

Summary
In stationary-phase Escherichia coli, Dps (DNA-binding protein from starved cells) is the most abundant protein component of the nucleoid. Dps compacts DNA into a dense complex and protects it from damage. Dps has also been proposed to act as a global regulator of transcription. Here, we directly examine the impact of Dps-induced compaction of DNA on the activity of RNA polymerase (RNAP). Strikingly, deleting the dps gene decompacted the nucleoid but did not significantly alter the transcriptome and only mildly altered the proteome during stationary phase. Complementary in vitro assays demonstrated that Dps blocks restriction endonucleases but not RNAP from binding DNA. Single-molecule assays demonstrated that Dps dynamically condenses DNA around elongating RNAP without impeding its progress. We conclude that Dps forms a dynamic structure that excludes some DNA-binding proteins yet allows RNAP free access to the buried genes, a behavior characteristic of phase-separated organelles.
Keywords: Dps, nucleoid, stationary phase, transcription, stress response, RNA polymerase, single-molecule biophysics, magnetic tweezers, DNA condensation
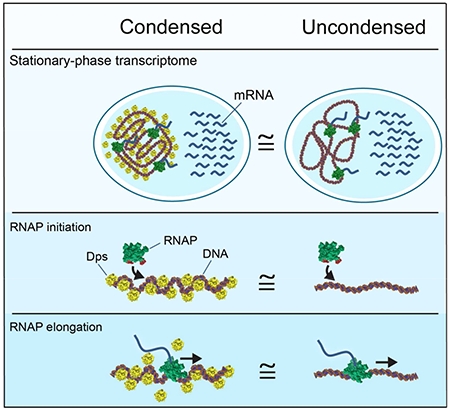
In Brief:
Despite markedly condensing the bacterial chromosome, the nucleoid-structuring protein Dps selectively allows access by RNA polymerase and transcription factors at normal rates while excluding other factors such as restriction endonucleases
Introduction
In all living cells, DNA is organized into compact structures that influence transcription, repair, and replication. In eukaryotic cells, the link between histone-induced DNA compaction and transcriptional activity is well established. Changes in histone occupancy have been shown to provide epigenetic control of transcription (Goldberg et al., 2007), to block the initiation of transcription (Hartley and Madhani, 2009), and to pause or arrest actively transcribing RNA polymerase II (Churchman and Weissman, 2011; Hodges et al., 2009). In addition to compaction due to histones, eukaryotic cells can sequester DNA in membrane-free, phase-separated organelles (Hyman et al., 2014). These structures provide a way to selectively enrich for or exclude proteins from accessing the enclosed DNA. Some organelles, such as heterochromatin, suppress gene expression, while others, such as the nucleolus, allow for transcription to occur on the condensed DNA. Phase separation may underpin genome restructuring into transcriptionally active and silent domains in eukaryotes (Strom et al., 2017).
In bacteria, DNA is organized and condensed by nucleoid-associated proteins (NAPs), a diverse and unevenly understood group (Dorman, 2013). Dps is a NAP that was first discovered in E. coli (Almiron et al., 1992), with homologues identified in over 1000 species of bacteria and Archaea (Calhoun and Kwon, 2011). As E. coli cells enter stationary phase, the expression of Dps increases dramatically (Almiron et al., 1992; Azam et al., 1999; Meyer and Grainger, 2013). Dps can bind DNA and condense it into a dense and compact structure both in vitro and in vivo (Kim et al., 2004; Wolf et al., 1999) (Fig. 1A). While deleting dps causes few detectable phenotypes during exponential phase, Δdps cells subjected to starvation or several other forms of stress exhibit sharp decreases in survival rates (Nair and Finkel, 2004). Importantly, the DNA-binding activity is essential for the stress protection conferred by Dps (Karas et al., 2015).
Figure 1. Dps compacts the nucleoid in stationary-phase E. coli.
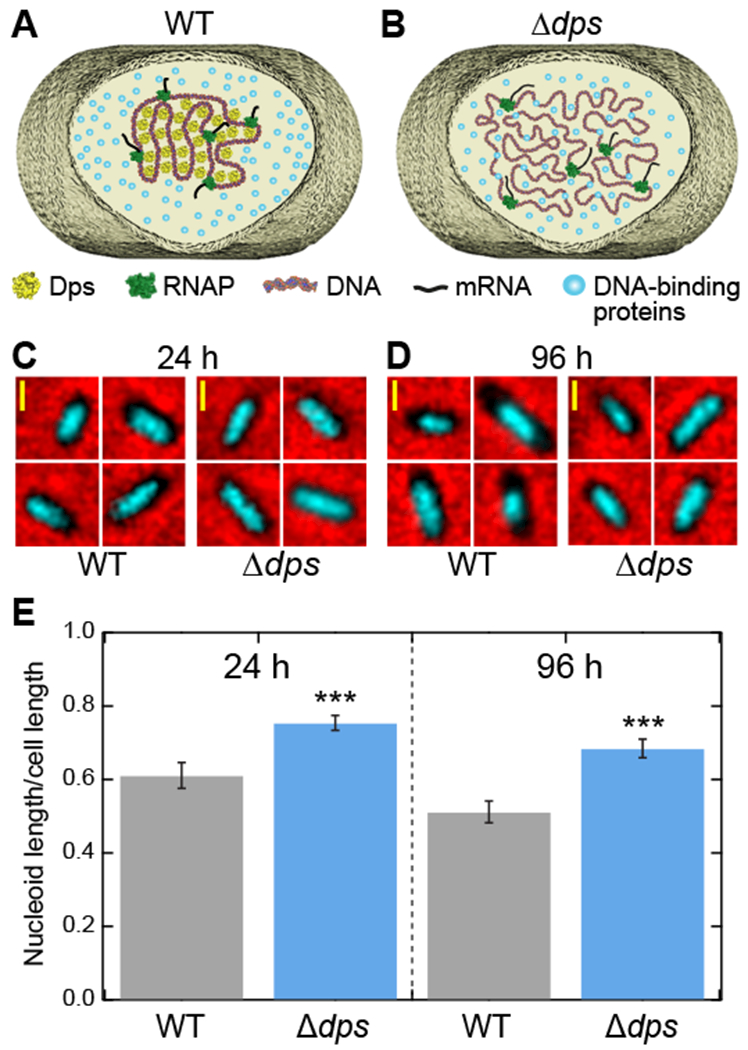
(A) Schematic of the structure of DNA in a wild-type cell during stationary phase. Dps condenses the cellular DNA. (B) In Δdps cells, Dps-mediated DNA compaction cannot occur. (C, D) Fluorescence images of the nucleoid from wild-type and Δdps cells stained with Hoechst 33258 (cyan) were superimposed onto phase-contrast images of the same cells (black on red) grown for (C) 24 hours or (D) 96 hours. (E) Ratios of nucleoid length to cell length, extracted from fluorescence images (n = 133 - 208 cells per condition). The error bars represent the estimate of the standard errors by bootstrapping. See also Figure S1.
The dramatic changes in DNA topology induced by Dps binding could potentially alter transcription, as has been observed for DNA condensed by eukaryotic histones and other prokaryotic NAPs including H-NS (Hommais et al., 2001) and HU (Kar et al., 2005). The density of observed Dps-DNA structures, along with differences in protein expression patterns between wild-type and Δdps cells reported during stationary phase (Almiron et al., 1992), have prompted suggestions that Dps may act as a pleiotropic regulator of transcription in stationary phase (Browning and Busby, 2004; Dame, 2005; Dorman, 2013). A recent study reported Dps-associated changes in the transcription of specific genes in exponential phase (Antipov et al., 2017), providing support for this hypothesis.
In this study, we examined how Dps influences the activity of RNA polymerase (RNAP) in stationary phase. Surprisingly, we found that deletion of dps caused no significant change of global transcriptional patterns in vivo during stationary phase, and produced only mild changes in the proteome. Investigation of the effect of Dps on the initiation of transcription in vitro found no changes in RNAP initiation activity at physiologically relevant Dps concentrations. To probe the effects of Dps on RNAP elongation in vitro, we used a single-molecule transcription assay to examine whether Dps-mediated DNA compaction could induce RNAP pausing or arrest during transcriptional elongation. We again found no significant change in RNAP transcription dynamics. We conclude that in contrast to histones and other specific NAPs, Dps does not affect transcription during stationary phase. Instead, Dps provides the first identified example of a DNA-binding protein that can completely decouple DNA condensation from transcriptional regulation, providing bacteria greater freedom to tailor transcriptional responses to various sources of stress while protecting the genome from damage. We propose Dps achieves this decoupling by creating a phase-separated organelle in bacteria that is permeable to RNAP.
Results
Dps significantly compacts the nucleoid in stationary phase
While isolated Dps biocrystals have been observed in vivo by electron microscopy (Frenkiel-Krispin et al., 2001; Wolf et al., 1999), the overall effect of Dps on the compaction of the nucleoid of intact cells has not been measured directly. We therefore set out to measure the size of the nucleoid in a wild-type E. coli strain as well as in its isogenic Δdps derivative (Karas et al., 2015). Our expectation, based on AFM studies on nucleoids extracted from cells (Kim et al., 2004), was that the formation of Dps complexes in the wild-type strain should lead to a compact nucleoid (Fig. 1A) compared to the Δdps strain (Fig. 1B).
Cultures of wild-type and Δdps strains were incubated for 24 (stationary phase) or 96 hours (late stationary phase). Western blot analysis confirmed that Dps protein levels in the wild-type strain increased dramatically upon entering stationary phase (Fig. S1A, B), consistent with previous results (Azam et al., 1999; De Martino et al., 2016). Nucleoids of the stationary-phase cells were labeled with the fluorescent nucleic acid stain Hoechst 33258. Cells were imaged using fluorescence and phase contrast microscopy, and both the length of the cell and the length of the nucleoid were measured along the long axis of individual cells (Fig. 1C, D). The ratio of these lengths was averaged over ≥130 individual cells for each condition tested (Fig. 1E). The presence of Dps caused a significant reduction in the fractional length of the nucleoid in both 24- and 96-hour starved cells, whereas cell length remained unchanged in all conditions (Fig. S1C). Compared to wild-type cells, Δdps nucleoids exhibited an increase in length of 24% at 24 hours and 34% at 96 hours. We conclude that a significant fraction of the genome is condensed by Dps in stationary phase, consistent with a previous estimate that Dps occupies a large but incomplete fraction of the stationary-phase nucleoid (Talukder and Ishihama, 2015).
Deletion of dps does not affect the transcriptome of stationary-phase bacteria
To test whether compaction of the nucleoid by Dps might influence transcription, we used RNA-Seq to survey the entire transcriptome (Wang et al., 2009). Cultures of wild-type and Δdps strains were again incubated for 24 or 96 hours, and RNA was isolated from the cells (Fig. S2A). In order to focus specifically on changes to mRNA levels, we depleted the rRNA, generated cDNA libraries, and sequenced the resulting fragments. For each condition, we collected and analyzed more than 10 million aligned read pairs (Fig. S2B, C, D). The isolated RNA samples were of high quality (Fig. S2A), suggesting negligible degradation. Quantification of the RNA levels demonstrated that Dps did not have a significant influence on either the total RNA extracted or the overall amount of mRNA recovered (Fig. 2A, Fig. S2E).
Figure 2. Dps has no influence on the transcriptome and mild influence on the proteome in stationary-phase E. coli cells.
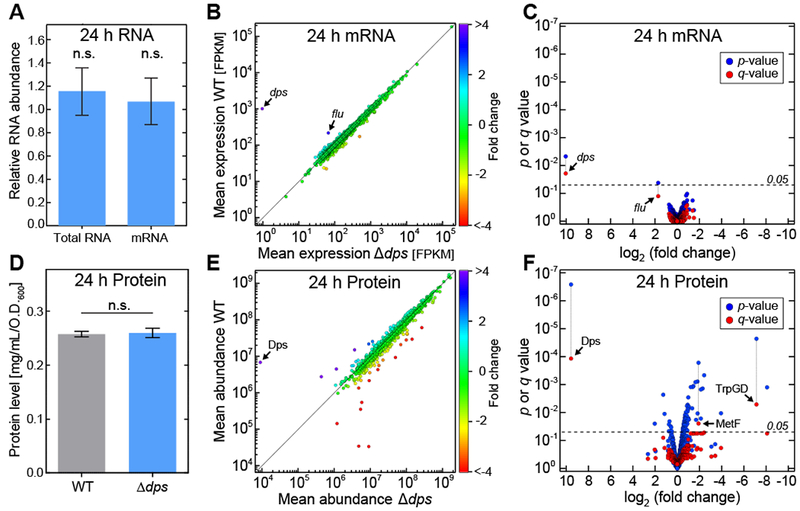
(A) The relative amounts of total RNA and mRNA (mean ± SE).. (B) Differential expression analysis of RNA sequencing. For each gene, the mean expression in the wild-type strain is plotted against the corresponding value in the Δdps strain. Colors represent the fold-difference between the two strains. (C) The significance of the shift in mean expression for each mRNA species (as determined by the p or q value) is plotted against the fold change. (D) Total protein levels (mean ± SE). (E) Differential expression profile of SILAC analysis. For each protein, the mean abundance in the wild-type strain (y-axis) is plotted against the corresponding value in the Δdps strain (x-axis). Colors represent the fold-difference between the two strains. (F) The significance of the shift in mean expression for each protein species (as determined by the p or q value) is plotted against the fold change. See also Figures S2 and S3.
Strikingly, the wild-type and Δdps strains showed a nearly one-to-one relationship in mRNA expression patterns after 24 hours of starvation, with 99.84% of genes exhibiting less than a two-fold change (Fig. 2B). Statistical analysis (Trapnell et al., 2010) showed that only two genes, dps and flu, show significant variation when analyzed individually (p < 0.05). In order to adjust for multiple testing, we derived the q-values (Benjamini and Hochberg, 1995), which left dps as the sole gene with significant variation (Fig. 2C). The same analysis was applied to the data obtained after 96 hours of starvation, and an almost identical pattern was observed (Fig. S2F).
