

Abstract
Macrolide antibiotics inhibit protein synthesis by targeting the bacterial ribosome. They bind at the nascent peptide exit tunnel and partially occlude it. Thus, macrolides have been viewed as ‘tunnel plugs’ that stop synthesis of every protein. More recent evidence, however, demonstrates that macrolides selectively inhibit translation of a subset of cellular proteins and that their action critically depends on the nascent protein sequence and on the antibiotic structure. Therefore, macrolides emerge as modulators of translation rather than global inhibitors of protein synthesis. The context-specific action of macrolides is the basis for regulation of the expression of resistance genes. Understanding the details of the mechanism of macrolide action may inform rational design of new drugs and unveil important principles of translation regulation.
Keywords: ribosome, macrolide, ketolide, antibiotic, translation, resistance
Understanding the mechanisms of antibiotic action is needed for development of better drugs and new tools for fundamental research
Antibiotics, the drugs that help cure infectious diseases caused by bacterial pathogens, have saved countless lives. Unfortunately, the excessive use of antibacterials in the clinic, veterinary medicine and farming has led to the development of resistance. Searching for new drugs is necessary to combat the spread of pathogens resistant to the available antibiotics. It is equally critical to understand the mechanistic basis of action of currently used antibiotics because this could lead to rational, innovative strategies for designing more efficient treatments. In addition, knowing how antibiotics work may contribute to their use as tools for unraveling basic mechanisms of translaiton.
The ribosome, which is responsible for the synthesis of all cellular proteins, is one of the best antibiotic targets. Many antibacterials inhibit cell growth by interfering with ribosome functions [1, 2]. Among them are macrolides, which have been used medically for more than six decades. However, we are only starting to understand the true mode of action of these antibiotics. Recent advances have revealed that, rather than simple inhibitors of protein synthesis, ribosome-targeting macrolides are modulators of translation. By deciphering how macrolides exploit the vulnerabilities of the protein synthesis apparatus we may find new ways to develop better drugs and learning fundamental aspects of the ribosomal response to environmental cues.
The classic model of macrolide action needs revision
Macrolide antibiotics (Box 1) inhibit protein synthesis by targeting the nascent peptide exit tunnel (NPET) (see Glossary) of the bacterial ribosome (Box 2). NPET, which is approximately 100Å long and 10–20Å wide, is a passageway through which the synthesized protein leaves the ribosome. Traditionally, it has been thought that macrolides stop translation by simply clogging the NPET, thereby blocking the passage of all the newly made polypeptides once they grow to the size of 3–10 amino acids [3–7]. To some extent, this view has been supported by structural studies showing that a macrolide molecule bound in the NPET significantly narrows the tunnel (Figure 1A–C). The “plug-in-the-bottle” model was also compatible with in vitro experiments showing that translation of some artificial mRNAs in the presence of erythromycin (ERY) resulted in accumulation of peptidyl-tRNAs carrying short peptides, indicative of interruption of translation at its early rounds [7, 8]. Peptidyl-tRNA accumulation was also observed in macrolide-treated cells [9].
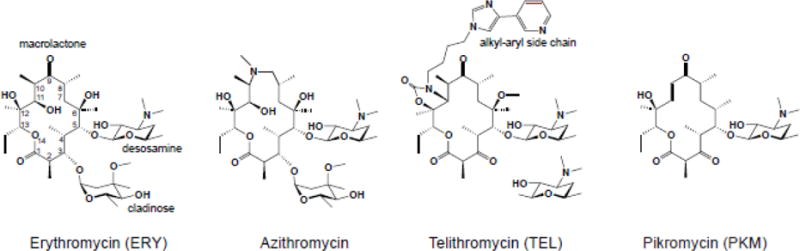
Box 1. Macrolides: chemical structure and brief history.
A macrolactone ring, which can range in size from 12 to 16 atoms, forms the core of the ribosome-binding macrolide antibiotics. Most of the clinically-relevant macrolides contain a 14-[e.g. erythromycin (ERY) or clarithromycin] or 15-atom (azithromycin) core (Figure I). The side chains appended to the macrolactone define many important biological and clinical properties of macrolides. Specific sugar residues are usually linked at the C3 and C5 positions of the ring. For example, ERY contains C3 cladinose and C5 desosamine. In clinical [e.g. telithromycin (TEL)] and natural [e.g. pikromycin (PKM)] ketolides (Figure I), the C3 sugar is substituted with a keto group (hence the name of the class). The most active semi-synthetic ketolides also carry an extended alkyl-aryl side chain, whose presence is important for activity and whose attachment site differs among different drugs [79–81].
Macrolides are among the oldest and most clinically-successful ribosome-targeting antibiotics. The prototype macrolide, erythromycin A (Figure I), was discovered more than 65 years ago and has been used clinically since the 1950s [82]. Macrolides of the second generation [e.g. clarithromycin, roxithromycin and azithromycin], developed in the 1980s, exhibited improved pharmacological properties [83]. Subsequent spread of resistance spurred the advancement of a newer generation of macrolides, called the ketolides, such as TEL (Figure I) or solithromycin, that showed enhanced activity against some of the resistant strains. Although side effects associated with their toxicity have so far precluded the broad clinical use of ketolides, their high antibacterial potency keeps them in the crosshair of the ongoing drug discovery efforts [84–86]. A recent breakthrough in the combinatorial chemical synthesis of macrolides raises new hopes for finding a diverse array of even more potent derivatives [87].
Box figure I.
Box 2. The target of macrolide antibiotics is the bacterial ribosome.
The ribosome is composed of two subunits, small and large (30S and 50S, respectively, in bacteria) (Figure II). The small subunit is in charge of decoding the genetic information encoded in mRNAs while the large subunit is responsible for polymerizing amino acids into proteins. The ribosome contains three tRNA binding sites: The growing protein chain is attached to the tRNA located in the P site, while the incoming amino acid is delivered to the ribosome by aminoacyl-tRNA that binds in the A site. On its way out of the ribosome, the deacylated tRNA resides in the E site (not shown in Figure II). Addition of individual amino acids to the growing protein is catalyzed in the peptidyl transferase center (PTC) of the large ribosomal subunit. Formation of the new peptide bonds occurs as the result of a nucleophilic attack of the A-site amino acid onto the carbonyl carbon atom of the ester bond linking the nascent peptide to the P-site tRNA (Figure II). The efficiency of peptide bond formation depends on the nature of the donor and acceptor substrates participating in the reaction [88, 89]. The elongating protein is threaded through the nascent peptide exit tunnel (NPET) on its way out to the cytoplasm (Figure II). Rather than being a passive passageway, the NPET is a functionally important compartment, capable of sensing the structure of the growing protein and modulating the ribosome functions in response not only to the peptide sequence but also to cues from the environment [18, 90, 91]. It is here in the NPET where macrolides bind, at a short distance from the PTC [6, 78, 92–94].
Box figure II.
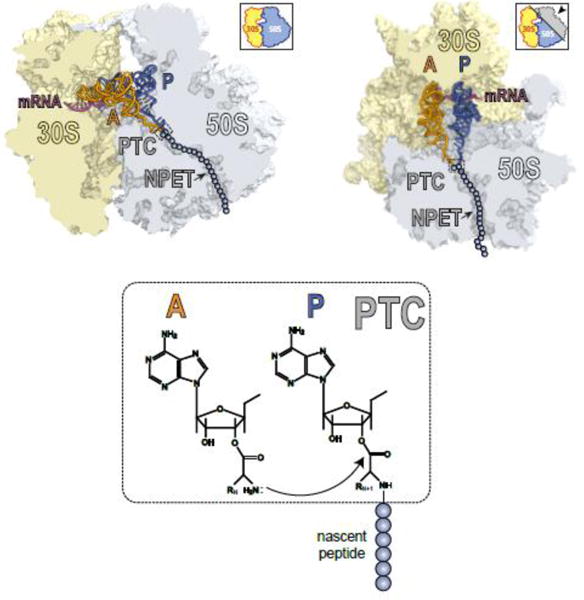
Figure 1. Macrolides narrow the nascent peptide exit tunnel but allow synthesis of certain proteins.
