

Abstract
Cerebral edema is a life-threatening neurological condition characterized by brain swelling due to accumulation of excess fluid both intracellularly and extracellularly. Fulminant hepatic failure (FHF) develops cerebral edema by disrupting blood brain barrier (BBB). However, the mechanisms by which mediator induces brain edema in FHF remain to be elucidated. Here, we assessed a linkage between brain edema and lysophosphatidic acid (LPA) signaling by utilizing an animal model of FHF and in vitro BBB model. Azoxymethane-treated mice developed FHF and hepatic encephalopathy, associated with higher autotaxin (ATX) activities in serum than controls. Using in vitro BBB model, LPA disrupted the structural integrity of tight junction proteins including claudin-5, occludin and ZO-1. Furthermore, LPA decreased transendothelial electrical resistances in in vitro BBB model, and induced cell contraction in brain endothelial monolayer cultures, both being inhibited by a Rho-associated protein kinase inhibitor, Y-27632. The brain capillary endothelial cells predominantly expressed LPA6 mRNA, whose knockdown blocked the LPA-induced endothelial cell contraction. Taken together, the up-regulation of serum ATX in hepatic encephalopathy may activate the LPA–LPA6–G12/13–Rho pathway in brain capillary endothelial cells, leading to enhancement of BBB permeability and brain edema.
Keywords: lipid mediator, lysophospholipid, neurological diseases, GPCR, ROCK, p2ry5
Graphical abstract:
Introduction
Blood-brain barrier (BBB) separates a brain from circulatory system to protect the central nervous system (CNS) from xenobiotic or toxic substances, while allowing entry of beneficial nutrients to the CNS [1]. This primary interface is composed of tight junction-based morphological barrier between endothelial cells, and also cell-cell interactions of endothelial cells with pericytes and astrocyte end-feet [1]. Disruption of BBB causes brain edema and serious complication frequently associated with neurological conditions such as stroke, brain tumors and meningitis [2]. Acute liver failure is a life-threatening condition most often caused by virus infections and drug intoxication [3, 4]. It produces hepatic encephalopathy and cytotoxic brain edema characterized by swelling of astrocytes and intact BBB [5, 6]. On the other hand, vasogenic mechanism in fulminant hepatic failure (FHF)-associated brain edema is proposed, since liver-derived matrix metalloproteinase-9 disrupts BBB worsening brain edema in a murine model of FHF [7].
Lysophosphatidic acid (LPA; 1- or 2-acyl-sn-glycero-3-phosphate) is a bioactive lysophospholipid that induces a variety of cellular responses such as angiogenesis, neurite retraction, cytoskeletal reorganization and cell proliferation [8]. LPA is produced from lysophosphatidylcholine (LPC) by the action of autotaxin (ATX) that is a major determinant of LPA levels in the blood [9]. Plasma LPA level and serum ATX activities are elevated in human and animal hepatitis [10, 11], due to impairment of ATX clearance [12]. This implies a possible linkage among ATX activities, brain edema, and BBB disruption. Biological function of LPA is elicited through six subtypes of G protein-coupled receptors (GPCRs), LPA1–6 [13, 14]. Several in vitro studies indicated the roles of LPA in endothelial barrier function [15–17], suggesting LPA as one of the important factor of vasogenic edema. However, it has not yet determined which LPA receptor subtypes are responsible for regulating endothelial BBB function. In the present study, we demonstrate that serum ATX activity increased in FHF mice, and LPA dysregulated endothelial BBB function via LPA6.
Materials and methods
Mice
Male C57BL/6J mice weighing 24–28 g were purchased from CLEA Japan. Mice were housed under specific pathogen-free conditions in an air-conditioned room and fed standard laboratory chow ad libitum (MF; Oriental Yeast), in accordance with institutional guidelines. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Tokyo.
Fulminant hepatitis model induced by azoxymethane (AOM)
The mouse model of FHF was induced with an intraperitoneal injection of AOM (Sigma-Aldrich) at 50 μg/g of body weight. Blood samples were collected from anesthetized mice 24 h after injection. Serum was used for measuring activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), choline esterase (ChE) and alkaline phosphate (ALP) as well as concentrations of ammonia and albumin (Alb). All blood tests were performed by SRL Inc. (Tokyo, Japan). Livers were harvested and fixed in 10% formalin. These livers were processed, embedded in paraffin and stained with hematoxylin and eosin. For the analysis of mice in hepatic encephalopathy, a solution of 10% dextrose in saline (quarter-normal saline) was orally given at a dose of 0.2 ml/mouse every 3 h during the precoma and comatose stages to maintain intravascular volume. When comatose stage of encephalopathy was confirmed, blood samples were collected to measure ammonia level and ATX activity (see below).
