

Abstract
Inducible heat shock protein 70 (HSP70; also known as HSPA1 or HSP72) is implicated in cancer. As a stress-inducible heat shock protein, HSP70 is highly expressed in a variety of cancers and correlates with metastasis, chemotherapy resistance and tumor prognosis. The present study demonstrated that suppression of HSP70 through the specific inhibitor pifithrin-µ or by HSP70 knockdown enhanced cisplatin-induced apoptosis in HGC-27 gastric cancer cells. By contrast, upregulation of HSP70 through transfection of a HSP70 overexpressing plasmid decreased cisplatin-induced HGC-27 cell apoptosis. In exploring the underlying molecular mechanisms, the present results revealed that HSP70 antagonized cisplatin-induced HGC-27 cell apoptosis by regulating the mitogen-activated protein kinase (MAPK) signaling pathway. In addition, suppressing the MAPK pathway enhanced cisplatin-induced HGC-27 cell apoptosis. Collectively, the present findings suggest that inhibition of HSP70 expression enhanced the sensitivity of HGC-27 cells to cisplatin via the MAPK signaling pathway, and that HSP70 may serve as a potential therapeutic target in gastric cancer.
Keywords: heat shock protein 70, gastric cancer, apoptosis, cisplatin, mitogen-activated protein kinase signaling pathway
Introduction
Gastric cancer is a common and frequently occurring malignancy worldwide. At present, systemic chemotherapy is the most common clinical treatment for gastric cancer. Cisplatin (DDP) is a well-known chemotherapeutic drug, that is widely used in chemotherapy of multiple types of cancer, including gastric cancer (1). However, development of chemotherapy resistance significantly reduces its therapeutic effect in gastric cancer. The regulatory molecules involved in resistance in gastric cancer remain not fully elucidated and require further study.
The heat shock proteins (HSPs) are a highly conserved protein family consisting of several members. As molecular chaperones, HSPs have been reported to be involved in the folding, modification and degradation of intracellular proteins (2,3). The stress-inducible HSP70 (also known as HSPA1 or HSP72) is an important member of the HSP family. Recent studies have demonstrated that HSP70 is expressed at extremely low levels in unstressed normal cells, but is highly expressed in a variety of cancers and correlated with tumor grade, metastasis, chemotherapy resistance and poor prognosis (4–6). Because of its tumor-related expression and important role in cancer progression, HSP70 has become an attractive target for cancer therapy (7,8).
The primary aim of the present study was to investigate the effect of HSP70 in the cisplatin-induced apoptosis of gastric cancer cells and to explore its molecular mechanism. The results demonstrated that downregulating HSP70 expression enhanced cisplatin-induced gastric cancer apoptosis via regulation of the mitogen-activated protein kinase (MAPK) signaling pathway. The present study provided an experimental basis and novel insights for HSP70 as a target for cancer therapy.
Materials and methods
Reagents and antibodies
Cisplatin (purity, ≥99.9) was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany), and pifithrin-µ (PES), SB203580, SP600125 and U0126 were purchased from Selleck Chemicals (Houston, TX, USA). All of the above reagents were dissolved in DMSO and then diluted with sterilized PBS. The final concentration of DMSO in cultures was <0.1%. Rabbit monoclonal antibodies against p38 (cat. no. 9219s; 1:1,000), phosphorylated (p)-p38 (Thr180/Tyr182; cat. no. 9215s; 1:1,000), extracellular signal-regulated kinase (ERK, cat. no. 9102s; 1:1,000), p-ERK (Thr202/Tyr204; cat. no. 4376s; 1:1,000), c-Jun N-terminal kinase (JNK; cat. no. 9252s; 1:1,000), p-JNK (Thr183/Tyr185; cat. no. 4671s; 1:1,000), p-SRC proto-oncogene non-receptor tyrosine kinase (Src, Tyr416; cat. no. 6943s; 1:500), p-AKT serine/threonine kinase 1 (Akt, Ser473; cat. no. 4060s; 1:500), p-inhibitor of κB (IκB, Ser32; cat. no. 2859s; 1:500), poly-ADP-ribose-polymerase (PARP, cat. no. 9532s; 1:1,000), pro-caspase-3 (cat. no. 9662s; 1:500), cleaved caspase-3 (cat. no. 9661s; 1:500), β-actin (cat. no. 4970s; 1:1,000), GAPDH (cat. no. 5174s; 1:1,000), and rat monoclonal antibody against HSP70 (cat. no. 4573s; 1:1,000) were all from Cell Signaling Technology, Inc. (Beverly, MA, USA). All secondary antibodies (cat. no. 926-32210 or 926-32211; 1:5,000) were purchased from LI-COR Biosciences (Lincoln, NE, USA).
