

Abstract
The pathophysiological functions of the signaling molecules matrix metalloproteinase-14 (MMP-14) and integrin αvβ3 in various types of cancer are believed to derive from their collaborative activity in promoting invasion, metastasis, and angiogenesis, as shown in vitro and in vivo. The two effectors act in concert in a cell-specific manner through the localization of pro-MMP-2 to the cell surface, where it is processed to intermediate and matured MMP-2. The matured MMP-2 product is localized to the cell surface via its binding to integrin αvβ3. The MMP-14/MMP-2/integrin αvβ3 axis thus constitutes an attractive putative target for therapeutic interventions, but the development of inhibitors that target this axis remains an unfulfilled task. To address the lack of such multitarget inhibitors, we have established a combinatorial approach that is based on flow cytometry screening of a yeast-displayed N-TIMP2 (N-terminal domain variant of tissue inhibitor of metalloproteinase-2) mutant library. On the basis of this screening, we generated protein monomers and a heterodimer that contain monovalent and bivalent binding epitopes to MMP-14 and integrin αvβ3. Among these proteins, the bi-specific heterodimer, which bound strongly to both MMP-14 and integrin αvβ3, exhibited superior ability to inhibit MMP-2 activation and displayed the highest inhibitory activity in cell-based models of a MMP-14-, MMP-2-, and integrin αvβ3-dependent glioblastoma and of endothelial cell invasiveness and endothelial capillary tube formation. These assays enabled us to show the superiority of the combined target effects of the inhibitors and to investigate separately the role each of the three signaling molecules in various malignant processes.
Keywords: protein engineering, directed evolution, proteolysis, protease inhibitor, metastasis, bi-specific proteins, enzyme inhibition, angiogenesis, metastasis
Introduction
A variety of cancers are believed to stem from the dysregulation of signaling pathways that mediate cell behavior, i.e. proliferation, survival, migration, and invasion. Among the signaling molecules that have been implicated in the etiology of different cancers are integrin αvβ3 and matrix metalloproteinase-14 (MMP-14, also known as membrane–type-1 matrix metalloproteinase). Integrin αvβ3, like all integrins, binds to extracellular matrix (ECM)2 proteins, thereby promoting cell adhesion to the ECM and activating the signaling pathways involved in the progression of the cell cycle (1) and in angiogenesis (2, 3). MMP-14 is associated with proteolysis, angiogenesis, and cell migration and invasion in the ECM, with elevated levels being correlated with malignancy in different types of cancer (4, 5). In those cancers, it is the catalytic site on the extracellular domain of MMP-14 that drives invasion by enabling MMP-14 to function as a pericellular collagenase (6) and as an activator of pro-MMP-2 (7, 8). A number of studies on a variety of human cancers have shown a correlation between the proteolytic activities of MMP-14 and activated MMP-2, on the one hand, and the extent of endothelial cell invasion, on the other; these studies have also shown a direct link between these two MMPs and pericellular degradation, leading to angiogenesis and metastasis (8–11). For many human tumors, poor prognosis has thus been correlated with the dysregulation and overexpression of integrin αvβ3 (12, 13) and MMP-14 and MMP-2 (10), indicating that this axis could constitute an important target for therapeutic intervention.
The concept for designing such a therapeutic intervention may be drawn from numerous papers demonstrating cross-talk between biological processes mediated by MMP-14, integrin αvβ3, and their ligands, particularly pathways responsible for angiogenesis (14, 15) and metastasis (16, 17). In addition, recent studies have demonstrated a functional interaction between MMP-14 and integrin αvβ3. For example, it is known that integrin αvβ3, which is highly expressed on vascular sprouts (endothelial cells) during angiogenesis and on tumor cells (e.g. breast cancer, glioblastoma, and melanoma), localizes MMP-14 at the cell migration front (18) and attracts secreted MMP-2 to the cell surface, thereby promoting cell invasiveness (19, 20). In addition, MMP-14 and integrin αvβ3 associate on primary endothelial cells and together play a role in endothelial cell migration (18). A cooperative role of MMP-14 and integrin αvβ3 in activating pro-MMP-2 has also been reported (21), as has the co-immunoprecipitation of an MMP-14/integrin αvβ3/MMP-2 complex from glioma cells (20, 22, 23). Finally, MMP-14 has been shown to participate catalytically in the maturation of the integrin αv subunit and to correlate with β3 chain proteolytic cleavage and processing, both of which lead to functional activation of integrin αvβ3, thus modulating the adhesive, migratory, and metastatic behavior of tumor cells (23, 24).
MMP-14 and integrin αvβ3 work in concert to facilitate the processing and maturation of MMP-2 (21). This maturation is initiated by activation of pro-MMP-2 into intermediate MMP-2 in a process that is facilitated by two molecules of MMP-14 and one molecule of FL-TIMP2, the full-length molecule of tissue inhibitor of metalloproteinases 2 (TIMP2). Through its N-terminal domain, FL-TIMP2 binds to the catalytic site of one MMP-14 molecule, leading to MMP-14 inhibition. Through its C-terminal domain, cell-surface localized FL-TIMP2 binds to pro-MMP-2, thereby bringing it into the proximity of a second (catalytically active) MMP-14 molecule, which processes pro-MMP-2 into the MMP-2 intermediate form (21, 25, 26). Conversion of the intermediate MMP-2 into matured MMP-2 takes place in an integrin αvβ3-dependent process, but the details of this specific maturation mechanism remain to be elucidated (21, 23). It is the latter two forms of MMP-2 (i.e. intermediate and matured) that are able to degrade ECM components and to promote invasiveness (27, 28). Importantly, although MMP-2 is a secreted protein, localization of matured MMP-2 on cancer and endothelial cell surfaces, via integrin αvβ3, was found to increase cell invasiveness and angiogenesis (19, 29).
Given the complexity and redundancy of the MMP-14, MMP-2, and integrin αvβ3 signaling networks that result from the cross-talk between these effectors, it is likely that multicomponent therapeutics capable of perturbing parallel nodes of these critical pathways that are associated with angiogenesis and metastasis would be a promising means to combat drug resistance in various cancers, including melanoma (22), glioma (20), and breast cancer (21). Indeed, such a notion has attracted considerable attention for other systems and has accelerated the development of mixture therapeutics targeted at other cross-reactive signaling networks, such as vascular endothelial growth factor–epidermal growth factor receptor inhibitors (30, 31), many of which have already been introduced into pre-clinical and clinical practice (30). Nonetheless, despite their significant clinical success, mixture therapeutics have several well known limitations, including additive on- and off-target toxicity (32, 33). Moreover, their use is limited by the requirement to provide preclinical data demonstrating that the mixture has greater than additive activity or more durable responses than those obtained with mono-therapy (34). These limitations may be overcome by exploiting the enormous potential of combinatorial approaches for engineering multispecificity into natural protein ligands (35–37).
In this paper, we demonstrate that concurrent targeting of MMP-14 and integrin αvβ3 with bi-specific inhibitors offers enhanced therapeutic benefits compared with the respective mono-treatments. We show that these bi-specific inhibitors are the first reported proteins that target all the known functions that result from cross-talk between MMP-14 and integrin αvβ3, namely cancer cell invasion, angiogenesis, and MMP-2 activation and localization to the cell surface. The bi-specific inhibitors were derived from the 127-amino acid N-terminal domain of WT TIMP-2 (N-TIMP2WT) and that of an N-TIMP2 variant, designated N-TIMP25M, which was previously designed by our group for high affinity and specificity toward MMP-14 in preference to other MMPs (38). An overriding consideration in the design of the above inhibitors was the need for specificity, because MMP family members are also found in nonmalignant cells, and some MMPs, such as MMP-8 and MMP-12, exhibit anti-tumorigenic functions (39–41). It is thus not surprising that inhibitors with narrow or single MMP specificity have greater therapeutic potential than nonspecific inhibitors (42–44). As we have shown previously, specificity toward MMP-14 in preference to other MMPs can be obtained by utilizing N-TIMP25M (38), but cell specificity can best be achieved by tailoring the inhibitors for binding specificity to both MMP-14 and its co-localized receptor integrin αvβ3.
One of the main challenges that we had to address was engineering specificity for integrin αvβ3 binding, because the Arg-Gly-Asp (RGD) tripeptide motif typical of many natural ECM ligands, including fibronectin, vitronectin, fibrinogen, and osteopontin, is also recognized by other integrins, including those containing the αv subunit and integrins α5β1 and αiibβ3 (45). To generate mutated N-TIMP2 variants with binding specificity toward integrin αvβ3, we exploited the substantial differences between the above ligands in the three-dimensional orientations of flexible solvent-exposed loops containing the RGD motif; these differences are a function of the particular amino acid residues flanking the RGD sequence and hence dictate the integrin-binding affinity and specificity of the ligands (46).
N-TIMP2 shows extensive sequence diversity both within and between species (47), as its exposed loops are highly tolerant to substitution or to incorporation of additional amino acids (38, 47–49). To achieve high target affinity and specificity, we exploited these attributes of N-TIMP2 for use as a versatile platform for the development of both bivalent single-domain and heterodimeric MMP-14/integrin αvβ3 inhibitors that were generated from affinity high-throughput screens of an N-TIMP2RGD–loop library with random amino acid changes. The engineered bi-specific proteins were expressed as soluble proteins and were shown to bind simultaneously to both MMP-14 and integrin αvβ3, with different inhibitory and antagonistic activities, both in vitro and in cells. The bi-specific monomeric and heterodimeric inhibitors displayed superior therapeutic potential to that of MMP-14 and integrin αvβ3 mono-treatments. In addition, the availability of both mono- and bi-specific proteins allowed us to dissect out the role of each target in cell invasion and angiogenesis and to design inhibitors with superior potencies for each of these two cancerous processes, separately and together.
Results
N-TIMP2RGD loop library construction and screening
An N-TIMP2RGD yeast surface display (YSD) loop library was designed in such a way that positions 51–59 at the apex of loop B–C in N-TIMP2, which naturally binds integrin α3β1, were replaced with a sequence comprising an RGD motif flanked on each side by three additional random amino acids (XXXRGDXXX) (Fig. 1A and Fig. S1). To isolate high-affinity bi-specific N-TIMP2 binders for the MMP-14 catalytic domain (MMP-14CAT, residues 112–292) (50) and integrin αvβ3, six rounds of flow-cytometry sorting were performed, the first one for high-expressing clones (expression sort, ES) and the other five (sorts 1–5) for binding to soluble integrin αvβ3 (Fig. 1, A and B). In each round of sorting (sorts 1–5), the concentration of integrin αvβ3 was reduced, and the cell population binding integrin αvβ3 was enriched (Fig. 1B). Nine different clones were identified after sort 5 (Table 1). One of the nine clones, namely clone 2, designated N-TIMP2WT, RGD (Fig. S2), was chosen for further purification as a soluble protein on the basis of its strong integrin αvβ3 binding. This clone was then purified as a free N-terminal protein (which is crucial for MMP inhibition; Fig. S3), having a His tag and a c-Myc tag, both at the C terminus of the protein.
Figure 1.

Strategy for screening the N-TIMP2RGD library. A, YSD system. N-TIMP2RGD proteins were displayed on yeast. Expression of the N-TIMP2RGD proteins was detected by targeting the c-Myc epitope tag, expressed at the C terminus of the protein, with an antibody labeled with PE, and binding to integrin αvβ3 was detected by FITC staining. B, flow cytometry analysis of the sorted library tested against 50 nm integrin αvβ3. In the expression sort (ES), the entire N-TIMP2RGD protein–expressing cell population was sorted (black gate). In sort 1 (S1), the yeast cell population binding to integrin αvβ3 was collected (black gate). In sort 2 (S2) to sort 5 (S5), the integrin αvβ3 high-affinity population was collected (black gate). The population binding integrin αvβ3 was enriched as the sorting progressed from S2 to S5. C, N-TIMP2 protein variants. N-TIMP2WT, RGD is the bi-specific protein isolated after sort 5. N-TIMP25M, RGD is N-TIMP2WT, RGD with five mutations (5M); this mutant showed improved binding to MMP-14 (38). N-TIMP2HD is a heterodimer composed of N-TIMP25M and N-TIMP2WT, RGD proteins conjugated via a peptide linker. N-TIMP2WT and N-TIMP25M are mono-specific binders for MMP-14. Ala–N-TIMP2WT, RGD has a reduced affinity toward MMP-14 and a high affinity toward integrin αvβ3.