To confirm the sensitivity of the RNA sequencing technique, we determined differential expression between samples taken from the Δdps strain at 24 and 96 h of starvation. Here, the q-values indicated significant variation in the expression of 67 genes (Fig. S2G). We further verified our RNA sequencing results by independently analyzing the changes in expression of several E. coli genes using qPCR (Fig. S3). With the exception of flu, none of these genes exhibited significant variation in expression level between the wild-type and Δdps strains. However, flu is a phase-variable gene with an ON/OFF heritable expression pattern that can persist for many generations until undergoing a spontaneous switch (Diderichsen, 1980). Changes in flu expression therefore cannot be unambiguously attributed to Dps. Our results clearly demonstrate that Dps does not influence mRNA levels in stationary-phase bacteria.
Protein expression is mildly influenced by Dps
Our transcriptome results were unexpected given that a two-dimensional PAGE analysis indicated that Dps influences the expression levels of several proteins in stationary phase (Almiron et al., 1992). To assess whether Dps can influence protein abundances in vivo, we directly measured changes in the proteome using SILAC (stable isotope labeling with amino acids in cell culture), a sensitive mass spectrometry technique (Ong et al., 2003). We constructed double Arg−/Lys− auxotroph derivatives of our wild-type and Δdps strains of E. coli and grew them in synthetic media containing arginine and lysine labeled with either light or heavy isotopes. This differential labeling allowed us to compare the protein levels of the two strains directly in a single MALDI (matrix-assisted laser desorption/ionization) mass spectrometry assay by determining the isotope ratios of individual proteins.
Although overall protein levels were not altered between the wild-type and Δdps strains (Fig. 2D), analysis of the SILAC data revealed greater variability between the protein expression levels of the two strains compared to the variability observed for the mRNA. Within the proteome, 4.9% of detected proteins exhibited more than a two-fold change (Fig. 2E). Statistical analysis (Kammers et al., 2015) revealed that 12% of the protein species could be assigned significant p-values, and three of these proteins were deemed significant using q-values (Fig. 2F). Those proteins were Dps itself along with two enzymes involved in amino acid synthesis, anthranilate synthase component 1 and methylenetetrahydrofolate reductase.
Our SILAC data demonstrate a mild but detectable change in the proteome. However, given the lack of any detectable change in mRNA levels using the more sensitive RNA-Seq assay, these changes in the proteome cannot be attributed to transcription. Instead, Dps must influence rates of protein synthesis or protein degradation during stationary phase.
RNAP holoenzyme can initiate transcription on Dps-condensed DNA in vitro
One possible way to reconcile our RNA-Seq results would be if Dps somehow avoided condensing promoter sequences. To test this hypothesis, we examined Dps binding in vitro to linear DNA fragments containing RNAP promoter sequences (Fig. S4A). We selected four promoters that control a diverse set of genes: the rrnB P1 promoter that regulates rRNA and tRNA expression, the recA promoter that participates in the SOS response, the bacteriophage λ PR promoter, and the promoter for flu, the only gene that was detectably up-regulated at the mRNA level in stationary-phase cells. We used a gel-shift assay to measure the fraction of DNA condensed by Dps (Fig. S4B). Similar concentrations of Dps were required to bind and condense DNA containing each promoter (Fig. 3A, Table S1), consistent with reports that Dps exhibits loose sequence specificity (Azam and Ishihama, 1999).
Figure 3. Dps allows RNAP to bind to promoters but excludes KpnI restriction enzyme from its target site.
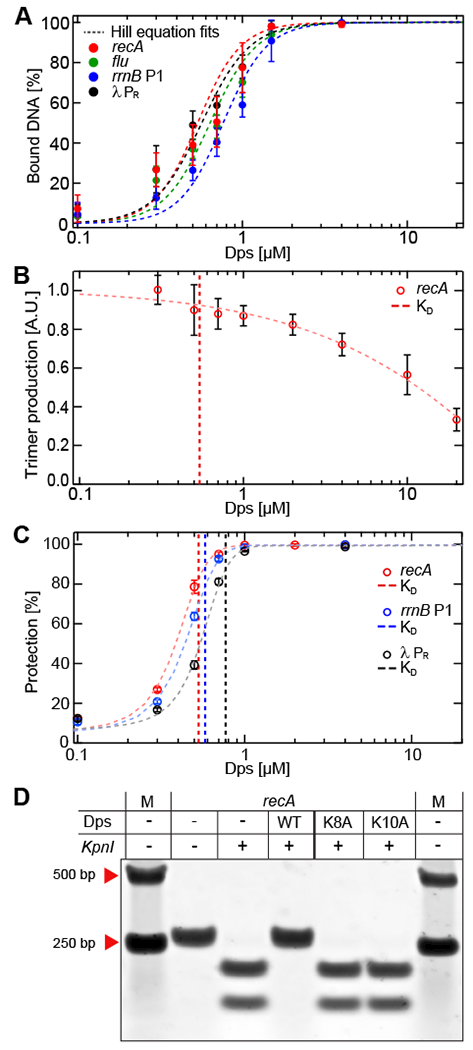
(A) Gel-shift analysis of Dps binding to linear promoter DNA fragments. The calculated KD and Hill coefficients resulting from fits to the Hill equation are summarized in Table S1. (B) Transcription initiation from the recA promoter. (C) Dps-mediated protection from DNA digestion. The vertical dashed lines in (B) and (C) indicate the KD of Dps for the different DNA templates shown in (A). The data in panels A-C are shown as mean ± SD from three biological replicates. (D) Wild-type, K8A, or K10A Dps proteins at 4 μM were bound to recA DNA, followed by incubation with or without KpnI. DNA:Dps complexes were dissociated by heparin. See also Figures S4 and S5.
Since Dps was capable of condensing these promoters, we next directly measured its effect on the first steps of transcript initiation in vitro. Linear DNA containing the recA promoter was incubated with various concentrations of Dps, spanning the critical DNA-condensing range. Next, RNAP holoenzyme was added, along with a dinucleotide RNA primer and a radiolabeled nucleotide triphosphate corresponding to the next position on the template. If the DNA remains accessible to RNAP in the presence of Dps, then an open complex should rapidly form, allowing RNAP to engage in multiple rounds of synthesis of abortive RNA trimers. At all Dps concentrations tested, we detected significant trimer production (Fig. 3B). Although high concentrations of Dps reduced trimer synthesis, no noticeable change occurred near the apparent KD of 0.54 μM when Dps first condenses the DNA. A similar pattern was observed for the λ PR promoter (Fig. S4C).
Even at high Dps concentrations where partial inhibition of trimer synthesis occurred, Dps did not interfere with binding of the RNAP holoenzyme to the promoter. When trimer production is measured as a function of time, the rate of production follows the functional form kss (1 – e−t/τoc), where τoc is a characteristic time constant associated with open complex formation and kss is the steady state rate of trimer production (McClure, 1980). We found τoc for the λ PR promoter to be indistinguishable in the absence and presence of 4 μM Dps, even though kss was reduced by 47% (Fig. S4D, E). We therefore conclude that high concentrations of Dps interfere with steps of initiation subsequent to open complex formation, e.g. the binding of nucleotide substrates.
Dps blocks the activity of restriction endonucleases but allows access to a transcriptional repressor
Since Dps did not block RNAP holoenzyme from binding promoters, we next asked whether Dps could block other enzymes from accessing bound DNA. Our recA, rrnB P1, and λ PR promoter sequences each contain recognition sites for restriction endonucleases (KpnI, HindIII, and HincII, respectively), allowing direct comparison of restriction enzyme activity to that of RNAP. Pre-incubating each DNA molecule with saturating Dps (4 μM) was sufficient to completely block the activity of the corresponding restriction enzyme (Fig. S4F, S4G). We then explored the effects of a range of Dps concentrations spanning the critical DNA-condensing concentrations for these promoter sequences (Fig. S4H). In contrast to RNAP holoenzyme, we found that the activity of restriction enzymes decreased sharply with increasing Dps concentrations, and significant protection was observed even at concentrations near the apparent KD (Fig. 3C).
To confirm that this protection was specifically associated with the binding of Dps to DNA, we probed the effect of two Dps point mutations (K8A and K10A) that lower the affinity of Dps for DNA (Karas et al., 2015). Wild-type or modified Dps was added to the three promoter DNA fragments to test their relative ability to protect against restriction enzyme cleavage (Fig. 3D, S4I, S4J). At a concentration of 4 μM, wild-type Dps bound nearly all the DNA template while K8A and K10A Dps bound almost none. We found that K8A and K10A Dps were unable to protect the promoter fragments from KpnI, HindIII, or HincII restriction activity, while the wild-type Dps again showed full protection. We conclude that Dps blocks the restriction enzymes tested specifically by binding and condensing DNA.
To measure the effect of Dps on other proteins related to transcriptional regulation, we looked at the activity of the transcriptional repressor LexA (Butala et al., 2009) using a run-off transcription assay on the recA template (Fig. S5). LexA inhibited transcription both in the presence and absence of 2 μM Dps. The σ70-dependent recA promoter contains canonical −35 and −10 elements that are also recognized by σs (Gaal et al., 2001), allowing us to explore the activity of σs holoenzyme using the same assay. We again observed that Dps did not block transcription by the σs holoenzyme or prevent the repression of transcription by LexA. Together, these experiments show that Dps fails to block either two different holoenzymes or a repressor from accessing DNA.
Individual transcription-elongation complexes are not affected by Dps-condensation of DNA
Transcription elongation by RNAP has been extensively studied using single-molecule force spectroscopy (Abbondanzieri et al., 2005; Shaevitz et al., 2003), which can identify strong pauses associated with specific regulatory sequences as well as weaker stochastic pauses that occur throughout genomic DNA. We therefore developed a single-molecule assay to investigate transcription elongation while controlling the compaction of DNA by Dps. Stalled transcription elongation complexes (TECs) were formed within a linear DNA template. The RNAP was then attached to a magnetic bead, and the DNA was attached to a glass coverslip. Force could be applied in different directions depending on whether the down- or upstream end of the DNA was tethered to the surface. In the assisting force (AF) configuration, RNAP was pulled in the downstream direction (Fig. 4A), while in the opposing force (OF) configuration, RNAP was pulled upstream on the DNA (Fig. 4B). Dps was then added to the flow cells, and transcription elongation was re-initiated by adding nucleotides.
Figure 4. Multiplexed single-molecule transcription-elongation assay and dwell time analysis of RNAP dynamics.
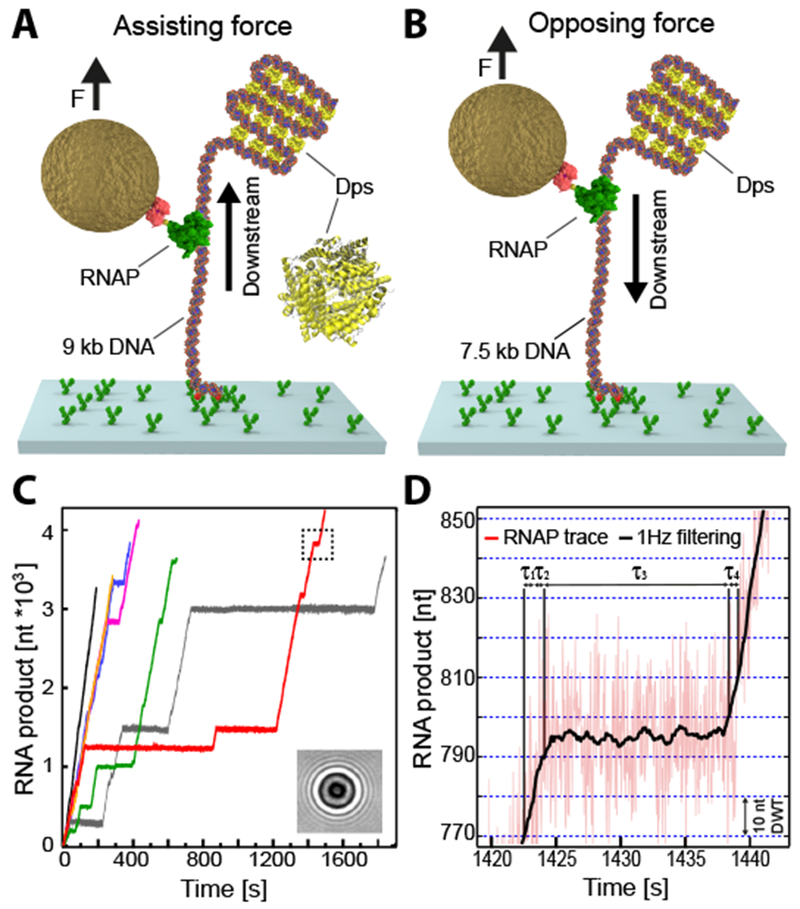
(A) Schematics of the single-molecule in vitro transcriptional assay in the assisting force (AF) configuration, showing a single RNAP bound to a surface-attached DNA template in the presence of Dps. A magnetic bead was attached to the RNAP and exerted a constant force of 5 pN on the ternary complex. (B) The opposing force (OF) experimental configuration (C) Individual RNAP trajectories over time measured at 25 Hz via the change of the diffraction pattern of the attached magnetic bead (inset). The dashed rectangle depicts the trace region magnified in (D). Dwell times (τn) associated with advancing 10 nt were extracted from 1 Hz-filtered elongation traces (black line). Boundaries denoted by blue dashed lines. The error bars represent the estimate of the standard errors by bootstrapping. See also Figure S6.
As the interaction of Dps and DNA is sensitive to tension and ion concentrations (Vtyurina et al., 2016), we independently determined the precise relationship between force and DNA extension in our transcription buffer in the presence of Dps (Fig. S6A). As previously reported, the force-extension curve of DNA exhibited reproducible hysteresis (Vtyurina et al., 2016). Forces below ~1 pN allowed extended DNA to be compacted by Dps, while forces greater than ~3 pN were necessary for compacted DNA to be pulled apart (Fig. S6B). We therefore selected a pulling force of 5 pN for our transcription assay to ensure that the DNA between RNAP and the surface remained under sufficient tension to prevent Dps condensation (Fig. S6C). The remaining DNA was under no tension and would therefore be compacted by Dps (Fig. 4A, B). In the AF configuration, any DNA:Dps complexes would lie downstream of RNAP and could potentially impede RNAP elongation (Fig. 4A). Conversely, in the OF configuration, the DNA:Dps complex would form upstream of RNAP and could therefore prevent reverse translocation (backtracking) of RNAP associated with pausing and arrest (Fig. 4B) (Shaevitz et al., 2003), potentially leading to higher overall rates of transcription (Nudler, 2012).