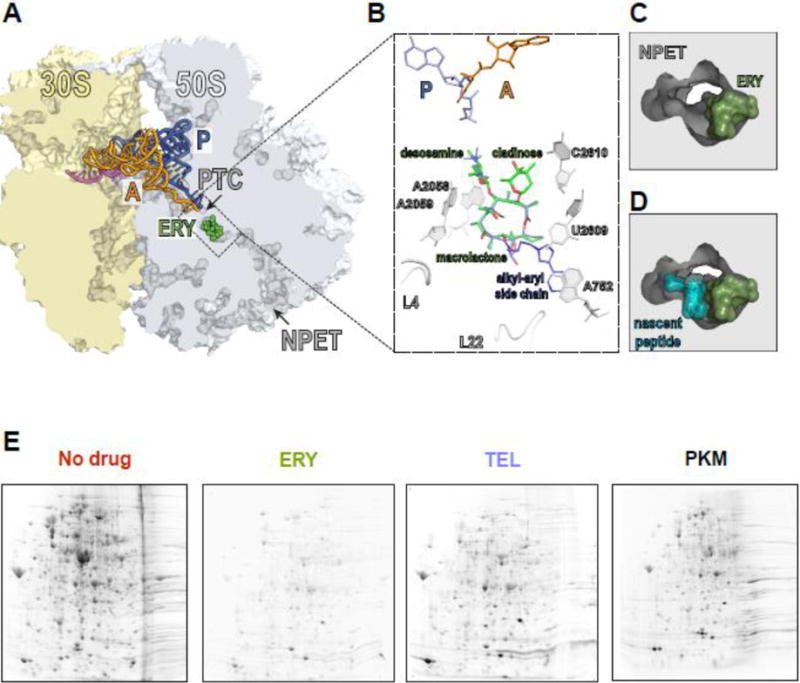
(A) The macrolide binding site in the bacterial ribosome. A cross-cut of the ribosome showing the A- and P-site tRNAs (orange and blue, respectively) and a segment of mRNA (magenta). ERY (green) and all the other macrolides bind in the NPET at a short distance from the PTC.
(B) The macrolide binding site is composed primarily of rRNA. The macrolactone ring of different macrolides (for comparison, the structures of ERY, green, and TEL, blue, have been superimposed) lays flat against the NPET wall and the C3 and C5 sugars protrude towards the PTC but do not reach its active site. C5 desosamine interacts with the splayed-out A2058 and A2059 rRNA residues in the NPET. C2610 nucleotide contacts C3 cladinose (present in ERY, but lacking in TEL). The alkyl-aryl side chain of ketolides, such as TEL, usually extends away from the PTC: In E. coli, it interacts with the A752-U2609 base pair, but its placement may differ in other bacteria [74, Dunkle, 2010 #7182, 75]. The loops of proteins L4 and L22, which form a constriction in the NPET, may directly interact with the side chains of some macrolides [76].
(C) and (D) View into the NPET from the PTC showing that the macrolide (ERY) narrows the tunnel’s aperture (C). The remaining opening of the NPET is nevertheless wide enough for a nascent peptide to be threaded through (D).
(E) Specific proteins are synthesized in macrolide-treated cells. Gel electrophoresis analysis of radiolabeled proteins translated in E. coli cells in the absence of antibiotics (No drug), or exposed to high concentrations of ERY, semi-synthetic ketolide TEL, or the natural ketolide pikromycin (PKM).
However, in the past several years, the simplistic view of macrolides acting like mere plugs of the NPET has been significantly transformed. New data have shown that these antibiotics allow passage of some nascent peptides through the NPET and can interfere with synthesis of a protein in a context-specific manner. Here, we highlight the recent findings that have formed the basis for our current understanding of how macrolides can selectively inhibit protein synthesis and act as modulators of translation.
Macrolides do not stop global translation but inhibit the synthesis of a subset of proteins
One striking observation that could not be easily explained by the traditional model of macrolide action was that protein synthesis is not completely inhibited even in cells exposed to very high concentrations of macrolides - exceeding by many fold the minimal inhibitory concentration (MIC) that prevents cell growth. For example, in Escherichia coli cells treated with 100-fold MIC of ERY, 5–7% of the total protein continues to be synthesized, whereas around 25% of translation persists in cells exposed to equivalent concentrations of telithromycin (TEL) [10], or as high as 40% with pikromycin (PKM) [11]. Remarkably, rather than equally curtailing synthesis of all proteins, macrolides virtually abolish the production of a number of polypeptides, whereas some others continue to be translated at levels comparable to those in untreated cells (Figure 1E) [10, 11]. These findings were in line with earlier observations that the extent of macrolide inhibition varied dramatically for different reporter proteins used in the in vitro experiments [4, 12].
A key feature of macrolide action emerged from these results: Rather than being global inhibitors of translation, macrolides selectively interfere with the production of a subset of proteins.
What distinguishes macrolide-sensitive and macrolide-resistant proteins?
It has been known that specific short peptides synthesized by the ribosome are able to co-translationally eject a macrolide molecule from its binding site in the NPET [13–16]. By analogy, if the N-terminal sequence of a cellular protein can dislodge the antibiotic from the ribosome, the evicted antibiotic would not be able to re-bind until translation of the protein is finished, because the NPET of the elongating ribosome is occupied by a growing polypeptide [4, 17]. The key postulate of this drug-eviction model is that resistant proteins are synthesized by the drug-free ribosome.
While the eviction model is attractive and may contribute to the selective escape of some polypeptides, biochemical evidence strongly argues that the synthesis of many, possibly most, of the cellular proteins resistant to macrolides occurs on the ribosomes that retain the antibiotic molecules [10]. Although ostensibly controversial, this possibility is compatible with the known X-ray structures of the macrolide-bound ribosome showing that the aperture of the drug-obstructed NPET is in fact wide enough to allow unfolded nascent proteins to be threaded through [6, 18]. The more recent structures of the ERY-bound translating ribosome carrying a nascent peptide clearly demonstrate that a protein chain can be fairly comfortably accommodated in the NPET together with the antibiotic [19–21] (Figure 1D). These findings have validated a seemingly heretic proposal, expressed by Weisblum more than two decades ago, that some nascent peptides could potentially slip through the macrolide-obstructed exit tunnel [22].
If the macrolide molecule does not stop the growing protein from advancing through the NPET, then why do macrolides prevent the translation of so many proteins? The answer to this question came from ribosome profiling (Ribo-seq) (see Glossary) experiments that provided the key breakthrough in our understanding of the mechanism that underlies the protein specificity of macrolides. Ribo-seq technology shows the distribution of ribosomes along translated mRNAs: peaks of ribosome density build up at the codons where translation slows down, whereas the codons that are traversed faster have fewer ribosomes associated with them [23] (Figure 2A). By comparing the distribution of ribosomes on mRNAs in untreated and drug-exposed cells, it is possible not only to determine whether the antibiotic abolishes translation of a gene but also at which specific mRNA codon(s) the translation is arrested.
Figure 2. Macrolides arrest the ribosome at specific mRNA codons.
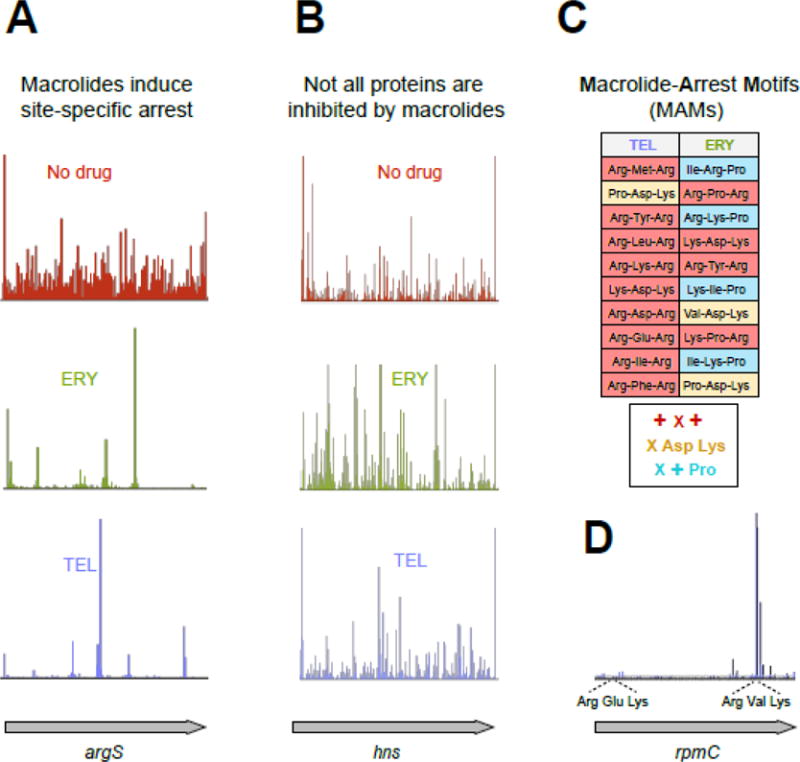
(A) Treatment of E. coli cells with high concentrations of ERY or TEL leads to dramatic redistribution of ribosomes along the genes (illustrated by the ribosome densities in the representative gene argS). The discrete peaks of ribosome density at specific codons of argS in the ERY- or TEL-treated cells reflect context-specific translation arrest [24, 25].
