

Abstract
Small ubiquitin-like modifier (SUMO) is commonly used as a protein fusion domain to facilitate expression and purification of recombinant proteins, and a SUMO-specific protease is then used to remove SUMO from these proteins. Although this protease is highly specific, its limited solubility and stability hamper its utility as an in vitro reagent. Here, we report improved SUMO protease enzymes obtained via two approaches. First, we developed a computational method and used it to re-engineer WT Ulp1 from Saccharomyces cerevisiae to improve protein solubility. Second, we discovered an improved SUMO protease via genomic mining of the thermophilic fungus Chaetomium thermophilum, as proteins from thermophilic organisms are commonly employed as reagent enzymes. Following expression in Escherichia coli, we found that these re-engineered enzymes can be more thermostable and up to 12 times more soluble, all while retaining WT-or-better levels of SUMO protease activity. The computational method we developed to design solubility-enhancing substitutions is based on the RosettaScripts application for the macromolecular modeling suite Rosetta, and it is broadly applicable for the improvement of solution properties of other proteins. Moreover, we determined the X-ray crystal structure of a SUMO protease from C. thermophilum to 1.44 Å resolution. This structure revealed that this enzyme exhibits structural and functional conservation with the S. cerevisiae SUMO protease, despite exhibiting only 28% sequence identity. In summary, by re-engineering the Ulp1 protease and discovering a SUMO protease from C. thermophilum, we have obtained proteases that are more soluble, more thermostable, and more efficient than the current commercially available Ulp1 enzyme.
Keywords: protein engineering, cysteine protease, enzyme kinetics, small ubiquitin-like modifier (SUMO), protein purification, fusion protein, protein design, protein solubility, Rosetta, thermostability
Introduction
The ability to express and purify recombinant proteins is critical to studying their structure and function. Genetic fusion to the C terminus of small ubiquitin-like modifier (SUMO)5 can chaperone folding and increase the soluble yield obtained from heterologous protein expression (1–4). An affinity tag, such as polyhistidine, is generally fused to the N terminus of SUMO to facilitate purification. The SUMO fusion is readily removed via digestion with the highly specific SUMO protease, which can leave the target protein intact without residual amino acids at its N terminus (i.e. no “scar” residues remain).
The most commonly employed SUMO fusion system utilizes the Saccharomyces cerevisiae SUMO protein Smt3 and SUMO protease Ulp1 (1, 2). In vivo, Ulp1 functions as an essential isopeptidase that is responsible for processing pre-Smt3 and generating mature Smt3 that ends with the canonical di-glycine motif. Ulp1 also functions by cleaving Smt3 off of proteins that have been conjugated post-translationally (5). Natively, Ulp1 is a 621-amino acid protein, and residues 403–621 constitute a conserved protease domain (2). This fragment can be purified without the N-terminal residues, is constitutively active, and can be used for removing genetically fused SUMO domains from recombinant protein in vitro in reactions that are analogous to the processing of pre-Smt3 (2); we will refer to this construct as Ulp1_WT. The active site is characteristic of papain-like cysteine proteases, and the catalytic triad consists of a cysteine nucleophile (Cys-580) coordinated by a histidine (His-514) and an acid (Asp-531). When functioning as a peptidase, Ulp1_WT is highly tolerant to sequence diversity at P′ residue positions; the only restriction is that the P1′ residue cannot be a proline (6).
Commercially available Ulp1_WT is prone to precipitation at room temperature and includes detergent to maintain solubility (Thermo Fisher Scientific, catalog no. 12588018). Precipitation of the enzyme can lead to incomplete digestion of SUMO fusion constructs as well as potentially nucleate aggregation of target proteins. To uncover determinants of Ulp1_WT that lead to poor solubility, we analyzed the structure of Ulp1_WT (PDB code 1EUV) using Rosetta and identified 10 hydrophobic residues that project outward toward solvent from the protein surface, not including residues involved in SUMO binding or the active site (Fig. 1A and Table 1). These residues are far enough from functional sites that they are unlikely to contribute to protease activity in vitro.
Figure 1.
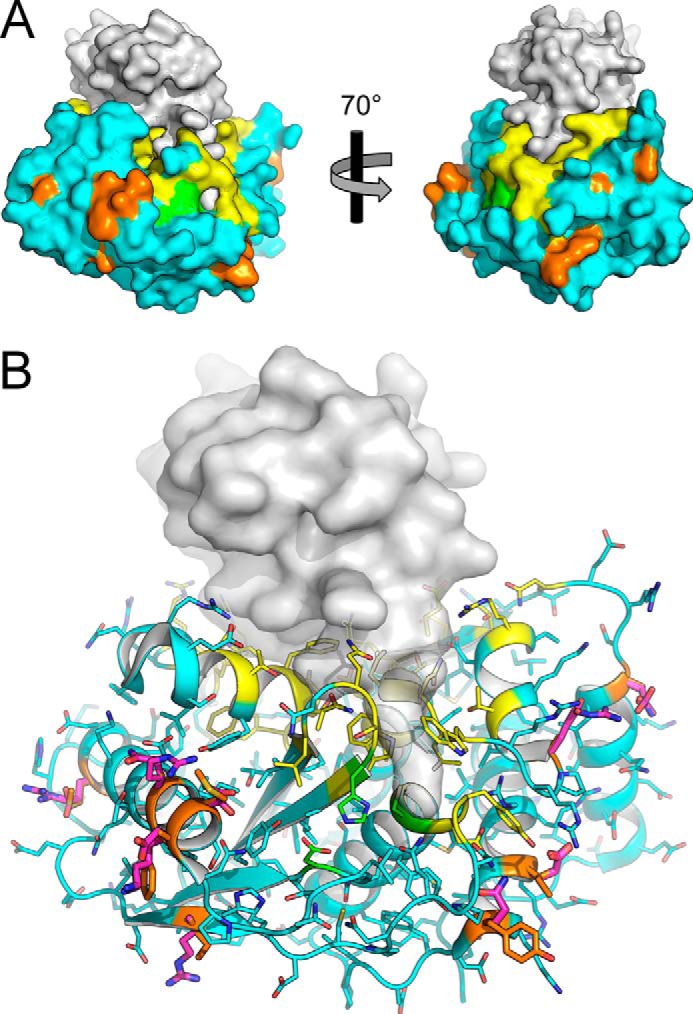
The SUMO protease Ulp1_WT (colored) is shown in complex with a substrate SUMO protein Smt3 (gray). Nonpolar amino acids of Ulp1_WT selected for computational design are colored orange, and residues that contact SUMO (yellow) or are part of the enzyme catalytic triad (green) were not permitted to change during design; all other residues are colored cyan. Models for Ulp1_WT and Smt3 are from PDB entry 1EUV. A, a molecular surface rendition. B, Smt3 is shown as a semitransparent molecular surface. The Ulp1_WT main chain is shown as a cartoon model, and amino acid side chains are shown as sticks. The side chains of the polar amino acid substitutions from Ulp1_R1–Ulp1_R4 are colored magenta; these and the corresponding side chains of Ulp1_WT are shown in boldface type for emphasis.