N-TIMP2 variants exhibit different affinities toward MMP-14 and integrin αvβ3
To optimize the inhibitory activity of the N-TIMP2 variants and to dissect out the contribution of each target receptor to tumor progression, we generated a pool of N-TIMP2 variants with different affinities for each target. We have recently shown that insertion of five point mutations, namely I35M, N38D, S68N, V71G, and H97R, into N-TIMP2 (generating N-TIMP25M) increases its affinity toward MMP-14 by 890-fold (38). In this study, we therefore incorporated these mutations into the bi-specific monomer, N-TIMP2WT, RGD, thereby generating the N-TIMP25M, RGD variant (Fig. 1C). We also engineered Ala–N-TIMP2WT, RGD, a protein variant having high affinity toward integrin αvβ3 and reduced inhibition activity toward MMP-14, by inserting an alanine residue before the cysteine residue at position 1 (Fig. 1C). We note that alanine insertion in TIMP2 was previously shown to abolish binding to MMP and its inhibition (51). N-TIMP2WT and N-TIMP25M served as mono-specific binders with different affinities to MMP-14 but no affinity to integrin αvβ3. In addition, an N-TIMP2 heterodimer (N-TIMP2HD), composed of N-TIMP25M at the N terminus fused via a flexible peptidic linker to N-TIMP2WT, RGD at the C terminus, was designed so as to reduce the mutual effects of the N-TIMP25M and N-TIMP2WT, RGD mutations on each other's activity.
The bi-specific monomer N-TIMP2WT, RGD was shown to bind both integrin αvβ3 and MMP-14CAT, with KD values of 64.8 nm (Fig. 2A) and 838 nm (Fig. 2C), respectively (Table 1). Moreover, N-TIMP2WT, RGD was capable of binding both receptors simultaneously, as determined by a co-injection surface plasmon resonance (SPR) assay (Fig. 2E). We have previously shown that the insertion of the five mutations into N-TIMP25M improved the affinity to MMP-14CAT compared with N-TIMP2WT (38). As expected, binding to integrin αvβ3 was not detected for the TIMP2WT and N-TIMP25M mono-specific MMP-14 variants (Fig. S4A). In agreement with these results, the insertion of the five mutations into N-TIMP25M, RGD significantly increased the affinity toward MMP-14CAT by ∼400-fold (KD of 2 nm), although the affinity toward integrin αvβ3 was decreased (KD of 531 nm) but not completely abolished (Table 1 and Fig. S4, C and D). The results of a comparison of the KD values obtained for the two monomeric bi-specific binders, N-TIMP2WT, RGD and N-TIMP25M, RGD, for both integrin αvβ3 and MMP-14CAT implied that by increasing the affinity to one target, the affinity to the other is reduced. However, by separating the two binding domains of each monomer in the engineered bi-specific heterodimer (N-TIMP2HD), we were able to generate a protein that had high affinity to both integrin αvβ3 and MMP-14CAT (KD of 130 and 0.78 nm, respectively, see Table 1 and Fig. 2, B and D). Similarly to N-TIMP2WT, RGD, N-TIMP2HD was able to bind both receptors simultaneously, as determined by a co-injection SPR assay (Fig. 2F).
Figure 2.

SPR sensorgrams and the inhibitory activity of the N-TIMP2WT, RGD and N-TIMP2HD variants. N-TIMP2WT, RGD (A) and N-TIMP2HD (B) binding to integrin αvβ3 is shown. N-TIMP2WT, RGD (C) and N-TIMP2HD (D) binding to MMP-14CAT is shown. Dual binding of N-TIMP2WT, RGD (E) and N-TIMP2HD (F) to integrin αvβ3 and MMP-14CAT is shown. E and F, variants were allowed to flow over integrin αvβ3 immobilized on the chip, followed by MMP-14CAT (blue) or buffer (control 1, red) injection at 300 s. There was an increase in response units (RU) for the variants after MMP-14CAT injection, which indicates that these variants bind simultaneously to both receptors. In control 2 (green), a buffer was allowed to flow over the integrin αvβ3 immobilized chip, followed by MMP-14 injection at 300 s. In control 3 (yellow), a buffer was allowed to flow over the integrin αvβ3 immobilized chip throughout the entire experiment. Substrate degradation velocity at different concentrations of N-TIMP2WT, RGD (0–100 nm) (G) and N-TIMP2HD (0–0.2 nm) (H) were fitted to Morrison's equation (Equation 2) to obtain Ki values.
Table 1.
Binding kinetics of N-TIMP2 variants
ND means binding not detected at 1 μm concentration; NA means not available; NT means not tested.
| N-TIMP2 variant | MMP-14 kinetics |
Integrin αvβ3 kinetics |
|||||
|---|---|---|---|---|---|---|---|
| Ki | KD | Kon | Koff | KD | Kon | Koff | |
| nm | nm | 104 ms−1 | 10−5 s−1 | nm | 104 ms−1 | 10−3 s−1 | |
| N-TIMP2WT | 2.01 ± 0.2 | 4.68 ± 0.01 | 11.4 | 53.6 | ND | ||
| N-TIMP25M | 0.011 ± 0.003 | 0.603 ± 0.001a | 6.2 | 3.74 | ND | ||
| N-TIMP2WT, RGD | 35.76 ± 2.28 | 838 ± 12.7 | NA | NA | 64.8 ± 0.46 | 1.71 ± 0.01 | 1.11 ± 0.01 |
| Ala–N-TIMP2WT, RGD | 132.6 ± 8.55 | NT | 197 ± 1.1 | 4.71 ± 0.08 | 9.28 ± 0.35 | ||
| N-TIMP25M, RGD | b | 2.08 ± 0.02 | 8.39 ± 0.21 | 17.4 ± 0.13 | 561 ± 276 | NA | NA |
| N-TIMP2HD | 0.077 ± 0.023 | 0.78 ± 0.01 | 7.6 ± 0.12 | 5.97 ± 0.09 | 130 ± 21.1 | NA | NA |
a Data were taken from Arkadash et al. (38).
b Ki values could not be determined accurately. Complete inhibition was detected at 0.8 nm.
In agreement with the SPR binding results, the ability of the N-TIMP2 variants to inhibit MMP-14CAT catalytic activity, as reflected by the Ki values, was as follows (best to worst): N-TIMP25M, N-TIMP2HD, N-TIMP2WT, N-TIMP2WT, RGD, and Ala–N-TIMP2WT, RGD (Table 1). Ala–N-TIMP2WT, RGD showed low inhibitory activity toward MMP-14CAT, although it retained its ability to bind to integrin αvβ3 (Table 1 and Fig. S4B).
In summary, three important points emerge from these results. 1) The three engineered bi-specific proteins bind to both targets but with different affinities: N-TIMP2WT, RGD binds strongly to integrin αvβ3 but weakly to MMP-14; N-TIMP25M, RGD binds strongly to MMP-14 but weakly to integrin αvβ3; and N-TIMP2HD binds strongly to both receptors. 2) N-TIMP2WT, RGD and N-TIMP2HD can bind simultaneously to both receptors. 3) There was good correlation between the KD (SPR) and Ki (inhibition of MMP-14 catalytic activity) potencies.
N-TIMP2 variants inhibit MMP-2 activation in the U87MG cell line
A major outcome of the cross-talk between MMP-14 and integrin αvβ3 is the processing/maturation of pro-MMP-2 to intermediate and subsequently fully matured MMP-2 forms. The MMP-2 processing/maturation mechanism takes place in two consecutive steps. In the first step, processing of the 68-kDa pro-MMP-2 by MMP-14 into the 64-kDa MMP-2 intermediate form is followed by conversion of the MMP-2 intermediate form to the matured 62-kDa MMP-2 form. In the second, more rapid, step, it is the interaction of the intermediate MMP-2 with integrin αvβ3 that facilitates, via an as-yet-unknown mechanism, the autocatalytic processing of the intermediate MMP-2 to the fully matured MMP-2 (21). Because MMP-2, MMP-14, and integrin αvβ3 work in concert, it is not a trivial task to separate out the specific and overall contributions of the intermediate and matured forms of MMP-2 to cancer cell invasion and angiogenesis (52). To address this problem, we evaluated the ability of the mono-specific (inhibiting MMP-14 or integrin αvβ3) versus the bi-specific (inhibiting both proteins) N-TIMP2 variants to inhibit the activation (maturation process) of MMP-2. For this purpose, we used a gelatin zymography assay, because all MMP-2 forms can cleave gelatin (53), but only the intermediate and the matured species can degrade the ECM components, such as collagen, gelatin, and fibronectin (53, 54), and promote cell invasion, with the matured form being more potent than the intermediate form (55).We utilized two controls for this assay as follows: (i) recombinant FL-TIMP2, a protein that is endogenously expressed in U87MG glioblastoma cells and is known to induce the formation of the matured MMP-2 and cell invasion (56), and (ii) cyclic RGD (cRGD), a known inhibitor of integrin αvβ3 (57). In this assay, U87MG cells (expressing both MMP-14 and integrin αvβ3, Fig. S5) were treated with 100 nm of each protein variant for 48 h. The culture supernatant, which contained the secreted pro/intermediate/matured MMP-2 forms, was loaded onto a gelatin gel, and the different MMP-2 forms were eluted as white bands on the gel. In addition, different concentrations of FL-TIMP2 and cRGD were tested for their pro-MMP-2 activation function. All the strong inhibitors of MMP-14, namely N-TIMP2WT, FL-TIMP2, N-TIMP25M, N-TIMP25M, RGD, and N-TIMP2HD, significantly inhibited the processing of endogenous pro-MMP-2 to the intermediate form (Fig. 3, A and B). However, FL-TIMP2, when added at a lower concentration (10 nm), activated processing of the pro-MMP-2 to the active forms (Fig. S6). Importantly, exposure to Ala–N-TIMP2WT, RGD, which binds strongly to integrin αvβ3 and possesses a lower MMP-14 inhibition potency, significantly increased the amount of the intermediate MMP-2 form by inhibiting its processing to the matured form; the same was true for cRGD at a concentration of 1 μm (Fig. S7). Thus, whereas Ala–N-TIMP2WT, RGD did not reduce the total amount of the ECM-reactive MMP-2 forms (i.e. both intermediate and matured forms that are able to degrade the ECM component) relative to the untreated control, it did increase the ratio between the intermediate (less ECM reactive) product and the matured product. Although N-TIMP2WT, RGD did not significantly inhibit the processing of the intermediate MMP-2, it did exhibit the same tendency to inhibit intermediate MMP-2 processing as the Ala–N-TIMP2WT, RGD variant, with the ratios between the intermediate and matured forms of MMP-2 being 2.1 and 2, for N-TIMP2WT, RGD and Ala–N-TIMP2WT, RGD, respectively, compared with 1.1 for the untreated control. Because the intermediate MMP-2 form is less ECM-reactive than the fully matured form, it was to be expected that the increase in the intermediate/matured MMP-2 ratio would be reflected in reduced ECM degradation and decreased invasion, as will indeed be shown in this paper. Among all the variants, N-TIMP2HD exhibited the strongest inhibition effect on MMP-2 activation, giving the largest decrease in both the intermediate and the matured forms relative to the other variants.
Figure 3.

Inhibition of pro-MMP-2 activation by N-TIMP2 variants. A, gelatin zymography gel results. The U87MG supernatant was loaded on gelatin zymography gel. Lane 1 represents untreated U87MG cells, and lanes 2–8, respectively, represent treatment with 100 nm of the following N-TIMP2 variants: N-TIMP2WT; FL-TIMP2; N-TIMP25M; N-TIMP2WT, RGD; N-TIMP25M, RGD; Ala–N-TIMP2WT, RGD; and N-TIMP2HD. Lane 9 represents treatment with cRGD (100 nm). B, quantification of the percentage of each MMP-2 form, i.e. pro/intermediate/matured (band intensity of each MMP-2 state, i.e. pro-MMP-2 (68 kDa), intermediate MMP-2 (64 kDa), and matured MMP-2 (62 kDa), was divided by the total band intensity of all the MMP-2 forms). Error bars represent S.E. Pro-MMP-2 processing and the total amount of the catalytically active MMP-2 were significantly inhibited by N-TIMP2WT (p < 0.05), FL-TIMP2 (p < 0.01), and N-TIMP5M, N-TIMP25M, RGD, and N-TIMP2HD (p < 0.001). Processing of the intermediate form was significantly inhibited by Ala–N-TIMP2WT, RGD (p < 0.05). The amount of the matured form of MMP-2 was significantly reduced by N-TIMP25M and N-TIMP25M, RGD (p < 0.05), N-TIMP2HD (p < 0.01), and N-TIMP2WT (p < 0.001). Statistical analysis was performed by Student's t test compared with the untreated control; n = 3.
