

Abstract
Constitutive NF-κB signaling represents a hallmark of chronic inflammation and autoimmune diseases. The E3 ligase TNF receptor–associated factor 6 (TRAF6) acts as a key regulator bridging innate immunity, pro-inflammatory cytokines, and antigen receptors to the canonical NF-κB pathway. Structural analysis and point mutations have unraveled the essential role of TRAF6 binding to the E2-conjugating enzyme ubiquitin-conjugating enzyme E2 N (Ubc13 or UBE2N) to generate Lys63-linked ubiquitin chains for inflammatory and immune signal propagation. Genetic mutations disrupting TRAF6–Ubc13 binding have been shown to reduce TRAF6 activity and, consequently, NF-κB activation. However, to date, no small-molecule modulator is available to inhibit the TRAF6–Ubc13 interaction and thereby counteract NF-κB signaling and associated diseases. Here, using a high-throughput small-molecule screening approach, we discovered an inhibitor of the TRAF6–Ubc13 interaction that reduces TRAF6–Ubc13 activity both in vitro and in cells. We found that this compound, C25-140, impedes NF-κB activation in various immune and inflammatory signaling pathways also in primary human and murine cells. Importantly, C25-140 ameliorated inflammation and improved disease outcomes of autoimmune psoriasis and rheumatoid arthritis in preclinical in vivo mouse models. Hence, the first-in-class TRAF6–Ubc13 inhibitor C25-140 expands the toolbox for studying the impact of the ubiquitin system on immune signaling and underscores the importance of TRAF6 E3 ligase activity in psoriasis and rheumatoid arthritis. We propose that inhibition of TRAF6 activity by small molecules represents a promising novel strategy for targeting autoimmune and chronic inflammatory diseases.
Keywords: NF-kappaB, small molecule, protein-protein interaction, inflammation, autoimmunity, psoriasis, chemical biology, drug discovery, rheumatoid arthritis, TRAF6, Ubc13
Introduction
Ubiquitination of proteins is a cellular process that either drives protein degradation by the proteasome in order to sustain protein homeostasis or regulates distinct cellular signaling pathways. Three enzymes, namely E1, E2, and E3, are needed to covalently attach ubiquitin to the substrate proteins. E1 is the activating enzyme initially binding the ubiquitin, E2 is the conjugating enzyme thereby determining the linkage type, and the E3 ligase is involved in substrate specificity and transfer of ubiquitin (1). Depending on the type of linkage, polyubiquitination can have various functions (1–3). Whereas, for example, Lys48-linked chains are known to target the substrate protein for proteasomal degradation via the 26S proteasome, Lys63-linked chains are associated with a variety of nonproteolytic functions, such as trafficking, DNA damage response, and NF-κB signaling in innate and adaptive immunity.
The eukaryotic transcription factor NF-κB plays a critical role in regulating the expression of a large variety of genes that are involved in several cellular processes, including innate and adaptive immune response, cell growth and survival, and cell development (4). Several signals, including cytokines, pathogens, injuries, and other stress conditions, induce activation of the NF-κB transcription factors that are tightly regulated via the formation of polyubiquitin chains. Deregulation of NF-κB signaling contributes to the pathogenesis of autoimmunity, chronic inflammation, and various cancers (5, 6). Within NF-κB signaling, approved ubiquitin-proteasome inhibitors mainly focus on the inhibition of the 26S proteasome (e.g. bortezomib and carfilzomib), thereby broadly influencing protein homeostasis. In contrast, regulators of the ubiquitination cascade might serve as more specific drug targets. Especially, E3 ligases are final regulators of the ubiquitination reaction, with more than 600 representatives; therefore, drugging E3 ligases might give rise to more specificity compared with the 26S proteasome (7).
The TRAF protein family comprises seven E3 ligase members, including TRAF6. Different members of this class are involved in distinct receptor signaling pathways, such as the IL-13 receptor family, T-cell receptor (TCR), IL-17 receptor, TNF receptor, and more (8, 9). TRAF-dependent signaling pathways contribute to the control of diverse cellular processes, including survival, proliferation, differentiation, and cytokine production (8). The TRAF6 protein belongs to the RING type of E3 ligases, which interacts with the heterodimeric E2 enzyme complex Ubc13–Uev1a to attach Lys63-linked ubiquitin chains to its substrate proteins (10–12). These Lys63-linked ubiquitin chains are necessary to activate innate and adaptive immune responses mainly through the NF-κB axis (13, 14). Of note, the TRAF6 ligase activity is strictly determined by its protein–protein interaction with a specific set of E2 enzymes, including Ubc13. Importantly, ubiquitin is directly transferred from ubiquitin-charged Ubc13 (E2) to the substrate, both proteins bound by the E3 ligase TRAF6 (15). Thus, targeting TRAF6 E3 activity requires the identification of protein–protein interaction inhibitors, which disrupt the TRAF6–Ubc13 interaction, because TRAF6 has no intrinsic catalytic activity (7).
Overexpression of TRAF6 as well as enhanced TRAF6 activity have been shown to promote chronic immune stimulation in a variety of disorders, including autoimmunity, inflammation, and cancer (8). For example, it has previously been demonstrated that TRAF6 expression is elevated in rheumatoid arthritis (RA) patients (16, 17) and in lupus cohorts (18). On a molecular level, disruption of TRAF6–Ubc13 binding by the cellular protein A20 (19) or by genetic mutations (20, 21) counteracts TRAF6 activity and NF-κB activation. To date, efforts on targeting TRAF6 activity have focused on disrupting TRAF6–substrate interaction (22), whereas inhibition of the TRAF6 activity by interfering with the TRAF6–Ubc13 association has not been attempted so far and represents an attractive novel strategy to combat TRAF6-dependent disease formation.
In this report, we demonstrate the identification of the first TRAF6–Ubc13 protein–protein interaction inhibitor that ameliorates disease outcome of two preclinical murine models of autoimmunity and chronic inflammation.
Results
Identification of TRAF6–Ubc13 inhibitors
To identify small-molecule inhibitors of the TRAF6–Ubc13 interaction, we established an AlphaScreen® binding assay with robust performance (Fig. S1A) and carried out a high-throughput screening campaign using 25,000 compounds (Fig. S1B). After removing AlphaScreen® frequent hitters using our own developed chemoinformatics filters (23, 24) and by employing strict selection criteria (dose-response and cell-based NF-κB activation assays), we were able to select the small molecule C25 as the most promising scaffold for further evaluation (Fig. S1, C–E). Subsequently, we performed a small structure–activity relationship analysis by testing a few commercially available analogs (Fig. S2A). Three analogs showed similar inhibitory effects on TRAF6–Ubc13 interaction when compared with C25 (Fig. S2B). Compound C25-140, however, had the best inhibitory potential on NF-κB activation after IL-1β stimulation (Fig. S2C), which led us to choose this compound for additional investigations. C25-140 (Fig. 1A and Fig. S2A) disrupted the TRAF6–Ubc13 binding in vitro (Fig. S2D) without affecting the interaction of Ubc13 and the deubiquitinase OTUB1 (25) (Fig. S2D), which competes with TRAF6 for Ubc13 binding (25). Furthermore, the binding of Ubc13 to Uev1a remained unimpaired upon compound treatment (Fig. S2E). Importantly, treatment of cells with C25-140 dose-dependently impeded TRAF6–Ubc13 interaction in co-immunoprecipitation experiments (Fig. 1B), and NMR studies proved that C25-140 directly binds to TRAF6 (Fig. 1C). Furthermore, C25-140 effectively reduced TRAF6-mediated ubiquitin chain formation in vitro (Fig. 1D). These data demonstrate that C25-140 directly binds to TRAF6, thereby blocking the interaction of TRAF6 with Ubc13, and as a consequence lowers TRAF6 activity.
Figure 1.

C25-140 binds TRAF6, inhibits TRAF6–Ubc13 interaction and TRAF6 activity. A, chemical structure of C25-140. B, ectopically expressed HA-TRAF6 RZ12 in HEK293T cells co-immunoprecipitates less endogenous Ubc13 after C25-140 treatment. Ubc13 levels in the IP fractions were densitometrically quantified in relation to precipitated TRAF6. Error bars, S.D.; n = 4 biological replicates were quantified; unpaired t test (two-tailed); ****, p < 0.0001; D, DMSO. C, heteronuclear correlation NMR experiments using 15N-labeled WT TRAF6 RZ1 either alone (blue) or in the presence of C25-140 (red; molar ratio of TRAF6/C25-140 = 1:5). Several NMR signals undergo intensity reduction or broadening beyond detection (arrows), indicating binding in the so-called “intermediate exchange regime.” This suggests that protein–compound complex lifetime is in millisecond time scales and confirms direct binding of the compound to TRAF6. D, C25-140 dose-dependently counteracts in vitro ubiquitination by untagged WT TRAF6 RZ1 together with the E2 complex Ubc13–Uev1a. WB, Western blotting.
Selectivity profiling of C25-140 against other E3 ligases and E2 enzymes
To better understand the selectivity of C25-140 toward other E3 ligases, we analyzed the activity of various E3 ligases upon C25-140 treatment. These studies revealed that only the activity of cIAP1, an E3 ligase generating Lys63-linked polyubiquitin chains (26) similar to TRAF6, was affected by C25-140 (Fig. 2A). Notably, several other E3 ligases (MDM2, TRIM63, ITCH, E6AP, and RNF4) building other types of poly-ubiquitin chains were not influenced by C25-140 (Fig. 2, B–D). In addition to these studies on a set of E3 ligases, we tested whether C25-140 inhibits E1–E2 reactions. To this end, we treated different E1–E2 pairs employing nine different E2 enzymes with compound C25-140. All reactions, including UbcH13–Uev1a, were not affected by C25-140 (Fig. 3), confirming that C25-140 acts on the E3 ligase side.
Figure 2.

