

Abstract
Fine-tuning of the activity of inositol 1,4,5-trisphosphate receptors (IP3R) by a diverse array of regulatory inputs results in intracellular Ca2+ signals with distinct characteristics. These events allow the activation of specific downstream effectors. We reported previously that region-specific proteolysis represents a novel regulatory event for type 1 IP3R (R1). Specifically, caspase-fragmented R1 display a marked increase in single-channel open probability. More importantly, the distinct characteristics of the Ca2+ signals elicited via fragmented R1 can activate alternate downstream effectors. In this report, we expand these studies to investigate whether all IP3R subtypes are regulated by proteolysis. We now show that type 2 and type 3 IP3R (R2 and R3, respectively) are proteolytically cleaved in rodent models of acute pancreatitis. Surprisingly, fragmented IP3R retained tetrameric architecture, remained embedded in endoplasmic reticulum membranes and were not functionally disabled. Proteolysis was associated with a marked attenuation of the frequency of Ca2+ signals in pancreatic lobules. Consistent with these data, expression of DNAs encoding complementary R2 and R3 peptides mimicking fragmented receptors at particular sites, resulted in a significant decrease in the frequency of agonist-stimulated Ca2+ oscillations. Further, proteolysis of R2 resulted in a marked decrease in single-channel open probability. Taken together, proteolytic fragmentation modulates R2 and R3 activity in a region-specific manner, and this event may contribute to the altered Ca2+ signals in pancreatic acinar cells during acute pancreatitis.
Keywords: inositol trisphosphate receptor (IP3R); inositol 1,4,5-trisphosphate (IP3); protease; calcium intracellular release; pancreas; proteolysis
Introduction
Inositol 1,4,5-trisphosphate receptors (IP3R)2 are ubiquitous, intracellular Ca2+ release channels expressed predominantly in endoplasmic reticulum (ER) membranes (1–4). IP3R can encode Ca2+ changes with distinct spatial and temporal characteristics, and these signals subsequently play essential roles in controlling a plethora of biological processes (1, 2, 5–7). The versatility of these signals is a consequence, in large part, of the regulation of IP3R activity at multiple levels. First, there are three isoforms of IP3R, termed R1, R2, and R3, encoded by three different genes (1). IP3R can either form homo- or heterotetrameric channels. The composition of the assembled tetramer either dictates or contributes to the channel activity (8). Further, binding of numerous molecules can regulate IP3R properties, including biophysical characteristics, together with receptor localization (9, 10). Another level of modulation occurs as a function of posttranslational modifications, including phosphorylation and ubiquitination events, which can either alter channel activity or determine channel abundance (2, 11). Recently, our laboratory has demonstrated that region-specific proteolytic fragmentation dramatically alters R1 activity and thereby allows the receptor to potentially activate alternative downstream effectors (12). However, whether this is a regulatory event specific to R1 or a general form of regulation relevant to all isoforms of IP3R requires further investigation. Moreover, whether proteolysis of R2 and R3 has similar effects on the biophysical properties of all subtypes remains to be established.
R2 and R3 play pivotal roles in exocrine secretory systems (13–15). For example, R2- and R3-mediated Ca2+ signals play a functionally dominant role in the exocytosis of stored zymogens from the pancreas, which, along with the production of an NaCl-rich fluid, is the primary function of acinar cells. The central role of R2/R3 is most strikingly demonstrated by the observation that R2 and R3 compound knockout mice failed to thrive after weaning because of a severe disruption of exocrine function. Indeed, Ca2+ transients and amylase secretion in response to secretagogue stimulation were reported to be completely abolished in pancreatic acinar cells isolated from R2 and R3 double knockouts (13).