At the comatose stage, 2% evans blue dye (EBD; Sigma-Aldrich) dissolved in PBS was administered intravenously at a dose of 4 ml/kg and allowed to circulate for 20–30 min in some mice. After mice were anesthetized with urethane (Sigma-Aldrich), the brains were removed and divided into right and left hemispheres. One hemisphere was weighed, homogenized in an eight-fold volume of 50% trichloroacetic acid to dissociate the dye from proteins. After centrifugation at 10,000 × g for 10 min, the supernatants containing EBD were diluted with a four-fold volume of ethanol. Tissue levels of EBD were quantitated using the Fusion system (PerkinElmer Life Sciences) at an excitation wavelength of 620 nm and an emission wavelength of 675 nm. Sample values were compared with those of EBD standards mixed with the solvent (50–10,000 ng/ml).
Measurement of ATX activity
ATX activity was measured as lysophospholipase D activity according to Watanabe et al. [10]. In brief, serum samples were incubated with 2 mM 1-myristoyl lysoPC (Avanti Polar Lipids) in the presence of 100 mM Tris-HCl (pH 9.0), 500 mM NaCl, 5 mM MgCl2, 5 mM CaCl2 and 0.05% Triton X-100 for 3 h at 37°C. The liberated choline was detected by an enzymatic photometric method using choline oxidase (Asahi Chemical), horseradish peroxidase (Toyobo), and TOOS reagent (N-ethyl-N-(2-hydoroxy-3-sulfoproryl)-3-methylaniline; Dojindo, Kumamoto) as a hydrogen donor. Absorbance was read at 560 nm and converted to nanomoles of choline by comparison to a choline standard curve.
Cell culture and construction of BBB model
Rat brain endothelial cells (RBECs), astrocytes and pericytes were isolated and the in vitro BBB model was constructed with these three cell types, as previously described [18, 19]. The co-cultured cells were maintained in DMEM/F12 supplemented with 10% plasma-derived serum (Animal Technologies), 1.5 ng/ml basic fibroblast growth factor (Roche Applied Sciences), 100 μg/ml heparin, 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml sodium selenite, 50 μg/ml gentamycin and 500 nM hydrocortisone (all from Sigma-Aldrich) and used for experiments 3–5 days after culture.
Immunocytochemistry
To observe expression patterns of brain endothelial junctional proteins, RBECs co-cultured with astrocytes and pericytes on the insert membrane of transwell were stained for ZO-1, occludin and claudin-5. The insert membrane was washed in PBS and fixed with 3% paraformaldehyde in PBS for 10 min, and cells were permeabilized with 0.01% Triton-X for 10 min. Following blocking with 3% BSA (Sigma-Aldrich), the cells were incubated with the primary antibody anti-ZO-1 (1:100), anti-occludin (1:125) or anti-claudin-5 (1:100) (all from Zymed Laboratories) for 30 min. After washing with PBS three times, the cells were incubated for 30 min with either Alexa Fluor 546-conjugated goat anti-rabbit IgG (Invitrogen; 1:200) for ZO-1 and occludin or Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen; 1:200) for claudin-5. After mounting, confocal microscopy was performed with a Zeiss LSM510 laser-scanning microscope.
Assessment of endothelial barrier function following LPA stimulation in vitro
For the measurement of LPA-induced changes in barrier function of the RBEC monolayer, we used the xCELLigence system (Roche Applied Sciences), which detects the electrical impedance of cells grown directly on a gold microelectrode plate (E-plate; Roch Applied Sciences). Changes in impedance (represented as “cell index”) reflect changes in the endothelial barrier function and permeability [20]. In brief, RBECs (20,000–30,000 cells/well) were seeded on 96-well microtiter E-plates coated with collagen IV (Sigma-Aldrich) and fibronectin (Sigma-Aldrich), followed by a 12-h culture in 5% CO2 at 37°C. Cells were serum-starved with DMEM/F12 containing 0.1% BSA for 4 h and stimulated with various concentrations of 1-oleoyl-LPA (Avanti Polar Lipids) dissolved in PBS containing 3% BSA. The cell index was continuously monitored every 15 s for the duration of the experiments. To evaluate LPA-induced changes in TEER of BBB model, we utilized Epithelial-Volt-Ohm Meter and EndOhm-12 chamber electrode (World Precision Instruments). The BBB model was cultured on transwell plates for several days to obtain initial TEER values above 150 ohm·cm2, and then subjected to LPA stimulation. TEER was measured at indicated times during the experimental procedure. In some experiments, cells were pretreated with 10 μM Y-27632 (Calbiochem) or 10 μM Ki16425 (Sigma-Aldrich) for 10 min before LPA stimulation.