MMP-2 docking on the U87MG cell surface is impaired by an integrin αvβ3 inhibitor
The catalytic activity of MMP-2 localized on the cell surface via integrin αvβ3 mediation is known to promote cell invasiveness and angiogenesis (19, 23, 29). We therefore tested the ability of our inhibitors to prevent the interaction between matured MMP-2 and integrin αvβ3. Because the SPR results indicated that our bi-specific proteins can simultaneously engage MMP-14 and integrin αvβ3, Ala–N-TIMP2WT, RGD (which binds strongly to integrin αvβ3 and has low MMP-14 inhibition activity) was selected as the variant to compete with MMP-2 for binding to membrane-anchored integrin αvβ3. In keeping with our expectations that the competitive binding would inhibit MMP-2–induced ECM degradation (19), flow cytometry showed that there was indeed a reduction of recombinant matured MMP-2 levels on the surface of the U87MG cells 2 h after treatment with 2 μm Ala–N-TIMP2WT, RGD (67% reduction, Fig. 4A). To confirm that the matured form of MMP-2 was indeed removed by Ala–N-TIMP2WT, RGD, gelatin zymography was performed on the U87MG cell lysate, which contained the cell-surface–bound MMP-2; it was indeed found that only matured MMP-2 was removed from the cell surface (Fig. 4, B–D). To determine whether Ala–N-TIMP2WT, RGD prevents matured MMP-2 binding specifically to integrin αvβ3, a solid-phase assay was performed. Purified recombinant integrin αvβ3 was immobilized on a 96-well ELISA plate, followed by overnight incubation with either Ala–N-TIMP2WT, RGD or cRGD (2 μm) and with a U87MG supernatant obtained after 48 h of growth, i.e. containing all the MMP-2 forms. Both Ala–N-TIMP2WT, RGD and cRGD reduced the docking levels of pro-MMP-2 on integrin αvβ3 to a similar extent (∼57%, Fig. 4, E and F), whereas Ala–N-TIMP2WT, RGD significantly inhibited the interaction of matured MMP-2 with integrin αvβ3 compared with cRGD treatment (66 and 42%, respectively, Fig. 4, E and G). The differences between the assay results can be explained by the fact that in the flow cytometry (Fig. 4A) recombinant matured MMP-2 was used, whereas in the zymography assays (Fig. 4, B–G) all the MMP-2 forms secreted from U87MG cells were present; among all these forms, matured MMP-2 constitutes only <5% (Fig. 3). Because the matured form binds integrin αvβ3, greater inhibition of MMP-2 cell-surface localization was observed in Fig. 4A compared with Fig. 4, B–G. In addition, Fig. 4, B–D, presents an assay performed on a U87MG cell lysate, which probably contains other binding receptors for MMP-2, whereas Fig. 4, E–G, shows an assay performed solely on the immobilized recombinant integrin αvβ3 receptor. Pro-MMP-2 can bind to other receptors on the cell surface, such as MMP-14 and lipoprotein receptor-related protein-2 (58), and therefore might not be affected by the binding of Ala–N-TIMP2WT, RGD to integrin αvβ3. For this reason, only a small reduction was observed in Fig. 4, C and D (several integrins and MMP-14 being present), versus Fig. 4, F and G (integrin αvβ3 alone). Moreover, as mentioned previously, it is known that the form of MMP-2 that binds integrin αvβ3 is the mature form and not the pro-MMP-2 form. In Fig. 4, E–G, only integrin αvβ3 was present as a receptor in the system. Thus, there were no alternative receptors with which pro-MMP-2 could interact. Furthermore, whereas pro-MMP-2 was the dominant form of secreted MMP-2 (∼95%, Fig. 3), its binding to immobilized integrin αvβ3 was slightly higher than that of the mature form, which comprised less than 5% of the secreted MMP-2 forms. The above data thus attest to the preference in binding affinity of integrin αvβ3 for matured MMP-2 over pro-MMP-2.
Figure 4.

The Ala–N-TIMP2WT, RGD variant disrupts matured MMP-2 cell-surface localization by binding to integrin αvβ3. A, Ala–N-TIMP2WT, RGD removes matured MMP-2 from the U87MG cell surface as detected by flow cytometry analysis. A specific antibody against MMP-2 (clone CA-4001, Abcam) was utilized to detect MMP-2 on the U87MG cell surface. B, cell lysate gelatin zymography gel of untreated cells (left) and 2 μm treatment of Ala–N-TIMP2WT, RGD (Ala, right). C, Ala–N-TIMP2WT, RGD does not affect pro-MMP-2 localization on the cell surface. D, Ala–N-TIMP2WT, RGD removes matured MMP-2 from the cell surface. E, integrin αvβ3–bound MMP-2 gelatin zymography gel of untreated cells and 2 μm treatment of Ala–N-TIMP2WT, RGD and cRGD. F and G, Ala–N-TIMP2WT, RGD and cRGD interfere with binding of pro-MMP-2 (F) and matured MMP-2 (G) to integrin αvβ3. The results were normalized to untreated cells. Error bars represent S.D. Statistical analysis was performed by Student's t test compared with the untreated control; ***, p < 0.0001, n = 3.
N-TIMP2 variants inhibit the invasion of U87MG cells
Because the invasiveness of cancer cells is influenced by the proteolytic activity of MMP-14 and by integrin αvβ3–mediated cell migration, the ability of the N-TIMP2 variants to inhibit the invasion of U87MG cells was evaluated in a Boyden chamber Matrigel® invasion assay (Fig. 5). In this assay, only the heterodimer, N-TIMP2HD, and the mixture of N-TIMP25M and Ala–N-TIMP2WT, RGD (Fig. 5, H, J and K) significantly inhibited the invasion of U87MG cells compared with untreated cells (Fig. 5, A and K), with the bi-specific N-TIMP2HD heterodimer exhibiting the strongest inhibition effect (33% inhibition, Fig. 5, H and K). When U87MG cells were treated with a mixture of N-TIMP25M and Ala–N-TIMP2WT, RGD, high inhibition activity was observed (26% inhibition, Fig. 5, I and K), although this inhibition activity was inferior to that of N-TIMP2HD, as shown in Fig. 5. This superior inhibitory activity of N-TIMP2HD over the mixture of N-TIMP25M and Ala–N-TIMP2WT, RGD was statistically significant when the comparison was performed with untreated cells. However, when the two treatments were compared with one another, no statistically significant difference was observed (p = 0.1089). Compared with Ala–N-TIMP2WT, RGD (Fig. 5G) and N-TIMP25M (Fig. 5D), each alone, N-TIMP2HD exhibited a significantly stronger inhibition effect (Fig. 5K), a finding that implies a synergistic inhibition effect between N-TIMP25M and Ala–N-TIMP2WT, RGD. This synergistic inhibition was also observed for the TIME cell line (Fig. 7, D, G, H, and J).
Figure 5.
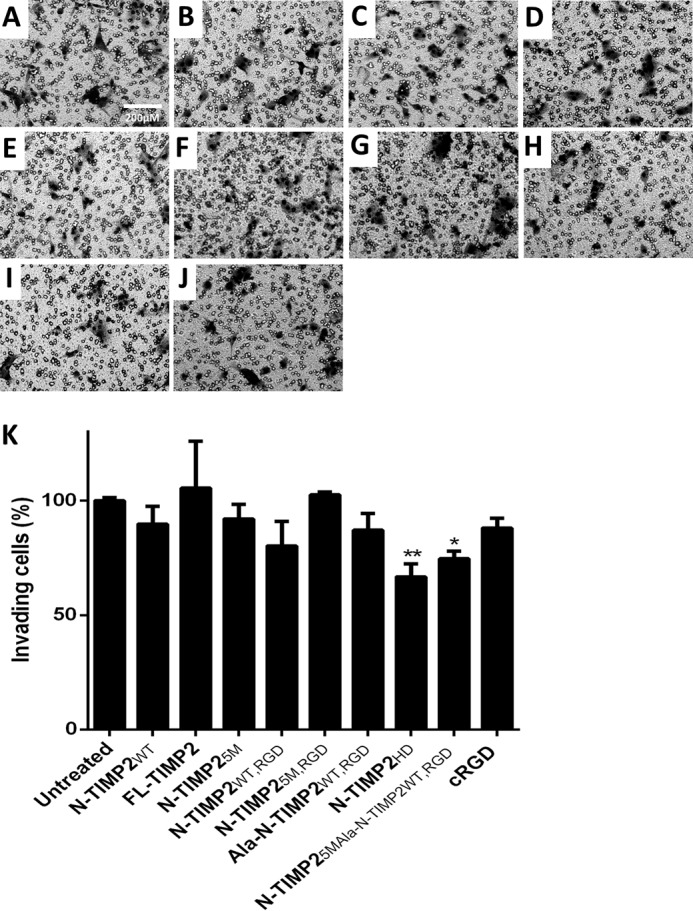
Inhibition of invasion of U87MG cells by N-TIMP2 variants. A, untreated cells. Cells were treated with the following: N-TIMP2WT (B); FL-TIMP2 (C); N-TIMP25M (D); N-TIMP2WT, RGD (E); N-TIMP25M, RGD (F); Ala–N-TIMP2WT, RGD (G); N-TIMP2HD (H); N-TIMP25M and Ala–N-TIMP2WT, RGD (I), and cRGD (J). K, quantification of invasive cells normalized to untreated cells. Error bars represent S.D. One-way ANOVA with Dunnett's multiple comparison with the untreated control was utilized for statistical analysis; *, p < 0.05; **, p < 0.005, n = 3. A comparison between treatment with N-TIMP2HD versus all other treatments showed the statistically significant superior inhibition ability of N-TIMP2HD when compared with N-TIMP2WT, FL-TIMP2, and Ala–N-TIMP2WT, RGD (p < 0.05), N-TIMP25M and cRGD (p < 0.005), and N-TIMP25M, RGD (p < 0.001).
Figure 7.
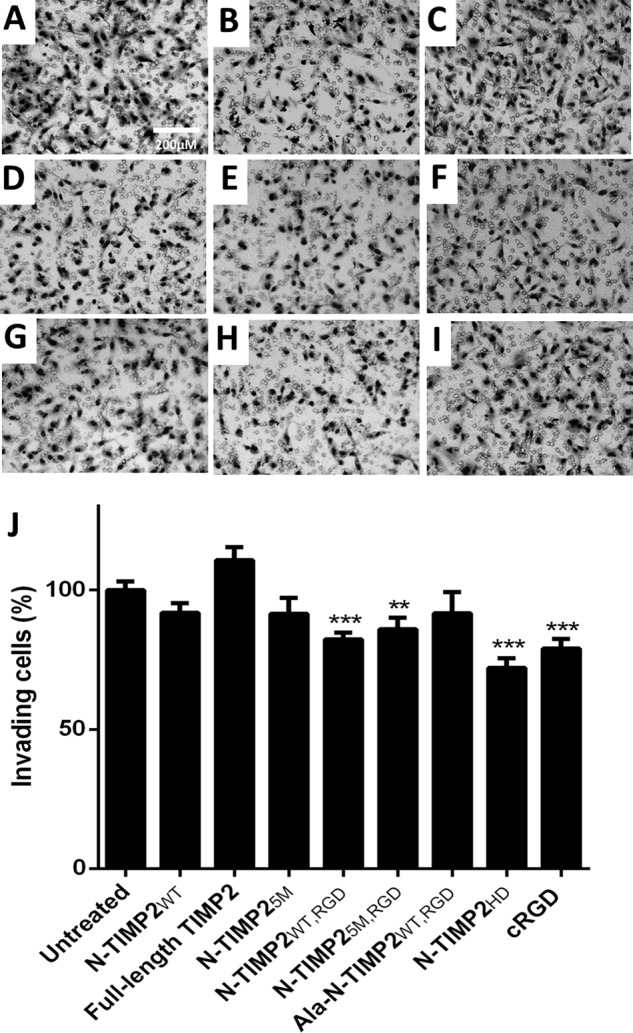
Inhibition of invasion of TIME cells by N-TIMP2 variants. A, untreated cells. B, N-TIMP2WT. C, FL-TIMP2. D, N-TIMP25M. E, N-TIMP2WT, RGD. F, N-TIMP25M, RGD. G, Ala–N-TIMP2WT, RGD. H, N-TIMP2HD. I, cRGD. J, quantification of invasive cells normalized to untreated cells. Error bars represent S.D. One-way ANOVA with Dunnett's multiple comparison with the untreated control was utilized for statistical analysis; **, p < 0.005; ***, p < 0.001, n = 3. A comparison between treatment with N-TIMP2HD versus all other treatments showed the statistically significant superior inhibition of N-TIMP2HD versus N-TIMP2WT, RGD, N-TIMP25M, RGD, and Ala–N-TIMP2WT, RGD (p < 0.05); N-TIMP2WT and N-TIMP25M (p < 0.005); and FL-TIMP2 (p < 0.001).
