

Abstract
BACKGROUND:
Fetal hypoxia–ischemia (H–I) results in significant morbidity and mortality. Little is known about the timing of death in human stillbirths. The vulnerability of the fetus varies with age at the time of insult, but it is unknown what happens to the timing of fetal death in relation to a fetal insult. We asked the question of whether the timing of fetal death was influenced by the age at which the insult occurred.
METHODS:
Fetal H–I was achieved at three ages by sustained uterine ischemia in rabbits, mimicking the acute placental insufficiency of placental abruption.
RESULTS:
H–I at 22 d gestation (E22) resulted in fewer perinatal deaths than at E25 and E29. Fetal deaths were grouped into early and late perinatal deaths. Early perinatal death mostly occurred immediately after H–I and these fetuses delivered before term. Late perinatal death occurred between the insult and delivery at term gestation. Early perinatal death occurred more often in the E25 hypoxic–ischemic group as compared with those of the E22 hypoxic–ischemic group.
CONCLUSION:
There is an increasing vulnerability to hypoxia with increasing gestational age. Perinatal deaths may occur long after the episode of H–I. The timing of an intrauterine hypoxic-ischemic event cannot be inferred from the detection of fetal death.
More than 3 million babies are stillborn worldwide each year (1) and there are approximately 26,000 stillbirths annually in the United States (2). This stillbirth rate has remained constant and surpasses the number of neonatal deaths in the United States (3). In addition, 3% of all pregnancies end with a second trimester miscarriage (4). The etiology is found in between 50 and 70% of cases (5–7) Understanding the etiology is the first step toward lowering this significant burden on society. Many of the risk factors for unexplained stillbirth such as maternal smoking and diabetes can be linked to poor placental function.
Fetal hypoxia–ischemia (H–I) results in miscarriages, stillbirths, cerebral palsy, mental retardation, and other neurodevelopmental disabilities, which represent a significant burden to society. There has been a presumption that most, if not all, cases of H–I-induced cerebral palsy occur around the time of birth as opposed to remote from delivery (8,9). We have developed an animal model of cerebral palsy following a single episode of antenatal H–I at preterm gestation in rabbit kits (10). The model is analogous to acute placental abruption/acute placental insufficiency at 70% gestation. A uterine ischemic insult results in global fetal H–I (10,11). Surviving postnatal rabbit kits manifest motor deficits including hypertonia and postural deficits characteristically observed in cerebral palsy.
In this model, there is a developmental susceptibility to H–Iinduced brain injury. Previous animal studies predominantly using umbilical cord occlusion in sheep at different gestational ages assessed the effect of gestational age on the susceptibility to H–I. These studies collectively indicate that the immature fetus survives a longer period of asphyxia than the near-term fetus (12,13). Few studies, however, have systematically examined the question of gestational susceptibility to death from a single sustained episode of H–I, especially from acute placental insufficiency as opposed to cord occlusion.
We hypothesized that timing of fetal death after fetal H–I from a model of acute placental insufficiency would be related to fetal age. In addition, that there would be less mortality after an insult at earlier gestational ages than at near-term gestation. Rabbits are perinatal brain developers (14,15), like humans, and white matter brain development is closer to humans than are those of rodents, sheep, or primates. The development of pro-oxidant systems is also closer to that of humans than it is to those of rodents. The multiparous nature of rabbits enabled a study with greater power than previous sheep and primate studies. The rabbit model was chosen because these advantages (10). Using this model, we investigated the incidence of intrauterine fetal demise (or stillbirths) following H–I at three gestational ages.
RESULTS
Fetal H–I from uterine ischemia for 40 min at 22 d gestation (E22) resulted in a mixture of stillbirths and survivors with and without hypertonia after delivery (10). We first determined whether sham uterine ischemia caused any mortality. The surgical controls that underwent fetal H–I for 30 min had only one fetal death and 96% of the fetuses survived to term (25 of 26). This showed that the effects of surgery were negligible as these results were not different from control naive rabbits.
We subsequently compared 40 min of uterine ischemia at three different gestational ages, E22, E25, and E29, corresponding to 70, 79, and 92% of term gestation, respectively. The number of live newborn rabbit kits decreased with H–I at increasing gestational ages (Figure 1) from the E22 group to the E29 group. The number of term stillbirths increased correspondingly with H–I at increasing gestational ages, from E22 to E29 (Figure 1). The number of preterm stillbirths also increased from E22 to E25. Comparisons between E22 and E25 were more valid than comparisons with E29. The immediate and acute dead population that constitutes the early perinatal death after E29 hypoxia was probably under-represented as compared with the E22 and E25 groups because there were only 2 d after E29 to count them by our definition of delivery before E31. The percentage of the live-born kits with severe hypertonia at E22, E25, and E29 were 30, 88, and 42%, respectively. There were only three litters of E29 kits that had any survivors.
