SOD1 switch in breast cancer. J Biol Chem. 2014;289:5412–5416. doi: 10.1074/jbc.C113.526475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C, Lu N, Zhou Y, Ni T, Dai Y, Li Z, Guo Q, Wei L. Oroxylin a modulates mitochondrial function and apoptosis in human colon cancer cells by inducing mitochondrial translocation of wild-type p53. Oncotarget. 2016 doi: 10.18632/oncotarget.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol. 2015;33:8–13. doi: 10.1016/j.ceb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi Amit R, Culotta Valeria C. SOD1 Integrates Signals from Oxygen and Glucose to Repress Respiration. Cell. 2013;152:224–235. doi: 10.1016/j.cell.2012.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Elazar Z. Chapter 8 Monitoring Starvation-Induced Reactive Oxygen Species Formation. Methods in Enzymology (Academic Press) 2009:119–130. doi: 10.1016/S0076-6879(08)03608-2. [DOI] [PubMed] [Google Scholar]
- Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Current biology : CB. 2014;24:R453–462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. Journal of Applied Physiology. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Soardo G, Donnini D, Domenis L, Catena C, De Silvestri D, Cappello D, Dibenedetto A, Carnelutti A, Bonasia V, Pagano C, et al. Oxidative stress is activated by free fatty acids in cultured human hepatocytes. Metabolic syndrome and related disorders. 2011;9:397–401. doi: 10.1089/met.2010.0140. [DOI] [PubMed] [Google Scholar]
- Somwar R, Erdjument-Bromage H, Larsson E, Shum D, Lockwood WW, Yang G, Sander C, Ouerfelli O, Tempst PJ, Djaballah H, et al. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proceedings of the National Academy of Sciences. 2011;108:16375–16380. doi: 10.1073/pnas.1113554108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A Fraction of Yeast Cu, Zn-Superoxide Dismutase and Its Metallochaperone, CCS, Localize to the Intermembrane Space of Mitochondria: A PHYSIOLOGICAL ROLE FOR SOD1 IN GUARDING AGAINST MITOCHONDRIAL OXIDATIVE DAMAGE. Journal of Biological Chemistry. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- Thomas Janice D, Zhang Y-J, Wei Y-H, Cho J-H, Morris Laura E, Wang H-Y, Zheng XFS. Rab1A Is an mTORC1 Activator and a Colorectal Oncogene. Cancer Cell. 2014;26:754–769. doi: 10.1016/j.ccell.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Tsang CK, Liu Y, Thomas J, Zhang Y, Zheng XFS. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat Commun. 2014;5 doi: 10.1038/ncomms4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang CK, Zheng XFS. Opposing Role of Condensin and Radiation-sensitive Gene RAD52 in Ribosomal DNA Stability Regulation. J Biol Chem. 2009;284:21908–21919. doi: 10.1074/jbc.M109.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine JS, Doucette PA, Zittin Potter S. COPPER-ZINC SUPEROXIDE DISMUTASE AND AMYOTROPHIC LATERAL SCLEROSIS. Annual Review of Biochemistry. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Wei Y, Tsang C, Zheng X. Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J. 2009;28:2220–2230. doi: 10.1038/emboj.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zheng X. Sch9 partially mediates TORC1 signaling to control ribosomal RNA synthesis. Cell Cycle. 2009;8:4085–4090. doi: 10.4161/cc.8.24.10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zheng X. Maf1 regulation: a model of signal transduction inside the nucleus. Nucleus. 2010;1:162–165. doi: 10.4161/nucl.1.2.11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nature protocols. 2010;5:51–66. doi: 10.1038/nprot.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Waggoner D, Subramaniam JR, Tessarollo L, Bartnikas TB, Culotta VC, Price DL, Rothstein J, Gitlin JD. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc Natl Acad Sci U S A. 2000;97:2886–2891. doi: 10.1073/pnas.040461197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T-J, Wang X, Zhang Y, Meng L, Kerrigan John E, Burley Stephen K, Zheng XFS. Identification of a Non-Gatekeeper Hot Spot for Drug-Resistant Mutations in mTOR Kinase. Cell Reports. 2015;11:446–459. doi: 10.1016/j.celrep.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall M. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zaman S, Lippman SI, Zhao X, Broach JR. How Saccharomyces responds to nutrients. Annu Rev Genet. 2008;42:27–81. doi: 10.1146/annurev.genet.41.110306.130206. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li X, Yang Y, Zhang Y, Wang HY, Zheng XFS. Significance and Mechanism of Androgen Receptor (AR) Overexpression and AR-mTOR Crosstalk in Hepatocellular Carcinoma. Hepatology (Baltimore, Md) 2017 doi: 10.1002/hep.29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ikeno Y, Bokov A, Gelfond J, Jaramillo C, Zhang H-M, Liu Y, Qi W, Hubbard G, Richardson A, et al. Dietary restriction attenuates the accelerated aging phenotype of Sod1(−/−) mice. Free radical biology & medicine. 2013;60:300–306. doi: 10.1016/j.freeradbiomed.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Florentino D, Chen J, Crabtree G, Schreiber S. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell. 1995;82:121–130. doi: 10.1016/0092-8674(95)90058-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.