

 This work is licensed under a
This work is licensed under a Summary
Adrenocortical carcinoma (ACC) during childhood is a rare malignant tumor that frequently results in glucocorticoid and/or androgen excess. When there are signs of microscopic or macroscopic residual disease, adjuvant therapy is recommended with mitotane, an adrenolytic and cytotoxic drug. In addition to the anticipated side effect of adrenal insufficiency, mitotane is known to cause gynecomastia and hypothyroidism in adults. It has never been reported to cause precocious puberty. A 4-year-old girl presented with a 6-week history of virilization and elevated androgen levels and 1-year advancement in bone age. Imaging revealed a right adrenal mass, which was subsequently surgically excised. Histology revealed ACC with multiple unfavorable features, including high mitotic index, capsular invasion and atypical mitoses. Adjuvant chemotherapy was started with mitotane, cisplatin, etoposide and doxorubicin. She experienced severe gastrointestinal side effects and symptomatic adrenal insufficiency, which occurred despite physiological-dose corticosteroid replacement. She also developed hypothyroidism that responded to treatment with levothyroxine and peripheral precocious puberty (PPP) with progressive breast development and rapidly advancing bone age. Five months after discontinuing mitotane, her adrenal insufficiency persisted and she developed secondary central precocious puberty (CPP). This case demonstrates the diverse endocrine complications associated with mitotane therapy, which contrast with the presentation of ACC itself. It also provides the first evidence that the known estrogenic effect of mitotane can manifest as PPP.
Learning points:
Adrenocortical carcinoma is an important differential diagnosis for virilization in young children
Mitotane is a chemotherapeutic agent that is used to treat adrenocortical carcinoma and causes adrenal necrosis
Mitotane is an endocrine disruptor. In addition to the intended effect of adrenal insufficiency, it can cause hypothyroidism, with gynecomastia also reported in adults.
Patients taking mitotane require very high doses of hydrocortisone replacement therapy because mitotane interferes with steroid metabolism. This effect persists after mitotane therapy is completed
In our case, mitotane caused peripheral precocious puberty, possibly through its estrogenic effect.
Background
Adrenocortical carcinoma (ACC) is a rare, malignant tumor of the adrenal glands with a bimodal distribution, affecting young children and adults. In children, it can present with virilization secondary to increased androgen levels. The primary treatment modality is surgery, but adjuvant chemotherapy is important (1), especially where there is evidence of residual disease or adverse histology.
Mitotane is an adrenal-selective chemotherapeutic agent derived from the insecticide DDT (2). It causes adrenal necrosis, particularly of cells in the zona glomerulosa and zona fasciculata and is associated with improved prognosis in both adults and children with ACC (2, 3). Despite its utility as an adrenal-selective drug, mitotane has a wide-ranging side effect profile including gastrointestinal disturbance, confusion, ataxia and raised liver enzymes (4). In terms of endocrine complications, adrenal insufficiency is near universal and hypothyroidism and gynecomastia have also been reported in adults (4, 5). There is scant data on the drug’s pediatric side effects and no data concerning frequency.
Case presentation
A 4-year-old girl was referred to pediatric endocrinology with a 6-week history of clitoromegaly and pubic hair development. She was otherwise well, with no clinical evidence of Cushing’s syndrome or estrogen exposure. No concerns were raised regarding her genitalia at birth. On examination, her clitoris was enlarged (length 2.6 cm), no gonads were palpable in the inguinal region and she had Tanner stage 3 pubic hair and prepubertal Tanner stage 1 breast development.
Investigation
Initial investigations showed elevated androgens, with estradiol and gonadotrophins in the prepubertal range. The cortisol response to synthetic ACTH was normal (Table 1a) and a pelvic ultrasound demonstrated a prepubertal uterus and ovaries. An abdominal ultrasound scan showed a right adrenal mass, which was subsequently confirmed with MRI. A laparoscopic transperitoneal right adrenalectomy was performed and tissue was sent for histological review at Boston Children’s Hospital resulting in the diagnosis of ACC. The tumor was classed as stage III due to spillage that occurred during surgery, indicating possible residual disease. Multiple unfavorable histological features were present including a high mitotic and proliferative index, atypical mitoses, anaplasia, vascular invasion, capsular invasion and necrosis.