Cell culture
The human gastric cancer HGC-27 cell line was obtained from GuangZhou Cellcook Biotech Co., Ltd. (Guangzhou, China). Cells were cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 10% non-essential amino acids, 100 µg/ml streptomycin and 100 U/ml penicillin and incubated at 37°C in an atmosphere of 5% CO2.
Plasmids and transfection
Green fluorescent protein (GFP)-labeled HSP70 overexpression plasmids and negative plasmids (empty vector control plasmid; CMV-MCS-EGFP-SV40-Neomycin), and HSP70 short hairpin (sh) RNA and negative plasmids (non-targeting shRNA plasmid; hU6-MCS-CMV-GFP-SV40-Neomycin), were purchased from GeneChem Co., Ltd. (Shanghai, China). HGC-27 cells were seeded into 12-well cell culture plates and cultured to 60–70% confluence, prior to transfection with plasmids for the indicated times using Lipofectamine® 3000 Reagent (Thermo Fisher Scientific, Inc.). Operations were performed according to the manufacturers instructions.
Annexin V/propidium iodide (PI) double staining assay
Apoptosis rate was detected by Annexin V/PI double staining (Jiancheng Bioengineering Institute, Nanjing, China). Briefly, following treatment, HGC-27 cells were collected and washed two times with PBS. Cells were resuspended in 500 µl of binding buffer, and 5 µl PI and 5 µl Annexin V-fluorescein isothiocyanate were added to the cells. Apoptosis rate was detected by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
DAPI staining
Following treatment, HGC-27 cells were washed three times with PBS and fixed with 4% paraformaldehyde for 20 min at room temperature. After rising with PBS, cells were stained with DAPI for 3 min in the dark. The morphology of apoptotic cells was examined using an inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Western blotting
HGC-27 cells were washed three times with cold PBS and then lysed in RIPA buffer containing protease inhibitors (Beyotime Institute of Biotechnology, Haimen, China) for 30 min on ice. Lysates were centrifuged (14,300 × g) at 4°C for 15 min, and the supernatant was collected. The bicinchoninic acid assay was used to measure protein concentration in the supernatant, and equal amounts of total protein (50 µg) were loaded onto 12% gels for SDS-PAGE. Separated proteins were transferred onto nitrocellulose membranes (Pall Corporation, Port Washington, NY, USA), which then were blocked with 5% skim milk for 1 h at room temperature and then incubated with the indicated primary antibodies overnight at 4°C. After washing three times with TBST, membranes were incubated with IRDye800 fluorophore-conjugated secondary antibody for 1 h at room temperature in the dark. Antigen-antibody complexes were detected using a LI-COR Odyssey Infrared Imaging System (LI-COR Biosciences) and LI-COR Odyssey analysis software was used to quantify the proteins.
Cell Counting Kit-8 (CCK-8) assay
HGC-27 cells were seeded into 96-well cell culture plates, and the following day they were treated with different doses of PES for 24 h. CCK-8 was then added at 10 µl/well. Following incubation for 2 h at 37°C and 5% CO2, absorbance at 450 nm was measured using a Multiskan GO plate reader (Thermo Fisher Scientific, Inc.). Each experiment was repeated three times.