mRNA expression profile of LPA receptors in cells composing BBB model
Total RNA was extracted from RBECs, pericytes and astrocytes using an RNeasy minikit (Qiagen). The first-strand cDNA was synthesized from 96 ng of total RNA by reverse transcription (RT) PCR using Superscript III (Invitrogen). An equal aliquot of the cDNA solution was used to amplify the different receptor transcripts using ExTaq DNA polymerase (Takara). The PCR program was as follows: denaturation at 96°C for 2 min and 35 cycles of amplification consisting of denaturation at 94°C for 10 sec, annealing at 56°C for 10 sec and extension at 72°C for 30 sec. The following primers were used (the expected size of the PCR products is shown in parentheses): LPA1-fw 5’-ttgtgctgggtgcctttattg-3’, LPA1-rv 5’-gatggggttcatagccgagtt-3’ (143 bp); LPA2-fw 5’-ccccgctaccgagagacaac-3’, LPA2-rv 5’-gtccaagagcagcaccacct-3’ (106 bp); LPA3-fw 5’-ctacaacaggagcaacacagacac-3’, LPA3-rv 5’-tgcgatgaccagggagttaga-3’ (121 bp); LPA4-fw 5’-tacccgattaccttgtgccttg-3’, LPA4-rv 5’-aacagcgactccatccttatgtgt-3’ (125 bp); LPA5-fw 5’-ggagtgctgatgataatggtgct-3’, LPA5-rv 5’-ggtgctttctgagggtggttc-3’ (183 bp); LPA6-fw 5’-agcagaagcaaaatgaacaaaacc-3’, LPA6-rv 5’-gttggagacagcaatgcagaga-3’ (198 bp); vWF-fw 5’-aagctttcatctcaaaggccaac-3’, vWF-rv 5’-catgaggtccaccaaactctgta-3’ (142bp); GFAP-fw 5’-atcgtggtaaagacggtggagat-3’, GFAP-rv 5’-tatctgaggagggagctttaggc-3’ (144 bp); NG2-fw 5’-ctggctgtgactgtgtctttcg-3’, NG2-rv 5’-ctggcacactggctaggaggt-3’ (152 bp); claudin-5 5’-ccctgctcagaacagactacagg-3’, claudin-5-rv 5’-ccccaggactcagtaggaactgt-3’ (148 bp). The PCR products were separated by electrophoresis on a 1% agarose gel.
siRNA transfection
RBECs (80,000 cells/cuvette) were transfected with 2 μM rat LPA6 siRNA (Silencer Select siRNA, ID number 228813; Ambion) or control siRNA (Silencer Select negative control 1; Ambion) using the Amaxa Nucleofector system under the T-005 program according to the manufacture’s instructions. Cells were stimulated with 30 μM of LPA or vehicle 48 h after siRNA transfection.
Statistical analyses
Data were expressed as the means ± SEM and analyzed statistically by Mann-Whitney U test or unpaired two-tailed t test using Prism 4 software (GraphPad Software). The differences were considered significant if P values were less than 0.05.
Results and Discussion
AOM induces FHF in C57BL/6 mice.
To investigate a possible link between LPA and BBB dysfunction in vivo, we used a mouse model of brain edema induced by FHF. AOM is an active metabolite of the cycad palm nuts, which can induce FHF and hepatic encephalopathy in C57BL/6 mice with a single injection [21]. Plasma ALT and AST levels increased in C57BL/6 mice 24 h after AOM injection, while plasma Alb, ALP and ChE levels decreased (Fig. 1A). Histopathological examination of liver sections from AOM-treated mice revealed centrilobular sinusoidal dilation, hemorrhage and hepatocyte necrosis (Fig. 1B). These results indicate that AOM induced FHF in C57BL/6 mice.
Fig. 1. AOM-injected mice developed FHF.
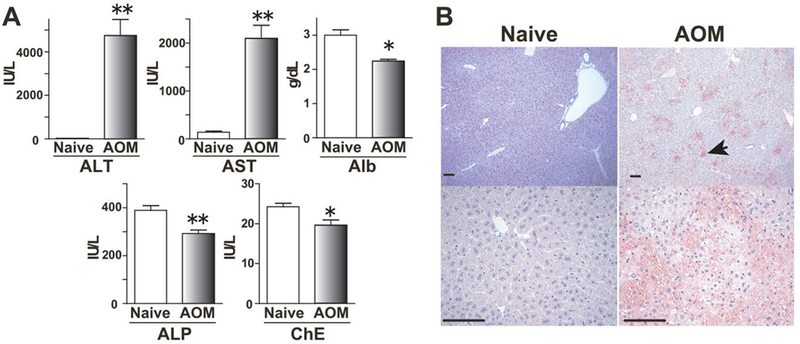
(A) Blood levels of ALT, AST, Alb, ALP and ChE at 24 h after AOM injection are shown (Control naive mice, n = 6; AOM-treated mice, n = 4). Mean + SEM, *, P< 0.05, **, P < 0.01 versus control mice by Mann-Whitney U test. (B) Histopathological changes of liver were observed 24 h after AOM exposure. Hematoxylin and eosin staining shows the severe hemorrhage (arrows), microvesicular steatosis and centrilobular necrosis in AOM-treated mice (Bars, 100 μm).
ATX activities correlate with FHF-associated brain edema.