Table 1.
Summary of solubility-enhancing mutations
| Residuea | Ulp1_WT | Ulp1_R1 | Ulp1_R2 | Ulp1_R3 | Ulp1_R4 |
|---|---|---|---|---|---|
| 420 | Ala | Asp | Asn | Asp | Glu |
| 444 | Ala | Glu | Glu | Glu | Arg |
| 539 | Ala | Asp | Asp | Asp | Asp |
| 540 | Met | Ser | Asp | Ser | Ser |
| 542 | Phe | Lys | Lys | Lys | Lys |
| 543 | Ala | Gln | Arg | Gln | Gln |
| 553 | Met | Lys | Arg | Glu | Lys |
| 567 | Ile | Arg | Arg | Arg | Arg |
| 601 | Tyr | Gln | Gln | Ser | Gln |
| 605 | Ile | Glu | Glu | Glu | Glu |
a Residues are numbered according to PDB entry 1EUV.
In this study, we sought to improve the current state-of-the-art protease by two orthogonal approaches. First, we used computational protein design to engineer Ulp1_WT and remove the solvent-exposed hydrophobic surfaces by mutating nonpolar amino acids to polar amino acids. Second, we mined the genome of a thermophilic organism for a homologous enzyme. We assessed these new proteases for solubility, thermostability, and enzyme activity. The enzymes reported here exhibit improved behavior as in vitro reagents when compared with Ulp1_WT.
Results and discussion
Computational design of solubility-enhancing mutations to Ulp1
Previous studies that leveraged computational methods to enhance protein solubility and minimize aggregation focused on restricting large contiguous hydrophobic patches during design of the entire protein surface (8) or on modulation of surface charge (9). For this application, our goal was to minimize the amount of nonessential hydrophobic surface exposed to solvent while introducing the fewest possible number of mutations, as preserving enzymatic activity was paramount.
Using Rosetta, we developed a generally applicable computational method that identifies hydrophobic residue positions on the surface of a protein and determines amino acid substitutions to polar residues that yield low-energy solutions (see supporting information for the RosettaScripts XML protocol). To do this, the algorithm performs iterative rounds of flexible backbone design (10), and the positions of all Cα atoms are constrained to favor retention of the starting coordinates. We utilized the previously reported crystal structure of Ulp1_WT in complex with Smt3 (PDB code 1EUV) as the starting model (2). In total, 10 hydrophobic residues that project toward solvent from the protein surface were detected and designed (Table 1). Residues on Ulp1_WT that form the interface with the substrate Smt3 were detected by the algorithm, and the catalytic triad residues were manually specified; these residues were not permitted to mutate (Fig. 1B).
We generated 10 designs and selected four for testing in the wet laboratory (which we have named Ulp1_R1–Ulp1_R4). These four variants were selected because they were maximally different from one another with regard to amino acid sequence and exhibited high sequence similarity at designed positions with human and mouse SUMO proteases (see supporting information for the designed structural models). The algorithm's results converged at four of the designed residue positions, and in each case, this amino acid is predicted to form a new salt bridge. Mutation of Ile-567 to Arg can form a salt bridge with Asp-565; mutation of Ile-605 to Glu enables a salt bridge with Arg-609; and mutation of Ala-539 and Phe-542 to Asp and Lys, respectively, enables these two residues to form a salt bridge. These charge–charge interactions are favored by the energy function over mutations that do not result in the formation of an ionic bond.
Identification of a SUMO protease from thermophilic fungi
The SUMO system is unique to eukaryotes, and whereas there are no known hyperthermophilic eukaryotes, the single-celled fungus Chaetomium thermophilum, originally isolated from compost, is a thermophile that can grow at temperatures up to 60 °C (11). This organism has generated considerable interest within the structural biology community, as proteins from organisms that grow at high temperature are generally more stable and more amenable to crystallization than proteins from mesophiles (12).
The genome of C. thermophilum was recently sequenced (13), and currently there is no annotated SUMO protease gene. To identify candidates, we performed a BLASTp search (14) with Ulp1_WT as the query sequence. The top scoring alignment was to UniProtKB accession number G0RZV7, which is annotated as “specific protease-like protein” and exhibits 28% sequence identity over the aligned region with Ulp1_WT (Fig. 2A). To further investigate, we predicted the three-dimensional structure of the domain that aligns to Ulp1_WT using Robetta (15). The predicted model indicates this sequence adopts a structure that is highly similar to Ulp1_WT (Fig. 2C). The catalytic triad and active site residues were structurally conserved, and surprisingly, many of the residues that mediate binding to the SUMO substrate were also conserved (see supporting information for the predicted model). This suggested that the sequence encoded a SUMO protease, which we will refer to henceforth as Cth SUMO protease (or Cth), and it seemed likely that this enzyme could recognize the S. cerevisiae SUMO protease substrate Smt3.
Figure 2.

