

Abstract
F1-ATPase is a rotary motor enzyme in which a single ATP molecule drives a 120° rotation of the central γ subunit relative to the surrounding α3β3 ring. Here, we show that the rotation of F1-ATPase spontaneously lapses into long (≈30 s) pauses during steady-state catalysis. The effects of ADP-Mg and mutation on the pauses, as well as kinetic comparison with bulk-phase catalysis, strongly indicate that the paused enzyme corresponds to the inactive state of F1-ATPase previously known as the ADP-Mg inhibited form in which F1-ATPase fails to release ADP-Mg from catalytic sites. The pausing position of the γ subunit deviates from the ATP-waiting position and is most likely the recently found intermediate 90° position.
ATP synthase of mitochondria, chloroplasts, and bacteria catalyzes ATP synthesis coupled with a transmembrane proton flow (1–4). The enzyme consists of a membrane-embedded, proton-conducting portion (F0) and a protruding portion (F1) in which catalytic sites for ATP synthesis/hydrolysis exist. The isolated F1 portion has ATPase activity; hence, it is often called F1-ATPase. It is composed of five different subunits with a stoichiometry of α3β3γδɛ. The α3β3γ subcomplex is the minimum ATPase-active complex, which has catalytic features similar to F1-ATPase. In the crystal structure (5), the central γ subunit is surrounded by an α3β3 cylinder where three α and three β subunits are arranged alternately, and the six nucleotide binding sites are located at the α/β subunit interfaces. Three of the binding sites are catalytic, and the β subunits provide most of the catalytic residues. The other three are noncatalytic, and the α subunits provide most residues contributing nucleotide binding.
It has been postulated that the energy of the proton flow liberated at F0 is transformed into the energy of ATP synthesis at F1 through rotation of the central γ subunit and vice versa—the energy of ATP hydrolysis can be converted into the energy of proton pumping through reverse rotation of the γ subunit (6). By using an α3β3γ subcomplex of thermophilic F1-ATPase (F1-ATPase) immobilized on a glass surface, we have observed ATP hydrolysis-driven rotation of the fluorescent actin filament attached to the γ subunit (7).
At nanomolar ATP concentration, F1-ATPase binds and hydrolyzes a single ATP molecule, makes a 120° rotation, and waits for the next ATP molecule. As the ATP concentration increases, the ATP-waiting period becomes shorter until it is finally undetectable, and rotation of the actin filament becomes apparently continuous over hundreds of revolutions (8). However, when the rotation was observed for long periods, occasional pauses of rotation were recognized, even at high ATP concentrations (7, 9). Here, we show that these pauses occur at an intermediate step of rotation and mostly correspond to the ADP-Mg inhibition, which has been observed in bulk-phase kinetics as a general feature of the F1-ATPases (and ATP synthases). Slow interconversion between rotating and pausing states thus contributes to the attenuation of ATPase during steady-state catalysis.
Materials and Methods
Protein Preparation.
Escherichia coli strains used were JM109 (10) for preparation of plasmids, CJ236 (11) for generating uracil-containing single-stranded plasmids for site-directed mutagenesis, and JM103Δ (uncB-uncD) for expression of the mutant complexes of F1 from the thermophilic Bacillus PS3. Plasmids M13mp18 and pKAGB1 (12), which carried genes for the α, β, and γ subunits of F1 from the thermophilic Bacillus PS3, were used for mutagenesis and for gene expression, respectively. Site-directed mutagenesis was accomplished as described by Kunkel et al. (11). The plasmid pKAGB1/αC193S/γS107C/βHis10tag has been described (7). The plasmids pKAGB1/αK175A/T176A and pKAGB1/βT165S, which have been described (13, 14), were used to generate plasmids for this study.
pKAGB1/ΔNC/βT165S/γS107C, βHis10-tags was prepared by removing the fragment containing the ΔNC (α K175A/T176A) substitution (EcoRI-BglII fragment) and ligating it into the pKABG1/βT165S/αC193S, γS107C, βHis10-tags (EcoRI-BglII fragment) from which the wild-type gene fragment had been removed. The plasmid pKAGB1/αC193S/γS107C/βHis10-tag was used to express protein α (C193S)3β3γ(S107C), which was considered to be wild type, and plasmid pKAGB1/ΔNC/βT165S/αC193S, γS107C, βHis10-tags was used to express protein α(ΔNC)3β(T165S)3γ(S107C), which was named ΔNC′ mutant. The α (C193S)3β3γ(S107C/I210C) was used for the measurement of angular position (15). These F1 subcomplexes were purified as described (12, 16). Purified F1 was passed through a DEAE column equilibrated with 100 mM potassium phosphate/0.2 mM EDTA (pH 7.0) to reduce the bound nucleotide to 0.01 mol per mol F1, which was confirmed with reverse-phase HPLC.