Increased blood ammonia level in FHF is known to induce brain edema, which is involved in cytotoxic edema [22]. We examined AOM-induced hepatic encephalopathy by measuring ammonia levels in blood. When mice showed a decreased spontaneous locomotive activity and became comatose (at the stage of encephalopathy), blood samples were harvested. The blood samples of AOM-treated mice had significantly higher ammonia level than those of control mice (Fig. 2A). To confirm that AOM-treated mice showed brain edema, EBD extravasation in brains was measured at the comatose stage. The mean concentration of EBD in brains of AOM-treated mice was about twice as high as that of control mice (12.8 ± 1.0 ng/mg brain vs. 6.7 ± 0.5 ng/mg brain, respectively; Fig 2B), suggesting that AOM-induced FHF mice developed cerebral edema.
Fig. 2. AOM-injected FHF mice developed brain edema and increased ATX activity.
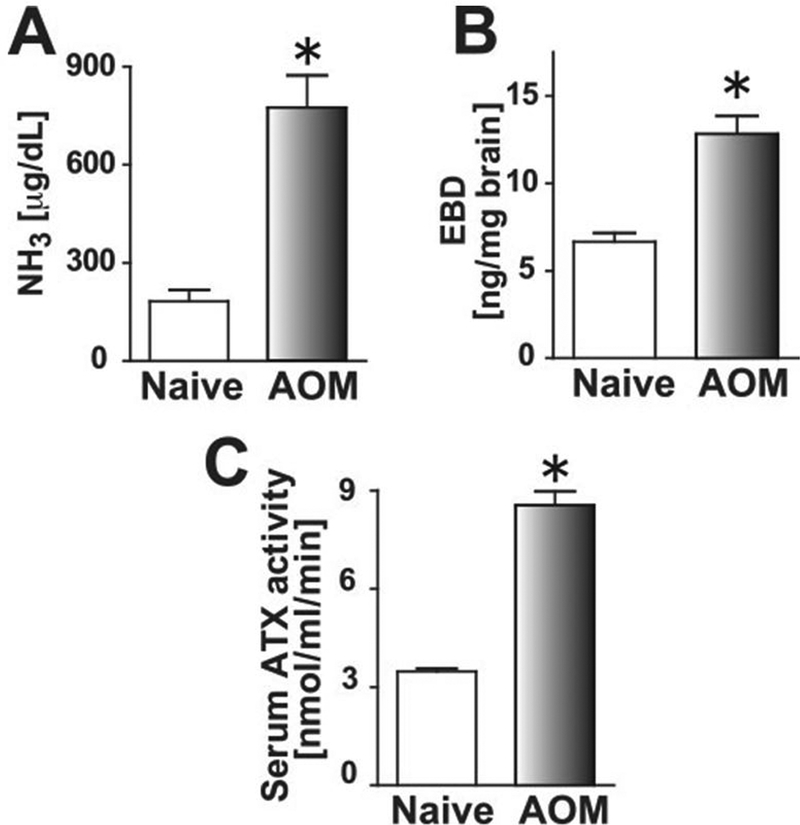
(A) Blood ammonia levels of control and comatose FHF mice were measured (Control, n = 5; AOM, n = 4). Mean + SEM, *, P < 0.05 versus control mice by two-tailed Student’s t test. (B) Brains from control and comatose FHF mice were harvested after intravenous injection of EBD. Extravasated brain EBD was quantified fluorometrically (Control, n = 6; AOM, n = 5). Mean + SEM, *, P < 0.01 versus control mice by two-tailed Student’s t test. (C) Serum ATX activities in control and comatose FHF mice were measured (Control, n = 10; AOM, n = 12). Mean + SEM, *, P < 0.0001 versus control mice by two-tailed Student’s t test.
Increased serum ATX activities were observed in patients with chronic hepatitis [10] and in rat models of acute and chronic hepatitis [11], presumably because hepatic failure delays the clearance of circulating ATX [12]. Therefore, we examined the ATX activity in mouse FHF serum to investigate whether LPA contributes to encephalopathy in FHF. The serum ATX activity was significantly increased in AOM-treated mice (8.5 ± 0.4 nmol/ml/min) compared with control mice (3.5 ± 0.1 nmol/ml/min) (Fig. 2C). The increased serum ATX activity, blood ammonia level, and extravasation of EBD indicated that AOM-induced FHF produced LPA-dependent vasogenic edema, which might be mediated through LPA signaling in BBB components.
LPA deranges barrier integrity in an in vitro BBB model.