A, structure-based sequence alignment of Ulp1/SENP family members. Residues belonging to the catalytic triad are marked with asterisks. Residues of Ulp1_WT that directly contact Smt3 (2) are marked above with blue dots. Vertical black lines in the SENP6 and SENP7 sequences denote gaps due to the absence of SENP6- and SENP7-specific loops in the other proteases. B, structure-based sequence alignment of Smt3/SUMO variants, including the potential SUMO ortholog from C. thermophilum (CTHT_0059470). Residues that contact Ulp1_WT in PDB entry 1EUV are marked above with blue dots. C, a cartoon model of Ulp1_WT (cyan) is shown aligned to the Robetta-predicted model of Cth SUMO protease (magenta). The substrate Smt3 from S. cerevisiae is shown as a transparent molecular surface (gray). Side chains for identically conserved active-site and substrate-binding residues are shown as sticks. Models for Ulp1_WT and Smt3 are from PDB entry 1EUV.
SUMO protease variants exhibit enhanced solubility and thermostability
We began characterization of the protease variants by expressing each protein in Escherichia coli with a polyhistidine tag, followed by purification via immobilized metal-affinity chromatography and size-exclusion chromatography. The overall expression yields were similar; 10–35 mg of purified protein were obtained per liter of culture.
Methods that can quantitatively assess the maximum solubility of a protein rely on the addition of a chemical precipitant to reduce protein solubility; we chose to use the nonionic surfactant PEG 8000 (16). The maximum theoretical protein solubility (S0) is obtained via log-linear regression analysis of protein solubility versus an increasing amount of precipitant (17, 18). It is important to note that such measurements of apparent solubility do not represent actual solubility, as this value is inherently dependent upon solution conditions (e. g. ionic strength, pH, temperature, etc.). However, this assay does enable a quantitative comparison of the relative solubility between different SUMO protease constructs.
Each of the SUMO protease variants exhibits improved solubility over Ulp1_WT; Ulp1_R3 is the most soluble, with over a 12-fold increase in maximum concentration (Fig. 3). The least soluble of the designed proteases is Ulp1_R4, which exhibits nearly a 2-fold increase in solubility (Table 2). Cth SUMO protease is also more soluble than Ulp1_WT, with a one-third increase in solubility. These data are consistent with the model that solvent-exposed hydrophobic residues drive lower solubility for Ulp1_WT in aqueous solution.
Figure 3.

Precipitation assays of SUMO proteases. Data points indicate the concentration of soluble protease remaining after equilibration at room temperature with increasing concentrations of the protein precipitant, PEG 8000. The y intercept of each log-linear trend line is S0, and this value represents the theoretical maximum solubility of the protein in the absence of precipitant. Error bars, S.D. from three or more replicates.
Table 2.
Summary of protease properties and activity
| Enzyme | S0 | Tma | Vmax | Km | Vmax/Km |
|---|---|---|---|---|---|
| g/liter | °C | s−1 enzyme−1 | nm | m−1 s−1 | |
| Ulp1_WT | 12.9 ± 1.5 | 39.9 | 30.3 ± 0.8 | 200 ± 30 | (1.52 ± 0.23) × 108 |
| Ulp1_R1 | 52.5 ± 1.3 | 25.4 ± 0.6 | 130 ± 20 | (1.95 ± 0.30) × 108 | |
| Ulp1_R2 | 107.2 ± 1.3 | 34.7 ± 0.8 | 170 ± 20 | (2.04 ± 0.25) × 108 | |
| Ulp1_R3 | 166.0 ± 1.5 | 34.5 ± 0.7 | 110 ± 20 | (3.14 ± 0.58) × 108 | |
| Ulp1_R4 | 21.4 ± 1.4 | 38.1 | 25.8 ± 0.5 | 70 ± 10 | (3.70 ± 0.53) × 108 |
| Cth SUMO protease | 17.4 ± 1.3 | 50.1 | 140. ± 7 | 6600 ± 1100 | (2.12 ± 0.37) × 107 |
a The reported Tm value is the average melting temperature obtained from CD spectroscopy measurements at 208-, 218-, and 222-nm wavelengths.
Next, we sought to determine the effect of design on the structure and folded state of the Rosetta-engineered Ulp1 variants and to further characterize the putative Cth SUMO protease. Steady-state CD spectra revealed that all of the proteases contain a mixed ɑ/β character at 20 °C (Fig. 4A). As expected, the Cth SUMO protease melts at higher temperature than its mesophilic homologue Ulp1_WT, and upon heating, Ulp1_WT and Cth SUMO protease both undergo a cooperative unfolding transition with melting temperatures at 40 and 50 °C, respectively (Fig. 4B). A visible protein precipitate was observable in the cuvette following thermal denaturation for Ulp1_WT and Cth SUMO protease. Similar to other Ulp1-like SUMO proteases, the full-length Cth SUMO protease includes a large N-terminal domain that may be required for protein stability at the higher temperatures at which C. thermophilum can grow. Curiously, only Ulp1_R4 of the Rosetta-designed variants undergoes a clear, cooperative unfolding transition upon heating. For all designs, some of the structural character reported by the far-UV wavelength measurements appears to melt with a Tm that ranges from 31 to 38 °C when monitoring the CD signal at 208 nm (Fig. 4B), and this may represent a soluble, partially unfolded state, as we did not observe precipitate in the cuvette for any of the designed variants after heating.
Figure 4.

CD of SUMO proteases. A, steady-state wavelength spectra. B, thermal denaturation.
Enzyme kinetics of SUMO protease variants
Sequence changes can impact enzymatic function, even when distal to the catalytic machinery (19). Thus, we sought to determine the effect of mutation on the rate of proteolysis for the Ulp_R1–4 variants and to determine whether Cth SUMO protease exhibited activity on the S. cerevisiae substrate protein Smt3. To accomplish this, we used an EGFP-Smt3-mCherry linear fusion construct whereby the cleavage after Smt3 is monitored by a loss of FRET (20, 21).