Rotation Assay.
The wild type and the ΔNC′ mutant were biotinylated at the cysteines of the γ subunit and conjugated with streptavidin (7). To visualize the rotation of the γ subunit under the microscope, a fluorescent actin filament or duplex of beads (φ = 440 nm, 517 nm) was attached to the γ subunit. A flow cell (10 μl) was made of two coverslips separated by two spacers of 50-μm thickness. The glass surface was coated with Ni-nitrilotriacetic acid. Beads (0.1–1 nM) or actin (1–10 nM) were mixed with 1/100× to 1/10× the molar concentration of F1 in buffer A [10 mM 4-morpholinepropanesulfonic acid (Mops)-KOH/50 mM KCl/1% (vol/vol) BSA, pH 7.0]. The mixture was applied to the flow cell. Unbound beads or actin filaments were removed from the flow cell by washing with buffer A, followed by applying buffer A or buffer B (100 mM potassium phosphate buffer, pH 7.0) containing 2 mM MgCl2, 0.2 μM–2 mM ATP, and the ATP-regenerating system (0.2 mg/ml creatine kinase and 2.5 mM creatine phosphate). In the presence of 0.1% lauryl dimethyl amine oxide (LDAO), the rotation of the F1 particles were observed in the presence of buffer B. The rotation of the ΔNC′ mutant also was observed in buffer B (17). Buffer exchange was performed as described. To visualize induction of ADP-Mg inhibition, after the observation of the rotation in ATP-containing buffer A (1 mM ATP-Mg), an ADP-containing buffer A (1 mM ATP-Mg, 1 mM ADP or 0.1 mM ADP) was infused into the same chamber for rotation observation (see Fig. 2 A and B). To visualize release from the ADP-Mg inhibited state, the ADP buffer was exchanged to ATP+re buffer (ATP-containing buffer and ATP-regenerating system; see Fig. 2C).
Figure 2.

Effect of load of attached probes on the durations of the pauses (longer than 10 s). Blue circles, actin filaments; red circles, pair of biotin-coated beads (diameter 440 nm or 517 nm); squares, the average for one molecule ± SE.
The fluorescent actin filament was observed with a fluorescence microscope (IX70; Olympus, New Hyde Park, NY). Photobleaching of the fluorescent actin filament was minimized by the use of a filter (ND20). The rotation of the actin filament could be observed for 40–50 min. The 440-nm and 517-nm beads were observed with a transmission light microscope.
Images from actin- or bead-labeled F1-ATPase were recorded with an intensified charge-coupled device (350 F; Videoscope, Dallas) camera on an 8-mm videotape. Analysis of rotational angle was performed as described (7).
Measurement of ATPase Activity.
ATPase activity was measured at 25°C in the presence of an ATP-regenerating system (18) consisting of 300 μg/ml pyruvate kinase, 300 μg/ml lactate dehydrogenase, 2.5 mM phosphoenolpyruvate, and 0.2 mM NADH in buffer A or buffer B containing 2 mM MgCl2 and the indicated ATP concentrations. Typically, the reaction was initiated by the addition of F1 to 1.2 ml of assay mixture. The rate of ATP hydrolysis was monitored as the rate of oxidation of the NADH, which was determined by the absorbance decrease at 340 nm. The spectrophotometer was equipped with a small stirrer to ensure rapid mixing. The maximum dead time of measurement was less than 0.75 sec after initiation of the reaction. The data from 2 to 300 s were usually used for analysis. The initial rapid activity (the activity of wild type is 187 s−1 and that of ΔNC′ mutant is 54 s−1) decreased to the steady-state activity at 2 mM ATPMg.
Materials.
LDAO (30% aqueous solution) was purchased from Calbiochem.
Results
Pauses of Rotation of F1-ATPase at High ATP.