Statistical analysis
Data were expressed as mean ± standard deviation. Statistical analysis was performed using SPSS17.0 software (SPSS, Inc., Chicago, IL, USA). The results were compared using one-way analysis of variance followed by a post hoc Tukey test for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
HSP70 overexpression antagonizes cisplatin-induced HGC-27 cell apoptosis
HGC-27 cells were transfected with GFP-HSP70 or control plasmid, and 24 h post-transfection they were stimulated with cisplatin (5 µg/ml) for 24 h. The morphology of apoptotic cells was observed with DAPI staining and the apoptosis rate was determined by flow cytometry. The expression of apoptosis-related proteins, including PARP, pro-caspase-3 and cleaved caspase-3, were detected by western blotting. HGC-27 cells transfected with control plasmid and not treated with cisplatin had light blue, round nuclear morphology as evident by DAPI staining (Fig. 1A). However, following cisplatin treatment, the nuclei exhibited morphological changes typical of apoptosis, including nuclear condensation and nuclear fragmentation (Fig. 1A). These typical cisplatin-induced morphological changes were clearly reversed in HSP70-overexpressing HGC-27 cells (Fig. 1A). In addition, cisplatin-induced expression of cleaved PARP and cleaved caspase-3 was decreased, while pro-caspase-3 levels were increased, in GFP-HSP70-overexpressing cells compared with cells transfected with control plasmid (Fig. 1B). Flow cytometry was also used to determine the apoptotic rate of HGC-27 cells induced by cisplatin treatment. The apoptotic rate was 31.38% in control plasmid-transfected cells, while it was reduced to 21.2% in HSP70-overexpressing cells (Fig. 1C).
Figure 1.

HSP70 overexpression antagonizes cisplatin-induced HGC-27 cell apoptosis. HGC-27 cells were transfected with GFP-HSP70 plasmid or control plasmid. After 24 h, cells were stimulated with 5 µg/ml cisplatin for the indicated times. (A) Nuclear morphology of apoptotic cells was detected by DAPI staining (magnification, ×100). (B) Expression levels of apoptosis-related proteins PARP, pro-caspase-3 and cleaved caspase-3 were detected by western blotting. (C) Apoptosis rate was determined by flow cytometry (representative plots and quantification is shown). **P<0.01, with comparisons indicated by lines. HSP70, heat shock protein 70; GFP, green fluorescent protein; PARP, poly-ADP-ribose-polymerase; DDP, cisplatin; CON, control.
Downregulation of HSP70 or suppression of its function enhances cisplatin-induced HGC-27 cell apoptosis
To further explore the role of HSP70 in cisplatin-induced apoptosis, a HSP70 inhibitor, PES, was used to treat the cells. PES interacts selectively with the stress-inducible HSP70 protein and inhibits its function (2,9,10). Firstly, the cytotoxicity of PES was determined using the CCK-8 assay. HGC-27 cells were treated with different concentrations of PES (1, 2, 4, 6, 8, 10 or 20 µM) for 24 h, and cell viability was analyzed by CCK-8 assay. PES did not affect cell viability, even at a dose of 10 µM (Fig. 2A); the dose of 8 µM was therefore selected for subsequent experiments. Secondly, the effects of PES on cisplatin-induced cell apoptosis were examined. Western blot analysis indicated that PES pretreatment increased the expression of cleaved PARP and cleaved-caspase-3, and decreased the levels of pro-caspase-3, compared with cells treated with cisplatin alone (Fig. 2B). An Annexin V/PI double staining assay indicated that the apoptotic rate in cisplatin-stimulated cells was 18.2%, while inhibition of HSP70 by PES increased this to 41.2% (Fig. 2C).
Figure 2.

Pretreatment with an HSP70 inhibitor enhances cisplatin-induced HGC-27 cells apoptosis. (A) HGC-27 cells were treated with different concentrations of PES for 24 h, and cell viability was measured using a Cell Counting Kit-8 assay. **P<0.01 compared with untreated cells. (B) HGC-27 cells were pretreated with PES (8 µM) for 2 h and then stimulated with cisplatin (5 µg/ml) for 24 h. Expression levels of apoptosis-related proteins PARP, cleaved-caspase-3 and pro-caspase-3 were detected by western blotting. (C) Apoptosis rate was determined by flow cytometry (representative plots and quantification is shown). **P<0.01 with comparisons indicated by lines. HSP70, heat shock protein 70; PES, pifithrin-µ; PARP, poly-ADP-ribose-polymerase; DDP, cisplatin.