N-TIMP2 variants inhibit in vitro angiogenesis in the TIME cell line
Endothelial cells form capillary-like structures when embedded in a Matrigel® matrix. This process, which simulates angiogenesis, involves endothelial cell migration, proliferation, and survival (59). Therefore, N-TIMP2 variants were evaluated for their ability to inhibit tube formation of TIME cells. Although FL-TIMP2 exhibited pro-angiogenic activity (Fig. 6, C, I, and J) and N-TIMP25M did not demonstrate any effect on angiogenesis (Fig. 6, D, I, and J) as compared with untreated cells (Fig. 6A), the other N-TIMP2 variants showed significant inhibition ability (Fig. 6, B and D–H). Compared with N-TIMP2WT, which exhibits good anti-angiogenic activity (61% reduction in of the number of tube segments (Fig. 6, B and I) and 22% reduction in tube total length (Fig. 6J)), the strong integrin αvβ3 binders N-TIMP2WT, RGD (82 and 35%, Fig. 6E), Ala–N-TIMP2WT, RGD (92 and 56%, Fig. 6G), and N-TIMP2HD (79 and 41%, Fig. 6H), exhibited a more pronounced inhibition effect. Moreover, whereas both N-TIMP2RGD variants and N-TIMP2WT did affect tube formation, at least in one parameter, i.e. tube total length or number of tube segments, cRGD (2 and 50 μm treatments) had no effect on tube formation (data not shown). In contrast to the strong MMP-14 inhibitor N-TIMP25M, N-TIMP2WT, although having lower inhibition activity, exhibited strong inhibition of tube formation in TIME cells, a function that may not be related to MMP-14 inhibition but rather to its ability to activate integrin α3β1, previously shown to lead to inhibition of angiogenesis (60). Because specificity to integrin αvβ3 compared with other integrins was not tested, it should be noted that inhibition of tube formation might also be mediated by other RGD-binding integrins or integrin α3β1. Notably, the strong inhibition of tube formation seen for the strong integrin αvβ3 binders (i.e. N-TIMP2WT, RGD, Ala–N-TIMP2WT, RGD, and N-TIMP2HD) indicates that integrin αvβ3 is a more important contributor than MMP-14 to tube formation. Furthermore, the agonistic effect of FL-TIMP2 may be due to pro-MMP-2 activation in TIME cells by FL-TIMP2 at a concentration of 2 μm, an effect previously shown by others (25, 56).
Figure 6.

Inhibition of tube formation by N-TIMP2 variants. A, untreated cells. B, N-TIMP2WT. C, FL-TIMP2. D, N-TIMP25M. E, N-TIMP2WT, RGD. F, N-TIMP25M, RGD. G, Ala–N-TIMP2WT, RGD. H, N-TIMP2HD. I, quantification of the number of tube segments normalized to untreated cells. J, quantification of tube total length normalized to untreated cells. Error bars represent S.D. Statistical analysis was performed by Student's t test compared with the untreated control; *, p < 0.05; ***, p < 0.0001, n = 6.
In angiogenesis, the invasion of endothelial cells is a crucial step that is mediated by both MMP-14 and integrin αvβ3. Thus, the ability of the N-TIMP2 variants to inhibit this process was evaluated in a Boyden chamber Matrigel® invasion assay (Fig. 7). Treatment with cRGD (Fig. 7I) significantly inhibited invasiveness in TIME cells, with 21% inhibition (Fig. 7J). Although none of the mono-specific variants significantly inhibited the invasion process, all the bi-specific variants, N-TIMP2WT, RGD (Fig. 7E), N-TIMP25M, RGD (Fig. 7E) and N-TIMP2HD (Fig. 7I), significantly inhibited this process, with 18, 14, and 28% of inhibition, respectively (Fig. 7J). Notably, in the invasion assay N-TIMP2HD proved to be a superior inhibitor to the bi-specific monomers (i.e. N-TIMP2WT, RGD and N-TIMP25M, RGD, p < 0.05) and to all the mono-specific proteins (i.e. N-TIMP2WT, N-TIMP25M, and Ala–N-TIMP2WT, RGD, p < 0.05).
N-TIMP2HD inhibits U87MG xenograft tumor growth in mice
Nude mice bearing subcutaneous U87MG xenografts were injected intra-tumor with N-TIMP2HD. The untreated control group was injected intra-tumor with PBS. The growth rate of the tumors was slower in the N-TIMP2HD–treated group, thereby demonstrating the anti-tumor activity of the bi-specific protein in an in vivo model (Fig. 8).
Figure 8.

N-TIMP2HD inhibits U87MG xenograft growth in mice. Tumor size in mice treated with 500 nm N-TIMP2HD and in untreated (PBS) mice was monitored over 25 days. Measurements from days 4, 11, and 25 are shown. Each group contained five mice. At day 18, one mouse from the N-TIMP2HD-treated group died during anesthesia. Error bars represent S.E. Statistical analysis was performed by Student's t test compared with the PBS-treated control; *, p < 0.05; **, p < 0.001.
Discussion
A potential risk in the therapeutic targeting of MMPs (and other enzymatic multifamilies) is that these proteins do not work in isolation but as part of complex enzymatic cascades, in which each MMP may cross-activate other MMPs and/or other proteins, which may compensate for their loss of function (61). An excellent example of such cross-reactivity is that of MMP-2 activation by MMP-14 (7, 21), a function that is facilitated by interaction of the former with integrin αvβ3 (21, 62, 63). Putative pathobiological roles have been ascribed to the MMP-14/MMP-2/integrin αvβ3 axis for a diverse array of cancers (20–22, 62), and this axis thus represents an attractive target for the development of novel cancer therapeutics.
We therefore evaluated the therapeutic potential of targeting this axis with novel bi-specific inhibitors acting against MMP-14 and integrin αvβ3. In parallel, studying these bi-specific inhibitors and their mono-specific counterparts allowed us to establish the roles of each target protein in important cancer pathways, such as MMP-2 activation, cancer, and endothelial cell invasion and tube formation.
From a practical point of view, the strength of dual-specificity targeting in anti-cancer therapeutics lies in its potential to abrogate the unwanted side effects of potent single-targeted MMP-14 or integrin αvβ3 inhibitors; such side effects are due to the roles that different MMPs and integrins play in homeostasis and other important normal biological functions, such as wound healing, cell adhesion, and cell differentiation (40, 64). It is likely that a dual-specific molecule would increase selectivity toward MMP-14 in preference to other MMPs via increased effective local concentrations due to the association of MMP-14 with integrin αvβ3. Moreover, a bi-specific inhibitor exhibiting synergistic effects only in cancerous tissues in which both MMP-14 and integrin αvβ3 are overexpressed and cross-react would contribute to target cell specificity. This notion is supported by the findings of Deryugina et al. (21) that MMP-14 and integrin αvβ3 are localized in close proximity on the cell surface, and their cross-reactivity and synergistic function strongly promote cancer cell invasion and maturation of pro-MMP-2 to active forms.
In keeping with the findings of Deryugina et al. (21), we showed by SPR that the bi-specific N-TIMP2 variants could simultaneously bind to MMP-14 and integrin αvβ3. We also showed that our wide range N-TIMP2 variants have different affinities and activities toward the two receptors (Table 1 and Fig. 2). A comparison of the inhibition of the two receptors separately and simultaneously aided us in defining the importance of the cross-talk between MMP-14 and integrin αvβ3 and in determining which of the two effectors plays the dominant role in a particular malignant process, namely MMP-2 activation, endothelial tube formation, and cancer cell invasion.
The superior inhibition of cancer and endothelial cell invasion by the heterodimeric variant N-TIMP2HD (versus the mono-specific and other bi-specific monomers) may be attributed to its ability to bind both receptors with the highest affinity relative to the other engineered proteins. In N-TIMP2HD (unlike in the other bi-specific N-TIMP2 monomers), the MMP-14 and integrin αvβ3 epitopes are relatively distant from one another, an arrangement that we believe allows optimal interaction with their respective targets. Indeed, the monomeric N-TIMP2WT, RGD, which was less potent than N-TIMP2HD in inhibiting cell invasion, showed decreased ability to simultaneously engage MMP-14 and integrin αvβ3 compared with N-TIMP2HD (Fig. 2, E and F). In addition, the bi-specific N-TIMP2HD was better than its mono-specific counterpart N-TIMP25M (solely a MMP-14 binder) in its ability to inhibit cellular MMP-2 activation and cell invasion (Figs. 3, 5, and 7), despite having MMP-14 binding and inhibition potencies similar to those of N-TIMP25M (Table 1). This finding is in agreement with the major role of integrin αvβ3 in MMP-2 activation, cell-surface localization, and cell invasion. The finding that N-TIMP2HD was also a better inhibitor than Ala–N-TIMP2WT, RGD (a strong integrin binder having reduced MMP-14 inhibition activity) illustrates the distinct contribution of MMP-14 to inhibiting MMP-2 activation and cell invasion. N-TIMP2HD was also superior to N-TIMP25M and comparable with Ala–N-TIMP2WT, RGD in its ability to inhibit tube formation; this finding indicates a more dominant role for integrin αvβ3 than MMP-14 in capillary tube formation. However, as described previously by Stratman et al. (65), in cellular three-dimensional and in vivo angiogenesis models, MMP-14 may also play a significant role in angiogenesis. Thus, it is possible that our engineered bi-specific variants could exhibit a superior effect to the mono-specific integrin αvβ3 binder in the inhibition of angiogenesis in vivo.
Compared with the bi-specific monomeric N-TIMP25M, RGD, Ala–N-TIMP2WT, RGD exhibited inferior ability to inhibit MMP-2 activation, and N-TIMP25M exhibited reduced ability to inhibit the intermediate MMP-2 processing to the matured form (Fig. 3), despite their higher binding to integrin αvβ3 and MMP-14, respectively. This finding again highlights the utility of integrin αvβ3 and MMP-14 dual targeting for superior inhibition of MMP-2 activation/processing. Notably, N-TIMP2HD exhibited better inhibition activity than N-TIMP25M, RGD in both MMP-2 activation/processing and biological assays, which supports our premise that distancing the mutated counterparts would result in better biochemical and biological inhibitory effects.
In contrast to N-TIMP2HD, FL-TIMP2 exhibited agonistic activity in the endothelial tube formation assay (Fig. 6), which is probably due to the interaction of its C-terminal domain with the hemopexin of pro-MMP-2, thereby bringing pro-MMP-2 to the cell surface where it is activated by MMP-14 (26). At low concentrations, FL-TIMP2 serves as an MMP-2 activator, although at high concentrations it acts as an inhibitor due to its interaction with and inhibition of most MMP-14 receptors (Fig. S6) (25, 27, 56). Notably, the C-terminal domain is absent in the engineered N-TIMP2 variants, which does not allow them to activate MMP-2 and to act as agonists (66).
The difference between the zymography results (Fig. 3), in which FL-TIMP2 inhibited MMP-2 processing, and its agonistic activity in the tube formation assay may result from the different incubation times for these experiments (48 h for zymography and 18–20 h for the other assays). In the 48-h incubation, significant amounts of endogenous FL-TIMP2 could be produced, and the full-length protein could thus act as an inhibitor for MMP-14–induced MMP-2 activation (56, 67, 68). Shorter incubation times might result in lower FL-TIMP2 production, and the protein would thus act as an activator, as shown previously (56, 67, 68). Because FL-TIMP2 is endogenously expressed by cancer cells, our inhibitors can compete with it in binding to MMP-14, a process that would (i) inhibit pro-cancerous activity resulting from FL-TIMP2 activity at low concentrations, and (ii) occupy MMP-14 receptors, thereby lowering the concentrations at which FL-TIMP2 acts as an inhibitor. These actions, together with the direct inhibition of MMP-14 and integrin αvβ3, may provide additional benefits for our engineered variants in the inhibition of endothelial tube formation and cell invasion.
The influence of our N-TIMP2 variants on MMP-2 is perhaps best reflected in our findings that Ala–N-TIMP2WT, RGD inhibited MMP-2 localization on the cell surface (Fig. 4). It is well known that matured MMP-2 is localized on the cell surface via integrin αvβ3 binding and that this docking increases cell invasiveness and angiogenesis (19, 29, 69). For technical reasons, we were not able to show that N-TIMP2WT, RGD and N-TIMP2HD have the same MMP-2 removal activity as Ala–N-TIMP2WT, RGD. Nonetheless, it seems reasonable to extrapolate the inhibition of MMP-2 localization on the cell surface by Ala–N-TIMP2WT, RGD to the bi-specific variants N-TIMP2HD and N-TIMP2WT, RGD, because these two variants are similar to Ala–N-TIMP2WT, RGD in terms of integrin αvβ3 binding. We may therefore presume that N-TIMP2HD will strongly inhibit MMP-2 activation and prevent the docking of the matured MMP-2 on the cell surface.