To test whether LPA modifies endothelial BBB integrity or not, LPA-induced TEER change was measured using in vitro BBB model with an epithelial-volt-ohm meter. This triple co-culture model comprised of monolayers of RBECs on the upper side of the transwell inserts, pericytes on the lower side of the inserts and astrocytes at the bottom of the culture dishes (Fig. 3A). By mimicking the anatomical situation in the cerebral microvessels, the in vitro BBB model is able to maintain the strong barrier integrity compared to the other in vitro BBB models tested [18, 19]. To observe LPA-induced change in morphological integrity, RBECs cultured on transwell insert in BBB model were stained for ZO-1, claudin-5, and occludin after 15 min following LPA stimulation (Fig. 3B). The expression of claudin-5 in non-treated RBECs was characterized by “belt-like” localization, whereas LPA treatment changed the localization from “belt-like” to “zipper-like”. Although tight junction proteins existed at cell-cell contacts, cell shapes became irregular after LPA treatment (Fig. 3B). In accordance with the LPA-induced morphorogical changes, LPA rapidly decreased TEER to 50% of the initial value within 15 min (Fig. 3C). Pertussis toxin (PTX) was used as a positive control, because it is reported to decrease TEER [23], resulting in severe TEER decrease (Fig. 3C). The vascular permeability is partly determined by balance between Rho and Rac1 activities [24], and activation of RhoA pathway is known to increase the permeability. Therefore, Y-27632, a Rho-associated protein kinase (ROCK) inhibitor, was added on the upper side of BBB model for 10 min before LPA stimulation. The LPA-induced TEER decrease was completely inhibited by Y-27632 (Fig. 3D), suggesting that LPA activates Rho-ROCK pathways mainly in RBECs. On the other hand, Ki16425, an antagonist for LPA1 and LPA3, had little or no effects on TEER decrease caused by LPA (Fig. 3D). These results suggest that G12/13-coupled LPA receptors, which activate Rho-ROCK pathway, in RBECs are responsible for regulating BBB integrity.
Fig. 3. LPA disrupted BBB barrier function in in vitro BBB model.
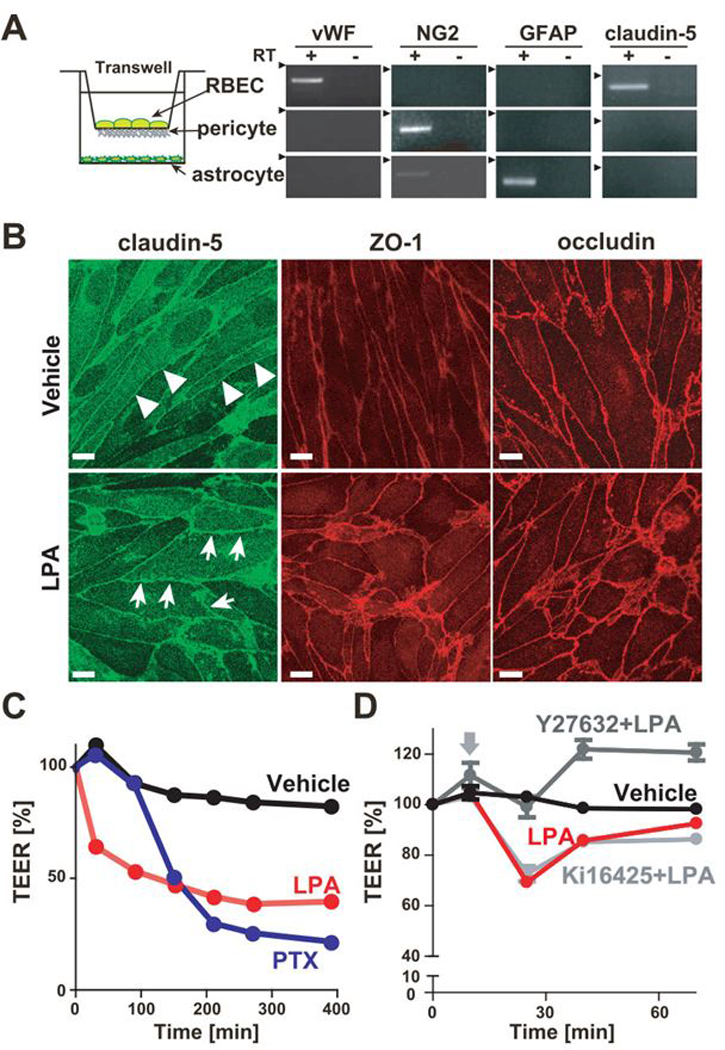
(A) Schematic representation of the in vitro BBB model is shown. mRNA expression in three primary cultured cells were examined by RT-PCR. (B) Immunofluorescent staining of confluent RBECs in BBB model for tight junction proteins ZO-1, claudin-5 and occludin. Cells were immunostained 15 min after 50 μM LPA stimulation. Arrows show zipper-like staining between LPA-treated RBECs, while the junctional immunostaining of RBECs forms a continuous, smooth and belt-like pattern (arrowheads) in vehicle-treated RBECs (Bar, 10 μm). (C) Change of TEER in BBB model treated with 50 μM LPA and 200 ng/ml PTX was evaluated by an epithelial-volt-ohm meter. Data is representative of three independent experiments with similar results. (D) Change of TEER in BBB model was evaluated by an epithelial-volt-ohm meter. Cells were pretreated with or without 10 μM Y-27632 and 10 μM Ki16425 for 10 min and then stimulated with 50 μM. Data is representative of three independent experiments with similar results.