R2 and R3 have the same general primary structure as R1 and, therefore, are predicted to be subject to cleavage by proteases at similar solvent-exposed sites (16). An early report suggested that R2 and R3 are substrates of caspase and calpain, respectively (17). In addition, our laboratory reported that, in a model of acute pancreatitis, R3 were cleaved into low-molecular-weight receptor fragments and that inhibition of proteasome activity failed to completely prevent receptor fragmentation (18). These observations imply that R3 were cleaved intracellularly by inappropriately activated digestive enzymes, such as trypsin and chymotrypsin, within pancreatic acinar cells during acute pancreatitis (16, 19, 20). Of note, both proteases are reported to cleave IP3R in vitro (16, 19–21). This evidence suggests that models of acute pancreatitis may represent an ideal experimental platform to investigate the in vivo consequences of R2 and R3 fragmentation and subsequent effects on agonist-stimulated [Ca2+]i signals.
In this study, utilizing both in vivo and in vitro rodent models of acute pancreatitis, we demonstrate that R2 and R3 are substrates for proteases. Moreover, proteolysis results in channels that retain a fundamental tetrameric architecture and remain in ER membranes but exhibit dramatically altered channel activity. Specifically, in both pancreatitis models and in expression systems mimicking fragmentation, agonist-stimulated Ca2+ oscillations are attenuated, and channel activity is reduced. This study therefore indicates that proteolysis is a novel mechanism for regulating R2 and R3 activity. Furthermore, although proteolysis modulates the activity of all IP3R isoforms, intriguingly, it does so in a subtype- and fragmentation pattern–specific manner.
Results
Generation of fragmented R2 and R3 in an in vivo acute pancreatitis model
To model acute pancreatitis, mice received three consecutive intraperitoneal (i.p.) injections of a supramaximal concentration of the secretagogue analogue caerulein (50 μg/kg/injection) or saline as a control. This approach represents a well-established, reversible, and relatively noninvasive mouse model of the disease (22, 23). Previous studies have reported that, although isolated pancreatic acinar cells (PAC) from healthy mice displayed robust Ca2+ oscillations in response to low concentrations of secretagogues, those from mice treated with this protocol evoked a single Ca2+ transient or repetitive Ca2+ transients with a significantly lower frequency (24). We first performed [Ca2+]i measurements in PAC within excised pancreatic lobules, as described previously (15). This paradigm allows analysis of Ca2+ signaling dynamics in PAC in a more native environment and without the time-consuming process of enzymatic digestion of pancreatic tissue needed to produce isolated acinar cells. Upon carbachol (CCh) stimulation (300 nm), PAC from control mice evoked robust Ca2+ oscillations, with each spike returning close to the basal Ca2+ level between each elevation. The oscillatory Ca2+ signals persisted beyond 30 min of the recording (Fig. 1, A and D). Consistent with previous studies (24, 25), PAC from acute pancreatitis mice elicited Ca2+ signals with a significantly lower frequency (Fig. 1, B and D). Moreover, a higher concentration of CCh (500 nm) failed to rescue the ability to induce strong Ca2+ oscillations in PAC from caerulein-treated mice (Fig. 1, C and D). This strongly indicates that the alteration of Ca2+ signals during acute pancreatitis is not likely due to desensitization to the stimuli but may result from modifications of the constituent components promoting Ca2+ signaling.
Figure 1.