Fig. 1A shows the typical time courses of the rotation of a fluorescent actin filament attached to the γ subunit of immobilized F1-ATPase in the presence of 2 mM ATP. At this ATP concentration, ATP binding should take place within 0.1 ms (8) and does not result in a pause of rotation. However, each F1-ATPase molecule made several distinct pauses during 500 s, some of which were longer than 60 s. Noticeably, paused filaments always stayed within one of three angular positions, consistent with the pseudo-3-fold symmetrical structure of F1-ATPase. There is no obvious preference among the three angular positions for pauses to occur. Sometimes, the time-averaged centers of three positions deviated slightly from the exact 3-fold symmetry, probably because of the oblique attachment of the F1-ATPase molecule to the glass surface. To confirm that these pauses are not due to obstruction by nearby proteins or surface, we observed the single molecule for a long period by using plastic beads of diameter 440 nm or 517 nm as a rotation probe under transmission light microscopy. Despite the difference in the observation system, the pauses in the rotation of a pair of biotin-coated beads attached to the γ subunit also occurred at three positions with a frequency and duration similar to those observed in the rotation of actin filament (Fig. 1B).
Figure 1.

Time courses of the rotation of F1-ATPase labeled with actin filaments or beads. (A) Rotation of the actin filament attached to the γ subunit in the presence of 2 mM ATP-Mg. Each colored line represents the pausing of rotation for longer than 10 s. The same colored pauses were derived from a single filament. The average images of each pause are trimmed in circles to identify the filament position; inner gray lines show average angular positions of the pauses made by that molecule. The rotations of the beads are shown in B. The position indicated by asterisks (*) marks when the actin filament or beads disappeared from the observation field.
The duration of all pauses longer than 10 s observed in the rotation of actin filaments and beads were plotted as a function of the length of the probes (Fig. 2). Values are scattered, but, on average, pauses continue for ≈30 s irrespective of the length of the probes. If pauses were caused by nearby obstacles, they should occur at random positions, and longer probes would generate more pauses because of an increased chance of encountering obstacles. Thus, the observed properties of pauses, the three angular-pause positions, and load-independence all suggest that the pause is not due to accidental obstruction by nearby proteins or by the surface, but that it reflects intrinsic properties of the F1 motor.
State of F1-ATPase Corresponding to the Pauses.
It is natural to assume that the F1-ATPase in the pausing phase should be in an inactive form that is not hydrolyzing ATP. Because rotation resumes after awhile, this inactive form should be activated again in the presence of ATP-Mg. The candidate for the inactive form of F1-ATPase that shows the above features is the ADP-Mg inhibited form, a state commonly found in F1-ATPases from mitochondria (19), chloroplasts (20), and bacteria (13, 18, 21). The ADP-Mg inhibited form is generated in a stochastic manner during catalytic turnover of ATP hydrolysis by stable entrapment of ADP-Mg at a catalytic site. The ADP-Mg can be either an immediate hydrolysis product that is left bound to the enzyme or it can be picked up from the bulk phase medium (21). If pausing F1-ATPase is really in the ADP-Mg inhibited form, inclusion of ADP-Mg in the solution during the rotation assay should result in an increase in the number of pauses. At first, rotation was observed in the presence of 1 mM ATP, and then a solution containing 0.1 mM ADP + 1 mM ATP (Fig. 3A) or 1 mM ADP + 1 mM ATP (Fig. 3B) was infused into the chamber. As expected, rotation was frequently (0.1 mM ADP) or almost completely (1 mM ADP) prevented after the infusion. Physical damage of the immobilized F1-ATPase by the infusion is unlikely to be the reason for stopping rotation, because the reverse order of addition produced the opposite result—the molecules were unable to rotate in the presence of 1 mM ADP + 1 mM ATP and began rotation after the ADP was removed (Fig. 3C). Further, the rotating molecules in 1 mM ATP lapsed into a pause after the infusion of the ADP-containing solution, and started rotation again after the second infusion by the ADP-free solution (Fig. 3D).
Figure 3.

Effect of ADP or ΔNC′ mutation on rotation. (A–D) Each line shows the rotation of wild-type F1-ATPase. Buffer containing 0.1 mM ADP (A) or 1 mM ADP (B) was infused into the chamber for rotation observation. (C) ADP containing buffer was exchanged to ATP+re buffer. (D) Buffer containing 1 mM ADP was slowly introduced into the chamber (the center trace). The chamber was again exchanged for the ATP+re buffer (the right trace). Each line shows the rotation of the ΔNC′ mutant in buffer B in the absence (E) or in the presence (F) of 0.1% LDAO. Each bar indicates 60 revolutions.