As a second method to suppress HSP70 function and explore its effect on cisplatin-induced apoptosis, HSP70-specific shRNA was transfected in the cells to down-regulate its expression. The nuclear condensation and nuclear fragmentation induced by cisplatin in HSP70 shRNA-transfected cells were obviously enhanced compared with control plasmid-transfected HGC-27 cells (Fig. 3A). The levels of cleaved PARP and cleaved caspase-3 following cisplatin stimulation were increased, while pro-caspase-3 levels were decreased, in the HSP70 shRNA-transfected cells compared with control (Fig. 3B). Concurrently, the apoptotic ratio was 45.8% in HSP70 shRNA-transfected cells and 27.15% in the control (Fig. 3C). Taken together, these results suggest that HSP70 serves a protective role in cisplatin-induced HGC-27 cell apoptosis.
Figure 3.

HSP70 downregulation enhances cisplatin-induced HGC-27 cell apoptosis. HGC-27 cells were transfected with HSP70 shRNA plasmid and control plasmid, and at 48 h post-transfection, cells were stimulated with 5 µg/ml cisplatin for the indicated times. (A) The morphology of apoptotic cell nuclei was detected by DAPI staining (magnification, ×100). (B) Expression levels of apoptosis-related proteins PARP, pro-caspase-3 and cleaved caspase-3 were detected by western blotting. (C) Flow cytometry was used to determine apoptosis rates (representative plots and quantification is shown). **P<0.01 with comparisons indicated by lines. HSP70, heat shock protein 70; sh, short hairpin; PARP, poly-ADP-ribose-polymerase; DDP, cisplatin; CON, control.
HSP70 affects cisplatin-induced MAPK signaling pathway activation
MAPK and survival-related signaling pathways, such as Akt and nuclear factor (NF)-κB, have an important role in the proliferation, differentiation and apoptosis of tumor cells (11–13). To investigate whether the molecular mechanism of HSP70 regulation of cisplatin-induced apoptosis is modulated by the MAPK, Akt or NF-κB signaling pathways, HGC-27 cells were treated with cisplatin for different amounts of time, and the phosphorylation of p38, ERK, JNK, Src, Akt and IκB were monitored by western blotting. Cisplatin stimulation resulted in a time-dependent increase in the phosphorylation of p38, ERK and JNK, but had no effect on the total levels of p38, ERK and JNK proteins (Fig. 4A). Phosphorylation of Src, Akt and IκB was also detected, but this was unaffected by cisplatin treatment (Fig. 4B). These results suggested that the MAPK signaling pathway may be involved in cisplatin-induced apoptosis.
Figure 4.

HSP70 affects cisplatin-induced MAPK signaling pathway activation. (A) HGC-27 cells were treated with cisplatin (5 µg/ml) for different amounts of time and the phosphorylation and total protein levels of (A) p38, ERK and JNK, and (B) Src, Akt and IκB, were monitored by western blotting. (C) HSP70-overexpressing and control plasmids were transfected into HGC-27 cells. (D) HGC-27 cells were pretreated with PES for 2 h and then stimulated with cisplatin for 6 or 8 h. (E) HSP70-shRNA and control plasmids were transfected into HGC-27 cells. After transfection, the cells were treated with cisplatin for 6 or 8 h and the phosphorylation and total protein levels of p38, ERK and JNK were monitored by western blotting. *P<0.05 compared with control cells at 6 h; #P<0.05 compared with control cells at 8 h. HSP70, heat shock protein 70; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; Src, SRC proto-oncogene non-receptor tyrosine kinase; Akt, AKT serine/threonine kinase 1; IκB, inhibitor of κB; sh, short hairpin; DDP, cisplatin; CON, control; p-, phosphorylated; PES, pifithrin-µ.
To further investigate the effects of HSP70 on cisplatin-induced phosphorylation in the MAPK signaling pathway, HSP70-overexpressing plasmids were transfected into HGC-27 cells. HSP70 overexpression did not exert any effects on p38, ERK or JNK phosphorylation compared with empty vector controls (Fig. 4C). Conversely, PES was used to inhibit HSP70 function, and HSP70 inhibition resulted in a striking reduction of cisplatin-induced phosphorylation of p38, ERK and JNK (Fig. 4D). HSP70 shRNA transfection also suppressed cisplatin-induced phosphorylation of p38, ERK and JNK (Fig. 4E). Collectively, these results suggest that HSP70 facilitates the activation of p38, ERK and JNK in HGC-27 cells.