Table 1.
Hormones before, during and after treatment.
| Baselinea | Post Opb | Cycle 7c | Cycle 8d | 1 month post cycle 8e | 2.5 month post cycle 8f | 2 month post M-Rxg | 4.5 month post M-Rxh | Reference range | |
|---|---|---|---|---|---|---|---|---|---|
| Weeks since diagnosis | 0 | 2 | 32 | 35 | 39 | 44 | 54 | 65 | |
| Thyroxine (µg/day) | 0 | 0 | 0 | 25 | 32 | 37.5 | 37.5 | 25 | |
| Testosterone | 6.7 | <0.4 | <0.4 | <0.4 | 0.0–0.5 nmol/L | ||||
| DHEA-S | 17.7 | 0.2 | <0.01 | <0.01 | 0.01–0.7 µmol/L | ||||
| Androstenedione | 10.9 | <1.0 | <1.0 | <1.0 | <2 nmol/L | ||||
| 17-Hydroxyprogesterone | 6.4 | <1.0 | <1.0 | <1.0 | <3.5 nmol/L | ||||
| ACTH | 3 | 133 | <1 | 93 | 2–11 pmol/L | ||||
| Cortisol | 151 | 255 | 1630 | 24 | 170–500 nmol/L | ||||
| Post-ACTH-cortisol | 430 | 517 | >400 nmol/L | ||||||
| Estradiol | <30 | <30 | <30 | <80 pmol/L | |||||
| LH (unstimulated) | <0.1 | <0.1 | 0–2.5 IU/L | ||||||
| FSH (unstimulated) | 0.3 | 0.3 | 0–6.5 IU/L | ||||||
| LH (stimulated) | 2.3 | 9 | 0–2.5 IU/L | ||||||
| FSH (stimulated) | 0.7 | 15 | 0–6.5 IU/L | ||||||
| Free T4 | 7.9 | 11 | 11.3 | 11 | 15 | 16 | 10–20 pmol/L | ||
| TSH | 4 | 7.8 | 0.91 | 2.2 | 1.3 | 3.8 | 0.5–4.5 pmol/L |
aBaseline tests at the time of initial presentation showing elevated androgens and normal response to synthetic ACTH; bTests from after surgery showing normal androgen levels and normal response to synthetic ACTH; cTests following early breast development showing GnRH-independent precocious puberty and central hypothyroidism; d,e,g,fTests showing return to normal thyroid function with increasing doses of thyroxine; g,hCortisol and ACTH results following cessation of M-Rx and continuing supra-physiological hydrocortisone; hElevated gonadotrophins demonstrating central precocious puberty. Androgen and thyroid function results are normal.
M-Rx, mitotane treatment.
Treatment
Given the tumor’s stage and histology, the patient was treated with a combination of oral mitotane and intravenous cytotoxic drugs (cisplatin, etoposide and doxorubicin) as per the ARAR0332 protocol [https://www.childrensoncologygroup.org/index.php/arar0332 -accessed 29/01/18]. As mitotane is known to cause adrenal insufficiency, the patient was started on a relatively low physiological replacement hydrocortisone (6 mg/m2/day, oral). The dose of mitotane was gradually increased to 2.5 g/m2/day, which was the maximal dose tolerated due to severe and continuous gastrointestinal side effects. Monitoring of levels was planned but given the low dose that our patient received, it was inferred that levels would be subtherapeutic and specific measurement was not clinically useful. Three months after starting mitotane, during admission for cycle 5, she was found to have hypochloremic hyponatremia in conjunction with hypomagnesemia, normal pH and potassium (Table 2). She was euvolemic and asymptomatic other than subtle periorbital edema. Both mineralocorticoid deficiency and cisplatin-induced renal salt wasting were considered; however, plasma renin was low.
Table 2.