Binding of ATP to the noncatalytic nucleotide-binding site on the α subunits stimulates the recovery of F1-ATPase from the ADP-Mg inhibited form (14). Therefore, the mutant F1-ATPase that has defective noncatalytic sites (ΔNC; ref. 13) is soon converted completely to the ADP-Mg inhibited form during catalysis. We examined whether the ΔNC mutant could rotate but were unable to find any rotating molecules. A second mutation, β-T165S, was introduced into the ΔNC mutant. The single β-T165S mutant is less susceptible to attaining the ADP-Mg inhibited form (14). The propensity of the ΔNC+β-T165S mutant (ΔNC′ mutant) to achieve the ADP-Mg inhibited form was intermediate, as it was greater than the wild-type F1-ATPase but less than the ΔNC mutant. When we observed rotation of the ΔNC′ mutant, the number of actively rotating molecules was much smaller than in wild-type F1-ATPase; even if they rotated, they stopped readily and stayed in the pausing phase for a long time (Fig. 3E). The inclusion of LDAO, which is known to be a potent activator of the ADP-Mg inhibited form (13, 21), resulted in a sharp decrease in the length of the long pauses (Fig. 3F). The same effect of LDAO was observed for the wild-type F1-ATPase (data not shown). These results support the contention that the state of F1-ATPase during the pause is the ADP-Mg inhibited form.
Kinetic Analysis of the Pause.
We collected the data from all pauses longer than 1 s from many rotating molecules at 2 mM ATP. The duration of each pause was plotted as the time-dependent decay of the number of pausing F1-ATPases, which remained in the pause and had not yet resumed rotation (Fig. 4A). The pauses observed for the rotation of probes of various lengths were analyzed together because the load, as shown in the previous section, does not affect the pausing (Fig. 2). The plot could not be fitted with a single exponential but was well fitted with the sum of two exponentials, suggesting that there are at least two kinds of pauses: the short-lived pause with a life-time (τsp) of 1.7 s, and the long-lived pause with a life-time (τlp) 32 s. On average, actively working F1 in the presence of 2 mM ATP spent 33.5% of its time in rotation, 4.5% in the short pause, and 62% in the long pause. A curve calculated from the long pause alone (the solid line in Fig. 4A) indicates that most (96% of the total) of the observed pauses longer than 10 s belong to the category of the long pauses. The frequency of the incidence for the rotating F1-ATPase to pause also was examined. For the short pauses, however, the incidence did not seem to be statistically random, and simple analysis was impossible. For the long pauses, the periods between one long pause (>10 s) and the next long pause (>10 s) were collected and analyzed. They were plotted as the time-dependent decay of the number of the “rotating” F1-ATPases that escaped from one long pause but had not lapsed into the next long pause yet (Fig. 4B). The data were fitted with a single exponential with a 34 s life-time (τr′). This value was obtained for the 72 pauses longer than 10 s and should be corrected by the uncounted long pauses shorter than 10 s (calculated to be 43). The corrected value of τr is 22 s; that is, rotating F1-ATPase lapses into the long pauses after 22 s, on average.
Figure 4.

Kinetic analyses of the rotation of single molecules. (A) The decay of the number of the pausing F1-ATPases in 2 mM ATP-Mg. The data are fitted with a double exponential function (dashed line) assuming two kinds of pauses, short pause and long pause, in the following manner: y = Nsp ⋅ exp (−t/τsp) + Nlp ⋅ exp (−t/τlp), where Nsp (number of the short pauses) = 340, Nlp (number of the long pauses) = 115, τsp (life-time of the short pause) = 1.7 s, τlp (life-time of the long pause) = 32.3 s, with an error of about ±15%. The solid line shows a single exponential curve calculated from Nlp = 115 and τlp′ = 32.2 s. (B) The decay of the number of the rotating F1-ATPase in 2 mM ATP-Mg. The rotation between pauses longer than 10 s were measured. The solid line shows a single-exponential function with kr−lp′ (= 1/τr′) (rate of conversion from rotation to the long pause) = 0.029 s−1.
Kinetic Analysis of the ADP-Mg Inhibited Form.