Effect of C25-140 on various E3 ligases/E2 enzyme reactions. A, cIAP1 generating Lys63-ubiquitin chains was also inhibited by C25-140. B, MDM2 and TRIM63 generating Lys48-ubiquitin chains were not impaired by C25-140. C, RNF4, a SUMO-targeted ubiquitin ligase, was also not affected by C25-140. D, activity of the HECT E3 ligases ITCH and E6AP was not inhibited by C25-140. The substrates and the compound concentrations are indicated; assays were purchased from Boston Biochem.
Figure 3.

Effect of C25-140 on E1 and E2 enzymes. Various E1–E2 ubiquitination reactions, including UbcH13–Uev1a, were carried out in the absence or presence of C25-140 (30 μm). None of the reactions is affected by the compound. C, control without ATP; D, DMSO; 140, compound C25-140.
Effects of compound C25-140 on pro-inflammatory signaling
Before moving into cell-based validation of C25-140, we analyzed whether C25-140 affects cell viability and cell cycle phases. C25-140 neither substantially influenced cell viability in MEF cells and Jurkat T cells at concentrations up to 50 μm (Fig. S3A) nor influenced cell cycle phases of Jurkat T cells (Fig. S3B).
We first tested the inhibitory potential of C25-140 in MEF cells that can be stimulated with pro-inflammatory cytokines IL-1β and TNFα to trigger NF-κB activation. Stimulation of these cells with IL-1β initially induces TRAF6 auto-ubiquitination that is required for signal progression through the IKK complex toward NF-κB activation and expression of NF-κB–dependent target genes (8). C25-140 treatment of MEF cells led to a dose-dependent reduction of TRAF6 auto-ubiquitination (Fig. 4A) followed by decreased levels of phosphorylated IκBα (Fig. 4B), which translated into diminished target gene expression (Fig. 4C). These data illustrate that inhibition of TRAF6 activity early during IL-1 receptor signaling hampers the signaling cascade toward NF-κB activation. Additionally, TNFα-induced phosphorylation of IκBα (Fig. S4A) as well as NF-κB–induced target gene expression (Fig. S4B) were affected by C25-140 treatment. Taken together, C25-140 efficiently inhibits IL-1β– and TNFα–mediated receptor signaling in the context of cytokine activation.
Figure 4.

C25-140 effect on proinflammatory signaling and T-cell activation. A, endogenous TRAF6 auto-ubiquitination (Ub) in MEF cells upon IL-1β stimulation is reduced after C25-140 treatment. B, C25-140 impairs IL-1β-induced IκBα phosphorylation. pIκBα levels were densitometrically quantified in relation to β-actin. Error bars, S.D.; n = 3 biological replicates were quantified; unpaired t test (two-tailed); ****, p < 0.0001. C, target gene (ICAM-1 and A20) expression is diminished after IL-1β stimulation and C25-140 treatment; error bars, S.D.; n ≥ 3 biological replicates; unpaired t test (two-tailed). *, p < 0.05; **, p < 0.01; ****, p < 0.0001. D, endogenous TRAF6 auto-ubiquitination after P/I stimulation is reduced after C25-140 treatment. E, C25-140 decreases IκBα phosphorylation after P/I stimulation in Jurkat T cells. pIκBα levels were densitometrically quantified in relation to β-actin. Error bars, S.D.; n = 3 biological replicates were quantified; unpaired t test (two-tailed). *, p < 0.05; **, p < 0.01. F, upon P/I stimulation, IL-2 and TNFα cytokine secretion is attenuated after C25-140 treatment. Error bars, S.D.; n = 3 biological replicates; unpaired t test (two-tailed). *, p < 0.05; **, p < 0.01; D, DMSO.
Effects of C25-140 on T-cell activation
In addition to IL-1β receptor signaling, we tested C25-140 on T-cell activation, as TCR-mediated NF-κB activation relies on TRAF6 activity (27, 28). In the human Jurkat T-cell line, T-cell activation by PMA/ionomycin (P/I) stimulation led to TRAF6 auto-ubiquitination that was diminished by C25-140 in a dose-dependent manner (Fig. 4D). Again, decreased TRAF6 auto-ubiquitination affected downstream signaling, as evident from reduced IκBα phosphorylation and cytokine secretion (Fig. 4, E and F). Moreover, P/I-induced c-Jun N-terminal kinase phosphorylation in mitogen-activated protein kinase signaling was diminished by C25-140 (Fig. S4C). All of these data emphasize that C25-140 also affects TCR-mediated immune response in a human-derived cell line.
C25-140 is effective in cytokine signaling of primary human and murine cells
Next, we used primary human peripheral blood mononuclear cells (PBMCs) from three healthy individuals to investigate whether C25-140 is also competing with TRAF6-dependent receptor signaling in primary human cells. In line with our data acquired in cell lines (Fig. 4), C25-140 reduced secretion of NF-κB–driven inflammatory cytokines, such as TNFα and IL-6, after IL-1β stimulation (Fig. 5A) or IL-1β and TNFα upon LPS stimulation (Fig. 5B), clearly indicating that C25-140 also reduces cytokine and innate immune responses in primary human blood cells. Similar inhibitory effects by C25-140 could be proven in human PBMCs after stimulating the adaptive immune response with anti-CD3/CD28 antibodies for TCR signaling (Fig. 5C). Therefore, C25-140 is capable of inhibiting cytokine signaling also in cells of primary human origin. In addition to primary human cells, we verified the inhibitory potential of C25-140 in ex vivo primary murine T cells and observed dose-dependent reduction of IL-2 mRNA and protein levels after CD3/CD28 stimulation (Fig. 5D).
Figure 5.

C25-140 impedes immune receptor signaling in primary human PBMC and primary murine T cells. A–C, PBMCs from healthy individuals were isolated and treated with C25-140 before stimulation with IL-1β (A), LPS (B), and CD3/CD28 (C). Secretion of various NF-κB–driven cytokines was measured by ELISA and showed reduction of distinct cytokine secretion by C25-140 in all donors. D, primary mouse CD4+ T cells were isolated and treated with C25-140 and subsequently stimulated with CD3/CD28. IL-2 mRNA expression and protein secretion were measured using qRT-PCR and ELISA, respectively. C25-140 dose-dependently reduced IL-2 expression; error bars, S.D.; n = 4 biological replicates; signals were normalized to DMSO; unpaired t test (two-tailed). **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; D, DMSO.
Absorption/distribution/metabolism/excretion (ADME) and pharmacokinetics (PK) assessment of C25-140
Before moving into in vivo mouse studies, we analyzed the compound's suitability for in vivo application. To this end, we performed ADME studies assessing key parameters (Fig. S5A). Here, C25-140 was stable in plasma as well as in microsomes, showed 96.8% plasma protein binding, and exhibited low binding to hERG. Only in terms of cytochrome P450 did the compound show some inhibitory potential on distinct CYPs (Fig. S5A). Next, we tested the PK of C25-140 after intravenous, peroral, and intraperitoneal injection of mice with 10 mg/kg C25-140 each. In general, a rapid initial distribution phase was observed, and C25-140 had good calculated oral bioavailability (Fig. S5, B and C). Altogether, results from ADME and pharmacokinetics measurements underscored that compound C25-140 had appropriate properties to be applied in mouse studies for in vivo efficacy testing.
C25-140 efficacy in a psoriasis mouse model
In a first in vivo mouse model, we aimed at investigating the effects of C25-140 on psoriasis, an autoimmune disease. In this model, psoriasis is induced by imiquimod (IMQ), which activates TLR7 signaling (29), a pathway employing TRAF6 for signal progression. IMQ was topically applied to the shaved back as well as the right ear of mice. On the same regions, C25-140 was topically applied twice daily at a final dose of ∼1.5 mg/kg per application. Parameters describing disease outcome were scored every day, and samples for IL-17 cytokine measurement were collected at day 6 (Fig. 6A). C25-140 was able to significantly reduce the “cumulative score” (Fig. 6B), the overall “thickness score” (Fig. 6C), “scaling” (Fig. 6D), and “erythema” (Fig. 6E). Moreover, analysis of IL-17 cytokine expression in the right ear tissue proved its down-regulation as a consequence of C25-140 treatment (Fig. 6F). This first efficacy study demonstrates that C25-140, an inhibitor of TRAF6 activity, is able to ameliorate symptoms of autoimmune psoriasis.
Figure 6.