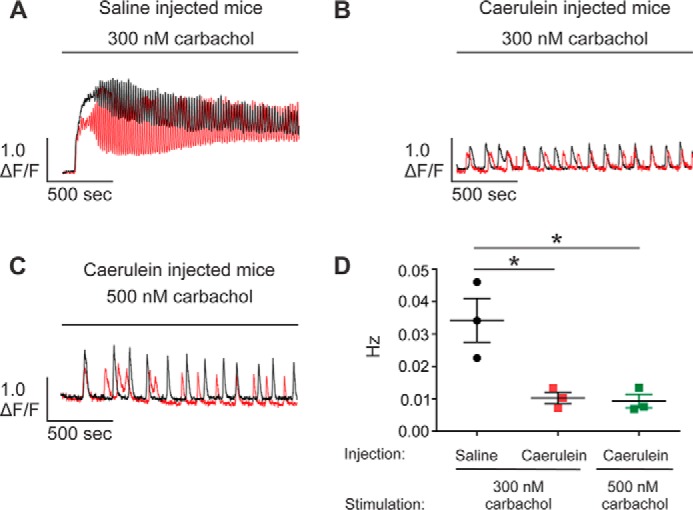
Altered temporal Ca2+ release profile in pancreatic lobules in the in vivo acute pancreatitis model. A–D, pancreatic lobules were excised from mice given three consecutive i.p. injections of either 0.9% saline or caerulein (50 μg/kg/injection), followed by loading with the Ca2+ indicator Fluo-2/AM (25 μm) for 1 h. [Ca2+]i dynamics were measured using a multiphoton microscope. PAC from mice treated with saline elicited robust Ca2+ oscillations in response to 300 nm carbachol (A and D). In contrast to the control saline injection condition, PAC from mice subjected to caerulein treatment elicited Ca2+ transients with a significantly lower frequency (B and D), which could not be rescued by increasing the concentration of the stimulus (C and D). Experiments for each condition were repeated in preparations from three different animals, with more than 20 calcium responses analyzed in each repeat. Two representative traces (black and red) were given for each of the conditions. *, statistical significance determined by one-way ANOVA followed by Tukey's multiple comparisons test.
It is well accepted that Ca2+ oscillations in pancreatic acinar cells are absolutely dependent on the activity of IP3R in the initial phase and, subsequently, on both IP3R and Orai-based channels during sustained stimulation (13, 26). We therefore postulated that the remarkable reduction in oscillatory Ca2+ signals observed (Fig. 1, B–D) resulted from altered regulation of IP3R-induced Ca2+ release. Although all three isoforms of IP3R are expressed in the pancreas, R2 and R3 are most abundant and functionally dominant (27, 28). Therefore, we performed Western blot assays to investigate whether R2 and R3 are modified in this model of acute pancreatitis. Consistent with our previous report, secretagogue-induced hyperstimulation significantly down-regulated both the full-length R2 and R3 (Fig. 2, A, B, F, and G) (18). In addition, we also observed the generation of both fragmented R2 and R3 receptors in mice treated with caerulein (Fig. 2, A, C, D, F, H, and I). Given the strong evidence that digestive enzymes such as trypsin and chymotrypsin are prematurely activated as an early event in PAC in models of acute pancreatitis (29–32), we speculated that IP3R were fragmented by intracellularly active digestive enzymes. Previous studies demonstrated that exposure of IP3R to low concentrations of trypsin and chymotrypsin in vitro results in five receptor fragments (16, 21, 33). These results have been interpreted to indicate that IP3R, structurally consists of five compact globular domains (IP3R fragments I, II, III, IV, and V) interconnected with four solvent-exposed linker regions. To estimate the relative sizes of receptor fragments and the cleavage sites on the IP3R in PAC from mice treated with caerulein, we constructed a library of cDNAs encoding R2- or R3-complementary receptor fragments based on the previously reported tryptic cleavage sites of the receptors (Fig. 3, A and D) (16). Each cDNA was then stably transfected into an IP3R-null background cell line, DT40–3KO (Fig. 3, B, C, E, and F). Fragmented R2 mimicking cleavage at the tryptic site in the second solvent-exposed region was named R2 I-II+III-V (tryp). According to this nomenclature, we generated R2 I-II+III-V (tryp) and R2 I-III+IV-V (tryp) for R2 and R3 I-II+III-V (tryp), R3 I-III+IV-V (tryp), and R3 I-IV+V (tryp) for R3. In addition, we also generated R2 I-IV+V (calpain) based on the sequence