To compare the kinetic parameters of the long pauses to those of the ADP-Mg inhibited form, we estimated from the bulk-phase experiments the rates of conversion from the active to the ADP-Mg inhibited form (ka-i) and those of reverse conversion (ki-a) during catalysis, with the equation:
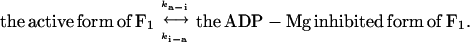 |
1 |
When the ATP hydrolysis assay was started by the addition of F1-ATPase to the solution, the initial rapid hydrolysis decelerated within 1 min and reached a slow steady-state of hydrolysis (Fig. 5A). This time course suggests that the initial rapid hydrolysis is catalyzed by the F1-ATPase free from ADP-Mg inhibition, and that the ADP-Mg inhibited form gradually accumulates, causing a decrease in the rate of hydrolysis. The final, steady-state hydrolysis at a stable rate is catalyzed by the active F1-ATPase that is in a dynamic equilibrium with the ADP-Mg inhibited form. Based on the above scheme, ka-i and ki-a at 2 mM ATP were calculated to be 0.034 s−1 and 0.018 s−1 from the apparent rate constant of deceleration (ka-i + ki-a) and the ratio of steady-state activity to the initial activity (ki-a/[ka-i + ki-a]) (9). A calculated curve using these rate constants can approximate the experimental data well (Fig. 5B, solid line). Thus, the life-time of the ADP-Mg inhibited form (τi = 1/ki-a) in 2 mM ATP is 56 s, and that of the active molecule (τa = 1/ka-i) is 29 s. These life-times coincide well with τlp and τr obtained from single-molecule analysis. Next, we compared the life-times of ADP-Mg inhibition from bulk-phase experiments and the life-times of pauses at several ATP concentrations. When the ATP concentration was decreased from 20 μM to 2 μM, both the life-times τi and τlp increased dramatically (≈25 times) (Table 1). The dependency of τa on ATP concentration is also very similar to that of τr (data not shown). The transitions at this concentration range are consistent with our previous observations that the rates of ADP-Mg inhibition were ATP-concentration dependent with an apparent Kd for ATP of 4 μM (13) or 13 ± 7 μM (22). By the same procedures, life-times in the presence of 100 mM potassium phosphate (Pi) and those of ΔNC′ mutant in 100 mM Pi with or without LDAO also were obtained. It is clear that the values of τi and τlp change in parallel and are always in the same range.
Figure 5.

Kinetic analyses of ADP-Mg inhibition as derived from the ATPase activity measurements. (A) The time course of ATP hydrolysis catalyzed by wild-type F1-ATPase (black line) in Buffer A at 2 mM ATP-Mg. Dashed line, the initial rate; blue line, steady-state rate. (B) Inactivation of ATPase activity (black line) was fitted with a single exponential function (red line): const. ⋅ exp (−kapp ⋅ t), where kapp (the apparent rate constant for inhibition) = 0.054 s−1.
Table 1.
Lifetimes obtained from single-molecule analysis and ATPase activity measurements under several conditions
| Protein | ATP, μM | Lifetimes, s
|
|
|---|---|---|---|
| Single molecule pausing F1-ATPase (τlp) | ATPase ADP-Mg inhibition (τi) | ||
| Wild type | 2000 | 32 | 56 |
| 31 | 71* | ||
| 200 | 48 | 30 | |
| 20 | 77 | 59 | |
| 2 | 694 | 1429 | |
| ΔNC′ | 2000 | 167 | 143* |
| 12 | 22† | ||
Buffer contains 100 mM potassium phosphate.
Buffer contains 100 mM potassium phosphate and 0.1% LDAO.
Orientation of γ Subunit in the ADP-Mg Inhibited Form.
The γ subunit makes a 120° rotation by using a single ATP and waits at this position (ATP-waiting position) for the next ATP. At low-ATP concentrations, this stepping rotation is well observed (8). As described, the pausing caused by ADP-Mg inhibition takes place in three angular positions that are separated by 120° from each other. To compare the relative positions of the long pause and the ATP-waiting state, we recorded the stepping rotation at 20 nM ATP at first, then infused the buffer containing 2 mM ATP into the observation chamber and observed the long pauses of the same molecule. As seen in Fig. 6, the positions of ADP-Mg inhibition and the ATP-waiting positions are significantly different and separated by 37.6° ± 2.5°. The same particle started stepwise rotation again at 0°, 120°, and 240° after exchange of the buffer for 200 nM ATP.
Figure 6.