Upon the addition of rNTPs, the stalled TECs resumed RNA synthesis. Measurements of the bead height over time were recorded and converted into the number of bases transcribed (Fig. 4C). RNAP exhibited periods of relatively constant rates of elongation punctuated by pauses of various lengths. Our magnetic tweezers apparatus achieves a spatiotemporal resolution of 1.5 nm over 1 second of sampling (Fig. 4D), allowing us to measure the dwell times needed for RNAP to transcribe successive 10 base pair segments of DNA. These dwell times allow us to analyze both pausing and elongation kinetics (summarized in Table S2).
A histogram of the dwell times of all AF traces showed a skewed distribution with a prominent peak at approximately 0.5 s and a long tail extending to larger dwell times (Fig. 5A). The peak corresponds to the pause-free transcriptional velocity, which we find to be ~25 nt/s. Dwell times in the tail correspond to broadly distributed pauses ranging from 1 second up to several minutes. A similar pattern is observed for the OF traces (Fig. 5B).
Figure 5. Dependence of transcription elongation dynamics on force and location of DNA:Dps complex.
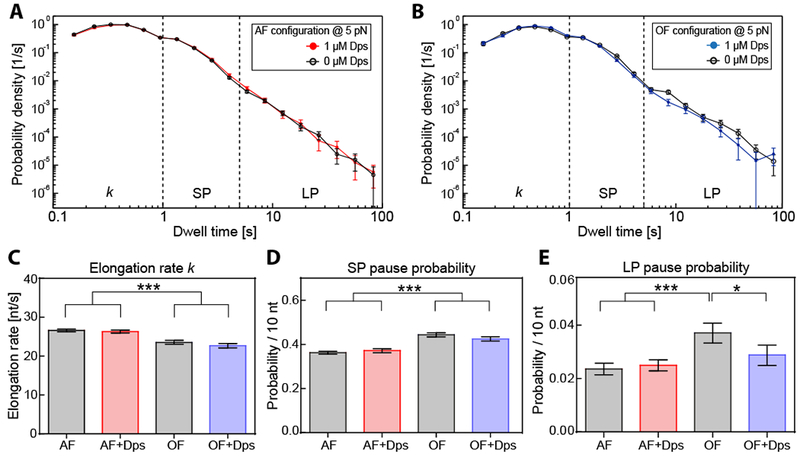
(A) Dwell time distributions for assisting force (AF) trajectories in the presence (red) and the absence (black) of 1 μM Dps at 20°C. (B) Dwell time distributions resulting from the opposing force (OF) experiments in the presence (blue) and the absence (black) of 1 μM Dps at 20°C. (C) Comparison of extracted RNAP elongation rates k for AF and OF experimental distributions shown in (A, B), determined by Galton distribution fits with an upper boundary of 1 s. (D, E) Calculated transcription pause probabilities (per 10 nt) for short (SP, D) and long pauses (LP, E) for the experimental configurations shown in (A, B). The error bars represent the SD. Statistical results, dwell times, and number of trajectories measured are summarized in Table S2. See also Figure S7.
We pooled the dwell-time measurements into three bins: 0-1 s to estimate the pause-free elongation, 1-5 s to measure the probability of entering short pauses, and >5 s to measure the probability of entering longer pauses. We found that pause-free elongation proceeded at a slightly increased rate in the AF configuration compared to the OF configuration, which our assay could readily distinguish (Fig. 5C). We also observed a slight decrease in the short pause probability for AF compared to OF, from 0.44 ± 0.01 to 0.36 ± 0.006 pauses per 10 nt (Fig. 5D). This result was again consistent with previous findings, suggesting that pauses result from a branched pathway that competes with nucleotide addition (Herbert et al., 2006).
The addition of Dps had little effect on elongation dynamics, either at 1 or 10 μM Dps (Figs. 5A, B, Fig. S7). Pause-free elongation proceeded at an indistinguishable rate whether or not Dps was present on either the upstream or downstream DNA (Fig. 5C). Similarly, the probability of entering a short pause did not change significantly when Dps was present (Fig. 5D).
The only significant effect of Dps on pausing was observed for long pauses (>5 s) in the opposing force configuration. Long pauses are more likely to be associated with backtracking and show a higher sensitivity to force (Shaevitz et al., 2003). The addition of Dps had no effect on the long pause probability for assisting forces but lowered the pause probability significantly for opposing forces (Fig. 5E). A sharp reduction in the incidence of backtracking can occur when other macromolecules are bound to the nascent RNA or upstream DNA (e.g. a trailing RNAP), creating a physical barrier that blocks reverse translocation of RNAP into a backtracked state (Epshtein et al., 2003). We find when Dps is bound to the upstream DNA it causes a slight reduction in backtracking, indicating Dps provides a mild barrier against reverse translocation. Taking all our data together, the overall effect of Dps on RNAP elongation and pausing is negligible in both the AF and OF configurations.
DNA:Dps complexes dynamically reorganize to accommodate transcription
In order to accurately and continuously measure the progress of RNAP in the previous experiments, we had to apply sufficient tension (5 pN) to eliminate Dps binding to either the upstream or downstream DNA. We next sought to examine elongation dynamics on fully condensed DNA by lowering the force to 0.7 pN in the OF configuration in the presence of 1 μM Dps (Figs. 6A, S5B). Under these loads, bare DNA is stretched to over 50% of its contour length, while DNA bound by Dps will be compacted to ~1% of its contour length (Vtyurina et al., 2016). In compacted DNA we observed minimal fluctuations in extension (Fig. 6B, upper panel). The addition of rNTPs caused the complexes to become more dynamic, as reflected by increases in the amplitude of fluctuations in the extension (Fig. 6B, lower panel). Individual traces exhibited bursts of rapid extension followed by a relatively steady motion in the downstream direction (Fig. 6C). We interpret this behavior as a local disruption of the DNA:Dps complex, followed by RNA chain extension as RNAP translocates forward on the DNA. An analysis of noise levels revealed that the NTP-induced fluctuations differed significantly from the dynamics of bare DNA or of DNA in the presence of Dps alone (Fig. 6D).
Figure 6. RNAP transcribes through a fully condensed DNA:Dps complex.
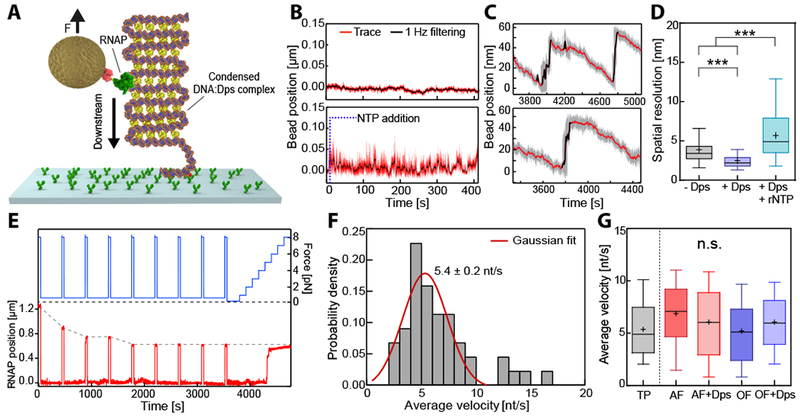
(A) The experimental configuration was similar to the OF configuration (see also Figure 4B) but at lower exerted force (0.7 pN), which allows Dps to condense the entire DNA tether. (B) Bead position traces for stalled RNAP on a condensed DNA:Dps complex, before (upper panel) and after (lower panel) transcription restarted upon the addition of rNTPs. (C) Two representative time traces of active RNAP on a condensed DNA:Dps complex. Steep upward jumps in bead position (black) were accompanied by gradual downward displacements (red). (D) Comparative box plot of noise levels (standard deviation) measured in the absence (grey) and in the presence of Dps prior to (purple) or following rNTP addition (cyan). (E) Example trace with transient pulling to 8 pN (blue) every 400 s to determine the absolute RNAP position (red) along the DNA tether. (F) Distribution of average velocities from (E). The red line indicates a Gaussian fit. (G) Comparative box plot of average velocities determined from the transient pulling (E) and transcription experiments for AF and OF configurations in the presence and the absence of Dps (see Figure 5). The outer confidence intervals of the box plots represent the 1.5 interquartile range. See also Figure S6.
To monitor the average velocity of RNAP on fully condensed DNA, we expanded upon the previous assay by introducing transient periods of high force (8 pN) interspersed by longer measurement periods at low force (0.5 pN) (Fig. 6E). Because Dps releases DNA rapidly at high load and quickly rebinds to DNA at low load (Fig. S6D), we could briefly assess the position of RNAP then return the DNA to a compacted state. We determined the average velocity (i.e. including pauses) of RNAP on condensed DNA under low load to be 5.4 ± 0.2 nt/s (Fig. 6F). This rate was consistent with the average velocities at high loads (Fig. 6G). Together, these results demonstrate that transcription is not impeded on DNA that has been fully condensed by Dps. Given that packed Dps arrays contain ~2 nm gaps just large enough to accommodate DNA (Grant et al., 1998), DNA:Dps complexes must therefore rapidly rearrange to allow for the passage of the significantly bulkier TEC.
Discussion
Using multiple independent lines of investigation (RNA-seq, in vitro initiation, and single-molecule elongation), we have examined the impact of Dps on transcription. In stark contrast to our expectations, we found that Dps did not measurably affect transcription by RNAP. Deletion of dps in E. coli did not significantly alter mRNA levels for a single gene during stationary phase, addition of Dps to DNA did not block open complex formation at promoter sequences, and the presence of Dps did not noticeably hinder the progress of TEC in single-molecule experiments. Furthermore, we provide evidence that Dps can compact DNA in all of these conditions, with measurable effects on other processes. The presence of Dps in vivo led to significant compression of the nucleoid of E. coli bacteria (Fig. 1E) and mild alteration of the proteome (Fig. 2F). Dps was observed to efficiently bind all promoter DNA sequences in vitro with similar affinities (Fig. 3A), preventing restriction enzymes from accessing the DNA (Fig. 3C). Finally, Dps induced dramatic compaction of DNA at low forces in our single-molecule assay (Fig. S6). We therefore conclude that transcription occurs freely in highly condensed DNA:Dps complexes.
Comparison to other nucleoid-associated proteins
The observation that Dps decouples DNA compaction from transcription stands in contrast not only to eukaryotic histones (Goldberg et al., 2007) but also other prokaryotic NAPs such as Fis, HU, and H-NS (Dorman, 2013). Each of these NAPs has been observed to condense generic DNA sequences in vitro much like Dps. Unlike Dps, these other NAPs have been shown to directly influence the transcription of specific genes. Dps is therefore the only NAP that has been shown to condense DNA without exhibiting any measurable effect on transcription.
It is possible that some NAPs regulate RNAP activity through mechanisms that are independent of their ability to compact DNA. However, for at least one NAP, the compaction of DNA has been directly linked to its effects on transcription: H-NS can compact DNA by forming bridged filaments that promote pausing of RNAP in vitro, leading to an increase in Rho-mediated termination (Kotlajich et al., 2015). These effects disappear when H-NS forms linear, non-compacted filaments on the DNA. The twin examples of Dps and H-NS show that DNA compaction may or may not influence transcription. Therefore, the specific mechanism used by each NAP to affect transcription must be directly confirmed rather than a priori assuming that compaction would naturally alter transcription.
Potential mechanisms of transcription in DNA:Dps complexes
Given the extensive and dense DNA:Dps complexes observed by electron microscopy (Wolf et al., 1999), it is not obvious how RNAP is able to navigate along the condensed DNA to specific promoters and to perform transcription initiation and elongation. Part of the explanation may lie in the unusual cooperative binding behavior of Dps. We have previously shown that DNA:Dps complexes can adopt long-lived metastable states over a range of tensions and buffer conditions, which can be explained by an Ising model (Vtyurina et al., 2016). This behavior requires multiple nearest-neighbor interactions between Dps dodecamers to stabilize the weak interaction with the DNA. As a result, Dps has a high avidity, or cumulative affinity, for DNA, despite the relatively low affinity of the individual contacts. A protein that establishes highly stable interactions with the DNA, such as RNAP (Vogel et al., 2002), could therefore displace Dps from a specific region of DNA without destabilizing the entire high-avidity complex.
However, this avidity/affinity argument may not be sufficient to explain our additional observation that Dps can interfere with the activity of restriction enzymes, since restriction enzymes also have high affinities for their target sequences (Hiller et al., 2003). The ability of Dps to selectively exclude access to nucleic acids is reminiscent of liquid-liquid phase-separated domains in eukaryotes (Hyman et al., 2014), such as the nucleolus (Mitrea et al., 2016), nuage in Drosophila germline cells (Nott et al., 2015), and heterochromatin domains (Strom et al., 2017). The proteins driving the formation of these domains typically have intrinsically disordered regions (IDRs). Like these eukaryotic proteins, Dps contains an IDR at the N-terminus (Grant et al., 1998) that has been shown to be necessary for DNA binding activity in vitro (Ceci et al., 2004; Karas et al., 2015).
Unlike these other examples of intracellular phase separation, Dps has been observed to form ordered crystalline arrays (Frenkiel-Krispin et al., 2001). Superficially, crystalline DNA complexes may seem incompatible with the need for RNAP holoenzyme to diffuse to its promoter, but lattice diffusion occurs in many solid systems via crystal vacancies (e.g., a substitutional alloy formed between two metals). Furthermore, our single-molecule experiments demonstrate that Dps complexes can rapidly rearrange (Fig. 6). This dynamic behavior indicates that Dps complexes may retain some features of a fluid. We therefore propose that, rather than forming static crystalline structures, Dps forms dynamic complexes with similar diffusive properties to liquid-liquid phase separated organelles (Fig. 7) such as the nucleolus (Mitrea et al., 2016). While RNAP can freely enter these organelles from the cytoplasm, other proteins (such as the restriction endonucleases used here) cannot cross this barrier. The differential solubility of various macromolecules in Dps complexes provides a simple mechanism for Dps to protect DNA while allowing transcription to continue. This testable physical model explains all the data collected above.
Figure 7. Proposed model of DNA protection by Dps.