MAPK pathway inhibition enhances cisplatin-induced HGC-27 cell apoptosis
To investigate the role of MAPK signaling in the process of cisplatin-induced HGC-27 cell apoptosis, cells were stimulated with cisplatin or cisplatin and a p38-specific inhibitor (SB203580), a JNK specific inhibitor (SP600125) or an ERK1/2 inhibitor (U0126) for 24 h. All inhibitor pretreatments enhanced the expression of cleaved PARP and cleaved caspase-3 and decreased the levels of pro-caspase-3 induced by cisplatin (Fig. 5A). Additionally, compared with cisplatin-only treated cells, apoptosis levels were significantly elevated in cells stimulated with cisplatin and the inhibitors (Fig. 5B). Finally, in order to confirm the inhibitory effects of each inhibitor, the phosphorylation levels of p38, ERK or JNK were respectively detected. Fig 5C illustrates that each inhibitor treatment suppressed its corresponding molecule, while having little effect on HSP70 protein expression levels. Collectively, these findings suggest that the MAPK signaling pathway antagonized cisplatin-induced apoptosis in HGC-27 cells.
Figure 5.

MAPK pathway inhibition enhances cisplatin-induced HGC-27 cell apoptosis. (A) HGC-27 cells were pretreated with specific inhibitors for p38, ERK or JNK for 2 h and then treated with cisplatin for 24 h. Expression levels of PARP, cleaved caspase-3 and pro-caspase-3 were detected by western blotting. (B) Apoptotic rate was determined by flow cytometry (representative plots and quantification is shown). (C) HGC-27 cells were pretreated with specific inhibitors for p38, ERK or JNK for 2 h and then treated with cisplatin for 6 h. Phosphorylation of p38, ERK, JNK and the levels of HSP70 were detected by western blotting. *P<0.05 and **P<0.01, with comparisons indicated by lines. MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; PARP, poly-ADP-ribose-polymerase; HSP70, heat shock protein 70; DDP, cisplatin; p-, phosphorylated.
Discussion
HSPs are chaperone proteins, which are induced by a variety of different stresses. As a member of this family, HSP70 has been reported to be highly expressed in different types of cancer (6) and is considered to be a negative prognostic factor in breast cancer, osteosarcoma and bladder cancer. By contrast, it has been reported as a positive prognostic factor in esophageal, pancreatic and renal cancers (14-16). Recently, multiple studies have demonstrated that HSP70 is involved in resistance to anticancer agents in tumors (14,17,18); however, the molecular mechanism of its action in resistance to chemotherapy remains unclear.
Several signaling pathways are reported to be involved in the regulation of tumor cells apoptosis, such as the MAPK, Akt and NF-κB signaling pathways (11–13). Ding et al (19) demonstrated that HSP70 expression decreases the sensitivity of osteosarcoma cells to baicalein via activation of the PI3K/Akt and MAPK/ERK signaling pathways. Other studies have also demonstrated that HSP70 regulates tumor cell apoptosis by inhibiting the activation of JNK and NF-κB signaling (20,21). Cisplatin, a commonly used chemotherapeutic drug, is used clinically to treat cancer, and HSP70 expression decreases the sensitivity of osteosarcoma cells to cisplatin (14). However, the question of whether HSP70 regulates the sensitivity of gastric cancer cells to cisplatin and its underlying molecular mechanism still remain to be explored.
Cisplatin induces cell death by activating the apoptosis pathway (22,23); however, HSP70 expression has been demonstrated to exert a strong anti-apoptotic effect (15). In the present study, the effect of HSP70 on cisplatin-induced HGC-27 gastric cancer cell apoptosis was examined. Western blotting was used to detect the expression levels of cleaved PARP, cleaved caspase-3 and pro-caspase-3, an Annexin V/PI assay was used to determine apoptotic rates, and DAPI staining was used to monitor the morphology of apoptotic nuclei. As illustrated in Figs. 1–3, HSP70 overexpression decreased cisplatin-induced HGC-27 cell apoptosis, while inhibition of HSP70 using a specific inhibitor or by transfection of specific shRNA enhanced the cellular apoptosis induced by cisplatin. The present results are consistent with the study of Mori et al (14). In addition, Liu et al (2) reported that an HSP70 inhibitor combined with cisplatin suppresses cervical cancer cell proliferation and transplanted tumor growth. Collectively, these results suggest that inhibition of HSP70 is beneficial to the antitumor activity of cisplatin.