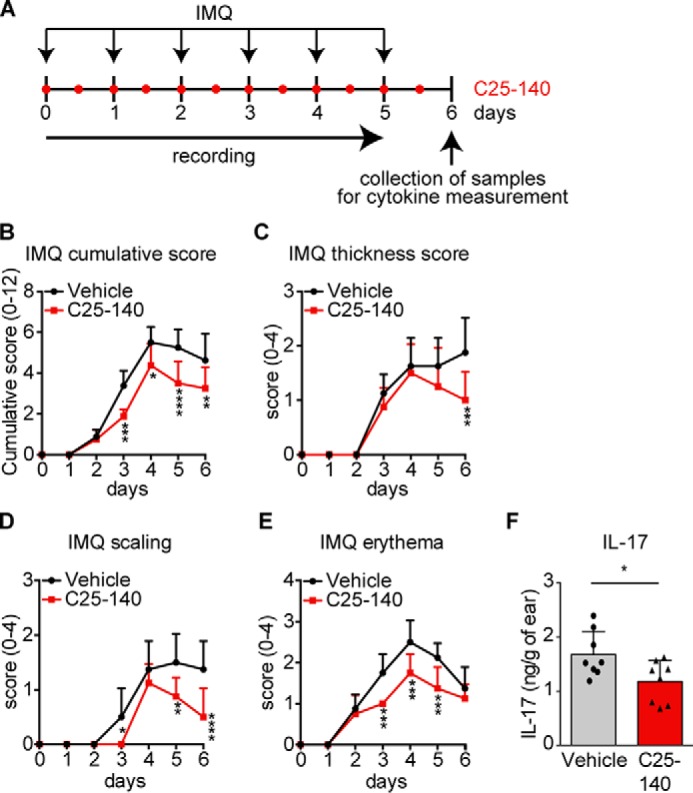
Topical application of the inhibitor C25-140 reduces in vivo outcome of imiquimod-induced psoriasis. A, preclinical mouse model for IMQ-induced psoriasis. Mice (n = 8/group) were topically treated with IMQ to the back and ear once a day. C25-140 was topically applied twice daily. Mice were scored throughout the study, and samples were collected at day 6 for cytokine measurement. B–E, mice were scored for cumulative score (B), thickness score (C), scaling (D), and erythema (E). All symptoms were improved by C25-140 treatment; error bars, S.D.; two-way ANOVA test (Sidak's multiple-comparison test). F, IL-17 cytokine levels were evaluated by ELISA and show a significant decrease after treatment; unpaired t test (two-tailed); error bars, S.D.; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
C25-140 efficacy in a preclinical mouse model for RA
As pointed out earlier, it has previously been reported that levels of TRAF6 are elevated in RA patients (16, 17). Therefore, we investigated whether C25-140 is effective in a preclinical mouse model of autoimmune rheumatoid arthritis. We chose the collagen-induced arthritis (CIA) mouse model (30) in which TRAF6 is up-regulated during disease progression and RA symptoms are reversed by the application of siRNA-based TRAF6 knockdown (17), stressing the biological relevance of this in vivo model for the TRAF6 E3 ligase functions. In our study, RA was induced by injection of collagen at day 0, followed by a collagen boost injection at day 21 to promote the development of arthritic symptoms. At day 28, compound C25-140 was intraperitoneally administered twice daily at three doses (6, 10, and 14 mg/kg) over a period of 14 days. In RA, patients usually receive treatment when symptoms become apparent. Thus, we started dosing C25-140 in the CIA model when mice already had developed symptoms of RA (arthritic index of ∼3), thereby reflecting the diseased patient. Prednisolone served as a positive control. Mice were daily scored for arthritic index and euthanized on day 42 to collect limbs for histopathology (Fig. 7A). Throughout the entire study, the body weight of all mice was monitored and did not show any reduction, indicating that there were no signs of obvious toxicity (Fig. S6). Intriguingly, C25-140 ameliorated the arthritic index to almost baseline levels in this efficacy model at doses of 10 and 14 mg/kg (Fig. 7B). Tissue sections of the limbs (Fig. 7C and Fig. S7) evidently verified that C25-140 dose-dependently improved symptoms of RA including inflammation and structural damage. In addition, all sections were quantified based on parameters of disease appearance, such as summed scores, inflammation, pannus, cartilage damage, and several more (Fig. 7 (D and E) and Fig. S8). These data also underlined the dose-dependent improvement of RA disease outcome. Taken together, we present a successful preclinical proof-of-concept study for C25-140 in an RA efficacy model.
Figure 7.

C25-140 ameliorates symptoms of RA in a preclinical mouse model. A, study design of a CIA preclinical mouse model for RA (n = 10/group). RA was induced by collagen application at day 0 and a booster injection at day 21. At day 28, C25-140 was intraperitoneally applied twice daily for 14 days. Mice were scored throughout the study, and limbs were collected at day 14 of treatment for histopathology. B, the average arthritic index was determined for each group to analyze efficacy of treatment. C25-140 and the control prednisolone significantly reduced the arthritic index; error bars, S.E.; two-way ANOVA test (Sidak's multiple-comparison test). C, in histopathology analyses, mice were scored for hematoxylin and eosin staining of sections for histopathology (ankle and hind paw). C25-140 at 14 mg/kg ameliorates disease outcome; more histology images are shown in Fig. S7. D, summed histology score. E, inflammation, pannus, and cartilage damage. All quantified parameters demonstrate a dose-dependent improvement of RA symptoms after C25-140 treatment; error bars, S.E.; one-way ANOVA test (Sidak's multiple-comparison test); more quantification of histology data is shown in Fig. S8; ns, not significant; ***, p < 0.001; ****, p < 0.0001; BID, twice daily.
Discussion
In this report, we present a high-throughput screening campaign that successfully discovered a novel PPI inhibitor, namely C25-140, which is capable of reducing TRAF6 E3 ligase activity by interfering with the TRAF6–Ubc13 interaction. Moreover, C25-140 exhibits inhibitory effects on various immune and inflammatory signaling pathways in cell lines as well as primary human and murine cells. Notably, inhibition of these pathways should be beneficial for treating a highly inflamed immune system. In line with this notion, C25-140 ameliorated symptoms of autoimmune diseases in in vivo preclinical efficacy models of psoriasis and RA. Hence, our reverse chemical genetics approach presented in this study underlines the importance of TRAF6 activity for the development and persistence of autoimmune diseases, such as psoriasis and RA.
TRAF6 belongs to the RING type of E3 ligases, where the RING domain makes a protein–protein interaction with the ubiquitin-charged Ubc13 protein. Here, the ubiquitin is transferred from Ubc13 (E2) to the substrate, and TRAF6 acts as a scaffold bridging these two proteins (15). Therefore, targeting the E3 activity of TRAF6 requires PPI inhibitors, which interfere with the binding of TRAF6 to Ubc13 (7). PPI inhibitors have been very difficult to target in drug discovery, as the interaction interfaces are usually large and contain many hydrophobic residues (31–33). However, PPIs comprise hot spot regions, and genetic point mutation can lead to complete disruption of binding, thereby suggesting that small molecules have the chance to interfere with these interfaces (33). Indeed, crystal structures of TRAF6–Ubc13 binding clearly imply that distinct amino acids of TRAF6 (e.g. Cys70 or Asp57) are crucial for its interaction with Ubc13 (10). Single point mutations of these amino acids entirely abrogated TRAF6–Ubc13 interaction (Fig. S1A) (10), emphasizing that there is a good chance to impede the TRAF6–Ubc13 PPI.
The goal of this study was to identify a selective inhibitor of TRAF6 activity that can be applied to chemical biology as well as drug discovery studies. Our selectivity profiling on other E3 ligases, including RING (e.g. cIAP1 and MDM2) and HECT (e.g. ITCH and E6AP) types of E3 ligases, revealed that C25-140 comprises good selectivity for TRAF6 over other E3 ligases with the exception that it also inhibits cIAP1, a RING E3 ligase generating Lys63 ubiquitin chains, similar to TRAF6. Importantly, C25-140 did not interfere with Lys48-linked polyubiquitin chain formation, which is critical to maintain cellular protein homeostasis. The compound also did not inhibit several other E1–E2 reactions including UbcH13, underlining that it acts on the E3 ligase side of the PPI. Hence, C25-140 is fairly selective and does not act as a broadband inhibitor of E3 ligases.
The inhibition of TNFα signaling by C25-140 was unexpected, as this particular receptor activation does not employ TRAF6 for signal progression toward NF-κB (34). This finding, however, can be explained by the fact that C25-140 also inhibits cIAP1 E3 ligase activity, which is a central regulator of TNFα receptor signaling (26). In fact, this C25-140 off-target activity may eventually be beneficial for the treatment of autoimmune and inflammatory diseases, as uncontrolled function of TNFα signaling has been shown to contribute greatly to the development of these diseases (35). Therefore, our study provides a novel small molecule that counteracts TRAF6–Ubc13 interaction as a target to reduce NF-κB signaling by various receptors (IL-1β receptor, TLR4, T-cell receptor, and TNFα receptor), thereby encompassing cumulative effects on signaling processes that drive chronic inflammation.
Current therapeutics for RA are mostly disease-modifying anti-rheumatic drugs and glucocorticoids with broad and diffuse immunosuppressive effects in the early stage (36–38) with the addition of antibody/biological therapy against specific surface targets (39, 40) at later stages. The only approved small molecule with target-based function against RA is tofacitinib, a Janus kinase inhibitor (41–43), which however causes serious side effects. Thus, the field relies on the discovery of additional approaches that use small molecules against novel disease-related targets. Accordingly, C25-140 is a novel small-molecule inhibitor that interferes with a distinct target (i.e. TRAF6 activity), which is critical for activation of various NF-κB signaling pathways. This novel strategy effectively leads to reduction of pro-inflammatory signaling to improve RA disease outcome.
In the past, some inhibitors of MYD88-mediated receptor signaling have been described, including IRAK4 activity (44), Ubc13–Uev1a interaction (45), MYD88 dimerization (46), and Ubc13 (47), but targeting the E3 ligase activity of TRAF6 has never been accomplished, as its ligase activity is determined by the E2–E3 protein–protein interaction. In general, inhibition of E2–E3 interactions have been challenging, and only inhibitors of an acetylated E2 together with a Cullin type E3 ligase have been reported (48). In contrast, our study describes the first-in-class protein–protein interaction inhibitor of a RING-E3 ligase (TRAF6)–E2 enzyme (Ubc13) binding and presents a novel strategy to treat autoimmune and chronic inflammatory diseases.
Experimental procedures
Plasmids, cell lines, antibodies, and compounds
Cell lines
HEK293T and MEF were grown in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 100 units/ml penicillin/streptomycin (Gibco). Jurkat cells were grown in RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco) and 100 units/ml penicillin/streptomycin (Gibco). Cells were rethawed every 3–4 weeks. Human PBMCs were cultured in RPMI supplemented with 10% FBS, 100 units/ml penicillin/streptomycin, and 50 μm β-mercaptoethanol (Gibco). Primary mouse CD4+ T cells were cultured as described previously (49).