In summary, our results suggest that MMP-14 inhibition is related to pro-MMP-2 processing, whereas inhibition of integrin αvβ3 contributes to the inhibition of intermediate MMP-2 activation (Fig. 3 and Table 2). Inhibition of integrin αvβ3 also plays a significant role in hindering tube formation, as reflected in the finding that Ala–N-TIMP2WT, RGD, N-TIMP2WT, RGD, and N-TIMP2HD were more active than N-TIMP25M, RGD and N-TIMP2WT (Fig. 6 and Table 2). As we have shown here, tube formation is thus more closely influenced by integrin αvβ3 than by MMP-14 or MMP-2. In contrast, cell invasion is coupled to both integrin αvβ3 and MMP-14 activities, and simultaneous inhibition of the two receptors by N-TIMP2HD results in superior inhibition (Figs. 5K and 7J and Table 2). In more specific terms, in addition to generating new molecules that bind MMP-14 and integrin αvβ3, this study has generated, for the first time, single molecules that successfully interfere with all the components of the MMP-14/MMP-2/integrin αvβ3 axis and inhibit their biological activity. The study therefore indicates the utility of initiating pre-clinical studies of our engineered MMP-14- and integrin αvβ3-binding N-TIMP2 variant proteins.
Table 2.
Summary of the variants' activities in different cellular assays
ND means inhibition was not detected.
| Variant | Tube formation inhibitiona,b | Invasion inhibitionb of TIME cells | Invasion inhibitionb of U87MG cells | Matured MMP-2 percentagec | Intermediate MMP-2 percentage | Total active MMP-2 |
|---|---|---|---|---|---|---|
| % | % | % | % | % | % | |
| FL-TIMP2 | −10d | −11d | −5d | 3 | 0.6 | 3.6 |
| N-TIMP2WT | 61 | 8 | 10 | 1.3 | 4.1 | 5.4 |
| N-TIMP25M | 1 | 8 | 8 | 2.5 | 0.5 | 3 |
| N-TIMP2WT, RGD | 82 | 18 | 20 | 2.3 | 4.7 | 7 |
| N-TIMP25M, RGD | 45 | 14 | −2.5 | 1.7 | 1.1 | 2.8 |
| Ala- N-TIMP2WT, RGD | 92 | 8 | 13 | 2.4 | 4.9 | 7.3 |
| N-TIMP2HD | 79 | 28 | 33 | 1.6 | 0.4 | 2 |
| cRGD | ND | 21 | 12 | 2.8 | 3.5 | 6.3 |
a Tube formation is measured as number of segments.
b Percent of inhibition was compared with the untreated control.
c For untreated cells, the percentages of the different MMP-2 forms are as follows: 3.9% for MMP-2 intermediate; 3.5% for MMP-2 matured; and 7.4% for total active MMP-2 (intermediate + matured).
d − represents activation.
Viewing the findings of this study within the broader context of the current drive to develop targeted cancer therapies reveals the implications of our approach for the future. In broad terms, our study illustrates a protein engineering strategy for producing bi-specific protein monomers and heterodimers as selective tumor-targeting agents with therapeutic potential. Even more importantly, we foresee that our strategy will generate a technology that can be broadly applied to targeting membrane receptors and soluble antigens in a variety of diseases.
Experimental procedures
MMP-14CAT protein production
The pET3a vector containing the human MMP-14 catalytic domain gene (MMP-14CAT, residues 112–292) with a C-terminal His6 tag (kindly provided by Irit Sagi, Weizmann Institute of Science, Israel) was expressed in Escherichia coli strain Bl21 (DE3) and purified as described previously (50). Following affinity chromatography using nickel-nitrilotriacetic acid-Sepharose beads (Invitrogen), CHAPS was added to a final concentration of 0.1% w/v, and the mixture was incubated for 3 days at 4 °C to digest the C-terminal hinge region of MMP-14 at residues 292–318. The protein was further purified on a gel-filtration Superdex 200 column (GE Healthcare) pre-equilibrated with buffer A (50 mm Tris, pH 7.5, 100 mm NaCl, 5 mm CaCl2). The protein concentration was determined by UV-visible absorbance at 280 nm (extinction coefficient (ϵ280) of 35,410 m−1 cm−1) using a NanoDrop spectrophotometer (ThermoFisher Scientific). The purity of the protein was determined by SDS-PAGE and MS analysis (MALDI-TOF Reflex-IV, Ilse Katz Institute for Nanoscale Science and Technology, BGU, Israel). MMP-14CAT was labeled for 1 h at room temperature with DyLight-488 amine-reactive dye (ThermoFisher Scientific) in 0.05 m sodium borate buffer, pH 8.5, at a molar ratio of 1:5 (protein/dye). The labeled protein was washed free of unbound dye with buffer A in a Vivaspin (GE Healthcare) with a 5-kDa cutoff until no color was observed in the flow-through.
Construction and expression of yeast surface–displayed N-TIMP2RGD library
Yeast surface display is a powerful directed evolution technology in which proteins are displayed on the yeast surface through genetic fusion to the yeast mating agglutinin protein Aga2p (70). Aga2p is disulfide-bonded to Aga1p, which is covalently linked to the yeast cell wall. The protein of interest is flanked by epitope tags, which are used to confirm expression of the construct on the yeast cell surface and to quantify surface expression levels. Fluorescence-activated cell sorting (FACS)-based high-throughput screening of tens of millions of yeast-displayed mutated proteins allows for the rapid isolation of variants with altered properties, such as increased target affinity. One of the benefits of yeast display over other protein engineering technologies, such as phage or mRNA display, is that multicolor FACS can be used to isolate clones with the desired target affinity by normalizing yeast expression levels with binding levels.
The pCTCON yeast surface display vector was digested and linearized with BamHI and Nhe-I. The N-TIMP2WT gene was amplified using PCR with homologous primers for pCTCON plasmids. PCR assembly with overlapping primers was used to prepare an N-TIMP2RGD library, in which positions 51–59 in N-TIMP2 were replaced with an RGD motif having three random amino acids flanking the motif on each side (underlined in the Ultra_RGD fragment, shown below). The assembly PCR products were amplified by PCR using homologous primers to the pCTCON construct: Fwd, 5′-GGTGGTTCTGGTGGTGGTGGTTCTGGTGGTGGTGGTTCTGCTAGCTGCAGCTGCTCCCCGGTG-3′, and Rev, 5′-GCTATTACAAGTCCTCTTCAGAAATAAGCTTTTGTTCAGATGGATCTTGGAAAGCCAATGGTTTATCTGGCAAGGATCCCTCGCAGCCCATCTGGTACC-3′.
The amplified gene along with the digested pCTCON linear vector were transformed by electroporation into Saccharomyces cerevisiae strain EBY100 by using recombinant homology as described previously (71). The library size was 1.75 × 107, as calculated by dilution plating on selective plates. The overlapping primers that were used for the assembly PCR were as follows: Ultra_1, 5′-TGCAGCTGCTCCCCGGTGCACCCGCAACAGGCGTTTTGCAATGCAGATGTAGTGATCAGGGCCAAAGCGGTCAGTGAGAAGGAAGTG-3′; Ultra_2, 5′-GCGGTCAGTGAGAAGGAAGTGGACTCTGGAAACGACATTTATGGCAACCCTATCAAGAGGATCCAGTATGAGATCAAGCAG-3′; Ultra_RGD, 5′-GAGGATCCAGTATGAGATCAAGCAGATANNSNNSNNSAGGGGGGACNNSNNSNNSATAGAGTTTATCTACACGGCCCCCTC-3′, where n = A, C, T, or G and S = C or G; Ultra_3, 5′-CCTTCTTTCCTCCAACGTCCAGCGAGACCCCACACACTGCCGAGGAGGGGGCCGTGTAGATAAACTC-3′; Ultra_4, 5′-CACGATGAAGTCACAGAGGGTGATGTGCATCTTGCCGTCCCCCTCGGCCTTTCCTGCAATGAGATATTCCTTCTTTCCTCCAACGTCCAG-3′; and Ultra_5, 5′-CTCGCAGCCCATCTGGTACCTGTGGTTCAGGCTCTTCTTCTGGGTGGTGCTCAGGGTGTCCCAGGGCACGATGAAGTCACAGAGGGTG-3′.
Screening of the N-TIMP2RGD library
The yeast-displayed N-TIMP2RGD library was grown in selective medium (yeast nitrogen base (6.7 g/liter), Na2HPO4 (5.4 g/liter), NaH2PO4·H2O (8.56 g/liter), dextrose (20 g/liter), Bacto Casamino acids (5 g/liter)) and induced for expression with galactose (20 g/liter), as described previously (72). Yeast cells, 2–20 × 106, were sorted in each round against different concentrations of integrin αvβ3, as described previously with some modifications (4). Briefly, yeast cells were labeled with integrin αvβ3 (R&D Systems) and a 1:200 dilution of chicken anti-c-Myc antibody (Abcam) in integrin-binding buffer (IBB, 20 mm Tris, pH 7.5, 100 mm NaCl, 1 mm MgCl2, 1 mm MnCl2, 2 mm CaCl2, and 1 mg/ml bovine serum albumin (BSA)) for 1 h at room temperature. Cells were washed and resuspended in ice-cold IBB containing a 1:25 dilution of FITC-labeled mouse anti-αv integrin (BioLegend) and a 1:100 dilution of phycoerythrin (PE)-conjugated anti-chicken–IgY (Santa Cruz Biotechnology). After 30 min on ice, the cells were washed with IBB and sorted using a iCyt Synergy FACS apparatus (Sony Biotechnology). In the expression sort, only yeast cells expressing the intact N-TIMP2RGD proteins were selected (using the anti-c-Myc antibody). For the subsequent integrin αvβ3 affinity sorts, the concentrations of integrin αvβ3 that were added to the yeast cells decreased with each sort, as follows: 500 nm (sorts 1 and 2), 200 nm (sort 3), 50 nm (sort 4), and 20 nm (sort 5). In each integrin–αvβ3–binding sort, 0.3–2% of the yeast high-affinity (normalized to expression) cell population was collected using a diagonal sorting gate. After each sort round, the yeast plasmid DNA was purified using the ZymoprepTM yeast plasmid miniprep I kit (ZymoResearch). Thereafter, the plasmid was transformed into electrocompetent E. coli and grown overnight on ampicillin LB agar plates. Then, 20–40 colonies were transferred to ampicillin/LB culture medium and grown overnight at 37 °C. The plasmid was extracted from the bacteria using HiYield plasmid mini kit (RBC Bioscience) and was sequenced (Genetics Unit, National Institute of Biotechnology in the Negev, Ben-Gurion University of the Negev, Israel).
Construction of monomeric and dimeric N-TIMP2 variants
Using PCR, the alanine residue was added before Cys-1 of N-TIMP2WT, RGD so as to generate a mono-specific integrin αvβ3-only binding variant (Ala–N-TIMP2WT, RGD). The following primers were applied: alanine pPic Fwd, 5′-GGTATCTCTCGAGAAAAGAGCATGCAGCTGCTCCCCG-3′, and pPic Rev, 5′-GCTGGCGGCCGCCTCGCAGCCCATCTGGTA-3′. N-TIMP25M, RGD was generated by the insertion of the following five mutations into the N-TIMP2WT, RGD gene: M35I, D38N, N68S, G71V, and R97H (Integrated DNA Technologies). N-TIMP2HD comprising N-TIMP25M linked to N-TIMP2WT, RGD with a flexible linker (SGGGGSGGGGSGGGGS) was generated by assembly PCR. First, the N-TIMP25M gene was amplified with the primers: pPic Fwd, 5′-GGTATCTCTCGAGAAAAGATGCAGCTGCTCCCCG-3′, and linker Rev, 5′-AGAACCACCACCACCAGAACCACCACCACCAGAACCACCACCACCACTCTCGCAGCCCATCTGGTACC-3′, generating the product N-TIMP25M-linker gene. The N-TIMP2WT, RGD gene was amplified with the primers: Fwd linker, 5′-GGTGGTTCTGGTGGTGGTGGTTCTGGTGGTGGTGGTTCTTGCAGCTGCTCCCCGGTG-3′, and pPic Rev, generating the product linker-N-TIMP2WT, RGD gene. Then, assembly PCR was performed, using the linker–N-TIMP2WT, RGD gene as the template and the N-TIMP25M–linker gene and pPic Rev as primers.
Production and purification of soluble N-TIMP2 proteins
N-TIMP2-soluble protein was produced by utilizing methylotrophic X33 Pichia pastoris according to the pPICZα protocol (Invitrogen) and as described previously with modifications (38). Briefly, the different N-TIMP2 genes were cloned into pPICZαA vector for expression in the X33 P. pastoris strain. Proteins were prepared with both c-Myc and His6 tags at the C terminus for protein detection and purification, respectively. N-TIMP2 proteins were purified from the yeast growth medium by affinity column chromatography using nickel-nitrilotriacetic acid–Sepharose beads (Invitrogen) equilibrated in a mixture of 50 mm Tris, pH 7.5, 300 mm NaCl, and 10 mm imidazole and eluted with a mixture of 50 mm Tris, pH 7.5, 300 mm NaCl, and 300 mm imidazole. Thereafter, separation on a gel-filtration Superdex 75 column (GE Healthcare), equilibrated in 50 mm Tris, pH 7.5, 300 mm NaCl, and 5 mm CaCl2 at a flow rate of 0.5 ml/min on an ÄKTA pure instrument, was performed. Protein size was verified by SDS-PAGE and MS analysis (Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Israel). Protein concentrations were determined by UV-visible absorbance at 280 nm, using a NanoDrop spectrophotometer (ϵ280 of 13,325 m−1 cm−1 for N-TIMP2WT, N-TIMP2WT, RGD, Ala–N-TIMP2WT, RGD, and N-TIMP25M, RGD, and 26,650 m−1 cm−1 for N-TIMP2HD). Purification yields for all proteins were 0.2–4 mg/liter.