Kinetic analysis revealed that the Rosetta design Ulp1 variants all exhibit Vmax and Km values comparable with those of the WT enzyme, indicating that solubility enhancement had little effect on enzyme activity (Fig. 5A). By extension, the Rosetta variants likely retain WT specificity for Smt3, because a change in substrate specificity should result in a change to the measured specificity constant (kcat/Km), whereas the Rosetta variants exhibit similar specificity constants for the cleavage of EGFP-Smt3-mCherry as compared with Ulp1_WT (Table 2). The Cth SUMO protease has roughly an order of magnitude higher Km but also a Vmax that is over 4-fold faster than Ulp1_WT (Fig. 5B, Table 2). Although the resulting lower specificity constant of the Cth SUMO protease may be due to intrinsic properties of the enzyme, it is more likely the result of S. cerevisiae Smt3 being a suboptimal substrate. When used in vitro with high substrate concentration, the weak Km may not be problematic, and the higher maximum rate could make Cth SUMO protease the superior reagent enzyme. At low substrate concentrations, or when the reaction needs to go to completion quickly, the Rosetta-designed Ulp1 variants would be the better reagents. Regardless, these data clearly demonstrate that Cth SUMO protease (UniProtKB accession number G0RZV7) encodes a bona fide SUMO protease.
Figure 5.
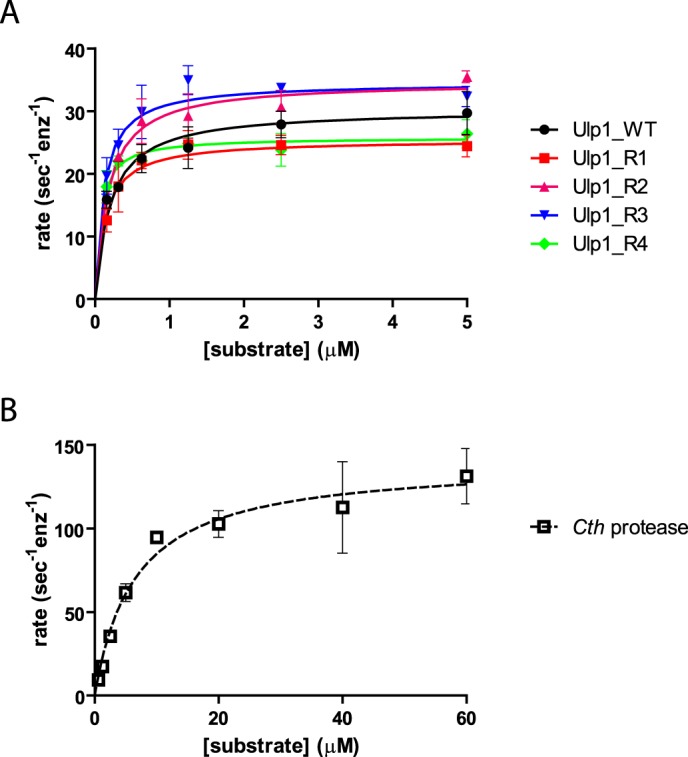
Kinetic characterization of SUMO proteases. Rates of cleavage of EGFP-Smt3GGGG-mCherry by Ulp1_WT and Rosetta-designed proteases (A) or the Cth SUMO protease (B). Rates were fit to the Michaelis–Menten equation to obtain Vmax and Km values (see Table 2). Error bars, S.D. from three or more replicates.
X-ray crystal structure of Cth SUMO protease
To determine the structural basis for Cth SUMO protease activity and to assess the accuracy of the Robetta structure prediction, we crystallized the protease domain of the Cth SUMO protease. We solved the X-ray crystal structure by molecular replacement using the structure of Ulp1_WT as the search model after removal of loops and side chains that differed based on sequence alignments (PDB code 1EUV). Molecular replacement yielded a solution that was improved by iterative rounds of building and refinement. The structure revealed one molecule of Cth SUMO protease per asymmetric unit. The model was refined to 1.44 Å with Rwork and Rfree values of 11.9 and 14.8%, respectively, and excellent geometry (Fig. 6A; also see Table 3).
Figure 6.

Structure of Cth SUMO protease. A, two orthogonal views of the Cth SUMO protease (maroon; PDB code 6DG4) superimposed onto the Ulp1_WT structure (cyan; PDB code 1EUV, chain A) with the SUMO-binding interfaces colored in yellow and cyan, respectively, and depicted in stick representation. The arrow points to the catalytic cysteine in the view on the left. B, superimposed catalytic pockets of Ulp1_WT (cyan) and the apo-Cth SUMO protease (maroon/yellow) with several key residues shown in stick representation. Numbering is for the Cth protease. C, C-terminal region of Smt3 (purple) and Smt3-binding surface of Ulp1_WT (cyan) superimposed onto apo-Cth structure (maroon/yellow). Several side chains that differ between Cth and Ulp1_WT in the Ulp1_WT-Smt3 interface are shown in stick representation.
Table 3.
X-ray crystallography data collection and refinement statistics for Cth SUMO protease
| Data collection | |
| Resolution rangea (Å) | 41.84–1.44 (1.49–1.44) |
| Space group | P 21 21 21 |
| Unit cell | |
| a, b, c (Å) | 45.9, 63.6, 83.7 |
| α, β, γ (degrees) | 90, 90, 90 |
| Total reflections | 311,011 (29,317) |
| Unique reflections | 44,252 (4318) |
| Multiplicity | 7.0 (6.8) |
| Completeness (%) | 98.4 (97.6) |
| Mean I/σI | 21.4 (4.8) |
| Wilson B-factor | 12.5 |
| Rmerge | 0.052 (0.206) |
| Rmeas | 0.056 (0.223) |
| Rpim | 0.021 (0.083) |
| CC½ | 0.999 (0.975) |
| CCa | 1 (0.994) |
| Refinement | |
| Reflections used in refinementa | 44,240 (4318) |
| Reflections used in the test set | 1999 (196) |
| Rwork | 0.119 (0.132) |
| Rfree | 0.148 (0.190) |
| CCwork | 0.971 (0.975) |
| CCfree | 0.973 (0.946) |
| Number of non-hydrogen atoms | 2562 |
| Macromolecules | 2073 |
| Ligands | 28 |
| Solvent | 461 |
| No. of protein residues | 253 |
| RMSD, bonds (Å) | 0.006 |
| RMSD, angles (degrees) | 1.19 |
| Ramachandran favored, allowed, outliers (%) | 98.4, 1.6, 0 |
| Rotamer outliers (%) | 1.4 |
| Clashscore | 3.08 |
| Average B-factor | 19.6 |
| Macromolecules | 15.6 |
| Ligands | 57.6 |
| Solvent | 34.9 |
a Statistics calculated using Phenix; highest shell is indicated in parentheses.