LPA increases barrier permeability of RBECs via LPA6-Rho–ROCK signaling pathway.
To exclude the possible effects from pericytes and astrocytes, we analyzed the monolayer culture of RBECs with the xCELLigence system, which is a highly sensitive system for real time measurement of endothelial permeability [25]. Upon LPA stimulation, the decrease in cell index was clearly observed in an LPA dose-dependent manner (Fig. 4A). Furthermore, Y-27632 completely blocked the LPA-induced cell index decrease (Fig. 4B). In contrast, the response of RBECs to LPA was only slightly affected by pretreatment with Ki16425 (Fig. 4B), implying that LPA1 and LPA3 are marginally involved in the decrease in cell index of RBECs upon LPA stimulation.
Fig. 4. LPA disrupted barrier function of RBEC monolayer cultures through LPA6.
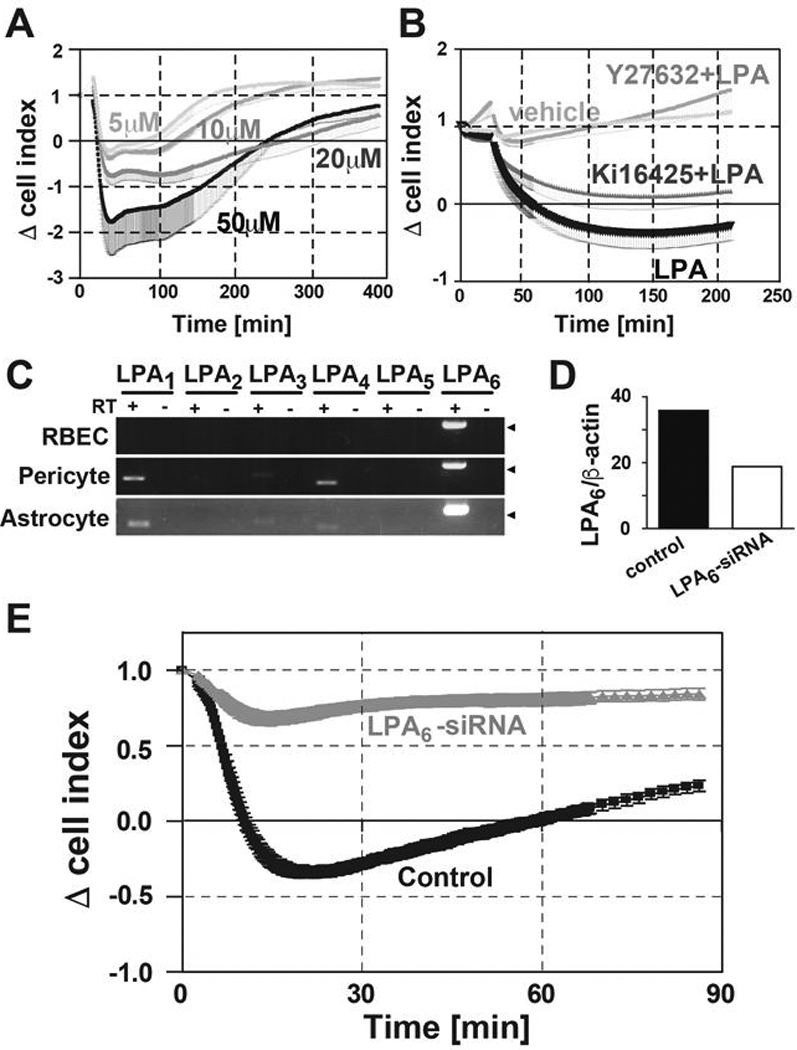
(A) Change of cell index in RBECs treated with 5, 10, 20 and 50 μM of LPA was evaluated by the xCELLigence system (n = 3). Data is representative of two independent experiments with similar results. Mean - SEM (B) RBECs were pretreated with or without 10 μM Y-27632 and 10 μM Ki16425 for 10 min. Then 10 μM LPA was added and the change of cell index was monitored (n = 3). Data are representative of two independent experiments with similar results. Mean - SEM. (C) mRNA expression profile of LPA receptors in RBECs pericytes and astrocytes were analyzed by RT-PCR. Arrowheads indicate the 200-bp DNA size marker. (D) Expression levels of LPA6 and β-actin mRNA are presented as a percentage of those with control siRNA (n = 2). Data is representative of three independent experiments with similar results. (E) Change of cell index in RBECs treated with 50 μM LPA was evaluated by xCELLigence system 48 h after siRNA transfection (n = 3). Data is representative of three independent experiments with similar results. Mean ± SEM.