Electrolytes and hormones at presentation of adrenal insufficiency.
| Cycle 5 week 1a | Febrile neutropeniab | Stress dose hydrocortisonec | Hydrocortisone reducedd | Hydrocortisone restored | Reference range | |
|---|---|---|---|---|---|---|
| Days since start of cycle 5 | 0 | 14 | 16 | 28 | 30 | |
| Sodium | 125 | 124 | 137 | 125 | 137 | 135–145 mmol/L |
| Potassium | 4.4 | 4.1 | 4.4 | 4.4 | 3.8 | 3.5–5.2 mmol/L |
| Chloride | 91 | 94 | 104 | 88 | 107 | 95–110 mmol/L |
| pH | 7.39 | 7.41 | 7.38 | 7.36–7.44 | ||
| Magnesium | 0.57 | 0.58 | 0.77 | 0.7–1.0 mmol/L | ||
| Renin | 4 | 3 | 4 | 94 | 9–34 U/L | |
| Aldosterone | 74 | 140–2200 pmol/L | ||||
| ACTH | 133 | 2–11 pmol/L |
aRoutine blood tests before cycle 5 of chemotherapy show hypochloremic hyponatremia with hypomagnesemia. Cisplatin-induced renal salt wasting was considered; bPatient developed febrile neutropenia and stress dose hydrocortisone was started; cElectrolytes improve following treatment with stress dose hydrocortisone; dPatient was readmitted with fatigue and hyperpigmentation following attempt to reduce the dose of hydrocortisone; eElectrolytes improve following permanent restoration of stress dose hydrocortisone.
The patient developed febrile neutropenia and was treated with stress doses of hydrocortisone (38.5 mg/m2/day, initially IV then oral). Attempting to reduce this dose over the next month resulted in severe fatigue, hypotension and recurrent hyponatremia. She also had increasing skin pigmentation –particularly on the hands, feet and existing scars (Fig. 1A and B) and ACTH levels were raised (Table 2). As it was likely that this was due to mitotane interfering with steroid metabolism (6), supra-physiological doses of hydrocortisone were continued.
Figure 1.
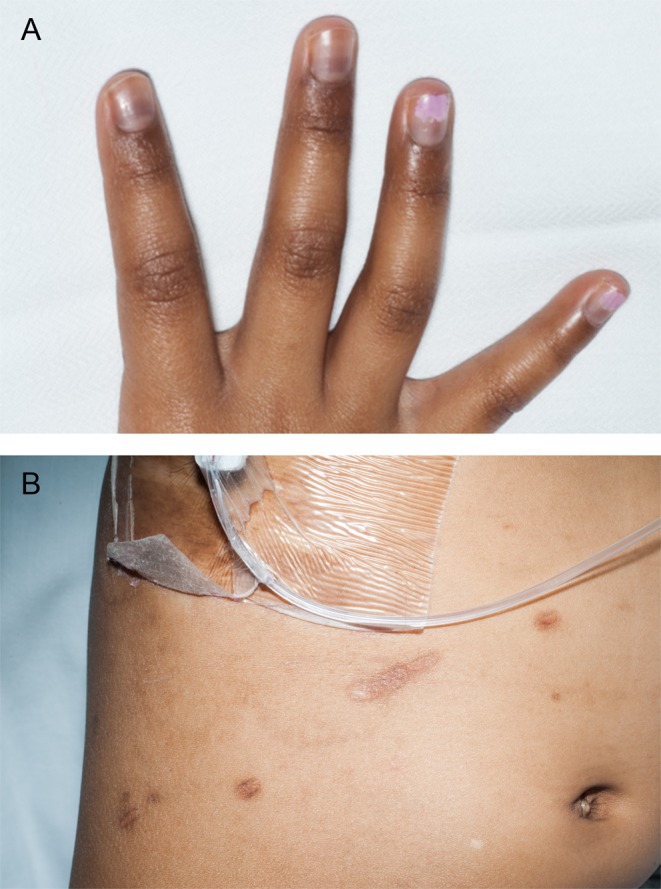
Increased skin pigmentation in our patient demonstrating adrenal insufficiency after an attempt to reduce the dose of replacement hydrocortisone. (A) Hyperpigmented nailbeds; (B) hyperpigmented existing abdominal scars.