Figure 1.
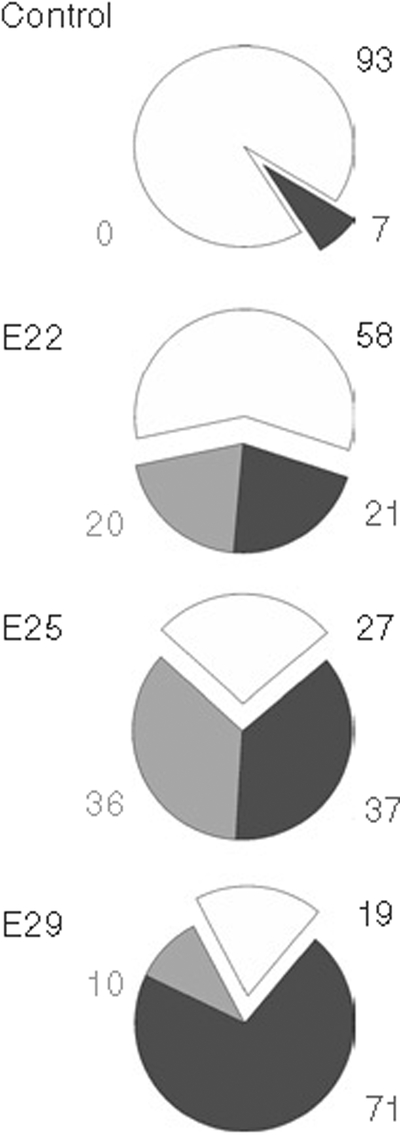
Delivery outcomes. Outcomes at 32 d gestation (E32) are shown as percentages of control animals and fetal hypoxia–ischemia for 40 minat E22, E25, and E29. Live-born kits are depicted as white area and black number. Early perinatal deaths (depicted as gray area and light gray number) are fetuses that die in utero and deliver before E31 (E22 20%, E25 36%, and E29 10%). Late perinatal deaths (black area and gray number) are all rabbits found dead at E32 after normal delivery or cesarean section (E22 21%, E25 37%, E29 71%). The difference between groups by Fisher’s exact test is highly significant (P < 0.0001). Hypoxia groups are also significantly different and each hypoxic group is significantly different from the other hypoxic groups (Kendall’s tau-b).
Normal fetal growth in rabbits was established from a population of naive animals, obtained by hysterotomy at E22, E25, or E29, or by spontaneous delivery at term. Growth in lategestation rabbits is linear (Figure 2). A regression line was calculated from the plot of weight and gestational age and the correlation was highly significant (R = 0.9554, P < 0.01).
Figure 2.

Fetal weight by gestational age. Rabbit fetal weight in grams is shown by gestational age. Naive fetuses at 22 d gestation (E22), E25, and E29 were delivered by hysterotomy and weighed. Newborn rabbit kits were weighed after birth (arrow) at E32. The growth is linear with R = 0.9554. The regression line is the solid black line and 95% confidence bands are gray dashed lines.
We next investigated all the miscarriages (Figure 3a). The regression line and confidence bands of the weight of miscarriages before E32 after H–I at E22, E25, and E29 was almost identical to that of the naive controls. This implies that death was very close to the initial H–I insult. In contrast to the miscarriages, the stillbirths had greater weights as an average, but the variance of the weights was higher (Figure 3b). Following E22 and E25 H–I, there was a bimodal distribution with the lower-weight animals being similar to the miscarriages and the larger animals closer to live-born kits. A clearer representation of this bimodal distribution is shown in frequency distribution of weights of the stillbirths from the E22, E25, and E29 groups (Figure 3c). The nonhypertonic live-born kits following H–I (E22 62.8 ± 2.7, E25 58.2 ± 3.6, E29 53.0 ± 9.0 g) are not significantly different from postnatal day (P)1 control kits (55.6 ± 0.5 g). The hypertonic kits (E22 52.6 ± 3.4, E25 52.0 ± 2.1, E29 46.0 ± 0.8 g) were not significantly smaller than either the nonhypertonic group or controls.
Figure 3.