To identify the responsible LPA receptors in controlling BBB integrity, the LPA receptor gene expression profiles in cells comprising BBB model were analyzed by RT-PCR. In RBECs, LPA6 was predominantly expressed (Fig. 4C), whereas pericytes and astrocytes expressed multiple LPA receptors LPA1, LPA3, LPA4 and LPA6. We previously reported that LPA6 induces ROCK-dependent morphological change in human umbilical vascular endothelial cells [25]. Following reports published by other research groups supported the notion that LPA6 couples to G12/13, leading to the activation of Rho and ROCK [26]. Therefore, to further investigate the role of endogenous LPA6 in RBECs, the monolayer RBECs were electroporated with LPA6 siRNA. The suppression of LPA6 gene expression was confirmed by quantitative real-time PCR 48 h after transfection, which showed about 50% reduction in LPA6 mRNA levels. Importantly, the response (Δcell index) to LPA was considerably inhibited in RBECs treated with LPA6 siRNA (Fig. 4D).
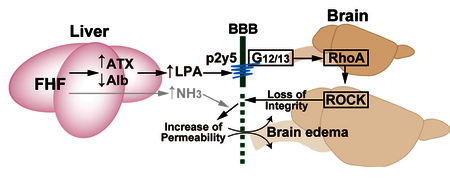
Taken together, we demonstrated the up-regulation of ATX activity in mice with FHF, which may be associated with hepatic encephalopathy and loss of BBB integrity. LPA6 is predominantly expressed in RBECs and the LPA–LPA6–G12/13–Rho pathway is involved in increase of endothelial BBB permeability. Osmotherapy using mannitol and glycerol is the most effective and rapid treatment for brain edema, reducing intracranial pressure and increasing cerebral blood flow. Currently, treatment of brain edema is limited. However, our findings combined with a recently solved LPA6 structure [27] have important implications for the design of new therapies for BBB disrupting disorders by targeting brain endothelial LPA6.
Highlights:
Serum autotaxin activity is elevated in the mouse with brain edema.
LPA6 is predominantly expressed in rat brain capillary endothelial cells.
LPA–LPA6–G12/13–Rho pathway regulates blood brain barrier permeability.
Acknowledgments
We thank Dr. Satoshi Ishii for generous support of this study, and Ms. Danielle Jones for editorial assistance. This work was supported, in part, by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Culture, Sports and Technology of Japan (to T.S.), and NIH R01NS103940 (to YK). K.Y. and Y.K. were supported by the Research Fellowships of Japanese Society for the Promotion of Science.
Abbreviations:
- LPA
lysophosphatidic acid
- GPCR
G-protein-coupled receptor
- Edg
endothelial cell differentiation gene
- PC
phosphatidylcholine
- lysoPLD
lysophospholipase D
- ATX
autotaxin
- BBB
blood-brain barrier
- TEER
transendothelial electrical resistances
- ROCK
Rho kinase
- FHF
fulminant hepatic failure
- AOM
azoxymethane
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ChE
choline esterase
- ALP
alkaline phosphatase
- Alb
albumin
- EBD
Evans blue dye
- RBEC
rat brain endothelial cell
- vWF
von Willebrand factor
- GFAP
glial fibrillary acidic protein
- PTX
pertussis toxin
- RT
reverse transcription
Footnotes
Conflict of interest
Masami Niwa and Shinsuke Nakagawa are founder of PharmaCo-Cell Company Ltd. Other authors have no conflicts of interest to declare.
References
- [1].Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ, Structure and function of the blood-brain barrier, Neurobiol Dis 37 (2010) 13–25. [DOI] [PubMed] [Google Scholar]
- [2].Walcott BP, Kahle KT, Simard JM, Novel treatment targets for cerebral edema, Neurotherapeutics 9 (2012) 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bernal W, Wendon J, Acute liver failure N Engl J Med 369 (2013) 2525–2534. [DOI] [PubMed] [Google Scholar]
- [4].Rama Rao KV, Reddy PV, Tong X, Norenberg MD, Brain edema in acute liver failure: inhibition by L-histidine, Am J Pathol 176 (2010) 1400–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bernal W, Wendon J, Rela M, Heaton N, Williams R, Use and outcome of liver transplantation in acetaminophen-induced acute liver failure, Hepatology 27 (1998) 1050–1055. [DOI] [PubMed] [Google Scholar]
- [6].Ranjan P, Mishra AM, Kale R, Saraswat VA, Gupta RK, Cytotoxic edema is responsible for raised intracranial pressure in fulminant hepatic failure: in vivo demonstration using diffusion-weighted MRI in human subjects, Metab Brain Dis 20 (2005) 181–192. [DOI] [PubMed] [Google Scholar]