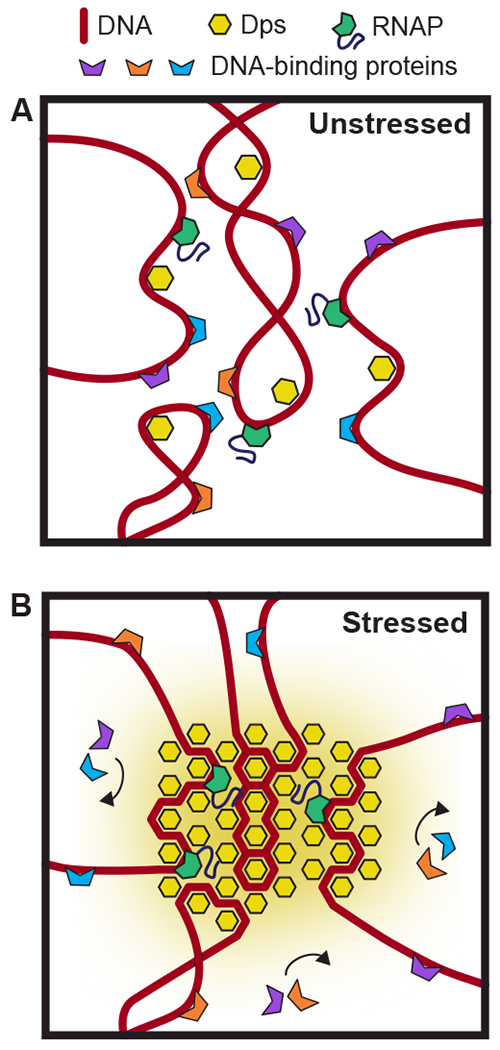
(A) In unstressed cells, Dps binds DNA transiently but is unable to condense the vast majority of the nucleoid. (B) Under conditions of high stress, dense complexes of Dps cover a large fraction of the nucleoid, creating phase-separated organelles. While RNAP can freely enter and diffuse inside these Dps complexes, other proteins are blocked from accessing the DNA.
Utility of a NAP that operates orthogonally to transcription
Given the crucial role that Dps plays in bacterial survival, the decoupling of bacterial transcription from nucleoid condensation may be important to maintain flexibility in the cellular response to stress. Rather than offering protection against any one specific form of stress, Dps has been shown to increase bacterial survival rates over a diverse range of stress conditions, including heat shock, osmotic shock, starvation, UV-exposure, antibiotics, and oxidative stress (Karas et al., 2015; Nair and Finkel, 2004). These different stresses trigger a variety of changes in the levels of alternative sigma factors, adjusting patterns of gene expression to mount an appropriate response (Gruber and Gross, 2003). By evolving a mechanism of Dps-induced compaction that is orthogonal to transcription, bacteria can protect their DNA with a “one-size fits all” approach while retaining maximum flexibility in tailoring their transcriptional response to the specific form of stress encountered. Our observations also suggest that the ability of Dps to increase bacterial survival rates during stress arises directly from DNA-binding and is not bolstered by the activation or repression of specific genes.
In addition to maintaining flexibility in the transcriptional response, Dps upregulation may also ensure that transcription can continue under conditions of extreme stress. In contrast to wild-type cells, DNA from Δdps cells has been shown to enter into a cholesteric phase after six days of starvation (Frenkiel-Krispin et al., 2001). In this phase, the DNA is placed into a dense liquid crystal which is enhanced by multivalent cations, yielding an even higher degree of compaction than observed in DNA:Dps complexes. This cholesteric phase of DNA can also be induced in vitro by solutions containing multivalent cations. While low concentrations of multivalent cations can enhance in vitro transcription, high concentrations of these cations have been shown to compact DNA and sharply inhibit transcription by E. coli RNAP in vitro (Luckel et al., 2005). Dps may therefore be needed in vivo to prevent the formation of cholesteric-phase DNA and its associated dampening effects on transcription.
Dps and the proteome
While our RNA-seq experiments reveal no effect of dps deletion on transcriptional levels, our SILAC results indicate that in stationary phase some protein levels do shift when Dps is present in stationary phase (Fig. 2). This finding is consistent with a previous study that observed a pleiotropic effect of dps deletion on protein synthesis levels in stationary phase (Almiron et al., 1992). A precedent for such differences between mRNA and protein levels is provided by H-NS, which has been shown to have vastly different effects on protein expression levels relative to mRNA levels for certain genes (Hommais et al., 2001). This discrepancy was attributed to post-transcriptional regulation, and H-NS was later demonstrated to stimulate translation of specific mRNAs (Park et al., 2010). A similar effect on translation may exist in the case of Dps. In support of this hypothesis, RNaseA was shown to disrupt the condensed structures formed by Dps in extracted mycobacterial nucleoids (Ghatak et al., 2011), indicating Dps does interact with RNA in vivo.
Another possible way for Dps to affect the proteome in stationary phase is that Dps could impact rates of protein degradation, which regulate the levels of many proteins during periods of stress (Meyer and Baker, 2011). Intracellular Dps levels are specifically regulated by the selective, ATP-dependent protease ClpXP (Stephani et al., 2003). Given the high overall concentrations of Dps in the cell during stationary phase, Dps might saturate the available ClpXP complexes, diverting them from other substrates. The resultant changes in protein lifetimes of ClpXP substrates could thereby potentially alter the relative abundances of the general pool of cellular proteins.
Implications for transcriptional regulation in bacteria
Several lines of evidence suggest that genome architecture might directly influence gene expression in bacteria (Dorman, 2013). However, the existence of an NAP that is capable of massively restructuring the nucleoid without affecting transcription complicates this view. A less dramatic reorganization of the nucleoid was achieved in Caulobacter crescentus by shifting the location of parM sites, resulting in a “rotated” chromosome (Umbarger et al., 2011). Similar to our study, the authors found no measurable changes in transcription as a consequence of the rotation. The precise nature of Dps-induced alterations in nucleoid structure is still incompletely understood, and additional studies using chromosome conformation capture techniques in Δdps strains are needed to determine how Dps affects internal contacts within the stationary-phase nucleoid. However, the dense Dps-DNA biocrystals observed in EM images (Wolf et al., 1999), the Dps-dependent nucleoid fibers observed by AFM (Kim et al., 2004), and the significant Dps-dependent compaction of the nucleoid reported here (Fig. 1) suggest that the nucleoid is reorganized at multiple scales. Further studies of Dps can therefore place constraints on which structural features of the nucleoid play a meaningful role in the regulation of transcription.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anne S. Meyer (anne@annemeyerlab.org).
Experimental Model and Subject Details
Bacterial strains and cell culture.
In this study, E. coli wild-type K-12 (W3110) and K-12 Δdps bacteria cells were used (Karas et al., 2015). For SILAC experiments, double ΔargE ΔlysA derivatives were constructed to maximize Arg and Lys isotope incorporation. The antibiotic-resistance-marked null alleles of argE (argE::tetR; (Singer et al., 1989)) and lysA (lysA::kanR;(Baba et al., 2006)) were transferred into the wild-type and Δdps E. coli strains by P1 transduction.
Strains were plated onto LB-agar plates and grown overnight at 37°C. For fluorescence microscopy, RNA sequencing, real-time qPCR, and Western blotting, single colonies were picked and grown overnight at 37°C in 2 mL rich Hi-Def Azure medium (Teknova) with 0.2% (m/v) glucose, while shaking at 250 rpm. Overnight cultures were diluted to O.D.600 = 0.03 in 15 mL rich Hi-Def Azure medium with 0.2% (m/v) glucose and then incubated at 37°C with shaking at 250 rpm. After 3, 24, and 96 h, samples were removed. For RNA sequencing, real-time qPCR, and Western blotting, an amount of the cultures corresponding to 1 mL of O.D.600 = 1 was transferred to Eppendorf tubes and centrifuged at 14,000 g for 2 min. The supernatant was removed, and the cell pellet was flash-frozen in liquid nitrogen.
For SILAC analysis, overnight cultures grown in complete Hi-Def Azure medium were diluted 1/100 into freshly prepared Azure medium supplemented with 0.2% glucose and either light (1.0 mM arginine, 0.4 mM lysine) or heavy (1.0 mM arginine-13C6, 0.4 mM lysine-4-4-5-5-d4) amino acids. Cultures were grown for 24 h at 37°C with shaking at 250 rpm. After 24 h, cultures were pelleted, resuspended in RIPA buffer (150 mM NaCl, 1.0% IGEPAL® CA-360, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-Cl, pH 8.0; Sigma), and lysed with sonication. Total protein concentration of lysates was determined via Bradford protein assay (Bio-Rad) using BSA as a standard. Samples for mass spectrometry analysis were prepared by mixing 50 μg of total protein each from samples grown with light and heavy amino acids in a final volume of 60 μL.
Method Details
Fluorescence microscopy
Per strain, five biological replicates were grown. After 3, 24, or 96 h of growth, 500 μL of cell culture was removed. Cells were washed twice with PBS buffer (VWR, composition: 137 mM NaCl, 2.7 mM KCl, 10 mM phosphate buffer). The cells were resuspended in 500 μL of 2.5% glutaraldehyde (Sigma Aldrich), followed by a 2-hour incubation at room temperature to fix the cells. Cells were washed twice with PBS, then permeabilized by resuspending the cells in 500 μL of PBS containing 0.1% Triton X-100 (Sigma-Aldrich). Cells were washed once with PBS containing 0.1% Triton X-100, then resuspended in 500 μL PBS. DNA staining was performed by adding 5 μL of 10 μg/mL Hoechst 33258 (Sigma-Aldrich) to the cells and incubating 20 min at room temperature. Samples were washed three times with PBS.
Cells were imaged using an Olympus IX81 microscope equipped with a 100× oil-immersion objective (UplanFL, N.A.1.30, Oil Ph3). A back-illuminated EM-CCD (Ixon, Andor) camera was used to record fluorescence and phase-contrast images with 1004 (H) by 1002 (V) pixels of 8 μm × 8 μm each. For imaging DNA stained with Hoechst 33258, the sample was illuminated using a 350 nm excitation laser and a 460/50 nm emission filter using a DAPI FilterCube (Chroma). To create phase contrast images, cells were illuminated by diffracted white light. The sample was alternately illuminated with the laser and white light to create both phase contrast and fluorescence images. Camera frames were acquired at a total rate of 12 Hz, with alternating exposure times and EM gains.
Images were analyzed using ImageJ (Schneider et al., 2012). Cell and nucleoid lengths were measured by plotting the pixel intensities along a line spanning the entire cell length in both the phase contrast and the fluorescence images. The nucleoid length was defined as the width of the region of the fluorescent peak with an intensity two times higher than the background, and the cell length was extracted from phase-contrast images as the width of the region with an intensity two times lower than the background. Cells less than 1.5 μm in length were excluded from data analysis. In order to estimate the standard errors associated with the mean cell length, mean nucleoid length, and mean relative nucleoid length, a bootstrap analysis was performed for each statistic. Resampling with replacement was performed both at the level of the five replicates and at the level of individual cells chosen within each replicate. 1,000 resampled data sets were created.
Western blotting
Per strain, three biological replicates were grown. Cell pellets were resuspended in 100 μL 2× SDS sample buffer (4% w/v SDS, 8% w/v glycerol, 80 mM Tris-HCl pH 6.8, 0.2% Bromophenol Blue) with 10 mM DTT to obtain an O.D.600 of 10. Samples were boiled at 95°C for 10 min. 10 μL sample per lane was analyzed on a 15% SDS-PAGE gel. Protein was transferred from the gel to a PVDF membrane (Thermo Scientific) through semi-dry blotting for 60 min at 15 V. Membrane was then blocked with 5% skim milk (powder for microbiology from Sigma) in TBS-T (10 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween-20) overnight. The membrane was incubated with primary anti-Dps antibody from rabbit in 5% milk TBS-T for 1 h at room temperature. The membrane was washed 4 times for 10 min with TBS-T and then incubated for 45 min with secondary antibody (Goat anti-Rabbit HRP, Thermo Scientific) in TBS-T. The membrane was washed 4 times for 10 min with TBS-T, and chemiluminescence was detected using the SuperSignal West Pico kit (Thermo Scientific) and a Biorad Imager. Detected bands were quantified using ImageQuant.
RNA sequencing
Total RNA was isolated with the High Pure total RNA isolation kit (Roche) and quantified using a Nanodrop (Thermo Scientific). Quality of total RNA was determined by gel electrophoresis, using a 1% agarose gel containing 0.5 μg/mL ethidium bromide. RNA was detected using UV with a Bio-Rad gel imager. Ribosomal RNA was depleted with the Ribo-Zero kit (Epicentre). The resulting mRNA was quantified with the Quant-iT RiboGreen RNA Assay Kit (Invitrogen) using a microplate reader (Tecan). To synthesize cDNA, 1 μL of 50 μM random hexamers (Invitrogen), 50 ng mRNA, 1 μL 10 mM dNTP mix (Promega), and nuclease-free water (Promega) up to 13 μL were mixed and subsequently heated to 65°C for 5 min and then cooled on ice for at least 1 min. After cooling, 4 μL of 5× reaction buffer of the high fidelity Reverse Transcriptase kit (Roche), 1 μL of 100 mM DTT, 1 μL RNasin® RNase inhibitor (Promega), and 1.1 μL of high fidelity Reverse Transcriptase (Roche) were added, and mixtures were incubated at 25°C for 5 min, at 50°C for 1h, and then at 70°C for 15 min. After the first-strand synthesis, the following components were added: 30 μL second-strand buffer (Invitrogen), 3 μL of 10 mM dNTP mix (Promega), 4 μL of E. coli DNA polymerase I (NEB), 1 μL DNA of E. coli ligase (New England Biolabs), 1 μL of 5 U/μL RNase H, and 91 μL nuclease-free water. Second-strand synthesis mixtures were incubated at 16°C for 2 h. The resulting double-stranded cDNA was then purified with a DNA purification kit (Promega). 1 ng of purified cDNA was prepared for sequencing using the Nextera XT DNA sample preparation kit (Illumina). Sequencing-ready cDNA libraries were pooled, loaded, and sequenced using the MiSeq (Illumina).
Per condition, more than 10 million sequenced reads were checked for quality and trimmed using trimmomatic software (Bolger et al., 2014). The base-calling accuracy was of high quality with average quality scores (Q scores) well above 30, allowing us to identify bases with more than 99.9% accuracy (Fig. S2D). The sequencing depth of more than 10 million reads in total per sample was sufficient to enable a robust analysis of the transcriptome, since 2-3 million reads per sample represents the lower threshold boundary to detect the majority of 2-fold differentially expressed genes with high (P < 0.001) statistical significance (Haas et al., 2012). Alignment of the reads to the E. coli K12 W3110 transcriptome was done using STAR (Dobin et al., 2013), transcript abundances were estimated by Cufflinks, and differential expression analysis was done by CuffDiff. Significance testing was done by Cuffdiff (Trapnell et al., 2013) based on the q value, which adjusts the p value to take into account the false discovery rate (Storey and Tibshirani, 2003). A significance level of q ≤ 0.05 was used.