(B) Some ORFs are completely translated in macrolide-treated cells. The occurrence of ribosome density throughout the representative gene hns in cells treated with TEL reveals HN-S as one of the proteins fully translated in the presence of this antibiotic (See Figure 1D).
(C) Ribosome with bound macrolide struggles to polymerize specific sequences, the Macrolide Problematic Motifs (MAMs). The table lists the 10 most prevalent MAMs enriched in the sites of translation arrest in the TEL- and ERY-treated cells [25]. The consensus sequences of the common MAMs are listed underneath the table (‘X’ indicates any amino acid, ‘+’ indicates Arg or Lys).
(D) Translation arrest at an MAM can be influenced by a more extended context, likely involving other segments of the nascent protein chain. The shown example illustrates how TEL-bound ribosomes could easily translate through the usually problematic Arg-Glu-Lys sequence (an MAM of the + X + type) present within the early codons of rpmC but became arrested at the second +X+ MAM (Arg-Val-Lys) located towards the end of the ORF.
Ribo-seq analysis showed that translation of nearly 80% of the genes in macrolide-treated cells proceeds beyond the early codons [24, 25], demonstrating that early interruption of protein elongation could be a contriubuting factor, but is clearly not the main mode of macrolide action. The most remarkable finding, however, was that translation of many genes was arrested at a few distinct sites through the length of the gene (Figure 2A, B). Analysis of the sites of the most pronounced drug-induced translation arrest showed that they are defined by specific sequence signatures [24, 25], which we will refer to as Macrolide-Arrest Motifs (MAMs) (see Glossary) (Figure 2C). The macrolide-bound ribosome stalls when it needs to polymerize the amino acid sequence of an MAM. Because the stalled ribosome peaks could be found anywhere within the open reading frames (ORFs), drug-arrested ribosomes often carry nascent polypeptides that span the entire length of the NPET. This result corroborated the structural and biochemical evidence that the growing protein can coexist with the macrolide molecule in the NPET of the translating ribosome (Figure 1D).
If a protein lacks MAMs, translation of its gene remains essentially impervious to the macrolide presence [10, 11] (Figure 1E) and high ribosome density is observed through the entire length of the ORF [24, 25] (Figure 2B).
Ribo-seq data pointed to another important aspect of macrolide action. Although all ribosome-targeting macrolides bind to the same site in the NPET (Figure 1B), the specificity of action critically depends on their chemical structure. As a result, the sites of translation arrests and the spectra of the affected proteins vary in cells treated with different macrolides. The Lys/Arg-X-Lys/Arg motif accounts for nearly 80% of the strongest ketolide arrest sites, but cladinose-containing ERY or AZI inhibit translation not only at this motif but at a wider array of MAMs (Figure 2C). The broader the variety of MAMs, the higher the chance that a protein will contain at least one of them. For this reason, ERY and AZI preclude the synthesis of more proteins (Figure 1C) whereas ketolides emerge as more selective inhibitors of translation that allow for more proteins to be synthesized. By rationally modifying the structural features of the drugs it is hypothetically possible to control not only the affinity or kinetics of drug-ribosome interactions [26], but also to modulate the spectrum of proteins whose translation would be inhibited by the antibiotic.
Implications of the dynamics of antibiotic-ribosome interactions for the mechanism of macrolide action
The understanding that the nascent chain can coexist in the NPET with the macrolide molecule (Figure 2A and D) changes our perception of the dynamics of the drug-ribosome interactions. Because the entrance into the NPET from the peptidyl transferase center (PTC) (see Glossary) (Box 2) side is too narrow, it has been suggested that macrolides access their binding site by diffusing through the tunnel from its exit [27]. When the NPET is vacant, the bound inhibitor can can freely exchang with the unbound drug and exist in dynamic equilibrium with the antibiotic in the cell cytoplasm. However, when the ribosome is engaged in translation, the nascent protein, advancing through the NPET, will eventually trap the macrolide molecule, making its departure impossible. The efficiency of antibiotic trapping is determined by the rates of the nascent peptide advancement through the NPET, drug dissociation, and peptidyl-tRNA drop-off [28]; all these kinetic parameters may depend on the nature of the synthesized protein. Once the N-terminus of the growing protein chain has bypassed the macrolide molecule, it should hinder antibiotic dissociation by narrowing the NPET constriction formed by the loops of ribosomal proteins L4 and L22 (Figure 1B). Only upon the release of the completed protein will the drug regain its ability to be exchanged with the cytoplasm. The duration of the antibiotic trapping could be significantly extended when translation is arrested at an MAM, possibly accounting, at least in part, for the prolonged post-antibiotic effects of some macrolides [29, 30]. Although Ribo-seq data and single-molecule fluorescence studies show that translation arrest at an MAM is transient [24, 25, 31], we know very little about the kinetics of macrolide-induced ribosome stalling.
The dynamics of the drug-ribosome interaction may have important practical implications. Depending on their structure, macrolides can signfiicantly vary in their ability to simply stop cells from growing (and thus act as bacteriostatic drugs) or actively kill bacteria preventing their re-growth upon removal of the antibiotic (thereby acting as bactericidal agents) [32]. Recent studies have shown that the kinetics of binding and dissociation from the ribosome rather than mere affinity is the critical parameter that distinguishes bacteriostatic and bactericidal macrolides [26]. Drugs that lack an extended alky-aryl side chain, as that seen in the TEL structure (Box 1), rapidly vacate the ribosome and exhibit primarily bacteriostatic action, whereas antibiotics that carry such an appendage exhibit a much slower dissociation kinetics that correlates with their bactericidal activity [26]. Therefore, taking into account not only the affinity of macrolides for the ribosome, but also the dynamics of their interaction with the target should guide future efforts for improving drug-treatment regimens.
Macrolides as modulators of peptide bond formation
One of the most unexpected aspects of macrolide action is that the MAM is not juxtaposed with the macrolide molecule in the NPET at the moment of translation arrest. Instead, the ribosome stalls when the MAM residues are positioned at the PTC and thus are too distant to establish extensive direct contacts with the antibiotic molecule (Figure 3). This means that the MAM sequence presents a problem not because it is simply stuck in the drug-obstructed NPET, but because the macrolide-bound ribosome is unable to polymerize it. Protein synthesis stops because macrolides prevent the ribosome from catalyzing peptide bond formation (Box 2) between the MAM residues [21, 33–35], as has been also shown previously for specific combinations of artificial donor and acceptor substrates [36–38] (Figure 3A–C). Therefore, instead of being simple tunnel plugs, macrolide antibiotics emerge as context-specific inhibitors of peptide bond formation.
Figure 3. Macrolides are selective modulators of the peptidyl transferase center.
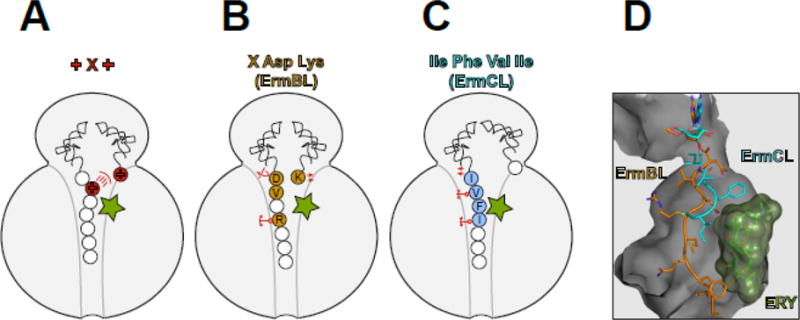
The interplay between the macrolide molecule and a MAM-containing nascent peptide alters the PTC properties and inhibits peptide bond formation. In (A–C), the residues critical for stalling are colored and in (B) and (C) are indicated with the single-letter code. (A) Stalling at the +X+ MAM may occur because the macrolide orients the lengthy, positively charged side chain of the penultimate amino acid of the nascent peptide towards the PTC A site, preventing the accommodation of the similarly long and positively charged acceptor amino acid. (B) The macrolide imposes an unfavorable orientation of the C-terminal Asp residue of the ErmBL peptide that contains the X-Asp-Lys MAM [19, 21]. The placement of the acceptor Lys in the A site is also suboptimal. The orientation of several key PTC nucleotides is altered in the stalled ribosome. The mobility of the nascent peptide in the NPET is restricted due to the antibiotic presence and specific interactions of the Arg residue of the nascent chain with rRNA of the tunnel wall. (C) Interactions of the ErmCL nascent chain with the NPET nucleotides and antibiotic misplace the peptide’s C-terminal residue in the PTC [20]. The adverse conformation of the PTC nucleotides prevents accommodation of the A site amino acid [20, 72]. (D). The different placement of the ErmBL and ErmCL nascent peptides in the NPET of the ERY-stalled ribosome shows that the peptide trajectory depends on the amino acid sequence.