Overall, the structure of Cth SUMO protease closely resembles previously determined Ulp1/SENP structures (Fig. 6A) (2, 21–25) and aligns to the S. cerevisiae Ulp1_WT-Smt3 structure (PDB code 1EUV, chain A) with an RMSD of 1.7 Å (Cα over 187 residues; Fig. 6A). The catalytic Asp-His-Cys triad and the glutamine that contributes to the formation of the oxyanion hole exhibit similar conformations when Cth and Ulp1_WT structures are compared (Fig. 6B). The conserved tryptophan and glycine residues (Trp-992, Trp-1071, and Gly-1132) that line the narrow catalytic tunnel occupy similar positions in the Cth structure as they do in several other structures of Ulp1/SENPs complexed with Smt3/SUMO. However, a notable exception occurs with Cth Trp-992, which would clash with the modeled C-terminal tail of Smt3 (Fig. 6B). This clash is most likely because SUMO is absent in our Cth structure, as the analogous Trp residue in SENP-1 (Trp-410) also rotates to accommodate SUMO-2 upon binding (24).
Although the SUMO-interacting surface of Cth SUMO protease resembles those of other Ulp1/SENPs, several substitutions in Cth indicate potential differences in interaction with its cognate SUMO(s), possibly explaining the weaker interaction with S. cerevisiae Smt3 that we observe in our FRET assay (as judged by the increase in Km). In Ulp1_WT, Glu-434 is within hydrogen-bonding distance of Smt3 Gln-73, whereas it is substituted to Pro-976 in the Cth SUMO protease (Fig. 6C). Whereas this residue is not strictly conserved in the Ulp1/SENP family, Ulp1_WT and SENP1, -2, and -6 have polar residues in this position (Fig. 2A). The probable C. thermophilum SUMO homolog (gene ID: CTHT_0059470) as well as human SUMO-2 and SUMO-3 also have a glutamine/asparagine in the corresponding position, although in human SUMO-1, this residue is substituted to alanine (Fig. 2B). The salt bridge between Ulp1_WT Glu-455 and Arg-71 of Smt3 appears absent in Cth as Glu-455 is substituted to Ile-999 (Fig. 6C). This residue is conserved as charged or polar side chains in all members of the Ulp1/SENP family (Fig. 2A). Arg-71 of Smt3 is conserved in human SUMO-1 and the Cth SUMO homolog but is substituted to a proline in SUMO-2 and SUMO-3 (Fig. 2B). Both Thr-477 and Asn-509 of Ulp1_WT are within hydrogen-bonding distance of Gln-95 of Smt3, with Asn-509 within hydrogen-bonding distance of the backbone of Ile-96 of Smt3 (Fig. 6C). Thr-477 is substituted to Pro-1031, and Asn-509 corresponds to Cys-1066 in Cth (Fig. 6C), thus suggesting that hydrogen-bonding interactions observed in Ulp1_WT-Smt3 are not present between Cth and its cognate SUMO substrate(s). Pro-1031 is conserved as charged or polar in all Ulp1/SENPs with the exception of SENP-2, where it is also a proline (Fig. 2A). Cth Cys-1066 is conserved as an asparagine or histidine in all Ulp1/SENPs (Fig. 2A). All SUMO variants, including that of Cth, have a glutamine in this position (Fig. 2B).
Several other differences between the SUMO-binding interfaces of Ulp1_WT and Cth SUMO protease are evident from the structure-based alignment. A universally conserved tryptophan in other Ulp1/SENPs is substituted to leucine in Cth (Leu-1044). In structures of Ulp1/SENP proteases, this tryptophan packs against a highly conserved glycine in Smt3 and contributes hydrogen bonds to the backbone of Smt3, SUMO-1, and SUMO-2/3 (Figs. 2 (A and B) and 6C) (2, 21–24). Mutation of this tryptophan to alanine in Ulp1_WT results in temperature-sensitive growth defects in S. cerevisiae (2). Thus, the presence of leucine in this position in Cth suggests the possibility of different packing interactions with cognate C. thermophilum SUMO variant(s), although at least one of these variants, CTHT_0059470, retains the conserved glycine (Fig. 2B). Another Ulp1_WT residue that presumably contributes to Smt3 recognition is Gln-512, which is within hydrogen-bonding distance to Arg-93 and the backbone of Smt3. In Cth, this residue is substituted to Ser-1068 and appears to face away from the modeled Smt3 C terminus (Fig. 6C). There is poor conservation of this residue among other SENPs, indicating that it may not be important for Smt3/SUMO recognition (Fig. 2A). Finally, it is worth noting that the Cth SUMO protease is more closely related to S. cerevisiae Ulp1 (i.e. Ulp1_WT) and human SENP1, -2, -3, and -5 and is quite different from SENP6 and SENP7, which have unique structural features and are more adept at deconjugating SUMO chains (Fig. 2A) (25).
Conclusion
We have obtained proteases that are more soluble, more thermostable, and more efficient than the current commercially available state-of-the-art Ulp1_WT enzyme both by successful re-engineering of Ulp1_WT and discovery of a new SUMO protease from C. thermophilum (Table 2). To accomplish this, we created an automated computational design method that is generally applicable to improving the solubility of proteins. Combining bioinformatic analysis with structure prediction allowed us to successfully identify a functional homologue to Ulp1_WT despite low sequence identity, and this could be a generalizable approach for assigning function to many unannotated genes.
The solubility of each protease we assessed roughly tracks with the amount of hydrophobic surface area that is exposed to solvent (Table 4). However, it is unclear what drives the 5-fold difference in S0 between Ulp1_R2 and Ulp1_R4, as these proteins exhibit the same amount of hydrophobic surface. The surface area calculations for Ulp1_WT and Cth SUMO protease are based on models determined via X-ray crystallography, whereas Ulp1_R1–4 are based on the Rosetta models, which may not represent the solution structure of these proteins as accurately as the experimentally determined models. However, we hypothesize that there are additional factors contributing to the observed solubility differences between these constructs, and the computational design protocol and assays described here will provide an ideal model system for future investigation.