Miscarriages and stillbirths. (a) Regression line and confidence bands (depicted as black dashed lines) of miscarriage weights (open circles). These lines are almost identical to the naive regression line and confidence bands (shown as gray solid lines), implying that the miscarried fetuses died at the time of fetal H–I insult. (b) Regression line and confidence bands (depicted as black dashed lines) of stillborn kits are disfferent from the miscarriages (gray solid lines). The average weight is significantly larger than the weights of naive fetuses at that age. This implies continued fetal growth and that death was delayed in some animals. (c) The bimodal distribution of stillbirths becomes evident when similar frequency distributions (spline curves in gray) are plotted for 22d of gestation (E22) (white bars) and E25 (gray bars), not so for the E29 (black bars) group. The majority of stillbirths survived the initial insult, continued to grow, and died at a later time point. The z-axis shows the age at H–I. H–I, hypoxia–ischemia.
To get a more precise time of death, we used the normal fetal growth rate shown in Figure 2 to generate time of death using the regression line (Figure 4a inset), given the assumption that H–I had no major impact on fetal growth. We then generated Kaplan–Meier survival curves for the stillbirth group. This assumption probably holds true for the E22 H–I as the liveborn kits weight were the same as control kits. Following E22 H–I, the early perinatal death population probably died within a day and half after fetal H–I (Figure 4a) and this constituted a quarter of the stillbirth deaths. The kits representing the rest of the perinatal deaths started dying after 4 d and continued into the last part of the pregnancy. This shows that the initial episode results in immediate and acute death in a portion of the litter and 78% of this early perinatal death population will deliver prematurely before E31.
Figure 4.

Estimated time of death for early and late perinatal deaths. The time of death of early (gray line) and late perinatal deaths (black line) was estimated from the weights and calculated regression slope from naive control fetuses and newborn kits (Figure 2) and the process shown by dashed arrow line in inset. Ordinate depicts in descending order the percentage of animals that died on each day. (a) At 22 d of gestation (E22), all the early perinatal deaths probably occurred within a day and a half after H–I. About a quarter of the late perinatal deaths also occurred within a day and a half of H–I, but the rest of the fetuses started dying after 4 d. (b) At E25, all the early perinatal deaths also occurred within 2 d after H–I; late perinatal deaths occurred throughout the remainder of the pregnancy with 47% of the late perinatal deaths occurring around birth (arrow). (c) At E29, there were few early perinatal deaths. Late perinatal deaths were the predominant form of death. H–I, hypoxia–ischemia.
For E25 H–I, the early perinatal death population behaved similarly to that after E22 hypoxia as all early perinatal deaths occurred within 2 d after fetal H–I. Only 15% of the late perinatal deaths occurred in the first 2 d and most of the E25 H–I group died afterwards (Figure 4b). The 82% of the population that died within 2 d were delivered before E31. The difference from E22 hypoxia was that a sizable portion of late perinatal deaths (47%) occurred around birth (from E31 onwards). Even accounting for the fact that a small amount of growth restriction followed H–I, the percentage of late perinatal deaths remains the same at 47%.
For E29 hypoxia (Figure 4c), the estimates of time of death spread out from H–I to the time of birth. Because of the closeness of H–I insult to time of birth, distinction between early and late perinatal deaths becomes difficult.
To confirm the timing of acute deaths after H–I, we performed hysterotomy 24 h after H–I at E22 or E25. We then compared these “actual” acute death rates with those previously estimated to occur in the first 24 h. Both after E22 (n = 29) and E25 (n = 21) H–I, the percentage of fetuses actually found dead, defined by an absence of fetal heart rate, (30 and 42%, respectively) was not significantly different from the estimated death by weight criteria (22 and 35%, respectively). The number of actual deaths at 24 h after E22 hypoxia was not significantly different from that after E25 hypoxia (χ2 P = 0.0769, power = 25%). The survivors in both groups had normal fetal heart rate. These studies also buttress our original assumption of normal fetal growth after H–I for estimating time of death, as the actual death rate, although not different, was slightly higher than the estimated death rate, and given the fact that with some intrauterine growth retardation, the actual death rates should have been lower than the estimated values.
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) diagnosed those fetuses that died by imaging the heart (Figure 5a). We also investigated the fetal brain apparent diffusion coefficient (ADC) changes in litters that underwent H–I under the magnet at E22, E25, or E29 and also obtained ADC values at 4, 24, and 72 h after H–I in a subset of E25 pregnant dams. Uterine position was confirmed by cesarean section at E31. The acute response to H–I showed that the later gestational ages had an earlier onset and deeper drop of ADC (Figure 5d). We were also able to obtain and analyze the ADC time course in 41 fetuses that were dead at cesarean section (12 dams). Immediate death could be diagnosed if fetal brain ADC did not recover above 0.7 μm2/ms by 20 min of reperfusion and remained below this threshold at the MRI session 4 h after H–I (Figure 5e). ADC in dead fetal brains remains low for at least 24 h after death. These studies show that time of death can be determined by the dynamic changes of ADC around an insult.