We are only starting to understand why polymerizing certain sequence motifs is difficult for the macrolide-bound ribosome. Biochemical experiments have shown that the positive charge of the key amino acids of the Lys/Arg-X-Lys/Arg motif accounts for its problematic nature; hence, this MAM is referred to as the +X+ motif [35]. Consistently, macrolides do not usually disrupt the polymerization of the sequence Asp/Glu-X-Asp/Glu, where the charges of the main residues of the motif are reversed [35]. However, not only the charge but also the length of the Arg and Lys side chains, the longest among the 20 canonical amino acids, matters for translation arrest. In fact, replacing the Lys residue of the Arg-X-Lys sequence with aminoalanine, which carries a positively charged but rather short side chain, lessens the ability of the macrolides to inhibit peptide bond formation [35].
In the absence of a high-resolution structure of the translation complex stalled at the +X+ MAM, we can only speculate why polymerizing this sequence is difficult for the macrolide-bound ribosome (Figure 3A). However, electron cryomicroscopy (cryo-EM) (see Glossary) has provided important insights into the macrolide-dependent ribosome stalling at some other MAMs (Figure 3B–D). Most of these studies have been carried out using regulatory ORFs of macrolide resistance genes that contain strategically placed MAMs directing programmed ribosome stalling (see section below). Analysis of the ERY-bound ribosome stalled at the MAMs encoded in the ermBL and ermCL ORFs showed that the nascent peptide, whose placement in the NPET is constrained by the antibiotic, could be actively involved in the stalling mechanism [19–21]. The idiosyncratic trajectory of ErmBL causes the misplacement and reorientation of its C-terminal residue in the PTC which is expected to impede the catalysis of peptide bond formation [19] (Figure 3B). Similarly, the C-terminal residue of ErmCL also appears to be displaced in the PTC of the macrolide-bound ribosome [20] (Figure 3B). Because the trajectory of the nascent chain in the drug-obstructed NPET is dictated by the peptide’s amino acid sequence (Figure 3D) [19–21] not only the MAM, which is mostly confined to the PTC, but also more distant segments of the growing protein likely modulate the efficiency of stalling. This conclusion resonates well with the results of Ribo-seq studies showing that not every potential MAM causes translation arrest [24, 25] (Figure 2D).
The analysis of the macrolide-stalled ribosomal complexes consistently showed rearrangement of nucleotides in the PTC active site (e.g. U2585), whose conformations in the drug-arrested ribosome are likely incompatible with the efficient catalysis of peptide bond formation [19–21] (Figure 3B,C). Importantly, even binding of a macrolide molecule to the vacant ribosome can already allosterically induce changes in the structure of the PTC [39]. Thus, the ribosome is likely able to integrate the signals generated by the nascent peptide and the antibiotic stalling cofactor [40, 41]. Although illuminating, the available cryo-EM reconstructions still lack an important control: the structure of a macrolide-bound ribosome with a nascent chain lacking MAM. In the absence of such a reference, it is impossible to conclude which of the many idyosyncrasies observed in the stalled ribosome complex are directly pertinent to the drug-induced translation arrest.
However, altogether, the understanding that macrolides are context-specific ribosome modulators leads to a concise model for the mechanism of action of these drugs, where the fate of the protein to be synthesized by the macrolide bound ribosome is defined by its amino acid sequence (Figure 4) (Key Figure).
Figure 4. The general model of macrolide action upon protein synthesis.
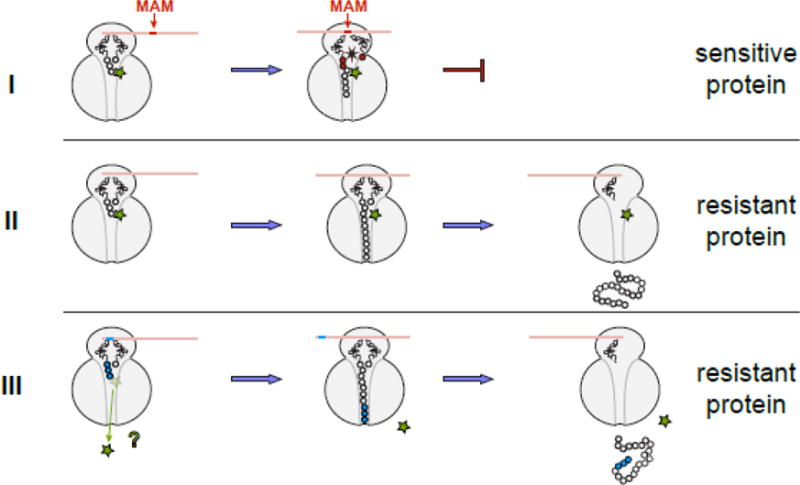
Translation of any protein can be initiated by the macrolide-bound ribosome and the N-termini of the majority of polypeptides can be threaded through the drug-obstructed NPET. The subsequent fate of the protein being made depends on its sequence and the structure of the bound antibiotic. (I) Translation of sensitive proteins is interrupted because the drug-bound ribosome is unable to efficiently catalyze peptide bond formation during synthesis of the MAM sequence. The structure of the macrolide molecule bound in the NPET dictates the spectrum of the MAM sequences and therefore, defines which proteins will be inhibited. If translation is arrested close to the start of the ORF, when the nascent peptide is short (<10 amino acids), peptidyl-tRNA likely dissociates from the ribosome (the effect known as peptidyl-tRNA drop-off) [7, 8, 77]. (II) If the protein sequence lacks MAMs, its translation will proceed unimpeded and the full-size protein will be produced in the cell exposed to the antibiotic. (III) Hypothetically, some proteins might be able to dislodge the antibiotic from the ribosome at the early stages of translation; in this case, the drug-free ribosome completes the synthesis of such protein. Antibiotic eviction has been observed with some artificial short peptides [13–16], but has not been demonstrated yet for the cellular polypeptides; yet this scenario remains a possibility for at least some of the bacterial proteins.
Context-specificity of macrolide action controls inducible resistance
One of the most important mechanisms of resistance to macrolide antibiotics is modification of a 23S rRNA adenine residue (A2058 in E. coli) in the macrolide binding site (Figure 1B) [42, 43]. Methylation of this nucleotide, catalyzed by Ery-resistance methyltransferases (Erm) (see Glossary), precludes antibiotic binding. However, the resistance conferred by A2058 methylation comes at a cost: it affects translation of some proteins, skews the cellular proteome, and results in reduced cell fitness [44].
An elegant mechanism, based on programmed translation arrest, reduces the fitness cost of resistance by allowing activation of the corresponding genes only when cells are under antibiotic threat [45, 46]. In the absence of the antibiotic, inducible macrolide resistance genes are translationally or transcriptionally repressed due to unfavorable mRNA folding (Figure 5A) [47, 48]. Presence of the macrolide directs programmed ribosome stalling at a specific codon of the short upstream ORF (leader ORF) (see Glossary), leading to mRNA re-folding and activation of expression of the resistance gene [42] (Figure 5A). The sequence of the leader ORFs and the spectra of the inducing macrolide antibiotics vary between different resistance genes [42, 49–51].
Figure 5. Inducible resistance exploits the context specific action of macrolides.