Table 4.
Computational measurement of solvent-accessible protein surface area
The values for Ulp1_WT and Cth SUMO protease were calculated from X-ray crystal structures (PDB entries 1EUV and 6DG4), and Ulp1_R1 to Ulp1_R4 were calculated from the Rosetta design models (included in the supporting information).
| Enzyme | Total surface | Hydrophobic surface |
Polar surface |
||
|---|---|---|---|---|---|
| Area | Percentage of total | Area | Percentage of total | ||
| Å2 | Å2 | % | Å2 | % | |
| Ulp1_WT | 11,154 | 6,132 | 55.0 | 5022 | 45.0 |
| Ulp1_R1 | 11,846 | 6,059 | 51.1 | 5787 | 48.9 |
| Ulp1_R2 | 11,734 | 5,974 | 50.9 | 5760 | 49.1 |
| Ulp1_R3 | 11,838 | 5,996 | 50.6 | 5843 | 49.4 |
| Ulp1_R4 | 11,749 | 5,977 | 50.9 | 5771 | 49.1 |
| Cth SUMO protease | 11,534 | 6,060 | 52.5 | 5475 | 47.5 |
For researchers looking to utilize an improved SUMO protease for preparation of recombinant protein, we recommend Ulp1_R3 or Cth SUMO protease. Expression plasmids for the engineered protease constructs reported in this study have been made available via Addgene.
Experimental procedures
Computational protein design
Generally applicable computational methods to design solubility-enhancing mutations and to calculate the solvent-accessible surface areas (total, hydrophobic, and nonpolar) were developed using the RosettaScripts application programming interface (26) for Rosetta (27). The xml protocols needed to run these methods are available in the supporting material.
Molecular cloning
E. coli codon optimization was performed using DNAworks (28). The gene for Ulp1_WT was cloned into vector pET28b (Novagen) with an N-terminal hexahistidine tag followed by a thrombin cleavage site. Ulp1_R1–4 were cloned into vector pET29b with a hexahistidine tag at both the N and C termini; these are plasmids pCDB325–328. Cth SUMO protease was cloned into vector pCDB98 with a C-terminal decahistidine tag; this plasmid is pCDB302.
Protein expression and purification
All Ulp1 variants and Cth SUMO protease were expressed and purified as follows. BL21(DE3)RIL cells were grown to optical density 0.6–0.8 at 37 °C and then induced by the addition of 0.5 mm isopropyl β-d-1-thiogalactopyranoside and grown at 18 °C for an additional 16–20 h (1 liter/construct). Cells were harvested by centrifugation and resuspended in 10 ml of buffer A (50 mm Tris, pH 7.5, 500 mm NaCl, 20 mm imidazole, 0.5 mm EDTA, 1 mm DTT) supplemented with 1.5 μl of benzonase (Sigma, E1014-25kU), lysed by sonication at 4 °C, and centrifuged at 39,000 × g at 4 °C for 25 min. The supernatant was applied to 2.5 ml of Ni2+-NTA beads prewashed with buffer B (50 mm Tris, pH 7.5, 500 mm NaCl, 20 mm imidazole, 0.25 mm EDTA, 0.5 mm DTT) and incubated at 4 °C for 20 min. The Ni2+-NTA beads were washed with 100 ml of buffer B, and sample was eluted with 10 ml of buffer C (50 mm Tris, pH 7.5, 500 mm NaCl, 250 mm imidazole, 0.25 mm EDTA, 0.5 mm DTT). The eluted sample was applied to a Superdex 75 26/60 column equilibrated in buffer D (50 mm Tris, pH 7.5, 200 mm NaCl, 5% (v/v) glycerol, 2 mm DTT, 1 mm EDTA). Fractions containing the highest purity of Ulp1 as judged by SDS-PAGE were concentrated down to 1–1.5 ml using spin columns, and concentration was measured by A280 using a NanoDrop (Thermo Scientific). Concentrated protein was aliquoted, flash-frozen in liquid N2, and stored at −80 °C for later use.
EGFP-Smt3GGGG-mCherry was expressed and purified as follows. BL21(DE3)RIL cells (4 liters) were grown to optical density 0.6–0.8 at 37 °C and then induced by the addition of isopropyl β-d-1-thiogalactopyranoside to 1 mm and grown for an additional 4 h at 30 °C. Cells were lysed and subjected to Ni2+-NTA purification as described above for Ulp1 variants, except 4 ml of Ni2+-NTA beads per 4 liters of culture were used. The EGFP-Smt3GGGG-mCherry Ni2+-NTA eluate was injected onto a Superdex 75 26/60 column equilibrated in buffer E (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm DTT). Appropriate fractions were pooled and diluted 3-fold with buffer F (50 mm MES, pH 6.0, 1 mm DTT, 1 mm EDTA). Diluted sample was applied to a MonoS HR 10/10 column equilibrated in 95% buffer F and 5% buffer G (50 mm MES, pH 6.0, 1 m NaCl, 1 mm DTT, 1 mm EDTA). The column was washed with 3 column volumes of 95% buffer F and 5% buffer G, and the sample was eluted in a linear gradient from 5 to 30% buffer G in 15 column volumes. The MonoS fractions were pooled, diluted 3-fold with buffer H (50 mm Tris, pH 7.5, 1 mm EDTA, 1 mm DTT), and applied to a MonoQ HR 10/10 column equilibrated with 95% buffer H and 5% buffer I (50 mm Tris, pH 7.5, 1 m NaCl, 1 mm EDTA, 1 mm DTT). The MonoQ column was washed with three column volumes of 95% buffer H and 5% buffer I, and the sample was eluted in a linear gradient from 5 to 30% buffer I in 15 column volumes. Fractions containing the highest purity of EGFP-smt3GGGG-mCherry as judged by SDS-PAGE were pooled, concentrated, and buffer-exchanged into buffer E on the concentrator. Buffer-exchanged sample was concentrated to a final volume of 0.5–1 ml, and the concentration was measured by A280 using the NanoDrop (ϵ280 = 56,270 m−1 cm−1). The sample was aliquoted and flash-frozen in liquid N2 and stored at −80 °C for later use.