homology between IP3R1 (R1) and R2 (Fig. 3, A–F). Notably, receptor fragments in PAC from caerulein-treated mice mainly migrated between 250 and 150 kDa, which were similar molecular weights as for R2 I-III (tryp), R2 I-IV (calpain), R3 I-III (tryp), and R3 I-IV (tryp), indicating that R2 and R3 were preferentially fragmented at the third and fourth solvent-exposed domains (Fig. 2, A and F). Moreover, the N-terminal fragments of cleaved R2 co-immunoprecipitated with the C-terminal fragments (Fig. 2E) in the pancreatic sample prepared from mice treated with caerulein, strongly suggesting that IP3R remained associated after proteolysis during the development of models of acute pancreatitis. Trypsin, one of the essential digestive enzymes, is activated during acute pancreatitis, which has been shown to cleave R2 and R3 in vitro (20, 34). To test the involvement of trypsin activity in receptor fragmentation, we used the in vivo acute pancreatitis model in trypsinogen 7 knockout mice. These animals are reported to have a 60% reduction in total trypsinogen content, the precursor of trypsin (35). Although genetic knockout of trypsinogen 7 had less of an effect on the generation of R2 fragments (Fig. 4, A and C), the elevation of R3 fragments was significantly reduced in this model of acute pancreatitis (Fig. 4, B and D). Therefore, although these data suggest a role of trypsin in receptor fragmentation, they also indicate that other proteases likely participate in receptor proteolysis.
Figure 2.

Generation of fragmented R2 and R3 in the in vivo acute pancreatitis model. A–I, mice received three injections of saline or caerulein hourly. Pancreata were then removed, homogenized, and prepared for Western blot (WB) detection. Samples from DT40–3KO cells stably expressing R2 I-III and R2 I-IV or R3 I-III and R3 I-IV were run on the same gels to indicate the relative sizes of the fragmented receptors. R2 and R3 were fragmented in pancreata from mice treated with caerulein (A and F). Statistics showed that there was a significant reduction in the full-length receptors (B and G) and a concomitant substantial increase in the fragmented receptors (B–D and G–I). The N-terminal fragments of R2 were co-immunoprecipitated (IP) with the C-terminal fragments using the C-terminal R2 antibody CT2, suggesting that the receptor remains associated after proteolysis (E). Each experiment was repeated four times. The arrows in A, E, and F indicate major receptor fragments. *, statistical significance determined by Student's t test. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 3.

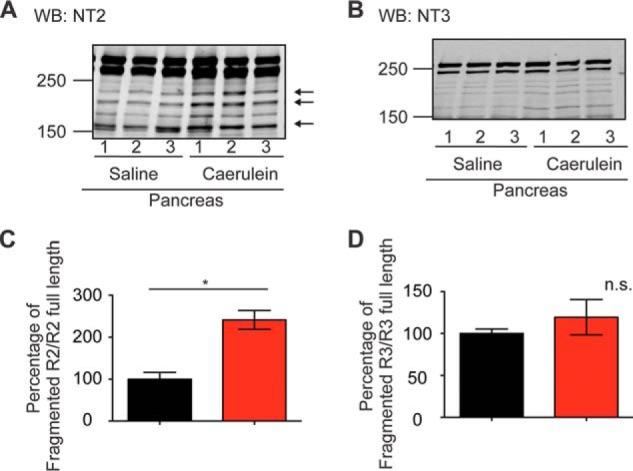
Generation of DT40–3KO cells stably expressing various types of fragmented R2 and R3. A–F, tryptic fragmentation sites (Arg-919, Arg-1583, and Arg-1884) and the predicted calpain fragmentation site (Glu-1869) on R2 and tryptic fragmentation sites (Arg-915, Arg-1574, Arg-1856, and Arg-1869) on R3 (A and D). The epitopes for the R2 N-terminal antibody NT2 (aa 320–338), the R2 C-terminal antibody CT2 (aa 2686–2701), the R3 N-terminal antibody NT3 (aa 22–230), and the R3 C-terminal antibody (aa 2658–2670) are labeled. cDNA encoding complementary pairs of polypeptide chains corresponding to various types of fragmented R2 and R3 were stably transfected into DT40–3KO cells. Western blot (WB) assays confirmed the expression of both the N-terminal fragment and the complementary C-terminal fragment of each type of fragmented R2 (B and C) or fragmented R3 (E and F) in DT40–3KO cells.