The present study also investigated the potential molecular mechanism of HSP70 antagonism of cisplatin-induced apoptosis. HGC-27 cells were treated with cisplatin for different amounts of time, and the phosphorylation states of p38 MAPK, ERK, JNK, Src, Akt and IκB were monitored by western blotting. The phosphorylation of p38, ERK and JNK were enhanced following cisplatin treatment, but Src, Akt and IκB were unaffected. This finding suggested that the MAPK signaling pathway is involved in cisplatin-induced apoptosis in gastric cancer cells. Therefore, to determine whether HSP70 regulates the activation of MAPK signaling, HSP70 was over-expressed in HGC-27 cells, and the results revealed no effect on the cisplatin-induced phosphorylation of p38, ERK and JNK compared with the empty plasmid-only transfected cells. However, knockdown of HSP70 by HSP70 shRNA transfection attenuated the phosphorylation of p38, ERK and JNK, similar to the HSP70 inhibition by PES treatment (Fig. 4). Taken together, these results demonstrated that HSP70 facilitates the activation of MAPK signaling in response to cisplatin in HGC-27 cells. Notably, overexpression of HSP70 in HGC-27 cell did not affect the phosphorylation of p38, ERK and JNK induced by cisplatin. A possible reason for this phenomenon might be the high expression levels of HSP70 in gastric cancer cells; the phosphorylation of p38, ERK and JNK induced by cisplatin may already be at a maximum level in these cells and therefore additional HSP70 expression may have no further effect on the activation of MAPK signaling pathway.
To establish that the MAPK pathway is involved in cisplatin-induced gastric cell apoptosis, specific inhibitors were used to suppress the activation of p38, ERK and JNK, and then cisplatin-induced apoptosis was examined. As expected, suppression of p38, ERK and JNK phosphorylation enhanced cisplatin-induced apoptosis (Fig. 5). It has been reported that ERK regulates cell apoptosis by promoting cell proliferation, while p38 and JNK are typically described as stress-activated kinases that mediate apoptotic signals (24–26). However, there are also some reports demonstrating that JNK and p38 act as anti-apoptotic signals (27,28). The present study tested the hypothesis that the MAPK signaling pathway acts as an anti-apoptotic signal in cisplatin-induced gastric cell apoptosis.
Collectively, the current results demonstrated that HSP70 protected cisplatin-induced gastric cancer cells from apoptosis by regulating the MAPK signaling pathway (Fig. 6). These findings present a novel insight into the anti-apoptotic mechanism of HSP70 and provide a theoretical basis for targeting HSP70 in clinical therapy of cancer drug resistance.
Figure 6.
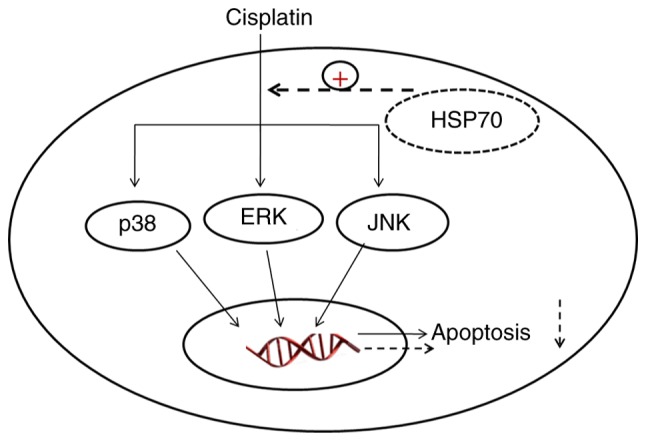
Schematic diagram illustrating the signaling pathway involved in the regulation of HSP70 in cisplatin-induced HGC-27 cell apoptosis. HSP70, heat shock protein 70; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase.
Acknowledgments
The authors thank teacher Deyu Dou for her help with the flow cytometry experiments. We appreciate the department of Parasitology for assisting with the experiments.