Comparison of angular positions of ATP-waiting state and ADP-Mg inhibition. (A) The stepwise rotation of F1-ATPase caused by waiting ATP was observed at 20 nM ATP. Each discrete 120° step was caused by ATP binding. (B) The pausing positions at 2 mM ATP. A reaction mixture containing 2 mM ATP was slowly introduced into the same chamber. Instead of the stepwise rotations, we could detect both continuous rotation and long pauses, as in Fig. 1. The angles of the long pauses, which are assumed to be caused by ADP-Mg inhibition, are shown. The positions of probes during the pausing period are presented at each of three angular positions.
At 200 nM ATP, both 120° steps caused by ATP-waiting and the pauses caused by the ADP-Mg inhibition were observed frequently enough for analysis (Fig. 7). Under these conditions, the ATP-waiting period is short (≈0.4 s), whereas ADP-Mg inhibition continues for a long period (≈30 min). Therefore, we can easily separate these two kinds of pauses; the result of the short ones define the ATP-waiting state, and long ones are the result of ADP-Mg inhibition. Moreover, the two paused states can be distinguished by the angle of rotation after the pause. Whereas steps between two adjacent ATP-waiting positions were 120°, the long pauses always started after ≈90° rotation.
Figure 7.

Motions of rotation probe stepping from the ATP-waiting position (A) to the next ATP waiting positions or (B) to the ADP-Mg inhibition. The ATP concentration was 200 nM. (A) The stepping motions (of which subsequent pauses were shorter than 5 s) are collected and overlayed (n = 13). (B) The stepping motions (of which subsequent pauses were longer than 2 min) are collected and overlayed (n = 13).
Discussion
Here, we showed that individual single molecules of active F1-ATPase alternate between at least three phases: continuous rotation, short pauses, and long pauses during catalysis at saturating ATP concentrations. The origin of the short pause is not yet known, but the long pause has been identified as the ADP-Mg inhibited state that has been recognized previously by bulk-phase kinetics. ADP-Mg inhibition is a common feature of the F1-ATPases and ATP synthases from various sources and, hence, likely to have functional significance. ATP hydrolysis and ATP-driven proton-pumping activity by ATP synthase are susceptible to this inhibition. Conversely, the ADP-Mg inhibition is relieved when membrane potential exists (23); ATP synthesis by ATP synthase is completely free from this inhibition (24, 25).
F1-ATPase rotates in discrete 120° steps, consistent with sequential ATP hydrolysis on the three β subunits. Recently, it was revealed that each 120° step is further divided to the 90° and 30° substeps, each taking only a fraction of a msec (15). ATP binding drives the 90° substep. The enzyme spent about 2 msec on average at the 90° position (the 90° dwell), irrespective of medium ATP concentrations. During the 90° dwell, at least two 1-msec events occur; the latter one, resetting the enzyme to start the next cycle, accompanies the 30° rotation. ADP-Mg inhibition arises at ≈83°, measured from the ATP-waiting position (Figs. 6 and 7), and therefore it is likely to be generated in a stochastic manner at the 90° dwell by some off-the-catalytic-pathway event.
Acknowledgments
We thank T. Nishizaka for actin preparation. We also thank Drs. J. Hardy and M. T. Stumpp for critical reading of the manuscript. Y.H.-H. and K.Y.H. are supported by Research Fellowships of Japan Society for the Promotion of Science for Young Scientists.