Figure 5.

MRI changes in fetal brains. (a) Normally beating fetal heart produces hypointense area on T2-weighted images due to MRI signal loss because of moving blood (dashed arrow). Arrowhead shows fetal liver. (b) Loss of hypointensity in heart area is shown in the same fetus at 20 min of reperfusion (dashed arrow), indicative of cessation of circulation and fetal demise. In comparison, no changes in intensity are observed in fetal liver (arrowhead). (c) At 72 h after H–I, loss of brain structure can be observed in dead fetuses on T2-weighted images, indicative of tissue decomposition (arrowhead), as compared with live fetal brains (dashed arrow). (d) Acute changes in fetal brain apparent diffusion coefficient (ADC) with H–I were compared at different gestational ages, 22 d of gestation (E22) (open circles, dashed line) and E25 (filled circles, solid line) with 40-min H–I, and E29 (gray squares, dotted line) with 32-min H–I. (e) Fetal brain ADC in stillborn fetuses (open squares, dotted line). Time course was compared with sham controls (open circles, solid line) and hypertonic survivors (black diamonds, solid line). Time of death was identified as immediate if fetal brain ADC did not recover above 0.7 m2/ms by 20 min of reperfusion and remained below this threshold at 4 h after H–I. H–I, hypoxia–ischemia; MRI, magnetic resonance imaging.
DISCUSSION
The principal findings of this study are that in utero fetal demise resulting from a fetal insult can occur either around the time of the insult or remote from the insult. The finding of late-onset deaths is especially important for issues related to the estimation of timing of insult in human deaths and has implications for newborn resuscitation. There are also malpractice implications for estimates of timing of insult from a newborn death. This study also showed that an insult at 70% gestation (E22) caused fewer fetal deaths than at later gestational ages in the rabbit (79% (E25) and 92% (E29) of term).
It is estimated that 12–18% of human stillbirths are due to abruptio placenta (16). The breakdown of the time of death from the acute placental insufficiency insult is not known in humans. Ours is also the first study to estimate the timing of deaths as worst-case consequences following global H–I in rabbits with enough power to make meaningful comparisons. We suspect that late deaths also occur remote from a global hypoxic–ischemic insult in humans. Longer exposure to slight decreases in global oxygen saturation can lead to later deaths. Supporting evidence for later-onset death is observed in premature infants as seen in the randomized study investigating two targets of oxygen saturation where differences in deaths occurred weeks after the onset of target oxygenation (17).
To explain the age-related vulnerability, sheep umbilical cord occlusion studies show that the immature fetus has a different metabolic response with increased anaerobic metabolism of glucose, resulting in less cardiovascular decompensation (18). Placental function may be more compromised in term gestation with the same amount of uterine ischemia (19) resulting in decreased fetal oxygenation, which is a critical determinant of fetal growth. Preterm rat fetuses have also been found to better maintain brain mitochondrial function in intrauterine ischemia than term rat fetuses (20). If mitochondrial function is better maintained for all vital organ systems, it may explain the better survival of preterm fetuses. The brain response to antenatal H–I, measured by the ADC of the fetal brain, shows that the same insult results in more injury to fetal brain at later gestational ages (Figure 5d). A subset of early prenatal deaths and miscarriages dies immediately after H–I and this can be detected by MRI imaging (Figure 5a,e). In these fetal brains, the ADC drop is similar initially to that of hypertonic kits but does not recover after H–I and remains low for a period of time (Figure 5e). Most interestingly, the ADC course shows “pseudonormalization” in fetal brain at later time points and even exceeds the values in control brains at 72 h, possibly due to tissue decomposition and loss of structural barriers to diffusion. This can be distinguished from the normalization of ADC, which is identified in surviving kits with a normal-looking heart.
We employed definitions of discovery for delineating two types of fetal death in our rabbit model to better understand the timing of the pathogenetic processes causing fetal death. The accepted definitions of stillbirth (fetal death after 20 wk of pregnancy) and miscarriage (fetal death before 20 wk) used in humans cannot be easily applied to rabbits. Also, unlike humans in whom the timing of the fetal insult is often obscure and most workups of stillbirth result in uncertain etiology (16), the animal model allowed us to compare an equal amount of fetal H–I at specific ages. The important point to note is that the same fetal H–I results in a cerebral palsy phenotype in the survivors following E22 H–I (10) and E25 H–I (21). A single insult could therefore result in a continuum of responses from relatively unaffected deliveries, mild growth restriction, to early and late fetal demise.