Real-time qPCR
Per strain, three biological replicates were grown. Total RNA was isolated from the cell pellets with the High Pure total RNA isolation kit (Roche). To synthesize cDNA, 1 μL 50 μM Random hexamers (Invitrogen), 1 μg total RNA, 1 μL 10 mM dNTP mix (Promega), and nuclease-free water (Promega) up to 13 μL were mixed and subsequently heated to 65°C for 5 min and then cooled on ice for at least 1 min. After cooling, 4 μL 5× first strand buffer (Invitrogen), 1 μL 0.1M DTT, 1 μL RNasin® RNase inhibitor (Promega) and 1μL of SuperScript III Reverse Transcriptase (Invitrogen) was added and mixtures were incubated at 25°C for 5 min, at 50°C for 1 h and then at 70°C for 15 min. After the first strand synthesis, the following components were added; 30 μL second strand buffer (Invitrogen), 3 μL of 10mM dNTP mix (Promega), 4 μL of E. coli DNA polymerase I (NEB), 1 μL E. coli DNA ligase (New England Biolabs), 1 μL 5 U/μL RNase H, and 91 μL nuclease-free water. Second strand synthesis mixtures were incubated at 16°C for 2 h. The resulting double-stranded cDNA was then purified with the SV DNA purification kit from Promega. For every RT reaction, a reaction was performed without reverse transcriptase to control for genomic DNA contamination. A qPCR reaction was then performed in duplicate on the purified cDNA. 1 μL of cDNA (corresponding to 20 ng of total RNA), 8 μL nuclease-free water, and 1 μL of 10 μM gene-specific primers were added to 10 μL of SsoFast EvaGreen Supermix. For every primer pair, one qPCR reaction was performed that did not contain any template cDNA (non-template control) to control for contamination and primer-dimers. qPCR reactions were performed using the Eco Real-Time PCR System with the following thermal profile: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of: 95°C for 10 s, 62°C for 10 s, 72°C for 10 s. The thermal profile ended with a melt curve of 95°C for 15 s, 55°C for 15 s, and 95°C for 15 s. Analysis of the results was done using the ΔΔCt Method (Schmittgen and Livak, 2008). Statistical analysis was performed using an unpaired, two-tailed t-test.
Sample preparation for SILAC analysis
Light and heavy samples were mixed in equal amounts (determined by Bradford), and 1 volume of trichloroacetic acid (TCA) was added to 4 volumes of sample. Following a 60-min incubation on ice, the samples were centrifuged at 13,000 g for 15 min. The pellets were washed twice with 200 μL of cold acetone, resuspended in 50 mM ammonium bicarbonate, and protein concentration was measured. Upon addition of dithiothreitol (DTT; 5 mM final), samples were heated at 60°C for 30 min, followed by addition of iodoacetic acid (IAA; 15 mM final) and a 15-min incubation at room temperature in the dark. Trypsin was added at 1:50 ratio, and samples were digested overnight at 37°C. The digestion was stopped with 0.5% trifluoroacetic acid (TFA), and samples were centrifuged at 13,000 g for 15 min. The supernatants were removed, dried in a SpeedVac concentrator, and stored at −80°C. Prior to mass spectrometry analysis, samples were resuspended in 50 mM acetic acid.
Orbitrap Fusion mass spectrometry for SILAC analysis
Capillary-liquid chromatography-nanospray tandem mass spectrometry (Capillary-LC/MS/MS) for protein identification was performed on a Thermo Scientific Orbitrap Fusion mass spectrometer equipped with an EASY-Spray™ Source and operated in positive ion mode. Samples were separated on an EASY-Spray nano column (Pepmap™ RSLC, C18 3μ 100 A, 75 μm X150 mm Thermo Scientific) using a 2D RSLC HPLC system (Thermo Scientific). Each sample was injected into the μ-Precolumn Cartridge (Thermo Scientific,) and desalted with 0.1% Formic Acid in water for 5 min. The injector port was then switched to inject, and the peptides were eluted off of the trap onto the column. Mobile phase A was 0.1% Formic Acid in water, and acetonitrile (with 0.1% formic acid) was used as mobile phase B. Flow rate was set at 300 nL/min. Mobile phase B was increased from 2% to 35% over 220 min, then increased from 35 - 55% over 50 min, then increased from 55%–90% over 8 min, and then kept at 90 % for another 5 min before being brought back quickly to 2% over 2 min. The column was equilibrated at 2% of mobile phase B (or 98% A) for 15 min before the next sample injection. MS/MS data was acquired with a spray voltage of 1.7 KV and a capillary temperature of 275°C. The scan sequence of the mass spectrometer was based on the preview mode data-dependent TopSpeed™ method: the analysis was programmed for a full scan recorded between m/z 400 - 1600 and a MS/MS scan to generate product ion spectra to determine amino acid sequence in consecutive scans starting from the most abundant peaks in the spectrum in the next 3 s. To achieve high mass accuracy MS determination, the full scan was performed at FT mode, and the resolution was set at 120,000. The AGC Target ion number for FT full scan was set at 2 × 105 ions, maximum ion injection time was set at 50 ms, and micro scan number was set at 1. MSn was performed using ion trap mode to ensure the highest signal intensity of MSn spectra using both CID (for 2+ to 4+ charges) and ETD (for 4+-7+ charges) methods. The AGC Target ion number for ion trap MSn scan was set at 1000 ions, maximum ion injection time was set at 100 ms, and micro scan number was set at 1. The CID fragmentation energy was set to 35 %. Dynamic exclusion was enabled with a repeat count of 1 within 60 s and a low mass width and high mass width of 10 ppm. Protein abundances were determined for all proteins identified by two or more peptides in all three replicates. The p values and q values were then calculated using an empirical Bayes method to adjust the estimate of variance of each protein species (Kammers et al., 2015).
Dps protein expression and purification
Wild-type and K8A Dps protein were expressed and purified as previously described (Karas et al., 2015; Vtyurina et al., 2016). Briefly, Dps was expressed from Escherichia coli BL21(DE3) cells carrying the pET17b-dps or pET17b-dpsK8A plasmid Cells were grown at 37°C with shaking at 250 rpm until O.D.600= 0.4 - 0.6. The expression of Dps was induced by addition of 0.3 mM isopropyl β-D-1-thiogalactopyranoside. The cells were disrupted with a French press, and cell lysates were passed through a DEAE Sepharose CL-6B column (GE Healthcare) equilibrated with 50 mM Hepes-KOH containing 100 mM NaCl, pH 7.3. Contaminating proteins were precipitated with 60% ammonium sulfate, while Dps protein remained soluble and was collected in the supernatant. Next, Dps was precipitated with 90% ammonium sulfate and collected in the pellet. Buffer exchange to 50 mM Hepes-KOH with 150 mM NaCl and 0.1 mM EDTA, pH 7.3 using a PD-10 column lowered the ionic strength. Sample was loaded onto an SP-Sepharose column (GE Healthcare), and Dps was eluted with a 50 mM-to-1 M NaCl gradient followed by concentration of Dps using centrifugal filter unit (Amicon Ultra Filtration Unit) with a 10K molecular weight cut-off and exchange into a storage buffer 50 mM Hepes-KOH containing 100 mM NaCl, pH 7.3. The monomer concentration of purified Dps sample was determined by measuring the absorbance at 280 nm, using a molar extinction coefficient of 15,470 M−1·cm−1.
LexA protein expression and purification
LexA was expressed from E. coli XJb(DE3) Autolysis™ cells (Zymo Research) carrying the pET21a-LexA plasmid (Zhang et al., 2010). Cells were grown in LB supplemented with 100 mg/L carbenicillin at 37°C with shaking at 250 rpm until O.D.600= 0.5. Expression of LexA was induced by addition of 0.4 mM isopropyl β-D-1-thiogalactopyranoside for 3 h, and autolysis was induced with 0.07 % arabinose supplemented 1 h prior to cell collection. The cells were resuspended in lysis buffer (500 mM NaCl, 50 mM Tris-HCl pH 6.9, 5% glycerol, 0.2 mM β-mercaptoethanol with 1 × Complete EDTA-free protease inhibitor (Roche)), disrupted with sonication, and clarified by centrifugation. The cell lysate was passed through a HisTrap HP column (GE Healthcare) equilibrated with lysis buffer. The column was washed in lysis buffer, and LexA was eluted with heparin column loading buffer (10 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 0.1 mM DTT, 5% glycerol, 100 mM NaCl) with a 0-500 mM Imidazole gradient. Fractions containing LexA were loaded onto a HiTrap Heparin HP column (GE Healthcare) equilibrated in heparin column loading buffer. LexA was eluted with a 0.1-1.5 M NaCl gradient, exchanged into 2× storage buffer (10 mM Tris-HCl, pH 7.5, 0.1 mM DTT, 0.1 mM DTT, 5% glycerol, 200 mM NaCl) with a HiPrep 26/10 Desalting column (GE Healthcare), mixed with one volume of glycerol, and stored at −20°C. The monomer concentration of purified LexA sample was determined by measuring the absorbance at 280 nm, using a molar extinction coefficient of 6,970 M−1·cm−1.
Dps-DNA binding assay
16.5 ng of linear DNA (recA: 252 bp, fluP 401 bp, λ PR 483 bp, rrnB 120 bp) and Dps (final monomer concentration between 0 to 4 μM Dps) were added to 1× PEG buffer (50 mM Hepes-KOH, pH 7.3, 100 mM KCl, 4 mM MgCl2, 5% PEG 8K). Ingredients were mixed and incubated for 15 min at 30°C. Samples were mixed with DNA loading dye (6× Blue/Orange Loading Dye; Promega), and loaded onto an unstained gel (0.7% agarose in 0.5× TB buffer, prerun for 30 min at 80 V and 4°C). Electrophoresis was performed for 3.5 h at 40 V and 4°C. The gel was post-stained with SYBR Gold dye (Invitrogen) for 30 min at room temperature. Imaging was performed on a Typhoon scanner (GE Healthcare) with an excitation wavelength (λex) of 488 nm, an emission wavelength (λem) of 520 nm, a photomultiplier tube (PMT) voltage of 300-400 V, and 100 μm pixel size. ImageQuant software was used for band intensity quantification. The fraction of bound DNA was calculated as 100% minus the fraction of unbound DNA, based on a no-Dps control lane. The data were fitted to the Hill equation (Θ = [Dps]n / KD + [Dps]n) to determine the apparent KD and n parameters of binding. Each experiment was performed at least in n = 4 replicates.
Restriction endonuclease digestion experiments
16.5 ng of a linear PCR-generated DNA templates containing recA, λ PR, and rrnB promoters was incubated without or with Dps (final monomer concentration between 0 to 4 μM Dps) in 1× PEG buffer (50 mM Hepes-KOH, pH 7.3, 100 mM KCl, 4 mM MgCl2, 5% PEG 8K) for 20 min at 30°C. Afterwards, restriction enzymes (0.05U KpnI for recA, 1U HindIII for rrnB P1, 2U HincII for λ PR; New England Biolabs) were added to the samples, followed by a 30 min incubation at 30°C (unless otherwise stated). Dps and/or restriction enzymes were removed from the DNA template by adding a final concentration of 120 μg/mL Heparin (Sigma-Aldrich) to the samples and mixing. Samples were mixed with DNA loading dye (6× Blue/Orange Loading Dye; Promega) and loaded onto a 0.7% (λ PR) or 1.5% (recA and rrnB P1) agarose gel in 0.5× TB buffer. Electrophoresis was performed for 2.5 h at 70 V at room temperature. The gel was post-stained with SYBR Gold dye (Invitrogen) for 30 min at room temperature. Imaging was performed on a Typhoon scanner (GE Healthcare) with an excitation wavelength (λex) of 488 nm, an emission wavelength (λem) of 520 nm, a photomultiplier tube (PMT) voltage of 400-500 V, and 50-100 μm pixel size. BioRad Image Lab™ software was used for band intensity quantification.
E. coli RNA polymerase holoenzyme
Wild-type E. coli RNA polymerase holoenzyme with pre-bound transcription factor σ70 was purified as described in (Svetlov and Artsimovitch, 2015). The enzyme contains a biotin-modification at the ß’-subunit as described previously (Abbondanzieri et al., 2005) that serves as an anchor to attach streptavidin-coated magnetic beads.
Bulk RNAP transcription experiments
Linear PCR-generated templates containing recA and λ PR promoters (15 nM) were incubated without or with Dps in 50 mM Hepes-KOH pH 7.3, 85 mM KCl, 15 mM NaCl, 4 mM MgCl2, 5% PEG 8K for 30 min at 30°C in a volume of 10 μL, followed by 2 min at 37°C. 5 μL of prewarmed (to 37°C) mixture containing RNAP holoenzyme (30 nM), dinucleotide primer (ApU or ApC, 300 μM), GTP (30 μM) and 1.5 μCi [α32P]-GTP (3000 Ci/mmol; Perkin Elmer) in 50 mM Hepes-KOH pH 7.3, 100 mM KCl, 4 mM MgCl2, 5% PEG 8K, 0.3 mM DTT were added, followed by 5 min incubation at 37°C. The final concentrations of RNAP, DNA template, and Dps were 10, 10, and 300 - 20,000 nM, respectively. Reactions were quenched at 2, 4, 8, 16, and 32 min by addition of an equal volume of STOP buffer (10 M urea, 20 mM EDTA, 45 mM Tris-borate; pH 8.3, 0.2% bromophenol blue, 0.2% xylene cyanol) and loaded onto a 10% denaturing urea-acrylamide gel in 0.5× TBE. The RNA products were analyzed using a Phosphorimaging System (GE Healthcare) and ImageQuant Software. In order to determine the values of τoc and kss, trimer production (TP) was measured at several time points and fit to the functional form:
For transcriptional run-off experiments, recA DNA template was incubated with Dps (or storage buffer) for 20 min at 37°C in 10 μL of 1× PEG buffer. LexA was added in 5 μL of PEG buffer and incubated for 20 min at 37°C, followed by the addition of a pre-heated (to 37°C) mix containing RNAP holoenzyme (σ70 or σS), GpU, NTPs, and [α32P]-GTP. The final concentrations were: DNA template, 20 nM; RNAP, 20 nM; Dps, 2 μM; LexA dimer, 20 or 100 nM; GpU, 125 μM; ATP, CTP and UTP, 100 μM; and GTP, 20 μM. After 15 min at 37°C, reactions were quenched and loaded onto a 6 % denaturing urea-acrylamide gel in 0.5× TBE.