Surface plasmon resonance experiments
The binding between MMP-14CAT and N-TIMP2 variants was detected using SPR spectroscopy on a ProteOnTM XPR36 (Bio-Rad), as described previously (73) with modifications. Briefly, for MMP-14CAT binding, an HTG chip (Bio-Rad) was loaded with 150 μl of 10 mm NiSO4 (pH 6.3) at a rate of 30 μl/min for 5 min. N-TIMP2 variants (0.0066–0.013 μg/μl, 150 μl) were then immobilized on the chip at a flow rate of 30 μl/min for 5 min; an empty channel was used as background. For determination of the affinity constants (KD) of either N-TIMP25M, RGD or N-TIMP2HD with MM-14CAT, MMP-14CAT was diluted in a mixture of 50 mm Tris, pH 7.5, 100 mm NaCl, and 5 mm CaCl2 to 3.1, 6.3, 12.5, 25, or 50 nm. For N-TIMP2WT, RGD and Ala–N-TIMP2WT, RGD, the following concentrations of MM-14CAT were used: 31.3, 62.5, 125, 250, and 500 nm. The analyte, MMP-14CAT, was allowed to flow over the chip at a flow rate of 25 μl/min for 981 s, followed by 20 min of dissociation. The response was monitored as a function of time at 25 °C, and the KD values between the N-TIMP2 variants and MMP-14CAT were determined. For measuring the binding constants between N-TIMP2 variants and integrin αvβ3, a GLC chip (Bio-Rad) was loaded with 150 μl of the amine-coupling reagents, 0.1 m N-hydroxysuccinimide and 0.4 m 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide, at a rate of 30 μl/min for 5 min. Thereafter, 150 μl of 0.067 and 0.02 μg/μl of integrin αvβ3 and BSA, respectively, were immobilized to different channels at a flow rate of 30 μl/min for 5 min. The BSA channel was used as a control. N-TIMP2WT, RGD was diluted in IBB, without BSA, to 12.5, 25, 50, 100, or 200 nm and allowed to flow over the chip at a flow rate of 30 μl/min for 818 s. N-TIMP25M, RGD was diluted to 9.4, 18.8, 37.5, 75, or 150 nm; Ala–N-TIMP2WT, RGD was diluted to 12.5, 25, 50, 100, or 200 nm, and N-TIMP2HD was diluted to 16.2, 32.4, 64.7, 129.4, or 258.8 nm. The three variants were allowed to flow over the chip at a rate of 40 μl/min for 613 s, followed by 20 min of dissociation. The KD value between the N-TIMP2 variants and integrin αvβ3 was determined; the χ2 values for all the analyses were less than 10%.
Dual receptor binding experiments with immobilized integrin αvβ3
Simultaneously, binding of N-TIMP2WT, RGD and N-TIMP2HDto both MMP-14 and integrin αvβ3 was tested using SPR. First, a GLC chip (Bio-Rad) was loaded with 0.02 μg/μl integrin αvβ3 and BSA as described previously. Then, IBB without BSA or 0.027 μg/μl of either N-TIMP2WT, RGD or N-TIMP2HD in IBB without BSA were allowed to flow over the chip at a flow rate of 30 μl/min for 300 s. Thereafter, 500 nm MMP-14 in IBB without BSA was allowed to flow over the chip at 30 μl/min for 437 s. The channel loaded with BSA served as a background control for integrin αvβ3 and MMP-14CAT binding. Injections of IBB without BSA buffer followed by MMP-14CAT in IBB without BSA served as a negative control. The dissociation of each complex in IBB without BSA was monitored for 600 s.
MMP-14CAT inhibition studies
The Km value for the purified MMP-14CAT protein was determined by measuring MMP-14CAT activity against different concentrations (0–25 μm) of the fluorogenic substrate Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2·TFA (Merck Millipore) and fitting the data to the Michaelis-Menten equation (Equation 1) using Prism (GraphPad Software). The calculated value of Km (16.79 ± 4.986 μm) and its standard error (S.E.) are the average values that were obtained from three independent experiments.
The Ki determination was performed as described previously (38). The inhibition of the catalytic activity of MMP-14CAT (0.005625 nm) against the fluorogenic substrate (7.5 μm) was tested using N-TIMP2WT at 0.2–25 nm, N-TIMP2WT, RGD and Ala–N-TIMP2WT, RGD at 3.9–500 nm, and N-TIMP25M, RGD and N-TIMP2HD at 0.0075–0.125 nm. The Ki calculation was performed by utilizing Morrison's equation (Equation 2), the classic competitive inhibition equation for tight binding, by using Prism (GraphPad Software). Ki and its S.E. values were the average values obtained from three independent experiments,
| (Eq. 1) |
where V is enzyme velocity; Vmax is enzyme maximum velocity achieved at maximum substrate concentration; S is substrate concentration, and Km is Michaelis-Menten constant,
| (Eq. 2) |
where Vi is enzyme velocity in the presence of inhibitor; V0 is enzyme velocity in the absence of inhibitor; E is enzyme concentration; I is inhibitor concentration; S is substrate concentration; Km is Michaelis-Menten constant, and Kiapp is the apparent inhibition constant, which is given by Equation 3,
| (Eq. 3) |
where Ki is the inhibition constant.
Cell cultures
TIME cells (ATCC) were maintained in vascular cell basal medium (ATCC) supplemented with 2% fetal bovine serum (FBS) and growth factor supplements (ATCC). The U87MG glioblastoma cell line (ATCC) was maintained in DMEM (Biological Industries) supplemented with 10% FBS (ThermoFisher Scientific), 1% l-glutamine (Biological Industries), and 1% penicillin/streptomycin (Biological Industries).
MMP-14 and integrin αvβ3 expression in U87MG and TIME cell lines
MMP-14 and integrin αvβ3 expression on U87MG and TIME cells was evaluated using an Accuri C6 flow cytometer (BD Biosciences). For MMP-14 detection, rabbit anti-MMP-14 antibody (Abgent) was applied, followed by goat anti-rabbit IgG-PE staining (Santa Cruz Biotechnology). Integrin αvβ3 was detected using anti-integrin αvβ3 antibody FITC-conjugated (LM609, Chemicon).
Inhibition of MMP-2 activation
A gelatin zymography assay was utilized to assess the ability of N-TIMP2 variants and FL-TIMP2 (R&D Systems) to inhibit cellular MMP-2 activation. U87MG cells (1.9 × 105) were grown on 96-well plates for 24 h. Then, cells were washed twice with serum-free DMEM and treated with 100 nm N-TIMP2 proteins in DMEM serum-free medium at 37 °C, 5% CO2. In addition, different concentrations of FL-TIMP2 (1, 10, and 100 nm) and of cRGD (100 nm and 1 μm) were tested for their pro-MMP-2 activation activity. After 48 h, the medium was collected and centrifuged for 5 min at 10,000 × g. The medium was loaded on gelatin gel, washed for 1 h in 2.5% Triton X-100, and incubated overnight in a developer buffer (50 mm Tris, pH 7.4, 10 mm CaCl2, and 0.02% NaN3) at 37 °C, as described previously (74). Gels were stained with SimplyBlueTM SafeStain (ThermoFisher Scientific) and captured with MiniBIS Pro (DNR Bio-Imaging Systems). The intensity of the white bands was quantified using ImageJ.
Inhibition of MMP-2 cell surface localization
Flow cytometry analysis was used to detect recombinant matured MMP-2 on the U87MG cell surface. U87MG cells were grown to 70–80% confluence. Cells (105) were washed three times with IBB + 0.1% BSA, followed by 2 h of incubation with 100 nm recombinant matured MMP-2 (BioLegend) and 2 μm Ala–N-TIMP2WT, RGD at 4 °C. Cells were washed three times with IBB + 0.1% BSA and then incubated with mouse monoclonal anti-MMP2 antibody (clone CA-4001, Abcam) for 1 h at 4 °C. Cells were washed three times with IBB + 0.1% BSA and were labeled for 30 min at 4 °C with PE-conjugated goat anti-mouse IgG (1:50 dilution, Sigma). Cells were washed three times with IBB + 0.1% BSA, and MMP-2 levels on cell surface were detected with an Accuri C6 flow cytometer (BD Biosciences).
To determine which form of MMP-2 was removed from the U87MG cell surface by Ala–N-TIMP2WT, RGD, a cell lysate was subjected to gelatin zymography, as described previously (21). Briefly, U87MG cells (2 × 105) were grown overnight on a 24-well plate. Then, the growth medium was replaced with a DMEM serum-free medium containing 2 μm Ala–N-TIMP2WT, RGD. After a 48-h incubation, cells were washed three times with PBS. Thereafter, PBS (100 μl) was added to each well, and cells were scraped from the plate. Scraped cells from two wells were centrifuged, and the cell pellet was lysed with 35 μl of SDS sample buffer (4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromphenol blue, and 0.125 m Tris HCl, pH 6.8) and incubated for 30 min at 37 °C. Thereafter, 50% glycerol (1:1 ratio) was added. The cell lysate (30 μl) was loaded on a gelatin gel, washed for 1 h in 2.5% Triton X-100, and incubated overnight in developer buffer at 37 °C.
To confirm that our inhibitor did indeed disrupt MMP-2 docking on integrin αvβ3, we utilized a solid-phase assay, as described previously (21). Wells of a 96-well ELISA plate were coated with 75 μl of integrin αvβ3 (2 μg/ml in PBS) and incubated overnight at 4 °C. The plate was washed three times with PBS, followed by blocking with 5% BSA. Then, 150 μl of U87MG serum-free medium obtained after 48 h of cell growth, which contained all the MMP-2 forms (pro/intermediate/matured), was added to each well, together with 2 μm of either Ala–N-TIMP2WT, RGD or cRGD, and the plate was incubated overnight at 4 °C. The plate was washed three times with PBS. Thereafter, the bound MMP-2 was solubilized with 25 μl of SDS sample buffer, and the contents of the three wells were combined and loaded on gelatin zymography gels (30 μl). Gels were stained with SimplyBlueTM SafeStain and captured with MiniBIS Pro. The bound MMP-2 was detected by the appearance of white bands, the intensity of which was quantified using ImageJ. All the assays of the inhibition of MMP-2 cell surface localization were performed in triplicate.
Boyden chamber invasion assay
An in vitro Boyden chamber assay was performed using ThinCertTM 24-well inserts (Greiner Bio-One), as described previously with minor modifications (75). ThinCertTM cell culture insert membranes were coated with Matrigel® diluted (1:30) with either DMEM without serum or vascular cell basal medium in the upper compartment. The lower compartment of the chamber was filled with 600 μl of either DMEM or vascular cell basal medium supplemented with 2% serum for U87MG or TIME cells, respectively. U87MG (1.5 × 104) or TIME (2 × 104) cells were treated with 100 nm N-TIMP2 variants in 200 μl of serum-free DMEM or 2 μm N-TIMP2 variants in 200 μl of vascular cell basal medium, respectively. A mixture of 100 nm of each N-TIMP2WT, RGD and N-TIMP25M was also used as a treatment for the U87MG cell line. The treatments were added to the pre-coated ThinCertTM cell culture inserts, which were then incubated for 20 h at 37 °C under 5% CO2. Invasive cells were stained with Dipp Kwik Differential Stain Kit (American Mastertech Scientific) and were detected by EVOS FL Cell Imaging System (ThermoFisher Scientific) at ×20 magnification. The experiment was performed in triplicate; 15 fields were counted for each treatment, and the average of cells per field was determined.
Tube formation assay
A tube formation assay was utilized as described previously with minor modifications (76). Briefly, growth factor-reduced Matrigel® (GFR-Matrigel, Corning) was thawed overnight at 4 °C on ice. Thereafter, 150 μl of GFR-Matrigel were inserted into each well of 48-well plates, followed by centrifugation at 300 × g for 15 min at 4 °C and incubation at 37 °C for 1 h. TIME cells (3.25 × 104) were then resuspended with 2 μm N-TIMP2 variants and FBS (2%) at a final volume of 200 μl, and the suspension was seeded on the Matrigel-coated plate. The plate was incubated at 37 °C under 5% CO2 for 18 h and was monitored using EVOS FL Cell Imaging System at ×2 magnification. Six repetitions were performed, and the tube total lengths and the numbers of segments were analyzed and quantified using ImageJ Angiogenesis plug-in.