Plasmids
cDNA of TRAF6 RZ 12 (residues 50–187) was cloned into pEF-HA backbone vector (50). cDNA of TRAF6 RZ1 (residues 50–159) or the TRAF6 D57K mutant was cloned into pASK IBA 3+ (IBA Lifesciences) and pGEX4T1 (GE Healthcare). Full-length Ubc13 was cloned into the pGEX4T1 vector with and without C-terminal FLAG-His tag. OTUB1 full-length cDNA was cloned into pGEX4T1, and Uev1a was cloned into pET28a vector.
Antibodies
These antibodies were used for Western blotting: β-actin I-19 (Santa Cruz Biotechnology, sc-1616; 1:1000); p-IκBα Ser32/36 5A5 (Cell Signaling, catalog no. 9246; 1:1000); IκBα C21 (Santa Cruz Biotechnology, Inc., sc-371; 1:1000) for MEF cells; IκBα L35A5 (Cell Signaling, catalog no. 4814; 1:1000) for Jurkat T cells; RNF4 (Abnova A01; 1:2000); TRAF6 (Abcam EP591Y; 1:1000); Ubc13 (Invitrogen, 37-1100; 1:1000); and ubiquitin P4D1 (Santa Cruz Biotechnology, sc-8017; 1:1000). Anti-HA affinity matrix (Roche Diagnostics; 1:40) was used for immunoprecipitation.
Compounds
All compounds were purchased from ChemDiv: C25 (catalog no. G827-0171), C25-140 (catalog no. G827-0140), C25-167 (catalog no. G827-0167), C25-189 (catalog no. G827-0189), C25-031 (catalog no. G827-0031), C25-166 (catalog no. G827-0166), C25-054 (catalog no. G827-0054), and C25-024 (catalog no. G827-0024).
For all experiments, C25-140 was resynthesized to ensure quality control for working with the correct molecule (synthesis procedure described below).
Recombinant protein expression and purification
Purification of Strep-tagged TRAF6 RZ1 protein
The plasmid was transformed into BL21 codon plus RIPL cells (Agilent Technologies) and cultured in Luria/Miller medium containing ampicillin and chloramphenicol. Protein production was induced at an A600 = 0.6–0.8 by adding 200 μg of anhydrotetracycline, 0.5 mm isopropyl-β-d-thiogalactopyranoside, and 100 μm ZnCl2. Cells were incubated at 18 °C for 16 h. Afterward, cells were centrifuged at 2600 × g for 20 min at 4 °C. The pellet was resuspended in 10 ml of Strep-wash buffer (IBA Lifesciences), including 1 pill of protease inhibitor Complete mini (Roche Diagnostics). The suspension was sonified (8 × 30 s) and subsequently centrifuged at 53,000 × g two times for 30 min each. For the purification of the StrepII-tagged proteins, the protein solution was applied to Strep-Tactin columns using the ÄKTA purifier (GE Healthcare). After washing, the StrepII-protein was eluted from the column using the Strep-elution buffer (IBA Lifesciences) containing desthiobiotin. The eluted protein was concentrated using Amicon filters (cutoff 3 kDa) down to 1 ml. Using the ÄKTA purifier and a HiTrap desalting 5-ml column (GE Healthcare), the protein was desalted and exchanged to storage buffer (20 mm Tris-HCl, pH 8.0, 20 mm NaCl, 1 mm 1,4-DTT, and 100 μm ZnCl2). The protein was concentrated in Amicon filters up to 2 μg/μl and stored in small aliquots at −80 °C.
Purification of untagged TRAF6 RZ1 protein
The cDNA sequence of WT TRAF6 RZ1 domain was cloned into the pGEX4T1 vector (GE Healthcare Life Sciences). For the purification of untagged TRAF6 RZ1, the protein was expressed in BL21 codon plus RIPL cells in M9 minimal medium. For 15N-labeled protein, the regular NH4 was replaced by 15NH4. For protein production, 1 mm isopropyl-β-d-thiogalactopyranoside and 100 μm ZnCl2 were added at A600 = 0.6–0.7. After 16 h at 18 °C, cells were centrifuged, and the pellet was resuspended in 10 ml of TRAF6 wash buffer (20 mm Tris, pH 8.0, 20 mm NaCl), including 1 Complete mini pill (Roche Diagnostics). After sonification (8 × 30 s) and centrifugation (2 × 30 min at 53,000 × g), the protein suspension was incubated with 1 ml of prewashed GSH Sepharose 4 Fast Flow beads (GE Healthcare) for 2 h at 4 °C. Subsequently, beads were washed with 100 ml of TRAF6 wash buffer and then incubated with 50 units of thrombin for 3 h at room temperature to cleave off the GST tag. The TRAF6 protein was eluted in 5 ml of wash buffer and cleared for remaining GST with 1 ml of GSH Sepharose 4 fast flow beads. Afterward, the solution was centrifuged for 5 min at 200 × g, and the supernatant was transferred into an Amicon filter for concentration, followed by centrifugation at 20,000 × g for 30 min. The supernatant was then applied to a HiLoad Superdex75 column (GE Healthcare) using the ÄKTA purifier for separating the untagged TRAF6 protein from uncleaved protein as well as buffer exchange in desalting buffer (2 mm Tris, pH 8.0, 20 mm NaCl, 1 mm DTT, and 100 μm ZnCl2). These proteins were used for 2D NMR and in vitro ubiquitination experiments. FLAG-His-tagged Ubc13 protein (Ubc13-FH) and GST proteins (including GST-OTUB1 and GST-Ubc13) were produced as described previously (51).
His-tagged Uev1a protein was purified according to the GST-OTUB1 protocol with the following buffers: lysis buffer (20 mm NaH2PO4, 20 mm NaCl, 50 mm imidazole), elution buffer (20 mm NaH2PO4, 20 mm NaCl, 500 mm imidazole), and desalting buffer (20 mm NaH2PO4, 20 mm NaCl). Ni-Sepharose 6 Fast Flow (GE Healthcare) was used instead.
AlphaScreen® assay for compound screening of TRAF6–Ubc13 PPI inhibitors
For performance of an automated AlphaScreen® assay, all of the following components were prediluted in AlphaScreen® buffer (1× PBS, 0.5% BSA, 0.01% Tween 20). First, the TRAF6 protein (end concentration 100 nm, 30-μl volume) was added to 384-well Opti-plates (PerkinElmer Life Sciences) involving the MultiFlo dispensing system (BioTek) followed by transfer of the compounds via the Sciclone G3 transfer station (PerkinElmer Life Sciences). After the addition of Ubc13-FH (end concentration 75 nm, 10-μl volume) with the MultiFlo system and incubation for 1 h at room temperature, both beads (Strep-Tactin Alpha Donor beads and nickel-chelate acceptor beads; end concentrations 4 μg each, 10-μl volume each, PerkinElmer Life Sciences) were added using the MultiFlo system in subdued light. Readout of the plates occurred after another hour of incubation at room temperature. Statistical parameters, including the coefficient of variation, Z′ factor, and signal window, were calculated to determine the quality of the assay. To evaluate the efficacy of the compounds, AlphaScreen® units of compound-treated samples were referred to DMSO control-treated samples. Thereby, the TRAF6D57K mutant served as the control for minimum signal and was included on every plate of the screening. Compounds that inhibited TRAF6WTStrepII-Ubc13-FH by more than 25% were considered active. After elimination of AlphaScreen® frequent hitter compounds and His-tag frequent hitters (23), 178 compounds were defined as primary hits and were subsequently tested in five-point serial dilution assays (2.5–40 μm) in TRAF6WTStrepII-Ubc13-FH AlphaScreen experiments. Only compounds with dose-dependent effects on TRAF6WTStrepII-Ubc13-FH (n = 27) were further taken.
AlphaScreen® counterassays involved the interactions of GST-OTUB1–Ubc13-FH and GST-Ubc13–Uev1a-His. For each assay, the GST-tagged protein was applied at a final concentration of 30 nm (30 μl), wherein the His-tagged protein was used at a concentration of 20 nm (10 μl). GSH-donor beads and nickel-chelate acceptor beads (PerkinElmer Life Sciences) were used at a final concentration of 3 μg/ml (10 μl each).
2D NMR
To determine whether the inhibitory compounds directly bind to TRAF6 rather than to Ubc13, 2D NMR experiments were conducted. Untagged 15N-labeled TRAF6 RZ1 protein (c = 120 μm) and compound (c = 20 mm stock) were incubated at a ratio of 1:5 in a 3-mm NMR tube for 10 min before acquisition of spectra. To observe chemical shift perturbations upon compound addition, two-dimensional SOFAST-HMQC spectra were acquired. All measurements were performed using the Bruker Avance 600-MHz spectrometer.
In vitro ubiquitination assays (Western blotting–based)
The ability of TRAF6 to form Lys63-linked polyubiquitin chains in conjunction with Ubc13 was analyzed after compound treatment. Therefore, untagged TRAF6WT as well as TRAF6D57K were recombinantly purified. 0.125 μm TRAF6 protein was pre-incubated with DMSO or compound in a total volume of 100 μl in Lys63 assay buffer (25 nm Tris-HCl, pH 7.6, 250 nm MgCl2, 500 nm creatine phosphate, 0.3 units/ml inorganic pyrophosphatase, 0.3 units/ml creatine phosphokinase) for 30 min at room temperature. Small aliquots for input samples were taken. A master mix containing 0.01 μm E1-activating enzyme (UBE1) (Boston Biochem), 0.2 μm E2-conjugating enzyme complex (Ubc13–Uev1a) (Boston Biochem), 1 mm ZnCl2, 2 mm ATP, and 4 μm mono-ubiquitin (Boston Biochem) was added to the TRAF6 protein, and the reaction mixture was incubated for 120 min at 37 °C. The reaction was stopped by adding 4× SDS loading buffer and boiling at 95 °C for 5 min. The input samples were analyzed using the Pierce silver stain kit (Thermo Fisher Scientific). Ubiquitination was detected in Western blot analysis using a ubiquitin antibody (52).