DNA constructs for single-molecule magnetic tweezers experiments
To create a digoxigenin (DIG)-enriched handle, a 643 bp fragment from pBluescript Sk+ (Stratagene, Agilent Technologies Inc., USA) was amplified by PCR in the presence of Digoxigenin-11-dUTP (Roche Diagnostics, Switzerland) using primers 1 and 2 (Table S3). Oligonucleotides (Table S3) were obtained from Ella Biotech GmbH, Germany.
For assisting force (AF) configuration, the digoxigenin-enriched DIG handle was ligated to a 4015 bp spacer consisting of lambda phage sequence from the plasmid pblue1,2,4 + pSFv1A using primers 3 and 4 (Table S3) followed by the T7A1 promotor in front of the RpoB coding sequence and the T7 terminator derived by PCR using plasmid pIA146 and primers 5 and 6 (Table S3). This resulted in a linear dsDNA construct of 9.2 kb.
For opposing force (OF) configuration, the T7 terminator site was removed from plasmid pIA146 by digesting the plasmid with HindIII and SphI (New England Biolabs, UK). Blunt ends were created using the Klenow fragment of DNA polymerase I (New England Biolabs, UK), and these blunt ends were ligated together with T4 DNA ligase (New England Biolabs, UK), resulting in plasmid pIA146Δterminator. DIG handles were ligated to a 1268 bp PCR fragment from plasmid pIA146Δterminator using primers 7 and 8 (Table S4) and a 5543 bp PCR fragment from plasmid pIA146 containing the T7A1 promotor and the E. coli RpoB coding sequence using primers 9 and 10. Prior to ligations, all amplicons were digested with the non-palindromic restriction enzyme BsaI-HF (New England Biolabs, UK). The ligation of the fragments was carried out with the T4 DNA ligase (New England Biolabs, UK). This resulted in a linear dsDNA construct of 7.5 kb.
For the linear 8.8 kb dsDNA construct used to perform force-extension experiments in presence of Dps (Figure S5A,B and Figure S6B), an additional biotin-enriched handle was created for attachment to the streptavidin-coated superparamagnetic beads. The digoxigenin- and biotin-enriched handles were created from a 1.2 kb fragment from pBluescript Sk+ (Stratagene, Agilent Technologies Inc., USA), amplified by PCR in presence of biotin-16-dUTP (Roche Diagnostics, Switzerland) and digoxigenin-11-dUTP (Roche Diagnostics, Switzerland) using primers 11 and 12 (Table S3). After digestion with BamHI, the handles were enzymatically ligated via T4 DNA ligase (New England Biolabs, UK) to a 1512 bp spacer consisting of lambda phage sequence from the plasmid pblue1,2,4 + pSFv1A using primers 13 and 14 (Table S3), and further to a 6083 bp PCR fragment from plasmid pIA146 containing the T7A1 promotor and the E. coli RpoB coding sequence using primers 15 and 16.
Superparamagnetic and polystyrene reference beads
Streptavidin-coated superparamagnetic beads (DynaBeads, #65601, LifeTechnologies) with a diameter of 1 μm were used in this study. Commercially available polystyrene beads (#17133, Polysciences GmbH) with a diameter of 1.5 μm were used as reference beads.
Magnetic tweezers experimental configuration
The magnetic tweezers implementation used in this study has been described previously (Dulin et al., 2015; Vtyurina et al., 2016). Briefly, light transmitted through the sample was collected by a 50× oil-immersion objective (CFI Plan 50XH, Achromat, 50×, NA = 0.9, Nikon) and projected onto a 12 megapixel CMOS camera (#FA-80-12M1H, Falcon2, Teledyne Dalsa) with a sampling frequency of 25 Hz. The applied magnetic field was generated by a pair of vertically aligned permanent neodymium-iron-boron magnets (SuperMagnete) separated by a distances of 1 mm, suspended on a motorized stage (#M-126.PD2, Physik Instrumente) above the flow cell. Additionally, the magnet pair can be rotated around the illumination axis by an applied DC servo step motor (C-150.PD, Physik Instrumente). Image processing of the collected light allowed us to track the real-time position of both surface attached reference beads and superparamagnetic beads coupled to RNAP in three dimensions over time. The bead position tracking was achieved using a cross-correlation algorithm realized with custom-written software (Cnossen et al., 2014) in LabView (2011, National Instruments Corporation). Bead positions were determined with spectral corrections to correct for camera blur and aliasing (Cnossen et al., 2014).
Single-molecule RNAP transcription assay
The flow cell preparation used in this study has been described in detail elsewhere (Dulin et al., 2015; Vtyurina et al., 2016). In short, polystyrene reference beads (Polysciences Europe) of 1.5 μm in diameter were diluted 1:4000 in PBS buffer (pH 7.4; Sigma Aldrich) and then adhered to the nitrocellulose-coated (Invitrogen) surface of the flow cell. Further, digoxigenin antibody Fab fragments (Roche Diagnostics) at a concentration of 0.5 mg/ml was incubated for 5 hours within the flow cell, following overnight incubation of 10 mg/ml BSA (New England Biolabs) diluted in buffer A containing 20 mM Tris, 100 mM KCl, 10 mM MgCl2, 0.05 % (v/v) Tween20 (Sigma Aldrich) and 40 μg/ml BSA (New England Biolabs), adjusted to pH 7.9.
The preparation of the RNAP ternary complex was performed as described previously (Abbondanzieri et al., 2005). Briefly, RNAP holoenzyme was stalled on the DNA constructs at position A29 after the T7A1 promoter sequence. To do so, 30 nM of RNAP holoenzyme was added to 3 nM linear DNA in buffer A and incubated 10 min at 37°C. Afterwards, 50 μM ATP, CTP, GTP (GE Healthcare Europe), and 100 μM ApU (IBA Lifesciences GmbH) were added to the solution and incubated for additional 10 min at 30°C. The ternary complex solution was diluted to a final concentration of 250 pM of the RNAP:DNA complex. The complex was flushed into the flow cell and incubated for 30 min at room temperature. The subsequent addition of 100 μL streptavidin-coated superparamagnetic beads (diluted 1:400 in PBS buffer; MyOne Dynabeads, Invitrogen/Life Technologies) with a diameter of 1 μm resulted in the attachment of the beads to biotinylated RNAP stalled on the DNA.
Before the re-initiation of transcription, 200 μL of buffer B containing 50 mM Hepes, 100 mM KCl, 4 mM MgCl2, 0.1 mg/ml BSA, pH 7.3 and 5% PEG 8K (Promega), was flushed through the flow cell At this step, for the experiments with Dps, different concentrations (1, 4, 7, or 10 μM) of Dps were added to buffer B while DNA was stretched at a force of 5 pN. Transcription was re-initiated by adding ATP, CTP, GTP, and UTP (GE Healthcare Europe) at a concentration of 1 mM to the stalled RNAP ternary complexes and immediately starting the single-molecule measurements. The experiments were conducted for 1 h at constant or transient pulling forces (details in corresponding manuscript text) with a camera acquisition rate of 25 Hz.
Quantification and statistical analysis
Dwell time analysis
Transcription traces were processed using custom-written Igor v6.39 and MatLab R2013b-based scripts. The absolute z-position of the RNAP during the transcription process was converted to transcribed RNA product as a function of time, using the end-to-end length determined by the extensible worm-like chain model (Odijk, 1995) with a stretch modulus of 800 pN and persistence length of 56 nm. To reduce the effect of Brownian noise in the dwell time analysis, all individual elongation traces were filtered prior to 1 Hz.
The transcription dynamics of E. coli RNAP were quantitatively assessed by a statistical analysis of elongation and transcriptional pausing. Pause distributions were evaluated using unbiased dwell time analysis (Dulin et al., 2015). The times needed for RNAP to transcribe through consecutive dwell time windows of 10 nt along the trace (prior filtered to 1 Hz using a mean filter) - defined as dwell times - were calculated for all RNAP trajectories and used to construct a dwell time probability distribution function. The dwell times were bootstrapped 1,000 times to estimate the standard deviation and confidence intervals of the distributions (Dulin et al., 2015).
To characterize the dwell time distribution, we divided it into three separate time ranges: the elongation region (0.1-1 s), which contained the elongation peak; the short pause region (1-5 s); and the long pause region (5-100 s). We fit a Galton distribution to the elongation region. The elongation rate is given by , where N fitted is the dwell time window size, and denotes the peak position of the fitted distribution. To calculate the probabilities of the short and long pauses, we integrated the dwell time distribution over the corresponding regions.
Statistical analysis
For single-molecule data (Figures 4, 5), we used Tukey’s outlier filter of leveraging the Interquartile Range for the data selection. This method is applicable to most ranges since it does not depend on distributional assumptions. It also ignores the mean and standard deviation, making it resistant to being influenced by the extreme values in the range (Frigge et al., 1989). The statistical test to analyze differences in RNAP elongation rates, pause probabilities, and average transcription velocities were performed using one-way two-tailed analysis of variance (ANOVA, significance level of α = 0.001) with subsequent Tukey post-hoc test for statistical comparison.
For the data obtained from bulk experiments, we employed a statistical analysis using an unpaired, two-tailed t-test with a significance level of α = 0.001.
Data and software availability
Custom-written Igor and MatLab scripts used for dwell time analysis of transcription traces will be provided upon request to the Lead Contact, Anne S. Meyer (anne@annemeyerlab.org).
Supplementary Material
Figure S1. Dps protein levels and DNA compaction increase dramatically in wild-type E. coli after 24 hours of growth. (A) Western blot using anti-Dps antibodies to indicate Dps levels in wild-type and Δdps E. coli cells in exponential phase and after 24 and 96 h of growth. The arrowhead indicates the position of Dps protein bands. (B) Quantified protein levels of three independent experiments in arbitrary units. The intensities of the bands were analyzed using ImageQuant. (C) Comparison of E. coli wild-type (grey) and Δdps (blue) cell lengths (dashed) and corresponding nucleoid lengths (solid) after 24 h (left) and 96 h (right) of growth, extracted from fluorescence images (n = 133 - 208 cells per condition, from 5 individual colonies). The error bars in (B), (C) represent the standard deviation. Statistical analyses were performed using unpaired, two-tailed t-test (*** = significance p ≤ 0.0001; n.s. = non-significant). Related to Figures 1 and 2.
Figure S2. RNA sequencing quality controls and gene expression in E. coli after 96 h of growth. (A) The total isolated RNA from exponential phase, 24 h, and 96 h cultures was run on an agarose gel for both E. coli wild-type and Δdps cells. The 23S and 16S rRNA are visible as distinct bands for all samples, indicating that the total RNA was intact in stationary phase cells. (B) Depth of sequencing. The total amount of aligned read pairs per strain for stationary phase cells after 24 h of growth and 96 h of growth is shown, pooled among four repeated experiments. The sequencing depth of more than 10 million reads in total per sample was sufficient to enable a robust analysis of the transcriptome, since 2-3 million reads per sample represents the lower threshold boundary to detect the majority of 2-fold differentially expressed genes with high (P < 0.001) statistical significance (Haas et al., 2012). (C, D) Quality of base calling for E. coli cells after (C) 24 h of growth and (D) 96 h of growth. Quality scores were generated per base position in the read, for each set of reads per sample. Higher scores indicate better base calls. The yellow box represents the inter-quartile range (25-75%), the red line depicts the median, the upper and lower whiskers represent the 10% and 90% confidence intervals, and the blue line displays the mean quality. The background of the graph divides the y-axis into very good quality calls (green), calls of reasonable quality (orange), and calls of poor quality (red). The sequenced reads all had a very high quality score (> 30), meaning that the sequences were determined with more than 99.9% accuracy. (E) The relative amounts of total RNA and mRNA isolated from E. coli wild-type and Δdps cells grown for 96 h. The statistical analysis was performed using an unpaired, two-tailed t-test with a significance level of α = 0.0001. No significant differences were observed. The error bars represent the mean standard error. (F) Differential expression analysis of RNA sequencing on wild-type and Δdps cells after 96 h of growth. Mean expression values were calculated from the fragments per kilobase of transcript per million mapped reads (FPKM). For each gene, the mean expression in the wild-type strain (y-axis) is plotted against the corresponding value in the Δdps strain (x-axis). Colors represent the fold-difference between the mRNA expression in the two strains. Besides dps and flu, all genes clustered close to the line x = y. (G) Differential expression analysis of RNA sequencing on Δdps cells after 24 and 96 h of growth. The mean expression value in FPKM of each mRNA species in the Δdps strain after 24 h of growth (y-axis) is plotted against the mean expression value in FPKM in the Δdps strain after 96 h of growth (x-axis). Comparison of the transcriptomes revealed 67 genes expressed at significantly different levels. The colors represent the fold change between the mRNA expression in cells. Filled circles represent genes that were significantly differentially expressed, and open circles represent genes that were not significantly different. Significance testing for (F) and (G) was performed using Cuffdiff, based on n = 4 independent experiments and a q- value significance level of q ≤ 0.05. Related to Figure 2.
Figure S3. RT-qPCR indicates that mRNA levels of E. coli genes are unaffected by the deletion of dps in cultures grown for 24 h. The delta threshold cycles (ΔCt) that represent the expression values of the target genes relative to that of three reference genes are depicted for six target genes of cells grown for 24 h. ΔΔCt analysis comparing the RT-qPCR results from E. coli wild-type and Δdps strains showed that, with the exception of flu, none of the genes was significantly differentially expressed. The expression of flu is significantly different with a p-value < 0.01. The error bars denote the standard error of the mean and are based on n = 3 independent experiments. Related to Figure 2.