Xenograft model
U87MG cells (5 × 106) in 100 μl of serum-free DMEM, in the presence or absence of 500 nm N-TIMP2HD, were mixed with 100 μl of Matrigel and injected subcutaneously into 6-week-old male Hsd/athymic nude–Foxn1nu mice (Envigo), as described previously (77, 78). Tumor growth was monitored over 25 days. Tumor size was measured with a caliper three times a week, and each measurement was followed by an additional intra-tumor injection of 50 μl of PBS containing 500 nm N-TIMP2HD to the treated group or 50 μl of PBS to the control group (a total of three injections per week for the entire experiment). Tumor volume was determined according to Equation 4,
| (Eq. 4) |
where L is length, and W is width.
All the animal experiments were conducted using protocols approved by the Committee of Use and Care of Animals at Ben-Gurion University of the Negev, and the animals were housed and handled according to the Unit for Laboratory Animal Medicine guidelines at Ben-Gurion University.
Author contributions
G. Y. and V. A. data curation; G. Y. and N. P. formal analysis; G. Y. and N. P. validation; G. Y. and N. P. investigation; G. Y. and V. A. methodology; G. Y. and N. P. writing-original draft; G. Y., V. A., and N. P. writing-review and editing; N. P. conceptualization; N. P. resources; N. P. supervision; N. P. funding acquisition; N. P. visualization.
Supplementary Material
Acknowledgments
We thank Dr. Alon Zilka, Lidan Aharon, and Yuval Zur for their technical assistance.
This work was supported by European Research Council “Ideas Program” ERC-2013-StG, Contract Grant 336041, and Israel Science Foundation Grant 615/14 (to N. P.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7 and Table S1.
- ECM
- extracellular matrix
- ANOVA
- analysis of variance
- MMP
- matrix metalloproteinase
- YSD
- yeast surface display
- ES
- expression sort
- TIME
- telomerase-immortalized microvascular endothelial
- PE
- phycoerythrin
- SPR
- surface plasmon resonance
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- IBB
- integrin-binding buffer
- Fwd
- forward
- Rev
- reverse
- FL
- full length.
References
- 1. Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- 2. Alghisi G. C., and Rüegg C. (2006) Vascular integrins in tumor angiogenesis: mediators and therapeutic targets. Endothelium 13, 113–135 10.1080/10623320600698037 [DOI] [PubMed] [Google Scholar]
- 3. Mizejewski G. J. (1999) Role of integrins in cancer: survey of expression patterns. Proc. Soc. Exp. Biol. Med. 222, 124–138 10.1046/j.1525-1373.1999.d01-122.x [DOI] [PubMed] [Google Scholar]
- 4. Nomura H., Sato H., Seiki M., Mai M., and Okada Y. (1995) Expression of membrane-type matrix metalloproteinase in human gastric carcinomas. Cancer Res. 55, 3263–3266 [PubMed] [Google Scholar]
- 5. Zhang W., Matrisian L. M., Holmbeck K., Vick C. C., and Rosenthal E. L. (2006) Fibroblast-derived MT1-MMP promotes tumor progression in vitro and in vivo. BMC Cancer 6, 52 10.1186/1471-2407-6-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li X.-Y., Ota I., Yana I., Sabeh F., and Weiss S. J. (2008) Molecular dissection of the structural machinery underlying the tissue-invasive activity of membrane type-1 matrix metalloproteinase. Mol. Biol. Cell 19, 3221–3233 10.1091/mbc.e08-01-0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stetler-Stevenson W. G. (1999) Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J. Clin. Invest. 103, 1237–1241 10.1172/JCI6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sato H., and Takino T. (2010) Coordinate action of membrane-type matrix metalloproteinase-1 (MT1-MMP) and MMP-2 enhances pericellular proteolysis and invasion. Cancer Sci. 101, 843–847 10.1111/j.1349-7006.2010.01498.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kikuchi R., Noguchi T., Takeno S., Kubo N., and Uchida Y. (2000) Immunohistochemical detection of membrane-type-1-matrix metalloproteinase in colorectal carcinoma. Br. J. Cancer 83, 215–218 10.1054/bjoc.2000.1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hofmann U. B., Westphal J. R., Zendman A. J., Becker J. C., Ruiter D. J., and van Muijen G. N. (2000) Expression and activation of matrix metalloproteinase-2 (MMP-2) and its co-localization with membrane-type 1 matrix metalloproteinase (MT1-MMP) correlate with melanoma progression. J. Pathol. 191, 245–256 10.1002/1096-9896(2000)9999:9999%3C::AID-PATH632%3E3.0.CO%3B2-# [DOI] [PubMed] [Google Scholar]
- 11. Nakamura H., Ueno H., Yamashita K., Shimada T., Yamamoto E., Noguchi M., Fujimoto N., Sato H., Seiki M., and Okada Y. (1999) Enhanced production and activation of progelatinase A mediated by membrane-type I matrix metalloproteinase in human papillary thyroid carcinomas. Cancer Res. 59, 467–473 [PubMed] [Google Scholar]
- 12. Kumar C. C., Malkowski M., Yin Z., Tanghetti E., Yaremko B., Nechuta T., Varner J., Liu M., Smith E. M., Neustadt B., Presta M., and Armstrong L. (2001) Inhibition of angiogenesis and tumor growth by SCH221153, a dual α(v)β3 and α(v)β5 integrin receptor antagonist. Cancer Res. 61, 2232–2238 [PubMed] [Google Scholar]
- 13. Albelda S. M., Mette S. A., Elder D. E., Stewart R., Damjanovich L., Herlyn M., and Buck C. A. (1990) Integrin distribution in malignant melanoma: association of the β3 subunit with tumor progression. Cancer Res. 50, 6757–6764 [PubMed] [Google Scholar]
- 14. Weis S. M., and Cheresh D. A. (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med. 17, 1359–1370 10.1038/nm.2537 [DOI] [PubMed] [Google Scholar]
- 15. Genís L., Gálvez B. G., Gonzalo P., and Arroyo A. G. (2006) MT1-MMP: universal or particular player in angiogenesis? Cancer Metastasis Rev. 25, 77–86 10.1007/s10555-006-7891-z [DOI] [PubMed] [Google Scholar]
- 16. Deryugina E. I., Ratnikov B. I., Yu Q., Baciu P. C., Rozanov D. V., and Strongin A. Y. (2004) Prointegrin maturation follows rapid trafficking and processing of MT1-MMP in Furin-negative colon carcinoma LoVo cells. Traffic 5, 627–641 10.1111/j.1600-0854.2004.00206.x [DOI] [PubMed] [Google Scholar]
- 17. Stefanidakis M., and Koivunen E. (2006) Cell-surface association between matrix metalloproteinases and integrins: role of the complexes in leukocyte migration and cancer progression. Blood 108, 1441–1450 10.1182/blood-2006-02-005363 [DOI] [PubMed] [Google Scholar]
- 18. Gálvez B. G., Matías-Román S., Yáñez-Mó M., Sánchez-Madrid F., and Arroyo A. G. (2002) ECM regulates MT1-MMP localization with β1 or αvβ3 integrins at distinct cell compartments modulating its internalization and activity on human endothelial cells. J. Cell Biol. 159, 509–521 10.1083/jcb.200205026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brooks P. C., Strömblad S., Sanders L. C., von Schalscha T. L., Aimes R. T., Stetler-Stevenson W. G., Quigley J. P., and Cheresh D. A. (1996) Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell 85, 683–693 10.1016/S0092-8674(00)81235-0 [DOI] [PubMed] [Google Scholar]
- 20. Deshane J., Garner C. C., and Sontheimer H. (2003) Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 278, 4135–4144 10.1074/jbc.M205662200 [DOI] [PubMed] [Google Scholar]
- 21. Deryugina E. I., Ratnikov B., Monosov E., Postnova T. I., DiScipio R., Smith J. W., and Strongin A. Y. (2001) MT1-MMP initiates activation of pro-MMP-2 and integrin αvβ3 promotes maturation of MMP-2 in breast carcinoma cells. Exp. Cell Res. 263, 209–223 10.1006/excr.2000.5118 [DOI] [PubMed] [Google Scholar]
- 22. Hofmann U. B., Westphal J. R., Van Kraats A. A., Ruiter D. J., and Van Muijen G. N. (2000) Expression of integrin α(v)β(3) correlates with activation of membrane-type matrix metalloproteinase-1 (MT1-MMP) and matrix metalloproteinase-2 (MMP-2) in human melanoma cells in vitro and in vivo. Int. J. Cancer 87, 12–19 10.1002/1097-0215(20000701)87:1%3C12::AID-IJC3%3E3.0.CO%3B2-A [DOI] [PubMed] [Google Scholar]
- 23. Deryugina E. I., Bourdon M. A., Jungwirth K., Smith J. W., and Strongin A. Y. (2000) Functional activation of integrin αvβ3 in tumor cells expressing membrane-type 1 matrix metalloproteinase. Int. J. Cancer 86, 15–23 10.1002/(SICI)1097-0215(20000401)86:1%3C15::AID-IJC3%3E3.0.CO%3B2-B [DOI] [PubMed] [Google Scholar]
- 24. Deryugina E. I., Ratnikov B. I., Postnova T. I., Rozanov D. V., and Strongin A. Y. (2002) Processing of integrin αv subunit by membrane type 1 matrix metalloproteinase stimulates migration of breast carcinoma cells on vitronectin and enhances tyrosine phosphorylation of focal adhesion kinase. J. Biol. Chem. 277, 9749–9756 10.1074/jbc.M110269200 [DOI] [PubMed] [Google Scholar]
- 25. Butler G. S., Butler M. J., Atkinson S. J., Will H., Tamura T., Schade van Westrum S., Crabbe T., Clements J., d'Ortho M. P., and Murphy G. (1998) The TIMP2 membrane type 1 metalloproteinase “receptor” regulates the concentration and efficient activation of progelatinase A. A kinetic study. J. Biol. Chem. 273, 871–880 10.1074/jbc.273.2.871 [DOI] [PubMed] [Google Scholar]
- 26. Itoh Y., and Seiki M. (2004) MT1-MMP: an enzyme with multidimensional regulation. Trends Biochem. Sci. 29, 285–289 10.1016/j.tibs.2004.04.001 [DOI] [PubMed] [Google Scholar]
- 27. Hernandez-Barrantes S., Toth M., Bernardo M. M., Yurkova M., Gervasi D. C., Raz Y., Sang Q. A., and Fridman R. (2000) Binding of active (57 kDa) membrane type 1-matrix metalloproteinase (MT1-MMP) to tissue inhibitor of metalloproteinase (TIMP)-2 regulates MT1-MMP processing and pro-MMP-2 activation. J. Biol. Chem. 275, 12080–12089 10.1074/jbc.275.16.12080 [DOI] [PubMed] [Google Scholar]
- 28. Pei D., and Weiss S. J. (1996) Transmembrane-deletion mutants of the membrane-type matrix metalloproteinase-1 process progelatinase A and express intrinsic matrix-degrading activity. J. Biol. Chem. 271, 9135–9140 10.1074/jbc.271.15.9135 [DOI] [PubMed] [Google Scholar]
- 29. Silletti S., Kessler T., Goldberg J., Boger D. L., and Cheresh D. A. (2001) Disruption of matrix metalloproteinase 2 binding to integrin αvβ3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 119–124 10.1073/pnas.011343298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kummar S., Chen H. X., Wright J., Holbeck S., Millin M. D., Tomaszewski J., Zweibel J., Collins J., and Doroshow J. H. (2010) Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat. Rev. Drug Discov. 9, 843–856 10.1038/nrd3216 [DOI] [PubMed] [Google Scholar]
- 31. Gridelli C., Rossi A., Ciardiello F., De Marinis F., Crinò L., Morabito A., Morgillo F., Montanino A., Daniele G., Piccirillo M. C., Normanno N., Gallo C., and Perrone F. (2016) BEVERLY: rationale and design of a randomized open-label phase III trial comparing Bevacizumab plus Erlotinib versus Erlotinib alone as first-line treatment of patients with EGFR-mutated advanced nonsquamous non-small-cell lung cancer. Clin. Lung Cancer 17, 461–465 10.1016/j.cllc.2016.04.001 [DOI] [PubMed] [Google Scholar]
- 32. Al-Lazikani B., Banerji U., and Workman P. (2012) Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 30, 679–692 10.1038/nbt.2284 [DOI] [PubMed] [Google Scholar]
- 33. Miles D., von Minckwitz G., and Seidman A. D. (2002) Combination versus sequential single-agent therapy in metastatic breast cancer. Oncologist 7, Suppl. 6, 13–19 [PubMed] [Google Scholar]
- 34. Yap T. A., Omlin A., and de Bono J. S. (2013) Development of therapeutic combinations targeting major cancer signaling pathways. J. Clin. Oncol. 31, 1592–1605 10.1200/JCO.2011.37.6418 [DOI] [PubMed] [Google Scholar]