Comparative solubility assay
Solubility measurements were performed by mixing each protease stock solution with increasing concentrations (1–2.5% (w/v) increments) of PEG 8000 (Rigaku). The reactions were allowed to equilibrate at room temperature for 10 min and then centrifuged in an Eppendorf 5430 microcentrifuge at 20,000 rpm for 10 min. The concentration of soluble protease was measured from the supernatant via Bradford assay (Bio-Rad) in a 96-well plate (Costar, catalog no. 3595) using a Synergy Neo2 plate reader (BioTek) with a minimum of three technical replicates. The resulting data were log10-transformed, plotted against the concentration of PEG, and fit to a least-squares regression line. The indicated errors in S0 are the S.E. of the y intercept as calculated by the S.E. of the regression (S), where S is found by dividing the sum of the squares of the deviation from the best-fit line by the number of data points beyond the minimum two required to fit the specified linear curve (29).
CD and thermal denaturation
CD measurements were performed using a JASCO J-1500 CD spectrophotometer and a quartz cuvette with a 1-mm light path. Protein samples were prepared in 10 mm sodium phosphate, pH 7.4. Steady-state wavelength spectra were recorded in 0.1-nm increments from 200 to 260 nm at 20 and 95 °C. Thermal denaturation was assessed by measuring the CD signal at 208-, 218-, and 222-nm wavelengths in 1 °C increments from 20 to 95 °C.
FRET assay for EGFP-Smt3GGGG-mCherry cleavage by Ulp1
All FRET assays were performed in buffer J (25 mm Tris, pH 7.5, 150 mm NaCl, 0.1% (v/v) Tween 20, 2 mm DTT, 0.5 mm EDTA) at 30 °C. Varying concentrations of EGFP-Smt3GGGG-mCherry were preincubated in 96-well half-area plates (Corning, catalog no. 3686) for 10 min at 30 °C, and SUMO protease was added to initiate the reaction (0.1–2 nm enzyme). The time course of the cleavage reaction was monitored by loss of FRET (excitation, 450 nm; emission, 655 nm) with 15 reads/well and 4–5-s read intervals on a SpectraMax M5 plate reader (Molecular Devices). Rates at each concentration of substrate were calculated by measuring the initial slope of reaction, dividing this value by the amplitude of the signal change, and multiplying by the concentration of substrate. Obtained values were then divided by the concentration of protease enzyme. Rates from three or more replicates for each concentration of substrate were averaged, and the S.D. was calculated to generate the error bars. The resulting data are plotted against the concentration of EGFP-Smt3GGGG-mCherry and fit to the Michaelis–Menten equation, rate = (Vmax × [S])/([S] + Km), using the nonlinear least-squares fit in Prism (GraphPad Software). The indicated errors in Vmax and Km values are S.E. of the fit as reported by Prism.
Structure determination by X-ray crystallography
His6-tagged Cth SUMO protease was crystallized via the hanging-drop method by mixing 2 μl of 10 mg/ml protein in buffer D with 2 μl of reservoir solution (0.1 m HEPES, pH 7.0, 2.1 m ammonium sulfate) and incubating over 500 μl of reservoir solution. Crystals were grown at 18 °C, harvested after 2 days, and cryo-protected by dipping into cryo-solution (0.1 m HEPES, pH 7.0, 2.5 m ammonium sulfate, 15% (v/v) glycerol) for 10–15 s before freezing in liquid N2. X-ray diffraction data were collected at the Advanced Photon Source 24-ID-E beam line at a wavelength of 0.97918 Å. Data were processed with HKL2000 (30), and a molecular replacement solution was obtained using Phaser (31) as part of the Phenix suite (32) with PDB entry 1EUV, chain A, as the search model. The molecular replacement solution was manually rebuilt using COOT (7) based on electron density and refined in Phenix.
Author contributions
Y.-T. K. L., V. B., C. D. L., and C. D. B. designed the experiments. B. M. F. and C. D. B. performed computational protein design with input from C. D. L. and D. B. V. B, Y.-T. K. L., T. A. H., L. P. C., and B. M. F. purified proteins. Y.-T. K. L. measured protein solubility. Y.-T. K. L. and J. M. J. performed CD spectroscopy. V. B. performed the enzyme assays. V. B. and C. D. L. determined the structure of Cth SUMO protease. Y.-T. K. L., V. B., C. D. L., and C. D. B. wrote the manuscript with input from all authors.
Supplementary Material
Acknowledgments
We thank Dr. Lance Stewart and Dr. Ruud van Deursen for helpful discussions. Work was based in part upon research conducted at NE-CAT beamlines (National Institutes of Health (NIH), NIGMS, Grant P41 GM103403 and NIH-ORIP HEI Grant S10 RR029205). Beamline research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
This work was supported in part by NIGMS, National Institutes of Health, Grants R01GM065872 (to C. D. L.), R35GM118080 (to C. D. L.), and P30 CA008748 (NCI-Cancer Center Support Grant). C. D. L. holds a patent for a rapidly cleavable SUMO fusion protein expression system for difficult to express proteins. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting RosettaScripts xml design protocols and models for Ulp1_R1 to Ulp1_R4 and Cth SUMO protease.
The atomic coordinates and structure factors (code 6DG4) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- SUMO
- small ubiquitin-like modifier
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- NTA
- nitrilotriacetic acid.