For analysis of cIAP1-, MDM2-, TRIM63-, RNF4-, ITCH-, and E6AP-mediated polyubiquitination, the respective ubiquitin ligase kit from Boston Biochem (K-102, K-260, K-200B, K-220, K-240, and K-270) was used. Here, reaction buffer, E1-activating enzyme, E2-conjugating enzyme, and E3 ligase and substrate (p53 for MDM2, S5A for TRIM63, and S5A for E6AP) were mixed and pre-incubated with compound or DMSO for 5 min at room temperature before adding Mg2+-ATP and mono-ubiquitin for the reaction start. Ubiquitination was visualized by Western blotting as described before (52).
E1–E2 reactions
UbcH (E2) enzyme kit (K-980B) and E1 enzyme (UBE1, e-305) were purchased from Boston Biochem. Ubiquitination assays were set as described above for cIAP1, but only involving E1–E2 enzymes. After 30 min of incubation at 37 °C, the reaction was stopped and analyzed by Western blotting, staining for ubiquitin P4D1.
AlphaSurefire
To detect protein levels of p-IκBα (Ser32/36) as well as total IκBα, 1 × 104 MEFs were seeded in 96-well plates, treated with compound for 6 h, and stimulated with 1 ng/ml IL-1β for 7 min. The p-IκBα and total IκBα protein levels were analyzed using the AlphaSurefire kits (PerkinElmer Life Sciences), following the manufacturer's instructions. The general two-plate protocol was conducted.
Co-immunoprecipitation studies in HEK293T cells
To study the interaction of TRAF6 and Ubc13 in cells, 1 × 106 293T cells were seeded, and after 18 h, cells were transfected with 1 μg of pEF-HA empty or pEF-HA-TRAF6 RZ12 plasmid using the X-tremeGENE HP DNA transfection reagent (Roche Diagnostics). 6 h after transfection, cells were treated with compound or DMSO. After 42 h of further incubation, cells were harvested in co-IP lysis buffer (150 mm NaCl, 25 mm Hepes (pH 7.5), 0.2% Nonidet P-40, 1 mm glycerol, 10 mm NaF, 8 mm β-glycerophosphate, 1 mm DTT, 300 μm sodium vanadate, complete protease inhibitor), and HA-TRAF6 was immunoprecipitated with anti-HA affinity matrix (Roche Diagnostics). Binding of Ubc13 to TRAF6 was examined by Western blotting.
Immunoprecipitation of TRAF6 for detection of endogenous ubiquitination
2.5 × 106 MEF cells were seeded, treated with compound for 6 h, and stimulated with IL-1β (3.5 ng/ml) for 10 min. For Jurkat T cells, 2.5 × 106 cells were seeded, treated with compound for 6 h, and stimulated with PMA (400 ng/ml) and ionomycin (600 ng/ml) (P/I) for 10 min. Cells were lysed in co-IP lysis buffer containing 1% SDS to eliminate all cellular interactions. After lysis, centrifugation, and collection of input samples, the samples were diluted to 0.1% SDS and incubated with 5 μl of TRAF6 antibody EP591Y (Abcam) and immunoprecipitated with Protein G–Sepharose beads (GE Healthcare). After washing of beads in co-IP wash buffer (150 mm NaCl, 25 mm Hepes (pH 7.5), 0.2% Nonidet P-40, 1 mm glycerol), TRAF6 protein was eluted, and levels of TRAF6 ubiquitination were detected by Western blotting.
Densitometric quantification of Western blotting bands
Western blots were densitometrically quantified using the LabImage1D software provided by Kapelan Bio-Imaging. Here, background-subtracted values of the protein of interest were calculated relative to the background-subtracted values of the control protein.
Analysis of mRNA expression
For MEF cells, 1.2 × 105 cells/sample were seeded, treated with compound for 6 h, and stimulated with either IL-1β (1 ng/ml) or TNFα (10 ng/ml) for 60 min. Jurkat T cells and primary mouse CD4+ T cells were seeded at 2 × 105 cells/sample, also treated with compound for 6 h, and stimulated with P/I (3h) or CD3/CD28 (4h). RNA was isolated according to the manufacturer's protocol of the Qiagen RNeasy kit (MEF cells) or the STRATEC INVITRAP® spin universal RNA minikit (T cells). To clear isolated RNA from genomic DNA, samples (up to 5 μg of RNA) were treated with RQ1 RNase-Free DNase I (Promega) as described in the technical manual. For the reverse transcription of RNA into cDNA, the DNase I–digested RNA samples were processed using the SuperScript III first strand cDNA synthesis system (Invitrogen/Thermo Fisher Scientific) following the manufacturer's protocol. Here, random hexamers provided with the kit were used for reverse transcription. cDNA samples were stored at −20 °C. For quantification of cDNA in the samples, the LightCylcer480 (Roche Applied Science) and the KAPA SYBR FAST quantitative PCR kit were used as described earlier (52). For relative quantification, the ΔΔCp method first described by Pfaffl (53) was used. The primers used for qRT-PCR are listed in Table 1.
Table 1.
Primers used for qRT-PCR
| Primer | Sequence | Sequence ID number |
|---|---|---|
| Mouse A20 for | 5′-GCT CAA CTG GTG TCG TGA AG-3′ | 5 |
| Mouse A20 rev | 5′-ATG AGG CAG TTT CCA TCA CC-3′ | 6 |
| Mouse β-actin for | 5′-CCT CTA TGC CAA CAC AGT GC-3′ | 7 |
| Mouse β-actin rev | 5′-GTA CTC CTG CTT GCT GAT CC-3′ | 8 |
| Mouse ICAM-1 for | 5′-CGC TCA GAA GAA CCA CCT TC-3′ | 9 |
| Mouse ICAM-1 rev | 5′-GGA GAC GCA GAG GAC CTT AAC-3′ | 10 |
| Mouse IL-2 for | 5′-GAG TGC CAA TTC GAT GAT GAG-3′ | 13 |
| Mouse IL-2 rev | 5′-AGG GCT TGT TGA GAT GAT GC-3′ | 14 |
| Human IL-2 for | 5′-CAC AGC TAC AAC TGG AGC ATT TAC-3′ | 17 |
| Human IL-2 rev | 5′-TGC TGA TTA AGT CCC TGG GTC-3′ | 18 |
| Human TNFα for | 5′-CCC AGG GAC CTC TCT CTA ATC-3′ | 19 |
| Human TNFα rev | 5′-GCT ACA GGC TTG TCA CTC GG-3′ | 20 |
| Human RNA polymerase II for | 5′-GCA CCA CGT CCA ATG ACA-3′ | 21 |
| Human RNA polymerase II rev | 5′-GTG CGG CTG CTT CCA TAA-3′ | 22 |
ELISA
For measurement of cytokine secretion, 2 × 105 Jurkat T cells or primary mouse CD4+ T cells or human PBMCs were seeded per sample, subsequently treated with compound for 6 h, and stimulated as indicated. 20 h after stimulation, supernatants were harvested and analyzed with the IL-2, IL-1β, IL-6, and TNFα Ready-Set-Go! ELISA (second generation) kits provided by Affymetrix eBioscience.
Viability, cell cycle, ADME, PK, and safety studies
Viability assay
For cell viability experiments, the CellTiter-Glo® 2.0 system (Promega) was used. MEF cells (1.2 × 103 cells/well) and Jurkat T cells (5 × 103 cells/well) were seeded in 384-well plates, and 18 h later, cells were treated with compounds using the Sciclone G3 automation system. After a 24-h incubation, the luciferase assay was performed according to the manufacturer's manual. Signals were measured on an Envision plate reader (PerkinElmer Life Sciences).
Cell cycle analyses
For cell cycle staining, cells were seeded at 2 × 105 cells/sample and treated with compound or DMSO. After the indicated incubation time (24 or 72 h), cells were fixed in 100% ethanol and subsequently stained with the propidium iodide staining solution (0.1% Triton X-100, 20 μg/ml propidium iodide, 200 μg/ml RNase A in PBS) for 40 min in the dark. The propidium iodide fluorescence was collected using the Attune flow cytometer (Thermo Fisher Scientific), and analysis of cell cycle phases was performed using the single-cell analysis software FlowJo as described previously (54).
ADME and pharmacokinetic studies
The Bienta Enamine Biology Services of CRO Bienta/Enamine Ltd. conducted all ADME (plasma stability, plasma protein binding, microsomal stability, log D, Caco-2 assay, CYP450 inhibition, and hERG predictor assay) as well as pharmacokinetic studies (Tmax, Cmax, AUC0–240 min, mean residence time, elimination t½, elimination rate constant, volume of distribution, and clearance) of C25-140 by using their standard protocols. Pharmacokinetic measurements were done by intraperitoneal, intravenous, or peroral application of 10 mg/kg C25-140. Each time point was investigated with n = 4 mice. Bioethical permission of pharmacokinetic measurements in mice was granted under the following approval numbers: IACUC number 1 from May 19, 2015 and IACUC number 6 from August 12, 2015 to Bienta.
Mouse studies
Isolation of primary mouse CD4-positive T cells (ex vivo)
CD4+ T cells from the peripheral lymph nodes of BALB/C mice were isolated by negative magnetic activation cell sorting selection using the CD4+ T-cell isolation kit II (Miltenyi). Per sample, 2 × 105 cells were seeded and treated with compound for 6 h. Cells were stimulated with anti-CD3 (0.5 mg/ml) and anti-CD28 (1 mg/ml) on plates that were pre-coated with rabbit anti-hamster IgG. Samples for qRT-PCR were harvested after 4 h whereas supernatants for analysis of cytokine secretion were harvested 20 h after stimulation.