Abbreviations
- F1-ATPase
α3β3γ subcomplex of thermophilic F1-ATPase
- LDAO
lauryl dimethyl amine oxide
- ΔNC
mutant F1-ATPase with defective noncatalytic sites
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Deckers-Hebestreit G, Altendlf K. Annu Rev Microbiol. 1996;50:791–824. doi: 10.1146/annurev.micro.50.1.791. [DOI] [PubMed] [Google Scholar]
- 2.Boyer P D. Biochim Biophys Acta. 2000;1458:252–262. doi: 10.1016/s0005-2728(00)00077-3. [DOI] [PubMed] [Google Scholar]
- 3.Weber J, Senior A E. Biochim Biophys Acta. 2000;1458:300–309. doi: 10.1016/s0005-2728(00)00082-7. [DOI] [PubMed] [Google Scholar]
- 4.Cross R L. Biochem Biophys Acta. 2000;1458:270–275. doi: 10.1016/s0005-2728(00)00079-7. [DOI] [PubMed] [Google Scholar]
- 5.Abrahams J P, Leslie A G, Lutter R, Walker J E. Nature (London) 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 6.Boyer P D. Biochim Biophys Acta. 1993;1140:215–250. doi: 10.1016/0005-2728(93)90063-l. [DOI] [PubMed] [Google Scholar]
- 7.Noji H, Yasuda R, Yoshida M, Kinosita K J. Nature (London) 1997;386:299–302. doi: 10.1038/386299a0. [DOI] [PubMed] [Google Scholar]
- 8.Yasuda R, Noji H, Kinosita K J, Yoshida M. Cell. 1998;93:1117–1124. doi: 10.1016/s0092-8674(00)81456-7. [DOI] [PubMed] [Google Scholar]
- 9.Masaike T, Mitome N, Noji H, Muneyuki E, Yasuda R, Kinosita K J, Yoshida M. J Exp Biol. 2000;203:1–8. doi: 10.1242/jeb.203.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Yanisch-Perron C, Vieria J, Messing J. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 11.Kunkel T A, Bebenek K, McClary J. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 12.Matsui T, Yoshida M. Biochim Biophys Acta. 1995;1231:139–146. doi: 10.1016/0005-2728(95)00070-y. [DOI] [PubMed] [Google Scholar]
- 13.Matsui T, Muneyuki E, Honda M, Allison W S, Dou C, Yoshida M. J Biol Chem. 1997;272:8215–8221. doi: 10.1074/jbc.272.13.8215. [DOI] [PubMed] [Google Scholar]
- 14.Jault J M, Dou C, Grodsky N B, Matsui T, Yoshida M, Allison W S. J Biol Chem. 1996;271:28818–28824. doi: 10.1074/jbc.271.46.28818. [DOI] [PubMed] [Google Scholar]
- 15.Yasuda R, Noji H, Yoshida M, Kinosita K J, Itoh H. Nature (London) 2001;410:898–904. doi: 10.1038/35073513. [DOI] [PubMed] [Google Scholar]
- 16.Amano T, Hisabori T, Muneyuki E, Yoshida M. J Biol Chem. 1996;271:18128–18133. doi: 10.1074/jbc.271.30.18128. [DOI] [PubMed] [Google Scholar]
- 17.Bald D, Muneyuki E, Amano T, Kruip J, Hisabori T, Yoshida M. Eur J Biochem. 1999;262:563–568. doi: 10.1046/j.1432-1327.1999.00410.x. [DOI] [PubMed] [Google Scholar]
- 18.Kato Y, Sasayama T, Muneyuki E, Yoshida M. Biochim Biophys Acta. 1995;1231:275–281. doi: 10.1016/0005-2728(95)00087-y. [DOI] [PubMed] [Google Scholar]
- 19.Milgrom Y M, Boyer P D. Biochim Biophys Acta. 1990;1020:43–48. doi: 10.1016/0005-2728(90)90091-h. [DOI] [PubMed] [Google Scholar]
- 20.Guerrero K J, Xue Z X, Boyer P D. J Biol Chem. 1990;265:16280–16287. [PubMed] [Google Scholar]
- 21.Jault J M, Matsui T, Jault F M, Kaibara C, Muneyuki E, Yoshida M, Kagawa Y, Allison W S. Biochemistry. 1995;34:16412–16418. doi: 10.1021/bi00050a023. [DOI] [PubMed] [Google Scholar]
- 22.Muneyuki E, Makino M, Kamata H, Kagawa Y, Yoshida M, Hirata H. Biochim Biophys Acta. 1993;1144:62–68. doi: 10.1016/0005-2728(93)90031-a. [DOI] [PubMed] [Google Scholar]
- 23.Galkin M A, Vinogradov A D. FEBS Lett. 1999;448:123–126. doi: 10.1016/s0014-5793(99)00347-6. [DOI] [PubMed] [Google Scholar]
- 24.Syroeshkin A V, Vasilyeva E A, Vinogradov A D. FEBS Lett. 1995;366:29–32. doi: 10.1016/0014-5793(95)00487-t. [DOI] [PubMed] [Google Scholar]
- 25.Bald D, Amano T, Muneyuki E, Pitard B, Rigaud J L, Kruip J, Hisabori T, Yoshida M, Shibata M. J Biol Chem. 1998;273:865–870. doi: 10.1074/jbc.273.2.865. [DOI] [PubMed] [Google Scholar]