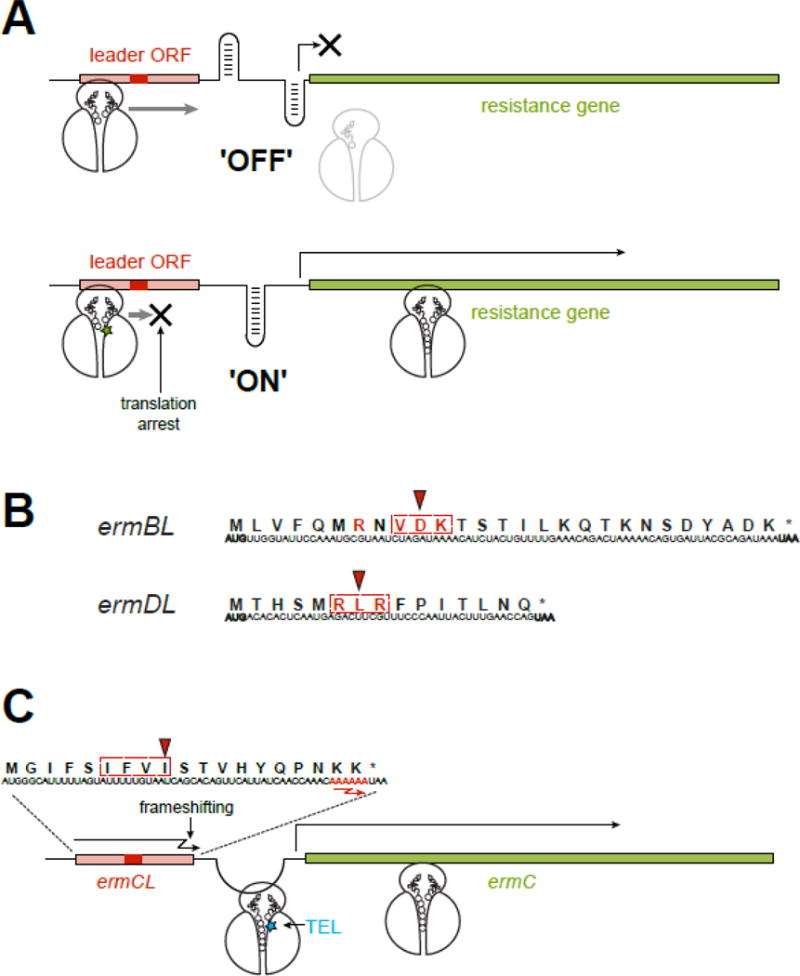
(A) In an inducible macrolide resistance operon, the resistance gene is preceded by a regulatory leader ORF. In the absence of antibiotic, the leader ORF is translated while the resistance gene is not expressed, because the ‘off’ conformation of the intergenic region of mRNA precludes the access to its translation initiation site. When macrolide is present, translation of the leader ORF is arrested at a specific codon within an MAM (red rectangle). The stalled ribosome re-arranges the mRNA structure into the ‘on’ conformation, releasing the initiation site of the resistance gene and activating its expression. The MAM location is optimal for the paused ribosome to activate the isomerization of the mRNA structure. In a similar scenario, ribosome stalling at the leader ORF can activate attenuated transcription of some of the resistance genes.
(B) Programmed ribosome arrest relies on the MAMs (dotted rectangle) encoded in the leader ORFs. The Val-Asp-Lys (the X-Asp-Lys MAM) is embedded in the ermBL gene, while the Arg-Lys-Arg (the +X+ MAM), is found in ermDL. The codon where the ribosome stalls in the presence of antibiotic is indicated by an arrowhead. The amino acids essential for programmed translation arrest are shown in red.
(C) Macrolide-induced miscoding accounts for an unorthodox induction of resistance. The ribosome with bound TEL (blue star) ignores the stall site within the ermCL ORF, where ERY would arrest translation at the Ile-9 codon (red arrowhead) of the Ile-Phe-Val-Ile MAM (dotted rectangle). Therefore, the TEL-bound ribosome traverses the entire ermCL ORF and reaches its last two Lys codons with the sequence AAA AAA (red) where TEL stimulates (−1) ribosomal frameshifting. Upon frameshifting, the 0-frame stop codon of the ermCL ORF is skipped and the ribosome continues translation through the intergenic region, dynamically unfolding the mRNA structure and releasing the translation initiation site of the resistance gene.
Although the general operation of the induction mechanism was elucidated several decades ago (reviewed in [42, 51]), key questions remained unanswered. Why is translation arrested at one specific codon of the leader ORF? How could the macrolide-bound ribosome even reach the site of productive translation arrest? Why are different Erm resistance genes induced by different antibiotics? The now understood context-specificity of macrolide action provides answers to these questions because the the leader ORFs encode peptides with strategically placed MAMs. The locations of MAMs with the leader peptide sequences have been evolutionarily optimized to arrest translation specifically at the codons where the stalled ribosome can induce the ‘ON’ conformation of the mRNA (Figure 5B). The leader ORFs of many macrolide resistance genes encode the +X+ motif [39, 50, 52]. Leader ORFs of other resistance genes may carry different MAMs identified by Ribo-seq experiments [20, 53] (Figure 4B). The nature of the MAM in the leader ORF-encoded peptide defines the spectrum of macrolides that can act as inducers of resistance [40, 49, 54]. It is hypothetically possible to engineer macrolides that, while being able to inhibit synthesis of some proteins, would not induce ribosome stalling at MAMs of regulatory genes, thereby avoiding activation of resistance.
Macrolides can induce miscoding and ribosomal frameshifting
Besides the ability of macrolides to stall the ribosome within specific sequence contexts, a lesser appreciated property of these drugs is their capacity to induce translation errors [55]. Although the nature of these effects is unclear, it is conceivable that inhibition of PTC functions (peptide bond formation or peptide release) resulting in an altered kinetis of translation could increase the chance of faulty events such as accommodation in the A site of a near-cognate aminoacyl-tRNA.
Macrolides can also stimulate ribosomal frameshifting. Interestingly, this activity accounts for an unconventional scenario of induction of resistance [56]. Cladinose-containing macrolides (e.g. ERY) activates expression of the ermC resistance gene via programmed translation arrest at the Ile-9 codon of the 19-codon ermCL leader ORF [33, 45, 46] (Figure 5A). Ketolides (e.g. TEL) do not direct stalling at ermCL MAM [33, 40, 57] but are nevertheless capable of inducing ermC, albeit with a lower efficiency compared to ERY [58]. This unconventional induction mechanism exploits the ability of the ribosome with bound TEL to reach the end of the ermCL leader ORF where it slips to the (−1) frame (Figure 5B). Continuous translation through the ermCL-ermC intergenic region results in the activation of the resistance gene [56]. Two aspects of macrolide action, the drug-specificity of the ribosomal response to MAMs and the ability of macrolides to provoke translation errors, make this unexpected induction scheme possible.
The overall contribution of the macrolide-induced miscoding to the antibacterial action of these antibiotics remains to be elucidated.
The mechanism of macrolide action reflects the ability of the ribosome to function as a small molecule sensor
Ribosome stalling at specific MAMs in response to different macrolides is just one manifestation of a more general phenomenon: modulation of translation by small molecules. Importantly, the capacity of the ribosome to recognize and respond to specific small molecules is uniquely modulated by the properties of the nascent protein. This ribosomal feature is vividly illustrated by mutations of the ErmBL peptide: by changing a single amino acid in the X-Asp-Lys MAM of ErmBL, it is possible to direct ribosome stalling, and hence activation of expression of the downstream gene, in response to the presence of only cladinose-containing macrolides, only ketolides, or both types of drugs [59] (Figure 6A).
Figure 6. Nascent peptides turn the ribosome into a small molecule sensor.
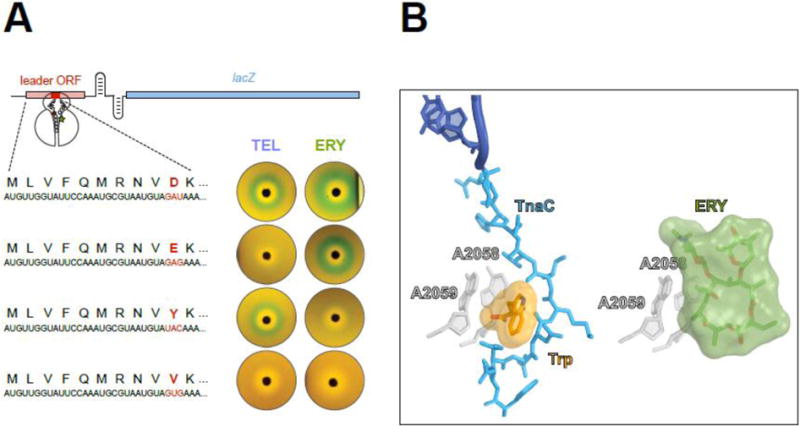
(A) Single amino acid changes in the ErmBL nascent peptide alters the ribosomal response to structurally different macrolides. The reporter cassette mimics an inducible erm resistance operon (see Figure 5), in which the resistance gene was replaced with lacZ. The reporter induction is visualized by the green color of the cell lawn in the vicinity of an antibiotic-containing disk placed on the agar plate. Both ketolide TEL or cladinose-containing ERY arrest ermBL translation and activate the reporter when the 10 th codon (red) of ermBL specifies Asp (wild type). Changing th the 10 codon to Glu preserves the response to ERY but eliminates the response to TEL. Tyr-10 allows the ribosome to respond exclusively to ketolides. Val-10 precludes the response to either TEL or ERY.
(B) The TnaC nascent peptide allows sensing of tryptophan. Activation of the tna operon depends on ribosome stalling on the tnaC leader ORF [60, 61]. Structural studies of the TnaC-stalled ribosome suggest that one of the two observed tryptophan molecules binds at the A2058/A2059 crevice (left image) [63], the same site that is exploited by macrolide antibiotics for binding to the ribosome (right image) [78]. Similar to the coordinated action of macrolides and nascent peptide in inducing translation arrest, the TnaC peptide cooperates with tryptophan to disrupt the PTC function and stall the ribosome at the last sense codon of the tnaC ORF.