Figure 4.
Fragmentation of R2 and R3 in T7 Mice. A–D, T7 mice received three injections of saline or caerulein hourly. Pancreata were then removed, homogenized, and prepared for Western blot (WB) detection. Although the ratio of the amount of R2 fragments to R2 full-length was still significantly increased (A and C), knockdown of trypsinogen 7 completely blocked the increase of the ratio for R3 (B and D). The arrows in A indicate the major receptor fragments. *, statistical significance determined by Student's t test; n.s., not significant.
Generation of fragmented R2 and R3 in an in vitro acute pancreatitis model
To further investigate R2 and R3 receptor fragmentation specifically in PAC, we next exploited a well-established in vitro pancreatitis model using isolated rat PAC (36). To mimic features of pancreatitis in vitro, isolated rat PAC were incubated with two supramaximal concentrations of CCh for different time periods followed by Western blot analysis (37). PAC had a relatively low level of fragmented R2 (Fig. 5A) and R3 (Fig. 5B) under control conditions, presumably as a result of prematurely activated proteolytic enzymes resulting from the inevitable cell damage occurring during pancreatic acinar cell preparation and manipulation. However, we cannot exclude the possibility that there is a basal level of fragmented R2 and R3 under physiological conditions. Nevertheless, in agreement with our in vivo findings, despite the appearance of low amounts of fragmented R2 and R3 without treatment, supramaximal concentrations of CCh consistently, in a concentration-dependent manner, further increased the levels of fragmented R2 (Fig. 5A) and R3 (Fig. 5B), accompanied by a concomitant reduction of full-length receptor. To demonstrate that fragmentation of IP3R in PAC was not limited to the hyperstimulation model of acute pancreatitis, PAC were exposed to the bile acid taurolithocholic acid 3-sulfate disodium salt (TLCS) (38–40). This paradigm is often utilized as a pathologically relevant stimulus to mimic pancreatic duct obstruction and subsequent bile reflux. TLCS exposure consistently resulted in increased fragmentation of R2 and R3 (Fig. 5, C–E). Importantly, the generation of receptor fragments could be reversed by preincubation of the cells with cell-permeable trypsin inhibitors (Fig. 5, C and D). Together with Fig. 4, these data confirm that trypsin activity, directly or indirectly, is responsible for R2 and R3 fragmentation during the development of acute pancreatitis.
Figure 5.

Generation of fragmented R2 and R3 in the in vitro acute pancreatitis model. A–E, rat pancreatic acinar cells were isolated and incubated with various supramaximal concentrations of carbachol for different amounts of time. Cells were then harvested and subjected to Western blot (WB) detection. Both R2 and R3 were cleaved in the in vitro acute pancreatitis model (A and B). The bile salt TLCS also resulted in receptor fragmentation, which can be blocked by preincubation of PAC with trypsin inhibitors (2 μm gabexate and 3 μm camostate) (C and D). Total protein staining was performed as a loading control for C and D in E. Each experiment was repeated three times. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Our previous studies investigating the consequences of R1 fragmentation have demonstrated that R1 fragments remain associated within the ER despite the loss of peptide continuity and retain the ability to be gated by IP3 (41). Thus, we next investigated whether R2 and R3 similarly retain tetrameric architecture and ER membrane localization after receptor fragmentation. Isolated rat PAC were incubated with supramaximal concentrations of CCh to induce acute pancreatitis in vitro. Membrane and cytosolic fractions were then prepared by differential centrifugation. Glyceraldehyde-3-phosphate dehydrogenase was used as a marker for the cytosol fraction. No detectable R2 and R3 signals were observed in the cytosol fraction, and both N- and C-terminal fragments of R2 (Fig. 6A) and R3 (Fig. 6B) were present in the membrane fraction, indicating that, like R1, R2 and R3 remained ER-associated after proteolytic fragmentation. Native nondenaturing gel analysis showed that fragmented R2 (Fig. 6C) and R3 (Fig. 6D) in PAC from rats subjected to the in vitro model of acute pancreatitis migrated at the same molecular weight as that of the full-length tetramers. Further, no detectable receptor fragments of lower molecular weights were present in any cell lysates under the pancreatitis conditions. In total, these data strongly suggest that all R2 and R3 exist as tetramers on the ER membrane after proteolytic fragmentation.