- 35. Papo N., Silverman A. P., Lahti J. L., and Cochran J. R. (2011) Antagonistic VEGF variants engineered to simultaneously bind to and inhibit VEGFR2 and αvβ3 integrin. Proc. Natl. Acad. Sci. U.S.A. 108, 14067–14072 10.1073/pnas.1016635108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. May C., Sapra P., and Gerber H. P. (2012) Advances in bispecific biotherapeutics for the treatment of cancer. Biochem. Pharmacol. 84, 1105–1112 10.1016/j.bcp.2012.07.011 [DOI] [PubMed] [Google Scholar]
- 37. McDonagh C. F., Huhalov A., Harms B. D., Adams S., Paragas V., Oyama S., Zhang B., Luus L., Overland R., Nguyen S., Gu J., Kohli N., Wallace M., Feldhaus M. J., Kudla A. J., et al. (2012) Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol. Cancer Ther. 11, 582–593 10.1158/1535-7163.MCT-11-0820 [DOI] [PubMed] [Google Scholar]
- 38. Arkadash V., Yosef G., Shirian J., Cohen I., Horev Y., Grossman M., Sagi I., Radisky E. S., Shifman J. M., and Papo N. (2017) Development of high affinity and high specificity inhibitors of matrix metalloproteinase 14 through computational design and directed evolution. J. Biol. Chem. 292, 3481–3495 10.1074/jbc.M116.756718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Folgueras A. R., Pendás A. M., Sánchez L. M., and López-Otin C. (2004) Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int. J. Dev. Biol. 48, 411–424 10.1387/ijdb.041811af [DOI] [PubMed] [Google Scholar]
- 40. Decock J., Thirkettle S., Wagstaff L., and Edwards D. R. (2011) Matrix metalloproteinases: protective roles in cancer. J. Cell. Mol. Med. 15, 1254–1265 10.1111/j.1582-4934.2011.01302.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. López-Otín C., Palavalli L. H., and Samuels Y. (2009) Protective roles of matrix metalloproteinases: from mouse models to human cancer. Cell Cycle 8, 3657–3662 10.4161/cc.8.22.9956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nuti E., Tuccinardi T., and Rossello A. (2007) Matrix metalloproteinase inhibitors: new challenges in the era of post broad-spectrum inhibitors. Curr. Pharm. Des. 13, 2087–2100 10.2174/138161207781039706 [DOI] [PubMed] [Google Scholar]
- 43. Hidalgo M., and Eckhardt S. G. (2001) Development of matrix metalloproteinase inhibitors in cancer therapy. J. Natl. Cancer Inst. 93, 178–193 10.1093/jnci/93.3.178 [DOI] [PubMed] [Google Scholar]
- 44. Devy L., Huang L., Naa L., Yanamandra N., Pieters H., Frans N., Chang E., Tao Q., Vanhove M., Lejeune A., van Gool R., Sexton D. J., Kuang G., Rank D., Hogan S., et al. (2009) Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 69, 1517–1526 10.1158/0008-5472.CAN-08-3255 [DOI] [PubMed] [Google Scholar]
- 45. Plow E. F., Haas T. A., Zhang L., Loftus J., and Smith J. W. (2000) Ligand binding to integrins. J. Biol. Chem. 275, 21785–21788 10.1074/jbc.R000003200 [DOI] [PubMed] [Google Scholar]
- 46. Li R., Hoess R. H., Bennett J. S., and DeGrado W. F. (2003) Use of phage display to probe the evolution of binding specificity and affinity in integrins. Protein Eng. 16, 65–72 10.1093/proeng/gzg002 [DOI] [PubMed] [Google Scholar]
- 47. Brew K., and Nagase H. (2010) The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta 1803, 55–71 10.1016/j.bbamcr.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sharabi O., Shirian J., Grossman M., Lebendiker M., Sagi I., and Shifman J. (2014) Affinity- and specificity-enhancing mutations are frequent in multispecific interactions between TIMP2 and MMPs. PLoS ONE 9, e93712 10.1371/journal.pone.0093712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bahudhanapati H., Zhang Y., Sidhu S. S., and Brew K. (2011) Phage display of tissue inhibitor of metalloproteinases-2. J. Biol. Chem. 286, 31761–31770 10.1074/jbc.M111.253328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ogata H., Decaneto E., Grossman M., Havenith M., Sagi I., Lubitz W., and Knipp M. (2014) Crystallization and preliminary X-ray crystallographic analysis of the catalytic domain of membrane type 1 matrix metalloproteinase. Acta Crystallogr. F Struct. Biol. Commun. 70, 232–235 10.1107/S2053230X13034857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wingfield P., Sax J. K., Stahl S. J., Kaufman J., Palmer I., Chung V., Corcoran M. L., Kleiner D. E., and Stetler-Stevenson W. G. (1999) Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli. J. Biol. Chem. 274, 21362–21368 10.1074/jbc.274.30.21362 [DOI] [PubMed] [Google Scholar]
- 52. Azzam H. S., Arand G., Lippman M. E., and Thompson E. W. (1993) Association of MMP-2 activation potential with metastatic progression in human breast cancer cell lines independent of MMP-2 production. J. Natl. Cancer Inst. 85, 1758–1764 10.1093/jnci/85.21.1758 [DOI] [PubMed] [Google Scholar]
- 53. Toth M., and Fridman R. (2001) Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods Mol. Med. 57, 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Werb Z. (1997) ECM and cell surface proteolysis: regulating cellular ecology. Cell 91, 439–442 10.1016/S0092-8674(00)80429-8 [DOI] [PubMed] [Google Scholar]
- 55. Lafleur M. A., Hollenberg M. D., Atkinson S. J., Knäuper V., Murphy G., and Edwards D. R. (2001) Activation of pro-(matrix metalloproteinase-2) (pro-MMP-2) by thrombin is membrane-type-MMP-dependent in human umbilical vein endothelial cells and generates a distinct 63 kDa active species. Biochem. J. 357, 107–115 10.1042/0264-6021:3570107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu K. V., Jong K. A., Rajasekaran A. K., Cloughesy T. F., and Mischel P. S. (2004) Upregulation of tissue inhibitor of metalloproteinases (TIMP)-2 promotes matrix metalloproteinase (MMP)-2 activation and cell invasion in a human glioblastoma cell line. Lab. Invest. 84, 8–20 10.1038/labinvest.3700003 [DOI] [PubMed] [Google Scholar]
- 57. Dechantsreiter M. A., Planker E., Mathä B., Lohof E., Hölzemann G., Jonczyk A., Goodman S. L., and Kessler H. (1999) N-Methylated cyclic RGD peptides as highly active and selective α(v)β(3) integrin antagonists. J. Med. Chem. 42, 3033–3040 10.1021/jm970832g [DOI] [PubMed] [Google Scholar]
- 58. Johanns M., Lemoine P., Janssens V., Grieco G., Moestrup S. K., Nielsen R., Christensen E. I., Courtoy P. J., Emonard H., Marbaix E., and Henriet P. (2017) Cellular uptake of proMMP-2:TIMP-2 complexes by the endocytic receptor megalin/LRP-2. Sci. Rep. 7, 4328 10.1038/s41598-017-04648-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Folkman J., and Haudenschild C. (1980) Angiogenesis in vitro. Nature 288, 551–556 10.1038/288551a0 [DOI] [PubMed] [Google Scholar]
- 60. Seo D. W., Li H., Guedez L., Wingfield P. T., Diaz T., Salloum R., Wei B. Y., and Stetler-Stevenson W. G. (2003) TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell 114, 171–180 10.1016/S0092-8674(03)00551-8 [DOI] [PubMed] [Google Scholar]
- 61. Ducharme A., Frantz S., Aikawa M., Rabkin E., Lindsey M., Rohde L. E., Schoen F. J., Kelly R. A., Werb Z., Libby P., and Lee R. T. (2000) Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 106, 55–62 10.1172/JCI8768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Baciu P. C., Suleiman E. A., Deryugina E. I., and Strongin A. Y. (2003) Membrane type-1 matrix metalloproteinase (MT1-MMP) processing of pro-αv integrin regulates cross-talk between αvβ3 and α2β1 integrins in breast carcinoma cells. Exp. Cell Res. 291, 167–175 10.1016/S0014-4827(03)00387-2 [DOI] [PubMed] [Google Scholar]
- 63. Gonzalo P., Moreno V., and Ga B. G. (2010) MT1-MMP and integrins: hand-to-hand in cell communication. Biofactors 36, 248–254 10.1002/biof.99 [DOI] [PubMed] [Google Scholar]
- 64. Srichai M. B., and Zent R. (2010) in Integrin Structure and Function (Zent R., and Srichai M. B., eds), Springer, New York [Google Scholar]
- 65. Stratman A. N., Saunders W. B., Sacharidou A., Koh W., Fisher K. E., Zawieja D. C., Davis M. J., and Davis G. E. (2009) Endothelial cell lumen and vascular guidance tunnel formation requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood 114, 237–247 10.1182/blood-2008-12-196451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morgunova E., Tuuttila A., Bergmann U., and Tryggvason K. (2002) Structural insight into the complex formation of latent matrix metalloproteinase 2 with tissue inhibitor of metalloproteinase 2. Proc. Natl. Acad. Sci. U.S.A. 99, 7414–7419 10.1073/pnas.102185399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Elliot S., Catanuto P., Stetler-Stevenson W., and Cousins S. W. (2006) Retinal pigment epithelium protection from oxidant-mediated loss of MMP-2 activation requires both MMP-14 and TIMP-2. Invest. Ophthalmol. Vis. Sci. 47, 1696–1702 10.1167/iovs.05-1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shen Q., Lee E. S., Pitts R. L., Wu M. H., and Yuan S. Y. (2010) Tissue inhibitor of metalloproteinase-2 regulates matrix metalloproteinase-2-mediated endothelial barrier dysfunction and breast cancer cell transmigration through lung microvascular endothelial cells. Mol. Cancer Res. 8, 939–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brooks P. C., Silletti S., von Schalscha T. L., Friedlander M., and Cheresh D. A. (1998) Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell 92, 391–400 10.1016/S0092-8674(00)80931-9 [DOI] [PubMed] [Google Scholar]
- 70. Boder E. T., and Wittrup K. D. (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 10.1038/nbt0697-553 [DOI] [PubMed] [Google Scholar]
- 71. Colby D. W., Kellogg B. A., Graff C. P., Yeung Y. A., Swers J. S., and Wittrup K. D. (2004) Engineering antibody affinity by yeast surface display. Methods Enzymol. 388, 348–358 10.1016/S0076-6879(04)88027-3 [DOI] [PubMed] [Google Scholar]
- 72. Boder E. T., and Wittrup K. D. (2000) Yeast surface display for directed evolution of protein expression, affinity, and stability. Methods Enzymol. 328, 430–444 10.1016/S0076-6879(00)28410-3 [DOI] [PubMed] [Google Scholar]
- 73. Anderson G. P., Glaven R. H., Algar W. R., Susumu K., Stewart M. H., Medintz I. L., and Goldman E. R. (2013) Single domain antibody–quantum dot conjugates for ricin detection by both fluoroimmunoassay and surface plasmon resonance. Anal. Chim. Acta 786, 132–138 10.1016/j.aca.2013.05.010 [DOI] [PubMed] [Google Scholar]
- 74. Frankowski H., Gu Y.-H., Heo J. H., Milner R., and Del Zoppo G. J. (2012) Use of gel zymography to examine matrix metalloproteinase (gelatinase) expression in brain tissue or in primary glial cultures. Methods Mol. Biol. 814, 221–233 10.1007/978-1-61779-452-0_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tamura M., Gu J., Takino T., and Yamada K. M. (1999) Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 59, 442–449 [PubMed] [Google Scholar]
- 76. Brouet A., Sonveaux P., Dessy C., Moniotte S., Balligand J. L., and Feron O. (2001) Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ. Res. 89, 866–873 10.1161/hh2201.100319 [DOI] [PubMed] [Google Scholar]
- 77. Babae N., Bourajjaj M., Liu Y., Van Beijnum J. R., Cerisoli F., Scaria P. V., Verheul M., Van Berkel M. P., Pieters E. H., Van Haastert R. J., Yousefi A., Mastrobattista E., Storm G., Berezikov E., Cuppen E., et al. (2014) Systemic miRNA-7 delivery inhibits tumor angiogenesis and growth in murine xenograft glioblastoma. Oncotarget 5, 6687–6700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Husain S. R., Joshi B. H., and Puri R. K. (2001) Interleukin-13 receptor as a unique target for anti-glioblastoma therapy. Int. J. Cancer 92, 168–175 10.1002/1097-0215(200102)9999:9999%3C::AID-IJC1182%3E3.0.CO%3B2-N [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.