The ribosomal property to act as a small molecule sensor is not limited to detecting only antibiotics: other molecules, which generally do not inhibit protein synthesis, can be recognized in a similar fashion. For example, expression of the tna operon in several bacterial species is based on recognition by the ribosome of elevated concentrations of L-tryptophan in the cell. Tryptophan sensing is aided by the TnaC nascent peptide and results in programmed translation arrest of the tnaC gene [60, 61]. Although many details of tryptophan-induced stalling are still unclear, the regulatory tryptophan molecule likely binds at (or close to) the site of macrolide binding in the NPET [62, 63] (Figure 6B), a crevice that has been proposed to serve as a binding pocket for different hydrophobic molecules [64]. Conceivably, specific amino acid sequences of nascent protein chains could facilitate binding and recognition of different effectors in this site. Furthermore, other binding sites in the NPET could be exploited by different small molecules known to cooperate with nascent peptides in inducing programmed translation arrest (reviewed in [65]).
Regulating translation via the interplay between small molecules and nascent peptides may extend beyond the bacterial ribosome. Although no antibioitcs binding in the NPET of the eukaryotic cytoplasmic ribosome are currently known [66], a recent report of an inhibitor of protein PCSK9 involved in cholesterol homeostasis may represent the first such example [67, 68]. The ribosome-targeting small molecule PF-06446846 selectively inhibits translation of only a handful of proteins in human cells, including PCSK9. Similar to macrolides, PF-06446846 binds in the NPET and cooperates with the nascent protein chain in inducing site-specific translation arrest [68, 69]. The future studies will likely reveal many more examples of gene control mechanisms involving nascent peptide-assisted small molecule sensing.
Concluding remarks
The NPET, whose existence was proposed decades ago [70], was initially viewed as a functionally inert crawlway. Research during the recent years has shown that the NPET is a dynamic, functionally important compartment that endows the ribosome with the ability to sense the nature of the protein it synthesizes and to respond to environmental cues, including the presence of specific small molecules. We now recognize the NPET as a hub for nascent protein-based translation regulation.
The closely examined stalling scenarios involving small molecules differ in several important details. However, a common theme is starting to emerge: the presence of a small molecule cofactor and a specific nascent peptide is sensed in the NPET [40, 60, 71–73]. The cofactor bound to the ribosome restricts the freedom of movement of the growing protein, enforcing it to adopt a specific trajectory. The nascent protein, locked in a defined conformation, and the cofactor molecule engage specific elements of the NPET and the integrated signal is then relayed to the PTC. Under the influence of the stalling signal, the properties of the PTC are altered in such way that formation of peptide bonds between specific donor and acceptor substrates becomes inefficient.
In this common scheme of events, macrolide antibiotics, rather than being simple, non-selective protein synthesis inhibitors, represent one example of context-specific translation arrest cofactors. The spectrum of known macrolides, the emerging approaches for synthesis of novel derivatives, and the new technologies for studying their effect on translation, makes this class of protein synthesis modulators an ideal model for unraveling the fundamental mechanisms of nascent peptide-based translation control.
Highlights.
Ribosome-targeting macrolide antibiotics were thought to simply plug the nascent peptide exit tunnel and interrupt the synthesis of any protein. However, structural, biochemical, and genome-wide studies have revealed macrolides as highly selective modulators of protein synthesis.
Macrolide molecules along with specific nascent peptides in the ribosomal tunnel allosterically affect the functional properties of the catalytic center of the ribosome.
The macrolide-bound ribosome is unable to polymerize specific amino acid sequences present in the proteins.
The programmed translation arrest the controls the expression of macrolide resistance genes exploits the context-specific action of macrolides.
The principles of the interplay between macrolides and nascent peptides that modulate the functions of the ribosome may apply to the translation control exerted by other small molecules that interact with the ribosome.
Outstanding questions.
What is the kinetics of the macrolide-induced translation arrest? For how long does the ribosome stall? Does kinetics of macrolide-induced stalling depend on the structure of the bound antibiotic?
How the extended context of the growing peptide in the NPET influences ribosome stalling at an MAM?
Can the N-terminal sequences of some proteins displace macrolide antibiotics from their binding site in the NPET?
What features of the N-terminal sequence of the growing polypeptide are conducive to the early translation arrest and peptidyl-tRNA drop-off?
Are there cellular factors able to rescue the macrolide-stalled ribosome with the nascent polypeptide threaded through the NPET?
Do MAMs represent the generally-problematic sequences for polymerization by the ribosome even in the absence of antibiotic?
How do the structural differences between NPETs of the ribosomes of different bacterial species affect the mode of macrolide action and the spectra of MAMs?
What is the effect of macrolides on mitochondrial translation? Does the context specificity remain the same as that seen in bacterial ribosome?
Is it possible to adapt macrolide molecules for context- and protein-specific inhibition of cytoplasmic translation in the eukaryotic (mammalian) cells?
Acknowledgments
We want to thank Yury Polikanov and Nikolay Aleksashin for help with preparation of some figures and analysis of the NPET characteristics. We thank Elizabeth Woods for proofreading the manuscript. We are in debt to the former and current members of our laboratory for their enthusiasm and dedication to studying the mechanisms of antibiotic action and for the energy they bring to these studies. The antibiotic work in our laboratory is supported by the NIH grants R01 AI125518 and R35 GM127134.
The research in our laboratory was previously supported by, among other sources, grants from pharmaceutical companies working on the development of macrolide antibiotics.
Glossary
- Cryo-EM
cryo-electron microscopy is a technique that allows to study structures of biomolecules by capturing them at cryogenic temperatures
- Erm
Erythromycin-resistance methyltransferases are enzymes that mono- or di-methylate nucleotide A2058 of the 23S ribosomal RNA (E. coli numbering), precluding binding of macrolides to the target site. Their activity in pathogenic bacterial strains constitutes one of the main resistance mechanisms against the macrolides and two other families of NPET-binding antibiotic
- Leader ORF
short open reading frame located upstream of an inducible resistance gene the expression of the resistance gene is often controlled by programmed translation arrest within this ORF
- MAMs
Macrolide-Arrest Motifs are amino acid sequences that the macrolide-bound ribosome is unable to efficiently polymerize
- NPET
the Nascent Peptide Exit Tunnel is a void spanning the body of the large ribosomal subunit through which the protein polymerized in the peptidyl transferase center is threaded to then leave the ribosome
- PTC
the Peptidyl Transferase Center located in the large ribosomal subunit. The formation of peptide bonds between the C-terminal amino acid of the nascent peptide and the incoming amino acid is catalyzed in the active site of the peptidyl transferase center
- Ribo-seq
a technique, also known as ‘ribosome profiling’, that allows to visualize the distribution of translating ribosomes along mRNAs in the living cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol. 2014;12:35–48. doi: 10.1038/nrmicro3155. [DOI] [PubMed] [Google Scholar]
- 2.Lin J, et al. Ribosome-targeting antibiotics: modes of action, mechanisms of resistance, and implications for drug design. Annu Rev Biochem. 2018;87:18.1–18.28. doi: 10.1146/annurev-biochem-062917-011942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Contreras A, Vazquez D. Cooperative and antagonistic interactions of peptidyl-tRNA and antibiotics with bacterial ribosomes. Eur J Biochem. 1977;74:539–547. doi: 10.1111/j.1432-1033.1977.tb11422.x. [DOI] [PubMed] [Google Scholar]
- 4.Odom OW, et al. The synthesis of polyphenylalanine on ribosomes to which erythromycin is bound. Eur J Biochem. 1991;198:713–722. doi: 10.1111/j.1432-1033.1991.tb16071.x. [DOI] [PubMed] [Google Scholar]
- 5.Arevalo MA, et al. Protein components of the erythromycin binding site in bacterial ribosomes. J Biol Chem. 1988;263:58–63. [PubMed] [Google Scholar]