Funding
This work was supported by the Anhui Province Natural Science Foundation (grant no. 1708085MH231), National Natural Science Foundation of China (grant no. 81601380), Natural Science Research Project of Anhui Colleges and Universities (grant no. KJ2016SD59), Outstanding Young Talent Support Program Key Projects in Anhui Colleges and Universities (grant no. gxyqZD2016173), research funding project for college students of Wannan Medical College (grant no. WK2016S24), College Students' Innovation and Entrepreneurship Training Program project (grant nos. 201710368002 and 201710368166) and Anhui Province Key Laboratory of Active Biological Macro-molecules (grant no. 1306C083008).
Availability of data and materials
The analyzed datasets generated during the study are available from the corresponding author on reasonable request.
Authors' contributions
ZQ and YZ conceived and designed the experiments. LS, TT, HT and YM performed the experiments. ZW, YL analyzed the data. LS and ZQ wrote the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J, Liu J, Guo SY, Liu HL, Li SZ. HSP70 inhibitor combined with cisplatin suppresses the cervical cancer proliferation in vitro and transplanted tumor growth: An experimental study. Asian Pac J Trop Med. 2017;10:184–188. doi: 10.1016/j.apjtm.2017.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Budina-Kolomets A, Webster MR, Leu JI, Jennis M, Krepler C, Guerrini A, Kossenkov AV, Xu W, Karakousis G, Schuchter L, et al. HSP70 inhibition limits FAK-dependent invasion and enhances the response to melanoma treatment with BRAF inhibitors. Cancer Res. 2016;76:2720–2730. doi: 10.1158/0008-5472.CAN-15-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshidomi K, Murakami A, Yakabe K, Sueoka K, Nawata S, Sugino N. Heat shock protein 70 is involved in malignant behaviors and chemosensitivities to cisplatin in cervical squamous cell carcinoma cells. J Obstet Gynaecol Res. 2014;40:1188–1196. doi: 10.1111/jog.12325. [DOI] [PubMed] [Google Scholar]
- 5.Daugaard M, Rohde M, Jäättelä M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 6.Kumar S, Stokes J, III, Singh UP, Scissum Gunn K, Acharya A, Manne U, Mishra M. Targeting Hsp70: A possible therapy for cancer. Cancer Lett. 2016;374:156–166. doi: 10.1016/j.canlet.2016.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radons J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones. 2016;21:379–404. doi: 10.1007/s12192-016-0676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Assimon VA, Gillies AT, Rauch JN, Gestwicki JE. Hsp70 protein complexes as drug targets. Curr Pharm Des. 2013;19:404–417. doi: 10.2174/138161213804143699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granato M, Lacconi V, Peddis M, Lotti LV, Di Renzo L, Gonnella R, Santarelli R, Trivedi P, Frati L, D'Orazi G, et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013;4:e730. doi: 10.1038/cddis.2013.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao J, Tong C, Liu Y, Wang J, Ni X, Xiong MM. Ginkgetin inhibits growth of breast carcinoma via regulating MAPKs pathway. Biomed Pharmacother. 2017;96:450–458. doi: 10.1016/j.biopha.2017.09.077. [DOI] [PubMed] [Google Scholar]
- 12.Mou S, Zhou Z, He Y, Liu F, Gong L. Curcumin inhibits cell proliferation and promotes apoptosis of laryngeal cancer cells through Bcl-2 and PI3K/Akt, and by upregulating miR-15a. Oncol Lett. 2017;14:4937–4942. doi: 10.3892/ol.2017.6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Zhou Y, Li Y, Zhou J, Wu Y, Cui Y, Yang G, Hong Y. Mutations of p53 and KRAS activate NF-κB to promote chemo-resistance and tumorigenesis via dysregulation of cell cycle and suppression of apoptosis in lung cancer cells. Cancer Lett. 2015;357:520–526. doi: 10.1016/j.canlet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Mori Y, Terauchi R, Shirai T, Tsuchida S, Mizoshiri N, Arai Y, Kishida T, Fujiwara H, Mazda O, Kubo T. Suppression of heat shock protein 70 by siRNA enhances the antitumor effects of cisplatin in cultured human osteosarcoma cells. Cell Stress Chaperones. 