In a large human population, late perinatal deaths from antepartum hemorrhage (that presumably included abruption) also were not different in weight from live-born neonates (22). We did not observe growth restriction from H–I at E22 mimicking the insult from abruptio placentae in our study, but did find mild growth restriction following H–I at E25 and E29.
An association between fetal growth restriction and stillbirths has been known in humans (23). Our study was not set up to answer the question of whether the fetuses that survived the immediate period following H–I at E25 or E29 develop growth restriction and become susceptible to death. In our analysis, we could not differentiate between the fetuses that grew normally and died early and those that had growth restriction and died late. Our estimate of time of death thus involved an assumption (of normal weight gain) that resulted in inferring a worst-case scenario. If there was growth restriction from the insult, our estimates of time of death actually reinforce the finding that a sizable portion of perinatal deaths are late. The method of estimating time of fetal deaths from the established gestational age–birth weight relationship appeared to be validated by the actual death counts 24 h after the H–I insult.
It is difficult to compare rabbit gestational age to that of humans as the rate of development for various organs and cells differ. From the previous study done on oligodendrocyte maturation (24), white matter development at E22 probably corresponds to 24–28 wk gestation in humans.
Our study confirmed our suspicion that all of the early perinatal deaths fetuses that delivered prematurely died within a short time frame (1 to 2 d) after the fetal H–I, even if the delivery was remote from H–I insult.
The percentage of rabbits that survived the process of birth decreased as fetal H–I neared term. The percentage of deaths before term delivery (24 h before E32 as shown in Figure 4) increased from 5% in the E22 group to 17% in the E25 group and 36% in the E29 group. Another implication of this finding is that the population of fetuses that is at high risk of dying at birth increases with gestational age following an identical insult. We speculate that this population could benefit from neuroprotective strategies just before birth or neonatal resuscitation at birth. There is a suggestion from historical trends that this may be true for humans as well. Increased intervention is believed to explain the decrease in fetal death risk at 40–43 wk as compared with an increase at 20–22 wk (in whites) from 1991 to 1997 (25).
Our study shows that it is perhaps inappropriate to time the fetal insult causing fetal or even neonatal death as proximate to the discovery of that death. Fetal deaths appear to occur days following an initial insult as evidenced by the timing of late perinatal deaths. Another important point from our study is that the cause of near-term fetal death may be from a fetal insult that occurred much earlier in gestational age. It is thus conceivable that infants who either die or come close to it after birth (e.g., babies with Apgars of 0 at 1 and 5 min who are resuscitated) may have suffered a debilitating insult significantly earlier in gestation. Notably, previous fetal hypoxic insults have been shown to markedly diminish cardiovascular responsiveness to subsequent hypoxia (26).
Most previous studies that show increased resistance to hypoxic damage in less mature rather than older fetuses have used the umbilical cord occlusion model. Umbilical cord occlusion results in a different pathophysiology from that of acute placental insufficiency (27). The afterload on the fetal heart is increased by changing the peripheral vascular resistance from very low to very high following cord occlusion. The initial fetal bradycardia response is more marked in uterine occlusion as compared with umbilical cord occlusion in rabbit embryos (E12) (28) and near-term rats (29). However, fetal deaths occur after longer periods of H–I in uterine ischemia as well. For example, fetal death can be provoked in late-gestation sheep (122–126 d) after 60 min of uterine ischemia (27), whereas only 10 min of umbilical cord occlusion yields similar results (30). Similarly, monkeys subjected to only 12–17 min of umbilical cord clamping at term require resuscitation to keep them alive (31). These studies however lack sufficient power to develop definite conclusions of different ages and survival to term.
The delivery of rabbits is unattended, and the rate of late perinatal deaths may be higher than that if delivery were attended with proper intensive care. In a study in developing countries, some essential newborn care results in a decrease of stillbirths but interestingly not in neonatal deaths (32). Training a group of caregivers in the Neonatal Resuscitation Program did not result in a significant decrease in either stillbirths or neonatal deaths (32). This study points to the need for early detection and possible treatment of antenatal insults.
In conclusion, there is an increasing vulnerability to death from H–I resulting from acute placental insufficiency with increasing gestational age. It also appears that the factors leading to peripartum fetal death may result from remote H–I events and that the timing of an intrauterine H–I event cannot be linked to the detection of fetal death, either in utero or at delivery.