Psoriasis mouse study (in vivo)
To evaluate the anti-inflammatory activity of the compound C25-140, an Imiquimod (IMQ)-induced psoriasis mouse model was carried out by the CRO Washington Biotechnology, Inc. (Baltimore, MD). Per group (vehicle– and C25-140–treated), eight male BALB/c mice were shaved in the back (1.5 × 2 cm), and IMQ and C25-140 were applied topically to the shaved back and the right ear every day for 6 days. Here, C25-140 was dissolved in acetone and applied at a total dosage of 500 μg/day (2 × 250 μg), representing a total of ∼1.5 mg/kg per application. Mice were monitored, and daily scores were independently collected on a scale from 0 to 4: 0 = none; 1 = slight; 2 = moderate; 3 = marked; and 4 = very marked. Ear thickness was measured by electronic calipers as an indicator of edema. IL-17 cytokine secretion of the right ear tissue was measured by ELISA. Bioethical permission for the IMQ-induced psoriasis study was granted under IACUC approval number 17-063 (to W. B.).
Collagen-induced arthritis model (in vivo)
To test the potential of C25-140 to ameliorate RA symptoms, a CIA model in DBA1/J mice was performed by the CRO Washington Biotechnology, Inc. (Baltimore, MD). Each group of 10 mice was subjected to a single subcutaneous injection of collagen/complete Freund's adjuvant emulsion (0.05 ml/mouse; 100 μg of collagen/CFA). After 20 days (day 21), mice were booster-injected with collagen (0.05 ml/mouse; 100 μg/mouse of collagen in incomplete Freund's adjuvant). On day 28, mice were scored for their arthritic index and for a subsequent 14 days intraperitoneally dosed twice daily with vehicle: 6 mg/kg C25-140, 10 mg/kg C25-140, and 14 mg/kg C25-140. 10 mg/kg prednisolone was applied once daily. Mice were daily scored for macroscopic signs of arthritis. Thereby, the following scoring index was applied to each individual paw: 0 = no visible effects of arthritis; 1 = edema and/or erythema of one digit; 2 = edema and/or erythema of two digits; 3 = edema and/or erythema of more than two digits; 4 = severe arthritis of entire paw and digits. Single paw scores were added and recorded. On day 42, animals were euthanized, limbs were collected, and histopathology was performed using toluidine blue staining with scoring (from 0 to 5) of several parameters (summed score, inflammation, pannus, bone resorption, cartilage damage, periosteal bone formation, and bone width). Bioethical permission for the collagen-induced arthritis study was granted under IACUC approval number 17-059 (to W. B.).
Experiments with human primary PBMCs
For isolation of PBMCs from 50 ml of human blood of three different donors, blood was mixed with 800 units of heparin (Sigma-Aldrich) and centrifuged at 300 × g for 10 min without brake. After removal of the plasma fraction, buffy coat was diluted in 2 volumes of PBS and added on top of 15 ml of Lymphoprep (STEMCELL Technologies). After centrifugation at 160 × g for 20 min at room temperature without brake, platelets were removed, and the cell suspension was centrifuged again at 350 × g for 20 min without brake. Mononuclear cells from the intermediate layer were transferred to a new Falcon tube and washed three times in PBS supplemented with 0.1% BSA and 2 mm EDTA. In the end, cells were resuspended in RPMI medium containing 10% fetal calf serum, 1% penicillin/streptomycin, and 50 μm β-mercaptoethanol. Per sample, 2 × 105 cells were seeded and treated with compound for 6 h. The cells were stimulated with either 1 μg/ml lipopolysaccharide (LPS) (Sigma-Aldrich); 20 ng/ml IL-1β (R&D Systems) or CD3/CD28 (1 μg of hCD3 (IgG2A), 4 μg of hCD28 (IgG1), 2 μg of anti-IgG2A, and 2 μg of anti-IgG2A; all from BD Pharmingen). Supernatants for analysis of cytokine secretion were harvested 20 h after stimulation. The permission to isolate, treat, and analyze human peripheral white blood cells ex vivo was granted by the ethics commission of the Technical University Munich (project number 358/15).
Ethical statement
Mouse studies
Bioethical permission for pharmacokinetic measurements in mice was granted to Bienta/Enamine under the following approval numbers: IACUC number 1 from May 19, 2015 and IACUC number 6 from August 12, 2015. Bioethical permission for the IMQ-induced psoriasis study was granted to Washington Biotechnology, Inc. under IACUC approval number 17-063. Bioethical permission for the CIA-induced RA study was granted to Washington Biotechechnology, Inc. under IACUC approval number 17-059.
Human ex vivo studies (PBMCs)
The permission to isolate, treat, and analyze peripheral white blood cells from healthy donors ex vivo was granted by the ethics commission of the Technical University Munich (project number 358/15). The studies abided by the Declaration of Helsinki principles.
Synthesis procedure for C25-140
All solvents were purchased from Sigma-Aldrich and were used as received; anhydrous solvents were used for reactions, and HPLC grade solvents were used for aqueous work-ups, recrystallizations, and chromatography. Other reagents were purchased from various vendors and were used as received. Glassware was dried by keeping it in an oven at 70 °C for 12 h before use. The pH of aqueous solutions was estimated using pH paper. Vacuum filtrations were carried out using a house vacuum line. In the individual procedures, the phrase “concentrated in vacuo” means that solvent was removed on a rotary evaporator using a diaphragm pump (with an automatic vacuum regulator) and then remaining traces of volatiles were removed on a high-vacuum (<1 torr) oil pump.
Reactions were monitored by TLC using EMD silica gel 60 F254 (250 μm) glass-backed plates (visualized by UV fluorescence) and by the LC-MS Waters Acquity H UPLC CLASS system, with an Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm), eluting at 0.8 ml/min, using a 3-min linear gradient method with a mobile phase consisting of water/acetonitrile (0.05% (v/v) TFA added to each): 95:5 → 5:95 (0–2.25 min), 95:5 (2.27–3 min). Sample runs were monitored using alternating positive/negative electrospray ionization (50–1000 atomic mass units) and UV detection at 254 nm. Automated preparative normal-phase chromatography was carried out on a Büchi Reveleris PREP purification system with three-channel viable UV-visible and evaporative light scattering detection (runs were monitored at 220–400 nm). Prepacked silica gel cartridges (12, 25, and 40 g) were employed for normal-phase chromatography, eluting at 20–30 ml/min. 1H NMR spectra were recorded at 400 MHz on a Bruker spectrometer and are reported in ppm using the residual solvent signal (DMSO-d6 = 2.50 ppm) as an internal standard. Data are reported as follows: ((δ shift), ((s = singlet, d = doublet, dd = doublet of doublets, br = broad, m = multiplet), (J = coupling constant in Hz) and (integration))) in Schemes 1–4.
Scheme 1.
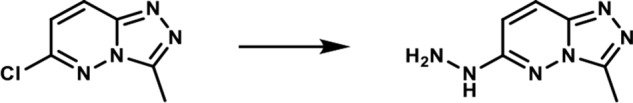
1-(3-Methyl-[1,2,4]-triazolo-[4,3-b]-pyridazin-6-yl)-hydrazine. A round-bottomed flask was charged with 6-chloro-3-methyl-[1,2,4]-triazolo-[4,3-b]-pyridazine (2.5 g, 0.015 mol) and ethanol (35 ml). To the resulting solution was added the hydrazine hydrate (2.6 ml, 0.053 mol). The reaction mixture was then heated at 80 °C for 3 h. After cooling to room temperature, the formed product slurry was filtered, washed with little ethanol, and air-dried to afford 2.43 g (98%) of the desired product as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (br s, 1H), 7.86 (d, J = 10 Hz; 1H), 6.78 (d, J = 10 Hz; 1H), 4.28 (br s, 2H), 2.55 (s, 3H); LC-MS (ESI+) m/z: [M + H]+ Calcd. for C6H9N6 165.17 found 165.20, tR = 0.17 min.
Scheme 2.
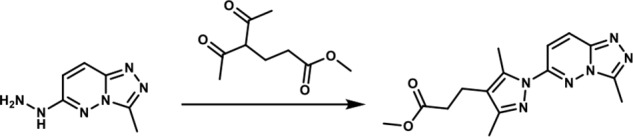
Methyl 3-(3,5-dimethyl-1-(3-methyl-[1,2,4]-triazolo-[4,3-b]-pyridazin-6-yl)-1H-pyrazol-4-yl)-propanoate. A round-bottomed flask filled with 1-(3-methyl-[1,2,4]-triazolo-[4,3-b]-pyridazin-6-yl)-hydrazine (190 mg, 1.066 mmol) and glacial acetic acid (1.90 ml) was added methyl 4-acetyl-5-oxohexanoate (0.22 mg, 1.17 mmol). The reaction mixture was stirred at room temperature for 15 min, and then the acetic acid was removed in vacuo. An aqueous solution of 5% sodium bicarbonate solution was added and the mixture was extracted with ethyl acetate (3×). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue obtained was purified by flash chromatography eluting with a gradient of ethyl acetate/methanol 100:00 → 90:10 as eluent. The combination of the appropriate fractions yielded 243 mg (66%) of the title compound as a light yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 8.37 (d, J = 10 Hz; 1H), 7.85 (d, J = 10 Hz; 1H), 3.6 (s, 3H), 2.73–2.66 (br m, 5H), 2.6 (s, 3H), 2.55–2.48 (br m, 2H), 2.24 (s, 3H); LC-MS (ESI+) m/z: [M + H]+ Calcd. for C15H19N6O2 315.35 found 315.40, tR = 0.98 min.
Scheme 3.