References
- 1. Lima C. D., and Mossessova E. (March 22, 2011) Rapidly cleavable sumo fusion protein expression system for difficult to express proteins. U. S. Patent US7910364B2 [Google Scholar]
- 2. Mossessova E., and Lima C. D. (2000) Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell. 5, 865–876 10.1016/S1097-2765(00)80326-3 [DOI] [PubMed] [Google Scholar]
- 3. Butt T. R., Edavettal S. C., Hall J. P., and Mattern M. R. (2005) SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 43, 1–9 10.1016/j.pep.2005.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marblestone J. G., Edavettal S. C., Lim Y., Lim P., Zuo X., and Butt T. R. (2006) Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 15, 182–189 10.1110/ps.051812706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li S. J., and Hochstrasser M. (1999) A new protease required for cell-cycle progression in yeast. Nature 398, 246–251 10.1038/18457 [DOI] [PubMed] [Google Scholar]
- 6. Owerbach D., McKay E. M., Yeh E. T. H., Gabbay K. H., and Bohren K. M. (2005) A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem. Biophys. Res. Commun. 337, 517–520 10.1016/j.bbrc.2005.09.090 [DOI] [PubMed] [Google Scholar]
- 7. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jacak R., Leaver-Fay A., and Kuhlman B. (2012) Computational protein design with explicit consideration of surface hydrophobic patches. Proteins 80, 825–838 10.1002/prot.23241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raghunathan G., Sokalingam S., Soundrarajan N., Madan B., Munussami G., and Lee S.-G. (2013) Modulation of protein stability and aggregation properties by surface charge engineering. Mol. Biosyst. 9, 2379–2389 10.1039/c3mb70068b [DOI] [PubMed] [Google Scholar]
- 10. Bhardwaj G., Mulligan V. K., Bahl C. D., Gilmore J. M., Harvey P. J., Cheneval O., Buchko G. W., Pulavarti S. V. S. R. K., Kaas Q., Eletsky A., Huang P.-S., Johnsen W. A., Greisen P. J., Rocklin G. J., Song Y., et al. (2016) Accurate de novo design of hyperstable constrained peptides. Nature 538, 329–335 10.1038/nature19791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amlacher S., Sarges P., Flemming D., van Noort V., Kunze R., Devos D. P., Arumugam M., Bork P., and Hurt E. (2011) Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 146, 277–289 10.1016/j.cell.2011.06.039 [DOI] [PubMed] [Google Scholar]
- 12. Kellner N., Schwarz J., Sturm M., Fernandez-Martinez J., Griesel S., Zhang W., Chait B. T., Rout M. P., Kück U., and Hurt E. (2016) Developing genetic tools to exploit Chaetomium thermophilum for biochemical analyses of eukaryotic macromolecular assemblies. Sci. Rep. 6, 20937 10.1038/srep20937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bock T., Chen W.-H., Ori A., Malik N., Silva-Martin N., Huerta-Cepas J., Powell S. T., Kastritis P. L., Smyshlyaev G., Vonkova I., Kirkpatrick J., Doerks T., Nesme L., Bassler J., Kos M., et al. (2014) An integrated approach for genome annotation of the eukaryotic thermophile Chaetomium thermophilum. Nucleic Acids Res. 42, 13525–13533 10.1093/nar/gku1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boratyn G. M., Schäffer A. A., Agarwala R., Altschul S. F., Lipman D. J., and Madden T. L. (2012) Domain enhanced lookup time accelerated BLAST. Biol. Direct. 7, 12 10.1186/1745-6150-7-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim D. E., Chivian D., and Baker D. (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531 10.1093/nar/gkh468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Middaugh C. R., Tisel W. A., Haire R. N., and Rosenberg A. (1979) Determination of the apparent thermodynamic activities of saturated protein solutions. J. Biol. Chem. 254, 367–370 [PubMed] [Google Scholar]
- 17. Kramer R. M., Shende V. R., Motl N., Pace C. N., and Scholtz J. M. (2012) Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys. J. 102, 1907–1915 10.1016/j.bpj.2012.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li L., Kantor A., and Warne N. (2013) Application of a PEG precipitation method for solubility screening: a tool for developing high protein concentration formulations. Protein Sci. 22, 1118–1123 10.1002/pro.2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fraser J. S., Clarkson M. W., Degnan S. C., Erion R., Kern D., and Alber T. (2009) Hidden alternative structures of proline isomerase essential for catalysis. Nature 462, 669–673 10.1038/nature08615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clegg R. M. (1995) Fluorescence resonance energy transfer. Curr. Opin. Biotechnol. 6, 103–110 10.1016/0958-1669(95)80016-6 [DOI] [PubMed] [Google Scholar]
- 21. Shen L., Tatham M. H., Dong C., Zagórska A., Naismith J. H., and Hay R. T. (2006) SUMO protease SENP1 induces isomerization of the scissile peptide bond. Nat. Struct. Mol. Biol. 13, 1069–1077 10.1038/nsmb1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reverter D., and Lima C. D. (2004) A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2-SUMO complex. Structure 12, 1519–1531 10.1016/j.str.2004.05.023 [DOI] [PubMed] [Google Scholar]
- 23. Reverter D., and Lima C. D. (2006) Structural basis for SENP2 protease interactions with SUMO precursors and conjugated substrates. Nat. Struct. Mol. Biol. 13, 1060–1068 10.1038/nsmb1168 [DOI] [PubMed] [Google Scholar]
- 24. Shen L. N., Dong C., Liu H., Naismith J. H., and Hay R. T. (2006) The structure of SENP1-SUMO-2 complex suggests a structural basis for discrimination between SUMO paralogues during processing. Biochem. J. 397, 279–288 10.1042/BJ20052030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lima C. D., and Reverter D. (2008) Structure of the human SENP7 catalytic domain and poly-SUMO deconjugation activities for SENP6 and SENP7. J. Biol. Chem. 283, 32045–32055 10.1074/jbc.M805655200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fleishman S. J., Leaver-Fay A., Corn J. E., Strauch E.-M., Khare S. D., Koga N., Ashworth J., Murphy P., Richter F., Lemmon G., Meiler J., and Baker D. (2011) RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One 6, e20161 10.1371/journal.pone.0020161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaufmann K. W., Lemmon G. H., Deluca S. L., Sheehan J. H., and Meiler J. (2010) Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry 49, 2987–2998 10.1021/bi902153g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoover D. M., and Lubkowski J. (2002) DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 30, e43 10.1093/nar/30.10.e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Devore J. L. (1987) Probability and Statistics for Engineering and the Sciences, 2nd Ed., Brooks/Cole Publishing Co., Boston [Google Scholar]
- 30. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 31. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.