MATERIALS AND METHODS
The surgical procedure has been described previously (10–11,21). Briefly, pregnant New Zealand white rabbits (Myrtle’s Rabbits, Thompson Station, TN) at 22, 25, and 29 d gestation (E22, E25, and E29), corresponding to 70, 79, and 92% of term gestation, respectively, were subjected to uterine ischemia. Development of rabbit white matter reaches certain key phases at these ages (24,33) and is analogous to that found in premature infants of 24–34 wk. The choice of 40-min fetal H–I was made because 40 min resulted in a reasonable balance of hypertonic survivors and late perinatal deaths at E32 (10). Shorter durations of H–I resulted in fewer hypertonic kits and at 50 min of H–I at E22 most of the newborn rabbit kits were found dead at E32. In pilot experiments starting before E21/E22, 40 min of H–I resulted in no detectable morbidity so the focus of the experiments started at E22.
The dams were briefly anesthetized using intravenous fentanyl (75 μg/kg/h) and droperidol (3.75 mg/kg/h). Bag and mask ventilation was used to maintain normal arterial pH (7.35–7.45). Spinal anesthesia was then administered with 0.3 cc of 0.75% bupivicaine using a 25-gauge spinal needle in the L2–L5 intervertebral space. The fentanyl and droperidol were then reduced to one-third the initial volume to allow the dam to breathe spontaneously. A 4F Fogarty arterial embolectomy catheter (Baxter Healthcare, Santa Ana, CA) was placed into the left femoral artery, inserted 10 cm into the descending aorta to above the uterine and below the renal arteries. The balloon was inflated with saline for 40 min. Blood pressure in the right leg was monitored noninvasively (Sensor Devices, Waukesha, WI) to confirm presence of uterine ischemia and/or reperfusion. The catheter was then removed and the animal was allowed to recover. The dams were allowed to give birth spontaneously and were monitored for deliveries. The weight of the newborn rabbit kits and gestational age at delivery were recorded for all deliveries. If the dam did not give birth by E32 or delivered prematurely, hysterotomy was performed at E32 to ensure that there were no remaining fetuses in utero. All kits that were dead after spontaneous birth were labeled as stillbirths.
Delivery Outcomes
Alive at term (E32) either after spontaneous delivery at 31.5 d or after hysterotomy at E32.
“Stillbirth”: Found dead at E32 after spontaneous delivery or after hysterotomy.
“Miscarriage”: Premature delivery before E31 and found to be dead.
These groups are named for convenience and are different from the strict definitions of human stillbirth and miscarriages. Dams that delivered the entire litter at E31 were not included for analysis for rigorous comparison between groups for this study. This population constituted only 6% of the total number of dams. Rabbit litter size in controls was 8.0 ± 0.3 kits (mean ± SEM). The litter sizes of the other groups were not statistically different after E22 (7.3 ± 0.7), E25 (7.3 ± 0.5), or E29 (8.8 ± 0.8) H–I. The miscarriage rate may be an underestimate as some of the fetuses from early deaths may have been eaten by the dam, especially in the E22 H–I group.
Groups: Assessed at P1.
Controls: P1 naive rabbits that were not subjected to fetal H–I, n = 385 kits.
Surgical controls: Fetal H–I for 30 min at E22, n = 26 fetuses.
E22 hypoxia: Fetal H–I for 40 min from uterine ischemia at E22, n = 123 fetuses.
E25 hypoxia: Fetal H–I for 40 min at E25, n = 129 fetuses.
E29 hypoxia: Fetal H–I for 40 min at E29, n = 109 fetuses.
Groups: Assessed 24 h after H–I to confirm early perinatal death rates.
E22 early: Fetal H–I for 40 min at E22, n = 97 fetuses, hysterotomy after 24 h.
E25 early: Fetal H–I for 40 min at E25, n = 100 fetuses, hysterotomy after 24 h.
The number of dead fetuses, as defined by an absence of fetal heartbeat, was determined using an 8-MHz Doppler transducer (Mini Dopplex D500; Huntleigh Technologies, Luton, UK).
Procedure for Estimating Time of Death of Early and Late Perinatal Deaths
Control animals were subjected to hysterotomy at E22, E25, and E29 (n = 20, 24, and 154 fetuses, respectively). Fetal weight was recorded and added to the database of weights of newborn rabbits at P1 (E32) in the control group. Two assumptions were made for estimating time of death: that the weights did not change much after death and that the rate of growth of fetuses after the episode of uterine ischemia in the hypoxic groups was similar to that of naive animals. A regression line was calculated from the plots for weights and age and the slope of the line was used to calculate the time of death from the weights of the in utero fetal demises and stillbirths. In timing of death we will refer to early perinatal deaths as occurring immediately and within 2 d after H–I and late perinatal deaths as occurring at E31 or later.