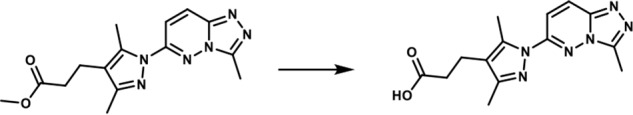
3-(3,5-Dimethyl-1-(3-methyl-[1,2,4]-triazolo-[4,3-b]-pyridazin-6-yl)-1H-pyrazol-4-yl)-propanoic acid. To a solution of methyl 3-(3,5-dimethyl-1-(3-methyl-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-1H-pyrazol-4-yl)propanoate (175 mg, 0.557 mmol) in THF (1.75 ml) and water (0.7 ml) was added LiOH·H2O (70.16 mg, 1.671 mmol). The reaction mixture was stirred at room temperature for 20 h. A 3 n HCl solution (8 ml) was added, and the mixture was extracted with a mixture of 20% isopropyl alcohol in dichloromethane (3×). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated in vacuo. The white solid obtained (140 mg, 84%) was used without further purification. 1H NMR (400 MHz, DMSO-d6): δ 12.19 (s, 1H), 8.36 (d, J = 10 Hz; 1H), 7.85 (d, J = 10 Hz; 1H), 2.76–2.66 (br m, 5H), 2.6 (s, 3H), 2.55–2.48 (br m, 2H), 2.24 (s, 3H); LC-MS (ESI+) m/z: [M + H]+ Calcd. for C14H17N6O2 301.32 found 301.40, tR = 0.84 min.
Scheme 4.
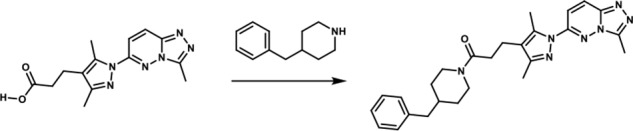
1-(4-Benzylpiperidin-1-yl)-3-(3,5-dimethyl-1-(3-methyl-[1,2,4]-triazolo-[4,3-b]-pyridazin-6-yl)-1H-pyrazol-4-yl)-propan-1-one. To a round-bottomed flask filled with a solution of 3-(3,5-dimethyl-1-(3-methyl-[1,2,4]-triazolo-[4,3-b]-pyridazin-6-yl)-1H-pyrazol-4-yl)-propanoic acid (50 mg, 0.17 mmol), in dimethylformamide (3 ml), were added 4-benzylpiperidine (0.06 ml, 0.33 mmol), HATU (68.52 mg, 0.18 mmol)), and triethyl amine (0.04 ml, 0.306 mmol). The resulting reaction mixture was stirred at room temperature until complete consumption of the starting material (17 h) was observed. It was then diluted with water (15 ml) and extracted with ethyl acetate (3 × 10 ml). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The product was purified by flash chromatography eluting with a linear gradient of ethyl acetate/methanol 100:00 → 50:50 as eluent. The combination of the appropriate fractions yielded 55 mg (70%) of the title product: 1H NMR (400 MHz, DMSO-d6): δ 8.4 (d, J = 10 Hz, 1H), 7.88 (d, J = 10 Hz, 1H), 7.33–7.09 (br m, 3H), 7.10–6.99 (br m, 2H), 4.39 (br d, 1H), 3.76 (br d, 1H), 2.86 (t, 1H), 2.71–2.61 (br m, 6H), 2.59 (s, 3H), 2.47–2.14 (br m, 4H), 2.24 (s, 3H), 1.77–1.63 (br m, 1H), 1.59–1.43 (br m, 2H), 0.99–0.73 (br m, 2H); LC-MS (ESI+) m/z: [M + H]+ Calcd. for C26H32N7O 458.58 found 458.61, tR = 1.16 min.
Author contributions
J. K. B., O. P., M. S., D. K., and K. H. conceptualization; J. K. B., G. M. P., K. S., I. R., and M. R. data curation; J. K. B. and I. R. software; J. K. B. and K. H. formal analysis; J. K. B. and M. R. validation; J. K. B., G. M. P., K. S., I. R., M. R., I. M., C. K., L. R., O. R., G. J., M. V., and K. H. investigation; J. K. B., G. M. P., K. S., and I. R. visualization; J. K. B., G. M. P., K. S., I. M., L. R., O. R., G. J., and M. V. methodology; J. K. B., D. K., and K. H. writing-original draft; J. K. B. and K. H. writing-review and editing; O. P., M. S., D. K., and K. H. supervision; D. K. and K. H. funding acquisition; K. H. project administration.
Supplementary Material
Acknowledgments
We thank Martin Göttlicher for helpful discussions. We further thank Scarlett Dornauer and Stefanie Brandner for excellent technical assistance and Sabrina Schreiner for providing the RNF4 antibody.
This work was supported by grants from the Life Science Foundation (to K. H.) and the SFB 1054 project A04 (to D. K.). A patent for the clinical use of C25-140 has been published by the European patent office.
This article contains Figs. S1–S8.
- IL
- interleukin
- TCR
- T-cell receptor
- RA
- rheumatoid arthritis
- SUMO
- small ubiquitin-like modifier
- MEF
- mouse embryo fibroblast
- TNF
- tumor necrosis factor
- PMA
- phorbol 12-myristate 13-acetate
- P/I
- PMA/ionomycin
- PBMC
- peripheral blood mononuclear cell
- ADME
- absorption/distribution/metabolism/excretion
- PK
- pharmacokinetics
- IMQ
- imiquimod
- CIA
- collagen-induced arthritis
- PPI
- protein–protein interaction
- FBS
- fetal bovine serum
- 2D
- two-dimensional
- IP
- immunoprecipitation
- qRT-PCR
- quantitative RT-PCR
- AUC
- area under the curve
- IACUC
- institutional animal care and use4 committee
- LPS
- lipopolysaccharide.
References
- 1. Husnjak K., and Dikic I. (2012) Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 81, 291–322 10.1146/annurev-biochem-051810-094654 [DOI] [PubMed] [Google Scholar]
- 2. Swatek K. N., and Komander D. (2016) Ubiquitin modifications. Cell Res. 26, 399–422 10.1038/cr.2016.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yau R., and Rape M. (2016) The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586 10.1038/ncb3358 [DOI] [PubMed] [Google Scholar]
- 4. Napetschnig J., and Wu H. (2013) Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 42, 443–468 10.1146/annurev-biophys-083012-130338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bassères D. S., and Baldwin A. S. (2006) Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 25, 6817–6830 10.1038/sj.onc.1209942 [DOI] [PubMed] [Google Scholar]
- 6. Toubi E., and Shoenfeld Y. (2004) Toll-like receptors and their role in the development of autoimmune diseases. Autoimmunity 37, 183–188 10.1080/08916930410001704944 [DOI] [PubMed] [Google Scholar]
- 7. Landré V., Rotblat B., Melino S., Bernassola F., and Melino G. (2014) Screening for E3-ubiquitin ligase inhibitors: challenges and opportunities. Oncotarget 5, 7988–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xie P. (2013) TRAF molecules in cell signaling and in human diseases. J. Mol. Signal. 8, 7 10.1186/1750-2187-8-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zotti T., Vito P., and Stilo R. (2012) The seventh ring: exploring TRAF7 functions. J. Cell Physiol. 227, 1280–1284 10.1002/jcp.24011 [DOI] [PubMed] [Google Scholar]
- 10. Yin Q., Lin S. C., Lamothe B., Lu M., Lo Y. C., Hura G., Zheng L., Rich R. L., Campos A. D., Myszka D. G., Lenardo M. J., Darnay B. G., and Wu H. (2009) E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16, 658–666 10.1038/nsmb.1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ye H., Arron J. R., Lamothe B., Cirilli M., Kobayashi T., Shevde N. K., Segal D., Dzivenu O. K., Vologodskaia M., Yim M., Du K., Singh S., Pike J. W., Darnay B. G., Choi Y., and Wu H. (2002) Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418, 443–447 10.1038/nature00888 [DOI] [PubMed] [Google Scholar]
- 12. Deng L., Wang C., Spencer E., Yang L., Braun A., You J., Slaughter C., Pickart C., and Chen Z. J. (2000) Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361 10.1016/S0092-8674(00)00126-4 [DOI] [PubMed] [Google Scholar]
- 13. Walsh M. C., Lee J., and Choi Y. (2015) Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 266, 72–92 10.1111/imr.12302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhoj V. G., and Chen Z. J. (2009) Ubiquitylation in innate and adaptive immunity. Nature 458, 430–437 10.1038/nature07959 [DOI] [PubMed] [Google Scholar]
- 15. Deshaies R. J., and Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 10.1146/annurev.biochem.78.101807.093809 [DOI] [PubMed] [Google Scholar]
- 16. Zhu L. J., Dai L., Zheng D. H., Mo Y. Q., Ou-Yang X., Wei X. N., Shen J., and Zhang B. Y. (2012) Upregulation of tumor necrosis factor receptor-associated factor 6 correlated with synovitis severity in rheumatoid arthritis. Arthritis Res. Ther. 14, R133 10.1186/ar3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H., Chen W., Wang L., Li F., Zhang C., and Xu L. (2015) Tumor necrosis factor receptor-associated factor 6 promotes migration of rheumatoid arthritis fibroblast-like synoviocytes. Mol. Med. Rep. 11, 2761–2766 10.3892/mmr.2014.3104 [DOI] [PubMed] [Google Scholar]
- 18. Namjou B., Choi C. B., Harley I. T., Alarcón-Riquelme M. E., BIOLUPUS Network B., Kelly J. A., Glenn S. B., Ojwang J. O., Adler A., Kim K., Gallant C. J., Boackle S. A., Criswell L. A., Kimberly R. P., Brown E. E., et al. (2012) Evaluation of TRAF6 in a large multiancestral lupus cohort. Arthritis Rheum. 64, 1960–1969 10.1002/art.34361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shembade N., Ma A., and Harhaj E. W. (2010) Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327, 1135–1139 10.1126/science.1182364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lamothe B., Besse A., Campos A. D., Webster W. K., Wu H., and Darnay B. G. (2007) Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of IκB kinase activation. J. Biol. Chem. 282, 4102–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kliza K., Taumer C., Pinzuti I., Franz-Wachtel M., Kunzelmann S., Stieglitz B., Macek B., and Husnjak K. (2017) Internally tagged ubiquitin: a tool to identify linear polyubiquitin-modified proteins by mass spectrometry. Nat. Methods 14, 504–512 10.1038/nmeth.4228 [DOI] [PubMed] [Google Scholar]