Outcome and follow-up
Three weeks later (cycle 7), the patient developed additional endocrine problems. After 4 months of treatment, at an age of 5 years and 3 months, she had early breast development with a rapidly advancing bone age (8 years and 10 months vs 5 years and 9 months performed 6 months previously). A GnRH (SA) stimulation test performed at this time confirmed GnRH-independent precocious puberty (Table 1c). Her estradiol and androgen levels were unrecordably low.
She developed probable central hypothyroidism (Table 1c) with a T4 of 7.9 pmol/L (11.0–22.0 pmol/L) and was started on treatment with oral levothyroxine (25 µg/day). This increased T4 to 10 mol/L over 1 month. Increasing the dose to 37.5 µg/day achieved a T4 of 15 pmol/L.
After completing her last cycle of chemotherapy, the patient’s thyroid function tests remained normal (Table 1h) and levothyroxine was discontinued. She continued to require hydrocortisone at supra-physiological doses and attempts to wean hydrocortisone produced increased fatigue. Her pubertal stage continued to advance to Tanner stage 3 breast development. She also had a single episode of vaginal bleeding 2 months after discontinuation of mitotane, which was thought to reflect a withdrawal bleed. An MRI scan showed that the patient’s uterus and ovaries remained prepubertal. Repeat GnRH stimulation testing revealed that she had developed secondary CPP (Table 1h). She was started on leuprolide 11.25 mg IM 3-monthly to suppress puberty.
Discussion
We report the case of a child with ACC who suffered multiple endocrinopathies as a result of mitotane chemotherapy. As a rare treatment for a rare condition, side effects in childhood ACC are poorly documented. Our case demonstrates the challenges of managing adrenal insufficiency and hypothyroidism in patients taking mitotane. Furthermore, we report the first case of mitotane causing PPP which subsequently triggered CPP.
Managing adrenal insufficiency during mitotane treatment is a challenge. Not only does mitotane decrease endogenous levels of glucocorticoid and mineralocorticoid through its cytotoxic effect on the adrenal cortex, it also reduces the efficacy of exogenous steroid replacement by increasing CYP34A-mediated hepatic clearance of glucocorticoids and production of cortisol-binding globulin (6, 7). In our patient, this resulted in symptomatic primary adrenal insufficiency as we did not anticipate the very high dose of hydrocortisone required (32 mg/m2/day). Five months after stopping mitotane, our patient continued to require supra-physiological doses of hydrocortisone to maintain near-normal ACTH levels.
Our patient also developed hypothyroidism. Typical changes in thyroid function during mitotane therapy include a reduced T4, with relatively normal levels of TSH and T3 (8). Explanations for this picture include increased thyroid-binding globulin and altered deiodinase activity (7). A recent study, using data from 10 adult, female patients treated with mitotane, demonstrated numerous changes in keeping with central hypothyroidism, possibly due to destruction of pituitary thyrotroph cells (5, 8). Given our patient’s normal TSH concentration, it appeared that central hypothyroidism was most likely. This was easily managed with levothyroxine, and resolved following cessation of treatment.
Although gynecomastia in men is a common side effect of mitotane treatment, PPP in girls appears to be undocumented (4). Our patient presented with virilization without signs of feminization that ceased to progress with excision of her ACC. However, while being treated with mitotane, she showed progressive breast development and a rapid advancement in bone age. Suppressed gonadotrophins during a GnRH stimulation test confirmed a peripheral cause. Mitotane is known to have estrogenic action. This has been demonstrated in cell lines, where mitotane increases SHBG production in an estrogen receptor alpha (ERα)-dependent manner (9). This is not surprising given mitotane’s similarity to DDT, another estrogenic endocrine disruptor. Thus, the development of feminization with advanced bone age can be explained by the estrogenic actions of mitotane.
Our patient’s precocious puberty continued to progress following withdrawal of mitotane. As confirmed by GnRH stimulation testing, this was due to development of CPP, which occurs following prolonged exposure to sex hormones or endocrine disruptors (10).