- 6.Schlunzen F, et al. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 7.Tenson T, et al. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J Mol Biol. 2003;330:1005–1014. doi: 10.1016/s0022-2836(03)00662-4. [DOI] [PubMed] [Google Scholar]
- 8.Otaka T, Kaji A. Release of (oligo) peptidyl-tRNA from ribosomes by erythromycin A. Proc Natl Acad Sci U S A. 1975;72:2649–2652. doi: 10.1073/pnas.72.7.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menninger JR, Otto DP. Erythromycin, carbomycin, and spiramycin inhibit protein synthesis by stimulating the dissociation of peptidyl-tRNA from ribosomes. Antimicrob Agents Chemother. 1982;21:810–818. doi: 10.1128/aac.21.5.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kannan K, et al. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 2012;151:508–520. doi: 10.1016/j.cell.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Almutairi MM, et al. Co-produced natural ketolides methymycin and pikromycin inhibit bacterial growth by preventing synthesis of a limited number of proteins. Nucleic Acids Res. 2017;45:9573–9582. doi: 10.1093/nar/gkx673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Starosta AL, et al. Interplay between the ribosomal tunnel, nascent chain, and macrolides influences drug inhibition. Chem Biol. 2010;17:504–514. doi: 10.1016/j.chembiol.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Tenson T, et al. Erythromycin resistance peptides selected from random peptide libraries. J Biol Chem. 1997;272:17425–17430. doi: 10.1074/jbc.272.28.17425. [DOI] [PubMed] [Google Scholar]
- 14.Tripathi S, et al. Ketolide resistance conferred by short peptides. J Biol Chem. 1998;273:20073–20077. doi: 10.1074/jbc.273.32.20073. [DOI] [PubMed] [Google Scholar]
- 15.Vimberg V, et al. Peptide-mediated macrolide resistance reveals possible specific interactions in the nascent peptide exit tunnel. Mol Microbiol. 2004;54:376–385. doi: 10.1111/j.1365-2958.2004.04290.x. [DOI] [PubMed] [Google Scholar]
- 16.Lovmar M, et al. The molecular mechanism of peptide-mediated erythromycin resistance. J Biol Chem. 2006;281:6742–6750. doi: 10.1074/jbc.M511918200. [DOI] [PubMed] [Google Scholar]
- 17.Tai PC, et al. Selective action of erythromycin on initiating ribosomes. Biochemistry. 1974;13:4653–4659. doi: 10.1021/bi00719a029. [DOI] [PubMed] [Google Scholar]
- 18.Tu D, et al. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell. 2005;121:257–270. doi: 10.1016/j.cell.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Arenz S, et al. A combined cryo-EM and molecular dynamics approach reveals the mechanism of ErmBL-mediated translation arrest. Nat Commun. 2016;7:12026. doi: 10.1038/ncomms12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arenz S, et al. Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol Cell. 2014;56:446–452. doi: 10.1016/j.molcel.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arenz S, et al. Molecular basis for erythromycin-dependent ribosome stalling during translation of the ErmBL leader peptide. Nat Commun. 2014;5:3501. doi: 10.1038/ncomms4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weisblum B. Insights into erythromycin action from studies of its activity as inducer of resistance. Antimicrob Agents Chemother. 1995;39:797–805. doi: 10.1128/aac.39.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ingolia NT, et al. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis AR, et al. Sequence selectivity of macrolide-induced translational attenuation. Proc Natl Acad Sci U S A. 2014;111:15379–15384. doi: 10.1073/pnas.1410356111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kannan K, et al. The general mode of translation inhibition by macrolide antibiotics. Proc Natl Acad Sci U S A. 2014;111:15958–15963. doi: 10.1073/pnas.1417334111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svetlov MS, et al. Kinetics of drug-ribosome interactions defines the cidality of macrolide antibiotics. Proc Natl Acad Sci U S A. 2017;114:13673–13678. doi: 10.1073/pnas.1717168115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lovmar M, et al. Erythromycin resistance by L4/L22 mutations and resistance masking by drug efflux pump deficiency. EMBO J. 2009;28:736–744. doi: 10.1038/emboj.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lovmar M, et al. Kinetics of macrolide action: The josamycin and erythromycin cases. J Biol Chem. 2004;279:53506–53515. doi: 10.1074/jbc.M401625200. [DOI] [PubMed] [Google Scholar]
- 29.Woosley LN, et al. CEM-101 activity against Gram-positive organisms. Antimicrob Agents Chemother. 2010;54:2182–2187. doi: 10.1128/AAC.01662-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhanel GG, et al. The ketolides: a critical review. Drugs. 2002;62:1771–1804. doi: 10.2165/00003495-200262120-00006. [DOI] [PubMed] [Google Scholar]
- 31.Johansson M, et al. Sequence-dependent elongation dynamics on macrolide-bound ribosomes. Cell Rep. 2014;7:1534–46. doi: 10.1016/j.celrep.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Credito KL, et al. Activity of telithromycin (HMR 3647) against anaerobic bacteria compared to those of eight other agents by time-kill methodology. Antimicrob Agents Chemother. 1999;43:2027–2031. doi: 10.1128/aac.43.8.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vazquez-Laslop N, et al. Molecular mechanism of drug-dependent ribosome stalling. Mol Cell. 2008;30:190–202. doi: 10.1016/j.molcel.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 34.Ramu H, et al. Nascent peptide in the ribosome exit tunnel affects functional properties of the A-site of the peptidyl transferase center. Mol Cell. 2011;41:321–330. doi: 10.1016/j.molcel.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 35.Sothiselvam S, et al. Binding of macrolide antibiotics leads to ribosomal selection against specific substrates based on their charge and size. Cell Rep. 2016;16:1–11. doi: 10.1016/j.celrep.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao JCH, Robishaw EE. Effects of macrolides on peptide-bond formation and translocation. Biochemistry. 1971;10:2054–2061. doi: 10.1021/bi00787a014. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka K, et al. Peptidyl puromycin synthesis; effect of several antibiotics which act on 50 S ribosomal subunits. FEBS Lett. 1971;13:65–67. doi: 10.1016/0014-5793(71)80666-x. [DOI] [PubMed] [Google Scholar]
- 38.Černa J, et al. Effects of macrolide antibiotics on the ribosomal peptidyl transferase in cell-free systems derived from Escherichia coli B and erythromycin-resistant mutant of Escherichia coli B. Biochim Biophys Acta. 1971;240:109–121. doi: 10.1016/0005-2787(71)90517-x. [DOI] [PubMed] [Google Scholar]
- 39.Sothiselvam S, et al. Macrolide antibiotics allosterically predispose the ribosome for translation arrest. Proc Natl Acad Sci U S A. 2014;111:9804–9809. doi: 10.1073/pnas.1403586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vazquez-Laslop N, et al. Role of antibiotic ligand in nascent peptide-dependent ribosome stalling. Proc Natl Acad Sci U S A. 2011;108:10496–10501. doi: 10.1073/pnas.1103474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koch M, et al. Critical 23S rRNA interactions for macrolide-dependent ribosome stalling on the ErmCL nascent peptide chain. Nucleic Acids Res. 2017;45:6717–6728. doi: 10.1093/nar/gkx195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weisblum B. Erythromycin resistance by ribosome modification. Antimicrob Agents Chemother. 1995;39:577–585. doi: 10.1128/AAC.39.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sutcliffe J, Leclercq R. Mechanisms of resistance to macrolides, lincosamides, and ketolides. In: Schönfeld W, Kirst HA, editors. Macrolide Antibiotics. Birkhäuser Verlag; 2002. pp. 281–318. [Google Scholar]
- 44.Gupta P, et al. Deregulation of translation due to post-transcriptional modification of rRNA explains why erm genes are inducible. Nat Commun. 2013;4:1984. doi: 10.1038/ncomms2984. [DOI] [PubMed] [Google Scholar]
- 45.Horinouchi S, Weisblum B. Posttranscriptional modification of mRNA conformation: mechanism that regulates erythromycin-induced resistance. Proc Natl Acad Sci U S A. 1980;77:7079–7083. doi: 10.1073/pnas.77.12.7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gryczan TJ, et al. Conformational alteration of mRNA structure and the posttranscriptional regulation of erythromycin-induced drug resistance. Nucleic Acids Res. 1980;8:6081–6097. doi: 10.1093/nar/8.24.6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Depardieu F, et al. Modes and modulations of antibiotic resistance gene expression. Clin Microbiol Rev. 2007;20:79–114. doi: 10.1128/CMR.00015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dar D, Sorek R. Regulation of antibiotic-resistance by non-coding RNAs in bacteria. Curr Opin Microbiol. 2017;36:111–117. doi: 10.1016/j.mib.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Mayford M, Weisblum B. The ermC leader peptide: amino acid alterations leading to differential efficiency of induction by macrolide-lincosamide-streptogramin B antibiotics. J Bacteriol. 1990;172:3772–3779. doi: 10.1128/jb.172.7.3772-3779.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramu H, et al. Programmed drug-dependent ribosome stalling. Mol Microbiol. 2009;71:811–824. doi: 10.1111/j.1365-2958.2008.06576.x. [DOI] [PubMed] [Google Scholar]