- 22. Zarzycka B., Seijkens T., Nabuurs S. B., Ritschel T., Grommes J., Soehnlein O., Schrijver R., van Tiel C. M., Hackeng T. M., Weber C., Giehler F., Kieser A., Lutgens E., Vriend G., and Nicolaes G. A. (2015) Discovery of small molecule CD40-TRAF6 inhibitors. J. Chem. Inf. Model. 55, 294–307 10.1021/ci500631e [DOI] [PubMed] [Google Scholar]
- 23. Schorpp K., Rothenaigner I., Salmina E., Reinshagen J., Low T., Brenke J. K., Gopalakrishnan J., Tetko I. V., Gul S., and Hadian K. (2014) Identification of small-molecule frequent hitters from AlphaScreen high-throughput screens. J. Biomol. Screen. 19, 715–726 10.1177/1087057113516861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brenke J. K., Salmina E. S., Ringelstetter L., Dornauer S., Kuzikov M., Rothenaigner I., Schorpp K., Giehler F., Gopalakrishnan J., Kieser A., Gul S., Tetko I. V., and Hadian K. (2016) Identification of small-molecule frequent hitters of glutathione S-transferase-glutathione interaction. J. Biomol. Screen. 21, 596–607 10.1177/1087057116639992 [DOI] [PubMed] [Google Scholar]
- 25. Wiener R., Zhang X., Wang T., and Wolberger C. (2012) The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 483, 618–622 10.1038/nature10911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bertrand M. J., Milutinovic S., Dickson K. M., Ho W. C., Boudreault A., Durkin J., Gillard J. W., Jaquith J. B., Morris S. J., and Barker P. A. (2008) cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 10.1016/j.molcel.2008.05.014 [DOI] [PubMed] [Google Scholar]
- 27. Oeckinghaus A., Wegener E., Welteke V., Ferch U., Arslan S. C., Ruland J., Scheidereit C., and Krappmann D. (2007) Malt1 ubiquitination triggers NF-κB signaling upon T-cell activation. EMBO J. 26, 4634–4645 10.1038/sj.emboj.7601897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun L., Deng L., Ea C. K., Xia Z. P., and Chen Z. J. (2004) The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301 10.1016/S1097-2765(04)00236-9 [DOI] [PubMed] [Google Scholar]
- 29. Gilliet M., Conrad C., Geiges M., Cozzio A., Thürlimann W., Burg G., Nestle F. O., and Dummer R. (2004) Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch. Dermatol. 140, 1490–1495 [DOI] [PubMed] [Google Scholar]
- 30. Brand D. D., Latham K. A., and Rosloniec E. F. (2007) Collagen-induced arthritis. Nat. Protoc. 2, 1269–1275 10.1038/nprot.2007.173 [DOI] [PubMed] [Google Scholar]
- 31. Gul S., and Hadian K. (2014) Protein-protein interaction modulator drug discovery: past efforts and future opportunities using a rich source of low- and high-throughput screening assays. Expert Opin. Drug Discov. 9, 1393–1404 10.1517/17460441.2014.954544 [DOI] [PubMed] [Google Scholar]
- 32. Scott D. E., Bayly A. R., Abell C., and Skidmore J. (2016) Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 15, 533–550 10.1038/nrd.2016.29 [DOI] [PubMed] [Google Scholar]
- 33. Wells J. A., and McClendon C. L. (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–1009 10.1038/nature06526 [DOI] [PubMed] [Google Scholar]
- 34. Lomaga M. A., Yeh W. C., Sarosi I., Duncan G. S., Furlonger C., Ho A., Morony S., Capparelli C., Van G., Kaufman S., van der Heiden A., Itie A., Wakeham A., Khoo W., Sasaki T., et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13, 1015–1024 10.1101/gad.13.8.1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kalliolias G. D., and Ivashkiv L. B. (2016) TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 12, 49–62 10.1038/nrrheum.2015.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sokka T., Envalds M., and Pincus T. (2008) Treatment of rheumatoid arthritis: a global perspective on the use of antirheumatic drugs. Mod. Rheumatol. 18, 228–239 10.3109/s10165-008-0056-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Visser K., and van der Heijde D. (2009) Optimal dosage and route of administration of methotrexate in rheumatoid arthritis: a systematic review of the literature. Ann. Rheum. Dis. 68, 1094–1099 10.1136/ard.2008.092668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wevers-de Boer K., Visser K., Heimans L., Ronday H. K., Molenaar E., Groenendael J. H., Peeters A. J., Westedt M. L., Collée G., de Sonnaville P. B., Grillet B. A., Huizinga T. W., and Allaart C. F. (2012) Remission induction therapy with methotrexate and prednisone in patients with early rheumatoid and undifferentiated arthritis (the IMPROVED study). Ann. Rheum. Dis. 71, 1472–1477 10.1136/annrheumdis-2011-200736 [DOI] [PubMed] [Google Scholar]
- 39. Choy E. H., Kavanaugh A. F., and Jones S. A. (2013) The problem of choice: current biologic agents and future prospects in RA. Nat. Rev. Rheumatol. 9, 154–163 10.1038/nrrheum.2013.8 [DOI] [PubMed] [Google Scholar]
- 40. Isaacs J. D. (2015) Decade in review-clinical rheumatology: 10 years of therapeutic advances in the rheumatic diseases. Nat. Rev. Rheumatol. 11, 628–630 10.1038/nrrheum.2015.138 [DOI] [PubMed] [Google Scholar]
- 41. Changelian P. S., Flanagan M. E., Ball D. J., Kent C. R., Magnuson K. S., Martin W. H., Rizzuti B. J., Sawyer P. S., Perry B. D., Brissette W. H., McCurdy S. P., Kudlacz E. M., Conklyn M. J., Elliott E. A., Koslov E. R., et al. (2003) Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 302, 875–878 10.1126/science.1087061 [DOI] [PubMed] [Google Scholar]
- 42. Ghoreschi K., Jesson M. I., Li X., Lee J. L., Ghosh S., Alsup J. W., Warner J. D., Tanaka M., Steward-Tharp S. M., Gadina M., Thomas C. J., Minnerly J. C., Storer C. E., LaBranche T. P., Radi Z. A., et al. (2011) Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 186, 4234–4243 10.4049/jimmunol.1003668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanaka Y., and Yamaoka K. (2013) JAK inhibitor tofacitinib for treating rheumatoid arthritis: from basic to clinical. Mod. Rheumatol. 23, 415–424 10.3109/s10165-012-0799-2 [DOI] [PubMed] [Google Scholar]
- 44. Kelly P. N., Romero D. L., Yang Y., Shaffer A. L. 3rd, Chaudhary D., Robinson S., Miao W., Rui L., Westlin W. F., Kapeller R., and Staudt L. M. (2015) Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 212, 2189–2201 10.1084/jem.20151074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pulvino M., Liang Y., Oleksyn D., DeRan M., Van Pelt E., Shapiro J., Sanz I., Chen L., and Zhao J. (2012) Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood 120, 1668–1677 10.1182/blood-2012-02-406074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Olson M. A., Lee M. S., Kissner T. L., Alam S., Waugh D. S., and Saikh K. U. (2015) Discovery of small molecule inhibitors of MyD88-dependent signaling pathways using a computational screen. Sci. Rep. 5, 14246 10.1038/srep14246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Strickson S., Campbell D. G., Emmerich C. H., Knebel A., Plater L., Ritorto M. S., Shpiro N., and Cohen P. (2013) The anti-inflammatory drug BAY 11–7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem. J. 451, 427–437 10.1042/BJ20121651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scott D. C., Hammill J. T., Min J., Rhee D. Y., Connelly M., Sviderskiy V. O., Bhasin D., Chen Y., Ong S. S., Chai S. C., Goktug A. N., Huang G., Monda J. K., Low J., Kim H. S., et al. (2017) Blocking an N-terminal acetylation-dependent protein interaction inhibits an E3 ligase. Nat. Chem. Biol. 13, 850–857 10.1038/nchembio.2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meininger I., Griesbach R. A., Hu D., Gehring T., Seeholzer T., Bertossi A., Kranich J., Oeckinghaus A., Eitelhuber A. C., Greczmiel U., Gewies A., Schmidt-Supprian M., Ruland J., Brocker T., Heissmeyer V., et al. (2016) Alternative splicing of MALT1 controls signalling and activation of CD4+ T cells. Nat. Commun. 7, 11292 10.1038/ncomms11292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eitelhuber A. C., Warth S., Schimmack G., Düwel M., Hadian K., Demski K., Beisker W., Shinohara H., Kurosaki T., Heissmeyer V., and Krappmann D. (2011) Dephosphorylation of Carma1 by PP2A negatively regulates T-cell activation. EMBO J. 30, 594–605 10.1038/emboj.2010.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weber E., Rothenaigner I., Brandner S., Hadian K., and Schorpp K. (2017) A high-throughput screening strategy for development of RNF8-Ubc13 protein-protein interaction inhibitors. SLAS Discov. 22, 316–323 [DOI] [PubMed] [Google Scholar]
- 52. Vincendeau M., Hadian K., Messias A. C., Brenke J. K., Halander J., Griesbach R., Greczmiel U., Bertossi A., Stehle R., Nagel D., Demski K., Velvarska H., Niessing D., Geerlof A., Sattler M., and Krappmann D. (2016) Inhibition of canonical NF-κB signaling by a small molecule targeting NEMO-ubiquitin interaction. Sci. Rep. 6, 18934 10.1038/srep18934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schorpp K., Rothenaigner I., Maier J., Traenkle B., Rothbauer U., and Hadian K. (2016) A multiplexed high-content screening approach using the chromobody technology to identify cell cycle modulators in living cells. J. Biomol. Screen 21, 965–977 10.1177/1087057116641935 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.