Magnetic Resonance Imaging
In a subset of dams, serial in utero MRI was performed using 3T magnet using single-shot fast-spin echo. T2-weighted images were taken for anatomical reference in axial, coronal, and sagittal planes, and diffusion-weighted images with b = 0, 0.8 ms/μm2 were taken during H–I and 20 min reperfusion, and at 4, 24, and 72 h after H–I as previously described (21).
Statistical Analysis
Data are presented as mean ± SEM. Power of the study was calculated as 90% to detect a difference of 25% in survival from E22 hypoxia for a group population of 68. Cross-tabulation analysis was done by Fisher’s exact test for all ages and Kendall’s tau-b and χ2 for comparison of hypoxic groups. Weights were analyzed by ANOVA with post hoc comparison of means by Bonferroni correction, or by regression. All analysis was done with SAS system for Windows, release 9.1.
The study was approved by the Animal Review Committee of the Evanston Northwestern Healthcare Research Institute. All animals received humane care in compliance with the “Guide for Care and Use of Laboratory Animals” (National Institutes of Health Publication No. 85–23, revised 1985).
Acknowledgments
STATEMENT OF FINANCIAL SUPPORT
This study was supported by National Institutes of Health grants NS43285 and NS41476 (to S.T.).
REFERENCES
- 1.Stanton C, Lawn JE, Rahman H, Wilczynska-Ketende K, Hill K. Stillbirthrates: delivering estimates in 190 countries. Lancet 2006;367:1487–94 [DOI] [PubMed] [Google Scholar]
- 2.Reddy UM, Goldenberg R, Silver R, et al. Stillbirth classification–developing an international consensus for research: executive summary of a National Institute of Child Health and Human Development workshop. Obstet Gynecol 2009;114:901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macdorman MF, Kirmeyer S. The challenge of fetal mortality. NCHS Data Brief 2009:1–8. [PubMed] [Google Scholar]
- 4.Dukhovny S, Zutshi P, Abbott JF. Recurrent second trimester pregnancy loss: evaluation and management. Curr Opin Endocrinol Diabetes Obes 2009;16:451–8. [DOI] [PubMed] [Google Scholar]
- 5.Walsh CA, Vallerie AM, Baxi LV. Etiology of stillbirth at term: a 10-year cohort study. J Matern Fetal Neonatal Med 2008;21:493–501. [DOI] [PubMed] [Google Scholar]
- 6.VanderWielen B, Zaleski C, Cold C, McPherson E. Wisconsin stillbirth services program: a multifocal approach to stillbirth analysis. Am J Med Genet A 2011;155A:1073–80. [DOI] [PubMed] [Google Scholar]
- 7.Reddy UM. Prediction and prevention of recurrent stillbirth. Obstet Gynecol 2007;110:1151–64. [DOI] [PubMed] [Google Scholar]
- 8.Draper ES, Kurinczuk JJ, Lamming CR, Clarke M, James D, Field D. A confidential enquiry into cases of neonatal encephalopathy. Arch Dis Child Fetal Neonatal Ed 2002;87:F176–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phelan JP, Martin GI, Korst LM. Birth asphyxia and cerebral palsy. Clin Perinatol 2005;32:61–76, vi. [DOI] [PubMed] [Google Scholar]
- 10.Derrick M, Luo NL, Bregman JC, et al. Preterm fetal hypoxia-ischemia causes hypertonia and motor deficits in the neonatal rabbit: a model for human cerebral palsy? J Neurosci 2004;24:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan S, Drobyshevsky A, Jilling T, et al. Model of cerebral palsy in the perinatal rabbit. J Child Neurol 2005;20:972–9. [DOI] [PubMed] [Google Scholar]
- 12.Mallard EC, Williams CE, Johnston BM, Gluckman PD. Increased vulnerability to neuronal damage after umbilical cord occlusion in fetal sheep with advancing gestation. Am J Obstet Gynecol 1994;170(1 Pt 1):206–14. [DOI] [PubMed] [Google Scholar]
- 13.Keunen H, Blanco CE, van Reempts JL, Hasaart TH. Absence of neuronal damage after umbilical cord occlusion of 10, 15, and 20 minutes in midgestation fetal sheep. Am J Obstet Gynecol 1997;176:515–20. [DOI] [PubMed] [Google Scholar]
- 14.van Marthens E, Harel S, Zamenshof S. Experimental intrauterine growth retardation. Biol Neonate 1975;26:221–31. [DOI] [PubMed] [Google Scholar]