This report demonstrates the wide ranging and potentially life-threatening side effects of mitotane in children. In addition to reinforcing the importance of prompt recognition of the complications of adrenal insufficiency and hypothyroidism, we provide the first evidence that the estrogenic effects of mitotane can cause feminization and subsequent development of PPP. Some side effects, such as the development of CPP, may occur after mitotane therapy has been completed and should be taken into account when treating children with ACC.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Signed, written consent has been obtained from the patient’s guardian, and consent form filed in the patient’s notes.
Author contribution statement
P D Oddie wrote the first draft of the manuscript. B B Albert, P L Hofman, C Jefferies, S Laughton and P J Carter provided clinical care to the patient. P D Oddie, P L Hofman and P J Carter edited and submitted the manuscript. P J Carter was the patient’s named physician and supervised the manuscript. All authors reviewed the final manuscript and approved it for publication.
References
- 1.Ribeiro RC, Figueiredo B. Childhood adrenocortical tumours. European Journal of Cancer 2004. 40 1117–1126. ( 10.1016/j.ejca.2004.01.031) [DOI] [PubMed] [Google Scholar]
- 2.Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, Rossetto R, Buci L, Sperone P, Grossrubatscher E. et al Adjuvant mitotane treatment for adrenocortical carcinoma. New England Journal of Medicine 2007. 356 2372–2380. ( 10.1056/NEJMoa063360) [DOI] [PubMed] [Google Scholar]
- 3.Redlich A, Boxberger N, Strugala D, Frühwald MC, Leuschner I, Kropf S, Bucsky P, Vorwerk P. Systemic treatment of adrenocortical carcinoma in children: data from the German GPOH-MET 97 trial. Klinische Pädiatrie 2012. 224 366–371. ( 10.1055/s-0032-1327579) [DOI] [PubMed] [Google Scholar]
- 4.Fassnacht M, Allolio B. Clinical management of adrenocortical carcinoma. Best Practice and Research Clinical Endocrinology and Metabolism 2009. 23 273–289. ( 10.1016/j.beem.2008.10.008) [DOI] [PubMed] [Google Scholar]
- 5.Russo M, Scollo C, Pellegriti G, Cotta OR, Squatrito S, Gullo D, Frasca F, Cannavò S. Mitotane treatment in patients with adrenocortical cancer causes central hypothyroidism. Clinical Endocrinology 2016. 84 614–619. ( 10.1111/cen.12868) [DOI] [PubMed] [Google Scholar]
- 6.Chortis V, Taylor AE, Schneider P, Tomlinson JW, Hughes BA, O’Neil DM, Libé R, Allolio B, Bertagna X, Beuschlein F, et al Mitotane therapy in adrenocortical cancer induces CYP3A4 and inhibits 5α-reductase, explaining the need for personalized glucocorticoid and androgen replacement. Journal of Clinical Endocrinology and Metabolism 2013. 98 161–171. ( 10.1210/jc.2012-2851) [DOI] [PubMed] [Google Scholar]
- 7.Van Seters AP, Moolenaar AJ. Mitotane increases the blood levels of hormone-binding proteins. Acta Endocrinologica 1991. 124 526–533. [DOI] [PubMed] [Google Scholar]
- 8.Zatelli MC, Gentilin E, Daffara F, Tagliati F, Reimondo G, Carandina G, Ambrosio MR, Terzolo M, Uberti EC. Therapeutic concentrations of mitotane (o,p’-DDD) inhibit thyrotroph cell viability and TSH expression and secretion in a mouse cell line model. Endocrinology 2010. 151 2453–2461. ( 10.1210/en.2009-1404) [DOI] [PubMed] [Google Scholar]
- 9.Nader N, Raverot G, Emptoz-Bonneton A, Déchaud H, Bonnay M, Baudin E, Pugeat M. Mitotane has an estrogenic effect on sex hormone- binding globulin and corticosteroid-binding globulin in humans. Journal of Clinical Endocrinology and Metabolism 2006. 91 2165–2170. ( 10.1210/jc.2005-2157) [DOI] [PubMed] [Google Scholar]
- 10.Latronico AC, Brito VN, Carel JC. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes and Endocrinology 2016. 4 265–274. ( 10.1016/S2213-8587(15)00380-0) [DOI] [PubMed] [Google Scholar]