2017;22:699–706. doi: 10.1007/s12192-017-0793-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goloudina AR, Demidov ON, Garrido C. Inhibition of HSP70: A challenging anti-cancer strategy. Cancer Lett. 2012;325:117–124. doi: 10.1016/j.canlet.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Daniel R. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10:86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Wang J, Zhou Y, Wang Y, Wang S, Zhang W. Hsp70 promotes chemoresistance by blocking Bax mitochondrial trans-location in ovarian cancer cells. Cancer Lett. 2012;321:137–143. doi: 10.1016/j.canlet.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 18.Behnsawy HM, Miyake H, Kusuda Y, Fujisawa M. Small interfering RNA targeting heat shock protein 70 enhances chemosensitivity in human bladder cancer cells. Urol Oncol. 2013;31:843–848. doi: 10.1016/j.urolonc.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Ding L, He S, Sun X. HSP70 desensitizes osteosarcoma cells to baicalein and protects cells from undergoing apoptosis. Apoptosis. 2014;19:1269–1280. doi: 10.1007/s10495-014-0995-y. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Liu L, Xing D, Chen WR. Inhibition of the JNK/Bim pathway by Hsp70 prevents Bax activation in UV-induced apoptosis. FEBS Lett. 2010;584:4672–4678. doi: 10.1016/j.febslet.2010.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CD, Chen SU, Chou CH, Chen MJ, Wen WF, Wu SY, Yang YS, Yang JH. High estradiol concentrations induce heat shock protein 70 expression and suppress nuclear factor kappa B activation in human endometrial epithelial cells. Biol Reprod. 2016;95:87. doi: 10.1095/biolreprod.116.140012. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Shao Z, Xiong L, Yang S. Inhibition of autophagy enhances cisplatin-induced apoptosis in the MG63 human osteosarcoma cell line. Oncol Lett. 2015;10:2941–2946. doi: 10.3892/ol.2015.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seki K, Yoshikawa H, Shiiki K, Hamada Y, Akamatsu N, Tasaka K. Cisplatin (CDDP) specifically induces apoptosis via sequential activation of caspase-8, -3 and -6 in osteosarcoma. Cancer Chemother Pharmacol. 2000;45:199–206. doi: 10.1007/s002800050030. [DOI] [PubMed] [Google Scholar]
- 24.Qi S, Kou X, Lv J, Qi Z, Yan L. Ampelopsin induces apoptosis in HepG2 human hepatoma cell line through extrinsic and intrinsic pathways: Involvement of P38 and ERK. Environ Toxicol Pharmacol. 2015;40:847–854. doi: 10.1016/j.etap.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 25.Dyari HRE, Rawling T, Chen Y, Sudarmana W, Bourget K, Dwyer JM, Allison SE, Murray M. A novel synthetic analogue of ω-3 17,18-epoxyeicosatetraenoic acid activates TNF receptor-1/ASK1/JNK signaling to promote apoptosis in human breast cancer cells. FASEB J. 2017;31:5246–5257. doi: 10.1096/fj.201700033R. [DOI] [PubMed] [Google Scholar]
- 26.Vethakanraj HS, Sesurajan BP, Padmanaban VP, Jayaprakasam M, Murali S, Sekar AK. Anticancer effect of acid ceramidase inhibitor ceranib-2 in human breast cancer cell lines MCF-7, MDA MB-231 by the activation of SAPK/JNK, p38 MAPK apoptotic pathways, inhibition of the Akt pathway, downregulation of ERα. Anticancer Drugs. 2018;29:50–60. doi: 10.1097/CAD.0000000000000566. [DOI] [PubMed] [Google Scholar]
- 27.Ruan J, Qi Z, Shen L, Jiang Y, Xu Y, Lan L, Luo L, Yin Z. Crosstalk between JNK and NF-κB signaling pathways via HSP27 phosphorylation in HepG2 cells. Biochem Biophys Res Commun. 2015;456:122–128. doi: 10.1016/j.bbrc.2014.11.045. [DOI] [PubMed] [Google Scholar]
- 28.Qi Z, Shen L, Zhou H, Jiang Y, Lan L, Luo L, Yin Z. Phosphorylation of heat shock protein 27 antagonizes TNF-α induced HeLa cell apoptosis via regulating TAK1 ubiquitination and activation of p38 and ERK signaling. Cell Signal. 2014;26:1616–1625. doi: 10.1016/j.cellsig.2014.03.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed datasets generated during the study are available from the corresponding author on reasonable request.