- [7].Nguyen JH, Yamamoto S, Steers J, Sevlever D, Lin W, Shimojima N, Castanedes-Casey M, Genco P, Golde T, Richelson E, Dickson D, McKinney M, Eckman CB, Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice, J Hepatol 44 (2006) 1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kihara Y, Mizuno H, Chun J, Lysophospholipid receptors in drug discovery, Experimental cell research 333 (2015) 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H, Serum lysophosphatidic acid is produced through diverse phospholipase pathways, J Biol Chem 277 (2002) 48737–48744. [DOI] [PubMed] [Google Scholar]
- [10].Watanabe N, Ikeda H, Nakamura K, Ohkawa R, Kume Y, Aoki J, Hama K, Okudaira S, Tanaka M, Tomiya T, Yanase M, Tejima K, Nishikawa T, Arai M, Arai H, Omata M, Fujiwara K, Yatomi Y, Both plasma lysophosphatidic acid and serum autotaxin levels are increased in chronic hepatitis C, J Clin Gastroenterol 41 (2007) 616–623. [DOI] [PubMed] [Google Scholar]
- [11].Watanabe N, Ikeda H, Nakamura K, Ohkawa R, Kume Y, Tomiya T, Tejima K, Nishikawa T, Arai M, Yanase M, Aoki J, Arai H, Omata M, Fujiwara K, Yatomi Y, Plasma lysophosphatidic acid level and serum autotaxin activity are increased in liver injury in rats in relation to its severity, Life Sci 81 (2007) 1009–1015. [DOI] [PubMed] [Google Scholar]
- [12].Jansen S, Andries M, Vekemans K, Vanbilloen H, Verbruggen A, Bollen M, Rapid clearance of the circulating metastatic factor autotaxin by the scavenger receptors of liver sinusoidal endothelial cells, Cancer Lett 284 (2009) 216–221. [DOI] [PubMed] [Google Scholar]
- [13].Kihara Y, Maceyka M, Spiegel S, Chun J, Lysophospholipid receptor nomenclature review: IUPHAR Review 8, British journal of pharmacology 171 (2014) 3575–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yanagida K, Ishii S, Non-Edg family LPA receptors: the cutting edge of LPA research, J Biochem 150 (2011) 223–232. [DOI] [PubMed] [Google Scholar]
- [15].Nitz T, Eisenblatter T, Psathaki K, Galla HJ, Serum-derived factors weaken the barrier properties of cultured porcine brain capillary endothelial cells in vitro, Brain Res 981 (2003) 30–40. [DOI] [PubMed] [Google Scholar]
- [16].Schulze C, Smales C, Rubin LL, Staddon JM, Lysophosphatidic acid increases tight junction permeability in cultured brain endothelial cells, J Neurochem 68 (1997) 991–1000. [DOI] [PubMed] [Google Scholar]
- [17].Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, Kaibuchi K, Persidsky Y, Ikezu T, Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells, Am J Pathol 172 (2008) 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, Tanaka K, Niwa M, A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes, Neurochem Int 54 (2009) 253–263. [DOI] [PubMed] [Google Scholar]
- [19].Nakagawa S, Deli MA, Nakao S, Honda M, Hayashi K, Nakaoke R, Kataoka Y, Niwa M, Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells, Cell Mol Neurobiol 27 (2007) 687–694. [DOI] [PubMed] [Google Scholar]
- [20].Atienza JM, Yu N, Kirstein SL, Xi B, Wang X, Xu X, Abassi YA, Dynamic and label-free cell-based assays using the real-time cell electronic sensing system, Assay Drug Dev Technol 4 (2006) 597–607. [DOI] [PubMed] [Google Scholar]
- [21].Matkowskyj KA, Marrero JA, Carroll RE, Danilkovich AV, Green RM, Benya RV, Azoxymethane-induced fulminant hepatic failure in C57BL/6J mice: characterization of a new animal model, Am J Physiol 277 (1999) G455–462. [DOI] [PubMed] [Google Scholar]
- [22].Butterworth RF, Pathophysiology of hepatic encephalopathy: a new look at ammonia, Metab Brain Dis 17 (2002) 221–227. [DOI] [PubMed] [Google Scholar]
- [23].Kugler S, Bocker K, Heusipp G, Greune L, Kim KS, Schmidt MA, Pertussis toxin transiently affects barrier integrity, organelle organization and transmigration of monocytes in a human brain microvascular endothelial cell barrier model, Cell Microbiol 9 (2007) 619–632. [DOI] [PubMed] [Google Scholar]
- [24].Komarova YA, Mehta D, Malik AB, Dual regulation of endothelial junctional permeability, Sci STKE 2007 (2007) re8. [DOI] [PubMed] [Google Scholar]
- [25].Yanagida K, Masago K, Nakanishi H, Kihara Y, Hamano F, Tajima Y, Taguchi R, Shimizu T, Ishii S, Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6, J Biol Chem 284 (2009) 17731–17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee M, Choi S, Hallden G, Yo SJ, Schichnes D, Aponte GW, P2Y5 is a G(alpha)i, G(alpha)12/13 G protein-coupled receptor activated by lysophosphatidic acid that reduces intestinal cell adhesion, Am J Physiol Gastrointest Liver Physiol 297 (2009) G641–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Taniguchi R, Inoue A, Sayama M, Uwamizu A, Yamashita K, Hirata K, Yoshida M, Tanaka Y, Kato HE, Nakada-Nakura Y, Otani Y, Nishizawa T, Doi T, Ohwada T, Ishitani R, Aoki J, Nureki O, Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6, Nature 548 (2017) 356–360. [DOI] [PubMed] [Google Scholar]