- 51.Subramanian SL, et al. Inducible resistance to macrolide antibiotics. In: Dougherty TJ, Pucci MJ, editors. Antibiotic Drug Discovery and Development. Springer Publishing Company; 2011. [Google Scholar]
- 52.Almutairi MM, et al. Resistance to ketolide antibiotics by coordinated expression of rRNA methyltransferases in a bacterial producer of natural ketolides. Proc Natl Acad Sci U S A. 2015;112(42):12956–12961. doi: 10.1073/pnas.1512090112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Min YH, et al. Translational attenuation and mRNA stabilization as mechanisms of erm(B) induction by erythromycin. Antimicrob Agents Chemother. 2008;52:1782–1789. doi: 10.1128/AAC.01376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamimiya S, Weisblum B. Induction of ermSV by 16-membered-ring macrolide antibiotics. Antimicrob Agents Chemother. 1997;41:530–534. doi: 10.1128/aac.41.3.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson J, et al. Effects of a number of classes of 50S inhibitors on stop codon readthrough during protein synthesis. Antimicrob Agents Chemother. 2004;48:4889–4891. doi: 10.1128/AAC.48.12.4889-4891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gupta P, et al. Regulation of gene expression by macrolide-induced ribosomal frameshifting. Mol Cell. 2013;52:629–642. doi: 10.1016/j.molcel.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmitz FJ, et al. Molecular analysis of constitutively expressed erm(C) genes selected in vitro by incubation in the presence of the noninducers quinupristin, telithromycin, or ABT-773. Microb Drug Resist. 2002;8:171–177. doi: 10.1089/107662902760326878. [DOI] [PubMed] [Google Scholar]
- 58.Bailey M, et al. Induction of ermC expression by ‘non-inducing’ antibiotics. Antimicrob Agents Chemother. 2008;52:866–874. doi: 10.1128/AAC.01266-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gupta P, et al. Nascent peptide assists the ribosome in recognizing chemically distinct small molecules. Nat Chem Biol. 2016;12:153–158. doi: 10.1038/nchembio.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gong F, Yanofsky C. Instruction of translating ribosome by nascent peptide. Science. 2002;297:1864–1867. doi: 10.1126/science.1073997. [DOI] [PubMed] [Google Scholar]
- 61.Cruz-Vera LR, et al. Features of ribosome-peptidyl-tRNA interactions essential for tryptophan induction of tna operon expression. Mol Cell. 2005;19:333–343. doi: 10.1016/j.molcel.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 62.Martinez AK, et al. Interactions of the TnaC nascent peptide with rRNA in the exit tunnel enable the ribosome to respond to free tryptophan. Nucleic Acids Res. 2014;42:1245–1256. doi: 10.1093/nar/gkt923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bischoff L, et al. Molecular basis for the ribosome functioning as an L-tryptophan sensor. Cell Rep. 2014;9:469–475. doi: 10.1016/j.celrep.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 64.Hansen JL, et al. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol. 2003;330(5):1061–1075. doi: 10.1016/s0022-2836(03)00668-5. [DOI] [PubMed] [Google Scholar]
- 65.Seip B, Innis CA. How widespread is metabolite sensing by ribosome-arresting nascent peptides? J Mol Biol. 2016;428(10 Pt B):2217–2227. doi: 10.1016/j.jmb.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 66.Garreau de Loubresse N, et al. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- 67.Petersen DN, et al. A small-molecule anti-secretagogue of PCSK9 targets the 80S ribosome to inhibit PCSK9 protein translation. Chem Biol. 2016;23:1362–1371. doi: 10.1016/j.chembiol.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 68.Lintner NG, et al. Selective stalling of human translation through small-molecule engagement of the ribosome nascent chain. PLoS Biol. 2017;15:e2001882. doi: 10.1371/journal.pbio.2001882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li W, McClure K, Montabana E, Liras S, Dullea R, Cate J. Structural basis for selective stalling of human ribosome nascent chain complexes by a drug-like molecule. bioRxiv. 2018 doi: 10.1101/315325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yonath A, et al. A tunnel in the large ribosomal subunit revealed by three-dimensional image reconstruction. Science. 1987;236:813–816. doi: 10.1126/science.3576200. [DOI] [PubMed] [Google Scholar]
- 71.Nakatogawa H, Ito K. The ribosomal exit tunnel functions as a discriminating gate. Cell. 2002;108:629–636. doi: 10.1016/s0092-8674(02)00649-9. [DOI] [PubMed] [Google Scholar]
- 72.Vazquez-Laslop N, Ramu H, Klepacki D, Kannan K, Mankin AS. The key role of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J. 2010;29:3108–3117. doi: 10.1038/emboj.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sohmen D, et al. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat Commun. 2015;6:6941. doi: 10.1038/ncomms7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berisio R, et al. Structural insight into the antibiotic action of telithromycin against resistant mutants. J Bacteriol. 2003;185:4276–4279. doi: 10.1128/JB.185.14.4276-4279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eyal Z, et al. Structural insights into species-specific features of the ribosome from the pathogen Staphylococcus aureus. Proc Natl Acad Sci U S A. 2015;112(43):E5805–E5814. doi: 10.1073/pnas.1517952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hansen JL, et al. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol Cell. 2002;10:117–128. doi: 10.1016/s1097-2765(02)00570-1. [DOI] [PubMed] [Google Scholar]
- 77.Menninger JR. Functional consequences of binding macrolides to ribosomes. J Antimicrob Chemother. 1985;16(Suppl A):23–34. doi: 10.1093/jac/16.suppl_a.23. [DOI] [PubMed] [Google Scholar]
- 78.Dunkle JA, et al. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Natl Acad Sci U S A. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiong L, et al. A ketolide resistance mutation in domain II of 23S rRNA reveals proximity of hairpin 35 to the peptidyl transferase centre. Mol Microbiol. 1999;31:633–639. doi: 10.1046/j.1365-2958.1999.01203.x. [DOI] [PubMed] [Google Scholar]
- 80.Hansen LH, et al. The macrolide-ketolide antibiotic binding site is formed by structures in domains II and V of 23S ribosomal RNA. Mol Microbiol. 1999;31:623–631. doi: 10.1046/j.1365-2958.1999.01202.x. [DOI] [PubMed] [Google Scholar]
- 81.Liu M, Douthwaite S. Activity of the ketolide telithromycin is refractory to Erm monomethylation of bacterial rRNA. Antimicrob Agents Chemother. 2002;46:1629–1633. doi: 10.1128/AAC.46.6.1629-1633.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McGuire JM, et al. Ilotycin, a new antibiotic. Antibiot Chemother (Northfield) 1952;2:281–283. [PubMed] [Google Scholar]
- 83.Bryskier A, et al. Macrolides - Chemistry, Pharmacology and Clinical Uses. Blackwell Science Ltd; 1993. [Google Scholar]
- 84.Bryskier A. Telithromycin–an innovative ketolide antimicrobials. Jpn J Antibiot. 2001;54(Suppl A):64–69. [PubMed] [Google Scholar]
- 85.Fernandes P. Use of antibiotic core structures to generate new and useful macrolide antibiotics. In: Sánchez S, Demain AL, editors. Antibiotics Current Innovations and Future Trends. Academic Press; 2015. [Google Scholar]
- 86.Metersky ML, Huang Y. Ketolide antibiotics: will they ever be used for community-acquired pneumonia? Ann Res Hosp. 2017;1:16. [Google Scholar]
- 87.Seiple IB, et al. A platform for the discovery of new macrolide antibiotics. Nature. 2016;533(7603):338–345. doi: 10.1038/nature17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wohlgemuth I, et al. Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J Biol Chem. 2008;283:32229–32235. doi: 10.1074/jbc.M805316200. [DOI] [PubMed] [Google Scholar]
- 89.Johansson M, et al. pH-sensitivity of the ribosomal peptidyl transfer reaction dependent on the identity of the A-site aminoacyl-tRNA. Proc Natl Acad Sci U S A. 2011;108:79–84. doi: 10.1073/pnas.1012612107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ito K, Chiba S. Arrest peptides: cis-acting modulators of translation. Annu Rev Biochem. 2013;82:171–202. doi: 10.1146/annurev-biochem-080211-105026. [DOI] [PubMed] [Google Scholar]
- 91.Wekselman I, et al. The ribosomal protein uL22 modulates the shape of the protein exit tunnel. Structure. 2017;25:1233–1241 e3. doi: 10.1016/j.str.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 92.Bulkley D, et al. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc Natl Acad Sci U S A. 2010;107:17158–17163. doi: 10.1073/pnas.1008685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schlunzen F, et al. Structural basis for the antibiotic activity of ketolides and azalides. Structure. 2003;11(3):329–338. doi: 10.1016/s0969-2126(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 94.Llano-Sotelo B, et al. Binding and action of CEM-101, a new fluoroketolide antibiotic that inhibits protein synthesis. Antimicrob Agents Chemother. 2010;54:4961–4970. doi: 10.1128/AAC.00860-10. [DOI] [PMC free article] [PubMed] [Google Scholar]