Figure S4. The effect of Dps on RNAP promoter binding and digestion with restriction endonucleases. (A) Promoters used in this study. The −35 and −10 hexamers are boxed (black), the transcription start site (+1) is indicated, and the specific recognition sequences for the restriction endonucleases are highlighted. (B) A representative gel-shift analysis of Dps binding to a linear DNA fragment (252 bp) that contains a recA promoter sequence, at a range of Dps concentrations (0 to 4 μM). DNA was visualized through gel electrophoresis and staining with SYBR Gold. (C) Transcription initiation assays in which Dps (at indicated concentrations) was pre-incubated with a linear bacteriophage λ PR promoter template. RNAP holoenzyme, ApC RNA primer, and radiolabeled GTP were then added, allowing open complexes to form, followed by repeated rounds of ApCpG synthesis. Transcription initiation is plotted for a range of Dps concentrations. The error bars represent the mean standard deviation. (D) Trimer production measured over time using the λ PR promoter as a template as in (B), in the presence and absence of 4 μM Dps. (E) Trimer synthesis ratio (4 μM Dps/0 μM Dps) over time, calculated from the trimer production over time shown in (D). (F) The linear recA promoter template (used also in Figure 3) can be digested by KpnI restriction endonuclease, which recognizes a sequence within the RNAP footprint in the promoter complex. Dps (0 or 4 μM) was pre-incubated with the recA fragment, followed by the addition of KpnI. DNA:Dps complexes as well DNA-bound KpnI were dissociated via incubation with heparin (where indicated). (G) Protection of the promoters recA, λ PR, and rrnB P1 against digestion by KpnI, HincII, and HindIII, respectively, at a Dps concentration of 0 and 4 μM is shown at a range of digestion reaction times (0 to 65 min). (H) Dps-based protection of the recA promoter fragment against digestion by KpnI at different Dps concentrations (0 to 4 μM). (I and J) Wild-type, K8A, and K10A Dps proteins at 4 μM were bound to (I) rrnB P1 or (J) λ PR promoter DNA as in (F) and (H), followed by incubation with or without HindIII or HincII, respectively (also shown for recA in Figure 3). The weakly DNA-binding Dps variants K8A and K10A did not protect DNA from cleavage by restriction endonucleases. DNA was visualized through gel electrophoresis. Related to Figure 3.
Figure S5: Dps-bound rec A promoter is accessible to σS RNAP holoenzyme and LexA repressor. (A) The recA DNA template schematic, with positions of the core promoter elements (−35 and −10), transcription start site (+1), and LexA recognition sequences (green) (Zhang et al., 2010) indicated. During in vitro run-off transcription experiments, RNAP is released at the end of the linear template (Run-off RNA; 177 nt) and at two hairpin-dependent termination sites (101 and 138 nt). (B) recA DNA template was incubated with Dps (or storage buffer) for 20 min at 37°C, after which LexA was added and incubated for 20 min at 37°C. Then, a mix containing RNAP holoenzyme (σ70 or σS), GpU, NTPs, and [α32P]-GTP was added and incubated for 20 min at 37°C to allow for run-off transcription. Reactions were quenched and visualized on a denaturing urea-acrylamide gel. (C) Run-off transcription gel for the recA template. Since the σ70 holoenzyme is more active on the recA template than the σS holoenzyme, different exposures of the same gel are shown for the two holoenzymes; for comparison, one lane from the σ70 set is shown alongside the σS reactions at the same exposure. (D) Production of run-off RNA, quantified for each RNAP holoenzyme separately, with RNA levels observed in the absence of Dps and LexA defined as 100%. The numbers shown are mean averages ± SD of n = 3 independent replicates. Related to Figure 3.
Figure S6: Dps-induced DNA condensation in presence of exerted force. (A) Extension of a 9-kb linear DNA template (red) under different force loads (blue) in the presence of 1 μM Dps. (B) Force-extension curves for a 9-kb linear DNA template (n = 7, mean ± SEM) in the absence (green) and presence (blue) of 1 μM Dps. Solid lines correspond to decreasing force and dashed lines to increasing force. The red-colored area between force-dependent DNA:Dps complex formation and disassembly depicts the boundaries of the mean transition points, indicating the hysteresis. (C) Extension of the RNAP ternary complex on a 7.5-kb linear DNA template in the presence of 1 μM Dps under high (5pN) and low (0.1 pN) force loads. (D) Temporal magnification of RNAP transcription on a 7.5-kb linear DNA template with transient pulling from low force to high force (8 pN) every 400 s in the presence of 1 μM Dps (Figure 6E). Related to Figures 4, 5 and 6.
Figure S7: RNAP transcription dynamics and DNA condensation at high Dps concentrations. (A) Dwell time distributions for assisting force (AF) trajectories in the presence (red) and the absence (black) of 10 μM Dps at 24°C. The distributions are separated with boundaries for the elongation region (0.1-1 s), short elemental pauses SP (1-5 s), and longer pauses LP (5-100 s). (B) Force-extension curves for a 8.8-kb linear DNA template (n = 7, mean ± SEM) in the absence (black) and presence (colored) of 4 μM (blue), 7 μM (cyan), and 10 μM (red) Dps. Solid lines correspond to decreasing force and dashed lines to increasing force. Related to Figure 5.
Highlights.
In E. coli, the condensation of DNA by Dps is decoupled from transcription.
Dps provides selective access of proteins to the encased DNA.
RNA polymerase readily initiates transcription on Dps-protected promoters.
Dps maintains a dynamic condensed structure during RNA chain elongation.
Acknowledgments
Funding to I.A. was provided by the National Institutes of Health (NIH, R01 GM67153). Funding to N.H.D. was provided by the Netherlands Organization for Scientific Research (NWO) via its TOP-GO program and by the European Union via an ERC Consolidator Grant (DynGenome, no 312221). Funding to E.A.A. and L.L. was provided by the Nanofront initiative of the NWO and the Department of Bionanoscience of Delft University of Technology. Funding to A.S.M. and B.E.-M. is part of the research program of the Foundation for Fundamental Research on Matter (FOM), which is financially supported by NWO. We thank Rick Gourse for a gift of RpoS protein and Phoebe Rice for a gift of LexA-expression plasmids. We are grateful to Martin Depken, Ilja Westerlaken, Michela De Martino, and Theo van Laar for fruitful discussions and assistance, to Michelle Gibbs and Kurt Fredrick for the gift of Arg and Lys isotopes for the SILAC analysis, and to Sophie Harvey, Liwen Zhang, and Arpad Somogyi at the Ohio State University Campus Chemical Instrument Center for mass spectrometry analysis and discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Abbondanzieri EA Greenleaf WJ Shaevitz JW Landick R Block SM Direct observation of base-pair stepping by RNA polymerase Nature 2005. 438 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almiron M Link AJ Furlong D Kolter R A novel DNA-binding protein with regulatory and protective roles in starved Escherichia coli Genes & development 1992. 6 2646–2654 [DOI] [PubMed] [Google Scholar]
- Antipov SS, Tutukina MN, Preobrazhenskaya EV, Kondrashov FA, Patrushev MV, Toshchakov SV, Dominova I, Shvyreva US, Vrublevskaya VV, Morenkov OS. et al. The nucleoid protein Dps binds genomic DNA of Escherichia coli in a non-random manner. PLoS One. 2017;12:e0182800. doi: 10.1371/journal.pone.0182800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam TA Ishihama A Twelve species of the nucleoid-associated protein from Escherichia coli. Sequence recognition specificity and DNA binding affinity J Biol Chem 1999. 274 33105–33113 [DOI] [PubMed] [Google Scholar]
- Azam TA Iwata A Nishimura A Ueda S Ishihama A Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid J Bacteriol 1999. 181 6361–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular systems biology. 2006;2 doi: 10.1038/msb4100050. 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing Journal of the Royal Statistical Society Series B (Methodological) 1995. 57 289–300 [Google Scholar]
- Blaby-Haas CE Furman R Rodionov DA Artsimovitch I de Crecy-Lagard V Role of a Zn-independent DksA in Zn homeostasis and stringent response Mol Microbiol 2011. 79 700–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM Lohse M Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data Bioinformatics 2014. 30 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning DF Busby SJ The regulation of bacterial transcription initiation Nat Rev Microbiol 2004. 2 57–65 [DOI] [PubMed] [Google Scholar]
- Butala M Zgur-Bertok D Busby SJ The bacterial LexA transcriptional repressor Cellular and molecular life sciences: CMLS 2009. 66 82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun LN Kwon YM Structure, function and regulation of the DNA-binding protein Dps and its role in acid and oxidative stress resistance in Escherichia coli: a review Journal of applied microbiology 2011. 110 375–386 [DOI] [PubMed] [Google Scholar]
- Ceci P Cellai S Falvo E Rivetti C Rossi GL Chiancone E DNA condensation and self-aggregation of Escherichia coli Dps are coupled phenomena related to the properties of the N-terminus Nucleic Acids Res 2004. 32 5935–5944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS Weissman JS Nascent transcript sequencing visualizes transcription at nucleotide resolution Nature 2011. 469 368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnossen JP, Dulin D, Dekker NH. An optimized software framework for real-time, high-throughput tracking of spherical beads. Rev Sci Instrum. 2014;85:103712. doi: 10.1063/1.4898178. [DOI] [PubMed] [Google Scholar]
- Dame RT The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin Mol Microbiol 2005. 56 858–870 [DOI] [PubMed] [Google Scholar]
- De Martino M Ershov D van den Berg PJ Tans SJ Meyer AS Single-Cell Analysis of the Dps Response to Oxidative Stress J Bacteriol 2016. 198 1662–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diderichsen B flu, a metastable gene controlling surface properties of Escherichia coli J Bacteriol 1980. 141 858–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A Davis CA Schlesinger F Drenkow J Zaleski C Jha S Batut P Chaisson M Gingeras TR STAR: ultrafast universal RNA-seq aligner Bioinformatics 2013. 29 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman CJ Genome architecture and global gene regulation in bacteria: making progress towards a unified model? Nat Rev Microbiol 2013. 11 349–355 [DOI] [PubMed] [Google Scholar]
- Dulin D Vilfan ID Berghuis BA Hage S Bamford DH Poranen MM Depken M Dekker NH Elongation-Competent Pauses Govern the Fidelity of a Viral RNA-Dependent RNA Polymerase Cell Rep 2015. 10 983–992 [DOI] [PubMed] [Google Scholar]
- Ederth J Artsimovitch I Isaksson LA Landick R The downstream DNA jaw of bacterial RNA polymerase facilitates both transcriptional initiation and pausing J Biol Chem 2002. 277 37456–37463 [DOI] [PubMed] [Google Scholar]
- Elgamal S, Artsimovitch I, Ibba M. Maintenance of Transcription-Translation Coupling by Elongation Factor P. mBio. 2016;7 doi: 10.1128/mBio.01373-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V Toulme F Rahmouni AR Borukhov S Nudler E Transcription through the roadblocks: the role of RNA polymerase cooperation Embo J 2003. 22 4719–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkiel-Krispin D Levin-Zaidman S Shimoni E Wolf SG Wachtel EJ Arad T Finkel SE Kolter R Minsky A Regulated phase transitions of bacterial chromatin: a non-enzymatic pathway for generic DNA protection Embo J 2001. 20 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigge M Hoaglin DC Iglewicz B Some Implementations of the Boxplot Am Stat 1989. 43 50–54 [Google Scholar]
- Gaal T Ross W Estrem ST Nguyen LH Burgess RR Gourse RL Promoter recognition and discrimination by EsigmaS RNA polymerase Mol Microbiol 2001. 42 939–954 [DOI] [PubMed] [Google Scholar]
- Ghatak P, Karmakar K, Kasetty S, Chatterji D. Unveiling the role of Dps in the organization of mycobacterial nucleoid. PLoS One. 2011;6:e16019. doi: 10.1371/journal.pone.0016019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD Allis CD Bernstein E Epigenetics: a landscape takes shape Cell 2007. 128 635–638 [DOI] [PubMed] [Google Scholar]
- Grant RA Filman DJ Finkel SE Kolter R Hogle JM The crystal structure of Dps, a ferritin homolog that binds and protects DNA Nat Struct Biol 1998. 5 294–303 [DOI] [PubMed] [Google Scholar]
- Gruber TM Gross CA Multiple sigma subunits and the partitioning of bacterial transcription space Annual review of microbiology 2003. 57 441–466 [DOI] [PubMed] [Google Scholar]
- Haas BJ, Chin M, Nusbaum C, Birren BW, Livny J. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes? BMC genomics. 2012;13:734. doi: 10.1186/1471-2164-13-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD Madhani HD Mechanisms that specify promoter nucleosome location and identity Cell 2009. 137 445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert KM La Porta A Wong BJ Mooney RA Neuman KC Landick R Block SM Sequence-resolved detection of pausing by single RNA polymerase molecules Cell 2006. 125 1083–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller DA Fogg JM Martin AM Beechem JM Reich NO Perona JJ Simultaneous DNA binding and bending by EcoRV endonuclease observed by real-time fluorescence Biochemistry 2003. 42 14375–14385 [DOI] [PubMed] [Google Scholar]
- Hodges C Bintu L Lubkowska L Kashlev M Bustamante C Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II Science 2009. 325 626–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommais F Krin E Laurent-Winter C Soutourina O Malpertuy A Le Caer JP Danchin A Bertin P Large-scale monitoring of pleiotropic regulation of gene expression by the prokaryotic nucleoid-associated protein, H-NS Mol Microbiol 2001. 40 20–36 [DOI] [PubMed] [Google Scholar]
- Huber W Carey VJ Gentleman R Anders S Carlson M Carvalho BS Bravo HC Davis S Gatto L Girke T et al. Orchestrating high-throughput genomic analysis with Bioconductor Nature methods 2015. 12 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA Weber CA Julicher F Liquid-liquid phase separation in biology Annual review of cell and developmental biology 2014. 30 39–58 [DOI] [PubMed] [Google Scholar]
- Kammers K Cole RN Tiengwe C Ruczinski I Detecting Significant Changes in Protein Abundance EuPA open proteomics 2015. 7 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S Edgar R Adhya S Nucleoid remodeling by an altered HU protein: reorganization of the transcription program Proceedings of the National Academy of Sciences of the United States of America 2005. 102 16397–16402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas VO Westerlaken I Meyer AS The DNA-Binding Protein from Starved Cells (Dps) Utilizes Dual Functions To Defend Cells against Multiple Stresses J Bacteriol 2015. 197 3206–3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J Yoshimura SH Hizume K Ohniwa RL Ishihama A Takeyasu K Fundamental structural units of the Escherichia coli nucleoid revealed by atomic force microscopy Nucleic Acids Res 2004. 32 1982–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlajich MV, Hron DR, Boudreau BA, Sun Z, Lyubchenko YL, Landick R. Bridged filaments of histone-like nucleoid structuring protein pause RNA polymerase and aid termination in bacteria. eLife. 2015;4 doi: 10.7554/eLife.04970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckel F Kubo K Tsumoto K Yoshikawa K Enhancement and inhibition of DNA transcriptional activity by spermine: a marked difference between linear and circular templates FEBS letters 2005. 579 5119–5122 [DOI] [PubMed] [Google Scholar]