- 15.Harel S, Shapira Y, Tomer A, Donahue MJ, Quilligan E. Vascular-induced intrauterine growth retardation: relations between birth weight and the development of biochemical parameters in young rabbits. Isr J Med Sci 1985;21:829–32. [PubMed] [Google Scholar]
- 16.Fretts RC. Etiology and prevention of stillbirth. Am J Obstet Gynecol2005;193:1923–35. [DOI] [PubMed] [Google Scholar]
- 17.Carlo WA, Finer NN, Walsh MC, et al. ; SUPPORT Study Group of the Eunice Kennedy Shriver NICHD Neonatal Research Network. Target ranges of oxygen saturation in extremely preterm infants. N Engl J Med 2010;362:1959–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Low JA. Determining the contribution of asphyxia to brain damage in the neonate. J Obstet Gynaecol Res 2004;30:276–86. [DOI] [PubMed] [Google Scholar]
- 19.Lackman F, Capewell V, Gagnon R, Richardson B. Fetal umbilical cord oxygen values and birth to placental weight ratio in relation to size at birth. Am J Obstet Gynecol 2001;185:674–82. [DOI] [PubMed] [Google Scholar]
- 20.Nakai A, Taniuchi Y, Asakura H, et al. Developmental changes in tolerance to transient intrauterine ischemia in rat cerebral mitochondria. Am J Obstet Gynecol 2001;184:731–5. [DOI] [PubMed] [Google Scholar]
- 21.Drobyshevsky A, Derrick M, Prasad PV, Ji X, Englof I, Tan S. Fetal brain magnetic resonance imaging response acutely to hypoxia-ischemia predicts postnatal outcome. Ann Neurol 2007;61:307–14. [DOI] [PubMed] [Google Scholar]
- 22.Chard T Does the fetus lose weight in utero following fetal death: a study in preterm infants. BJOG 2001;108:1113–5. [DOI] [PubMed] [Google Scholar]
- 23.M Kady S, Gardosi J. Perinatal mortality and fetal growth restriction. Best Pract Res Clin Obstet Gynaecol 2004;18:397–410. [DOI] [PubMed] [Google Scholar]
- 24.Drobyshevsky A, Song SK, Gamkrelidze G, et al. Developmental changes in diffusion anisotropy coincide with immature oligodendrocyte progression and maturation of compound action potential. J Neurosci 2005;25:5988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan H, Platt RW, Morin L, Joseph KS, Kramer MS. Fetal deaths in the United States, 1997 vs 1991. Am J Obstet Gynecol 2005;193:489–95. [DOI] [PubMed] [Google Scholar]
- 26.Gardner DS, Fowden AL, Giussani DA. Adverse intrauterine conditions diminish the fetal defense against acute hypoxia by increasing nitric oxide activity. Circulation 2002;106:2278–83. [DOI] [PubMed] [Google Scholar]
- 27.Ball RH, Espinoza MI, Parer JT, Alon E, Vertommen J, Johnson J. Regional blood flow in asphyxiated fetuses with seizures. Am J Obstet Gynecol 1994;170(1 Pt 1):156–61. [DOI] [PubMed] [Google Scholar]
- 28.Millicovsky G, DeSesso JM. Differential embryonic cardiovascular responses to acute maternal uterine ischemia: an in vivo microscopic study of rabbit embryos with either intact or clamped umbilical cords. Teratology 1980;22:335–43. [DOI] [PubMed] [Google Scholar]
- 29.Robinson KS, Kavlock RJ, Watkinson WP. Electrocardiographic responses of rat fetuses with clamped or intact umbilical cords to acute maternal uterine ischemia. Am J Obstet Gynecol 1983;147:795–8. [DOI] [PubMed] [Google Scholar]
- 30.Mallard EC, Gunn AJ, Williams CE, Johnston BM, Gluckman PD. Transient umbilical cord occlusion causes hippocampal damage in the fetal sheep. Am J Obstet Gynecol 1992;167:1423–30. [DOI] [PubMed] [Google Scholar]
- 31.Windle WF. Brain damage at birth. Functional and structural modifications with time. JAMA 1968;206:1967–72. [PubMed] [Google Scholar]
- 32.Carlo WA, Goudar SS, Jehan I, et al. ; First Breath Study Group. Newborn-care training and perinatal mortality in developing countries. N Engl J Med 2010;362:614–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buser JR, Segovia KN, Dean JM, et al. Timing of appearance of late oligodendrocyte progenitors coincides with enhanced susceptibility of pre-term rabbit cerebral white matter to hypoxia-ischemia. J Cereb Blood Flow Metab 2010;30:1053–65. [DOI] [PMC free article] [PubMed] [Google Scholar]