- McClure WR Rate-limiting steps in RNA chain initiation Proceedings of the National Academy of Sciences of the United States of America 1980. 77 5634–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AS Baker TA Proteolysis in the Escherichia coli heat shock response: a player at many levels Current opinion in microbiology 2011. 14 194–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AS Grainger DC The Escherichia coli Nucleoid in Stationary Phase Adv Appl Microbiol 2013. 83 69–86 [DOI] [PubMed] [Google Scholar]
- Mitrea DM, Cika JA, Guy CS, Ban D, Banerjee PR, Stanley CB, Nourse A, Deniz AA, Kriwacki RW. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife. 2016;5 doi: 10.7554/eLife.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S Finkel SE Dps protects cells against multiple stresses during stationary phase J Bacteriol 2004. 186 4192–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ Petsalaki E Farber P Jervis D Fussner E Plochowietz A Craggs TD Bazett-Jones DP Pawson T Forman-Kay JD et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles Molecular cell 2015. 57 936–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler E RNA polymerase backtracking in gene regulation and genome instability Cell 2012. 149 1438–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odijk T Stiff Chains and Filaments under Tension Macromolecules 1995. 28 7016–7018 [Google Scholar]
- Ong SE Foster LJ Mann M Mass spectrometric-based approaches in quantitative proteomics Methods 2003. 29 124–130 [DOI] [PubMed] [Google Scholar]
- Park HS Ostberg Y Johansson J Wagner EG Uhlin BE Novel role for a bacterial nucleoid protein in translation of mRNAs with suboptimal ribosome-binding sites Genes & development 2010. 24 1345–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD Livak KJ Analyzing real-time PCR data by the comparative C(T) method Nature protocols 2008. 3 1101–1108 [DOI] [PubMed] [Google Scholar]
- Schneider CA Rasband WS Eliceiri KW NIH Image to ImageJ: 25 years of image analysis Nature methods 2012. 9 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaevitz JW Abbondanzieri EA Landick R Block SM Backtracking by single RNA polymerase molecules observed at near-base-pair resolution Nature 2003. 426 684–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M Baker TA Schnitzler G Deischel SM Goel M Dove W Jaacks KJ Grossman AD Erickson JW Gross CA A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli Microbiological reviews 1989. 53 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephani K Weichart D Hengge R Dynamic control of Dps protein levels by ClpXP and ClpAP proteases in Escherichia coli Mol Microbiol 2003. 49 1605–1614 [DOI] [PubMed] [Google Scholar]
- Storey JD Tibshirani R Statistical significance for genomewide studies Proceedings of the National Academy of Sciences of the United States of America 2003. 100 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR Emelyanov AV Mir M Fyodorov DV Darzacq X Karpen GH Phase separation drives heterochromatin domain formation Nature 2017. 547 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetlov V Artsimovitch I Purification of bacterial RNA polymerase: tools and protocols Methods Mol Biol 2015. 1276 13–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukder A Ishihama A Growth phase dependent changes in the structure and protein composition of nucleoid in Escherichia coli Science China Life sciences 2015. 58 902–911 [DOI] [PubMed] [Google Scholar]
- Trapnell C Hendrickson DG Sauvageau M Goff L Rinn JL Pachter L Differential analysis of gene regulation at transcript resolution with RNA-seq Nature biotechnology 2013. 31 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C Williams BA Pertea G Mortazavi A Kwan G van Baren MJ Salzberg SL Wold BJ Pachter L Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation Nature biotechnology 2010. 28 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbarger MA Toro E Wright MA Porreca GJ Bau D Hong SH Fero MJ Zhu LJ Marti-Renom MA McAdams HH et al. The three-dimensional architecture of a bacterial genome and its alteration by genetic perturbation Molecular cell 2011. 44 252–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel SK Schulz A Rippe K Binding affinity of Escherichia coli RNA polymerase*sigma54 holoenzyme for the glnAp2, nifH and nifL promoters Nucleic Acids Res 2002. 30 4094–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vtyurina NN Dulin D Docter MW Meyer AS Dekker NH Abbondanzieri EA Hysteresis in DNA compaction by Dps is described by an Ising model Proceedings of the National Academy of Sciences of the United States of America 2016. 113 4982–4987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z Gerstein M Snyder M RNA-Seq: a revolutionary tool for transcriptomics Nature reviews Genetics 2009. 10 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SG Frenkiel D Arad T Finkel SE Kolter R Minsky A DNA protection by stress-induced biocrystallization Nature 1999. 400 83–85 [DOI] [PubMed] [Google Scholar]
- Zhang AP Pigli YZ Rice PA Structure of the LexA-DNA complex and implications for SOS box measurement Nature 2010. 466 883–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dps protein levels and DNA compaction increase dramatically in wild-type E. coli after 24 hours of growth. (A) Western blot using anti-Dps antibodies to indicate Dps levels in wild-type and Δdps E. coli cells in exponential phase and after 24 and 96 h of growth. The arrowhead indicates the position of Dps protein bands. (B) Quantified protein levels of three independent experiments in arbitrary units. The intensities of the bands were analyzed using ImageQuant. (C) Comparison of E. coli wild-type (grey) and Δdps (blue) cell lengths (dashed) and corresponding nucleoid lengths (solid) after 24 h (left) and 96 h (right) of growth, extracted from fluorescence images (n = 133 - 208 cells per condition, from 5 individual colonies). The error bars in (B), (C) represent the standard deviation. Statistical analyses were performed using unpaired, two-tailed t-test (*** = significance p ≤ 0.0001; n.s. = non-significant). Related to Figures 1 and 2.
Figure S2. RNA sequencing quality controls and gene expression in E. coli after 96 h of growth. (A) The total isolated RNA from exponential phase, 24 h, and 96 h cultures was run on an agarose gel for both E. coli wild-type and Δdps cells. The 23S and 16S rRNA are visible as distinct bands for all samples, indicating that the total RNA was intact in stationary phase cells. (B) Depth of sequencing. The total amount of aligned read pairs per strain for stationary phase cells after 24 h of growth and 96 h of growth is shown, pooled among four repeated experiments. The sequencing depth of more than 10 million reads in total per sample was sufficient to enable a robust analysis of the transcriptome, since 2-3 million reads per sample represents the lower threshold boundary to detect the majority of 2-fold differentially expressed genes with high (P < 0.001) statistical significance (Haas et al., 2012). (C, D) Quality of base calling for E. coli cells after (C) 24 h of growth and (D) 96 h of growth. Quality scores were generated per base position in the read, for each set of reads per sample. Higher scores indicate better base calls. The yellow box represents the inter-quartile range (25-75%), the red line depicts the median, the upper and lower whiskers represent the 10% and 90% confidence intervals, and the blue line displays the mean quality. The background of the graph divides the y-axis into very good quality calls (green), calls of reasonable quality (orange), and calls of poor quality (red). The sequenced reads all had a very high quality score (> 30), meaning that the sequences were determined with more than 99.9% accuracy. (E) The relative amounts of total RNA and mRNA isolated from E. coli wild-type and Δdps cells grown for 96 h. The statistical analysis was performed using an unpaired, two-tailed t-test with a significance level of α = 0.0001. No significant differences were observed. The error bars represent the mean standard error. (F) Differential expression analysis of RNA sequencing on wild-type and Δdps cells after 96 h of growth. Mean expression values were calculated from the fragments per kilobase of transcript per million mapped reads (FPKM). For each gene, the mean expression in the wild-type strain (y-axis) is plotted against the corresponding value in the Δdps strain (x-axis). Colors represent the fold-difference between the mRNA expression in the two strains. Besides dps and flu, all genes clustered close to the line x = y. (G) Differential expression analysis of RNA sequencing on Δdps cells after 24 and 96 h of growth. The mean expression value in FPKM of each mRNA species in the Δdps strain after 24 h of growth (y-axis) is plotted against the mean expression value in FPKM in the Δdps strain after 96 h of growth (x-axis). Comparison of the transcriptomes revealed 67 genes expressed at significantly different levels. The colors represent the fold change between the mRNA expression in cells. Filled circles represent genes that were significantly differentially expressed, and open circles represent genes that were not significantly different. Significance testing for (F) and (G) was performed using Cuffdiff, based on n = 4 independent experiments and a q- value significance level of q ≤ 0.05. Related to Figure 2.
Figure S3. RT-qPCR indicates that mRNA levels of E. coli genes are unaffected by the deletion of dps in cultures grown for 24 h. The delta threshold cycles (ΔCt) that represent the expression values of the target genes relative to that of three reference genes are depicted for six target genes of cells grown for 24 h. ΔΔCt analysis comparing the RT-qPCR results from E. coli wild-type and Δdps strains showed that, with the exception of flu, none of the genes was significantly differentially expressed. The expression of flu is significantly different with a p-value < 0.01. The error bars denote the standard error of the mean and are based on n = 3 independent experiments. Related to Figure 2.
Figure S4. The effect of Dps on RNAP promoter binding and digestion with restriction endonucleases. (A) Promoters used in this study. The −35 and −10 hexamers are boxed (black), the transcription start site (+1) is indicated, and the specific recognition sequences for the restriction endonucleases are highlighted. (B) A representative gel-shift analysis of Dps binding to a linear DNA fragment (252 bp) that contains a recA promoter sequence, at a range of Dps concentrations (0 to 4 μM). DNA was visualized through gel electrophoresis and staining with SYBR Gold. (C) Transcription initiation assays in which Dps (at indicated concentrations) was pre-incubated with a linear bacteriophage λ PR promoter template. RNAP holoenzyme, ApC RNA primer, and radiolabeled GTP were then added, allowing open complexes to form, followed by repeated rounds of ApCpG synthesis. Transcription initiation is plotted for a range of Dps concentrations. The error bars represent the mean standard deviation. (D) Trimer production measured over time using the λ PR promoter as a template as in (B), in the presence and absence of 4 μM Dps. (E) Trimer synthesis ratio (4 μM Dps/0 μM Dps) over time, calculated from the trimer production over time shown in (D). (F) The linear recA promoter template (used also in Figure 3) can be digested by KpnI restriction endonuclease, which recognizes a sequence within the RNAP footprint in the promoter complex. Dps (0 or 4 μM) was pre-incubated with the recA fragment, followed by the addition of KpnI. DNA:Dps complexes as well DNA-bound KpnI were dissociated via incubation with heparin (where indicated). (G) Protection of the promoters recA, λ PR, and rrnB P1 against digestion by KpnI, HincII, and HindIII, respectively, at a Dps concentration of 0 and 4 μM is shown at a range of digestion reaction times (0 to 65 min). (H) Dps-based protection of the recA promoter fragment against digestion by KpnI at different Dps concentrations (0 to 4 μM). (I and J) Wild-type, K8A, and K10A Dps proteins at 4 μM were bound to (I) rrnB P1 or (J) λ PR promoter DNA as in (F) and (H), followed by incubation with or without HindIII or HincII, respectively (also shown for recA in Figure 3). The weakly DNA-binding Dps variants K8A and K10A did not protect DNA from cleavage by restriction endonucleases. DNA was visualized through gel electrophoresis. Related to Figure 3.
Figure S5: Dps-bound rec A promoter is accessible to σS RNAP holoenzyme and LexA repressor. (A) The recA DNA template schematic, with positions of the core promoter elements (−35 and −10), transcription start site (+1), and LexA recognition sequences (green) (Zhang et al., 2010) indicated. During in vitro run-off transcription experiments, RNAP is released at the end of the linear template (Run-off RNA; 177 nt) and at two hairpin-dependent termination sites (101 and 138 nt). (B) recA DNA template was incubated with Dps (or storage buffer) for 20 min at 37°C, after which LexA was added and incubated for 20 min at 37°C. Then, a mix containing RNAP holoenzyme (σ70 or σS), GpU, NTPs, and [α32P]-GTP was added and incubated for 20 min at 37°C to allow for run-off transcription. Reactions were quenched and visualized on a denaturing urea-acrylamide gel. (C) Run-off transcription gel for the recA template. Since the σ70 holoenzyme is more active on the recA template than the σS holoenzyme, different exposures of the same gel are shown for the two holoenzymes; for comparison, one lane from the σ70 set is shown alongside the σS reactions at the same exposure. (D) Production of run-off RNA, quantified for each RNAP holoenzyme separately, with RNA levels observed in the absence of Dps and LexA defined as 100%. The numbers shown are mean averages ± SD of n = 3 independent replicates. Related to Figure 3.
Figure S6: Dps-induced DNA condensation in presence of exerted force. (A) Extension of a 9-kb linear DNA template (red) under different force loads (blue) in the presence of 1 μM Dps. (B) Force-extension curves for a 9-kb linear DNA template (n = 7, mean ± SEM) in the absence (green) and presence (blue) of 1 μM Dps. Solid lines correspond to decreasing force and dashed lines to increasing force. The red-colored area between force-dependent DNA:Dps complex formation and disassembly depicts the boundaries of the mean transition points, indicating the hysteresis. (C) Extension of the RNAP ternary complex on a 7.5-kb linear DNA template in the presence of 1 μM Dps under high (5pN) and low (0.1 pN) force loads. (D) Temporal magnification of RNAP transcription on a 7.5-kb linear DNA template with transient pulling from low force to high force (8 pN) every 400 s in the presence of 1 μM Dps (Figure 6E). Related to Figures 4, 5 and 6.
Figure S7: RNAP transcription dynamics and DNA condensation at high Dps concentrations. (A) Dwell time distributions for assisting force (AF) trajectories in the presence (red) and the absence (black) of 10 μM Dps at 24°C. The distributions are separated with boundaries for the elongation region (0.1-1 s), short elemental pauses SP (1-5 s), and longer pauses LP (5-100 s). (B) Force-extension curves for a 8.8-kb linear DNA template (n = 7, mean ± SEM) in the absence (black) and presence (colored) of 4 μM (blue), 7 μM (cyan), and 10 μM (red) Dps. Solid lines correspond to decreasing force and dashed lines to increasing force. Related to Figure 5.