Figure 6.

R2 and R3 retain a tetrameric architecture and ER membrane localization after receptor fragmentation. A–D, isolated rat PAC were incubated with various supramaximal concentrations of carbachol for different amounts of time, followed by membrane fractionation. Full-length receptors, the N-terminal receptor fragments, and the C-terminal receptor fragments all remained membrane-associated for both R2 (A) and R3 (B). Further, native gel analysis showed that fragmented R2 and R3 migrated at the same molecular weight as full-length receptors, indicating that R2 and R3 retain a tetrameric architecture after receptor fragmentation (C and D). R3 I-V represents the migration pattern of the dissociated monomeric N-terminal fragment. Each experiment was repeated three times. WB, Western blot; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Region-specific fragmentation regulates the temporal characteristics of Ca2+ signals mediated by R2 and R3
Given the critical roles of IP3R in Ca2+ signaling in PAC (13), our data so far led us to posit that proteolytic fragmentation mediates the alteration of R2 and R3 activities and, consequently, contributes to the altered Ca2+ signals in pancreatic acinar cells during the development of acute pancreatitis. To unambiguously study the function of only fragmented R2 and R3 without the potential confounding impact of contaminating signals from endogenous receptors, we performed single-cell Ca2+ imaging assays using DT40–3KO cells stably expressing complementary peptides to mimic R2 or R3 fragmented at different sites (Fig. 3). We have previously demonstrated that, after expression, these fragments are assembled into functional tetrameric IP3R on the ER membrane and are properly gated by IP3 (41). Consistent with our previous findings, all R2 and R3 generated from cDNAs encoding complementary fragments were capable of supporting Ca2+ signals in response to maximal protease-activated receptor 2 (PAR2) activation with trypsin (Fig. 7, A–H).
Figure 7.

All fragmented R2 and R3 were functional. A–H, a panel of DT40–3KO cells, each stably expressing one pair of complementary polypeptides representing fragmented R2 or R3 at particular sites, were loaded with Fura-2/AM, followed by stimulation of PAR2 receptor activation with trypsin. Single-cell imaging assays showed that all types of fragmented R2 (A–D) and fragmented R3 (E–H) were capable of eliciting a Ca2+ response in response to PAR2 activation. Each experiment was repeated more than three times with more than 30 cells in each run.
Next we investigated whether receptor fragmentation has an impact on the temporal characteristics of IP3R-mediated Ca2+ signals. Anti-IgM cross-links B-cell receptors on the cell surface of DT40–3KO cells and results in continuous production of IP3. Upon anti-IgM stimulation, DT40–3KO cells stably expressing either full-length R2 or R3 elicited robust Ca2+ oscillations in an isoform-specific manner (Fig. 8, A and G) (42–44). When cDNAs encoding complementary polypeptides to mimic receptor fragmentation introduced at the second solvent-exposed region were expressed, R2 I-II+III-V (tryp) and R3 I-II+III-V (tryp) were still capable of evoking strong oscillatory Ca2+ signals (Fig. 8, B, E, H, and K). However, anti-IgM stimulation of cells expressing constructs mimicking receptor fragmentation in R2 and R3 at solvent-exposed sites more toward to the C terminus resulted in Ca2+ oscillations with a significantly lower frequency (Fig. 8, C–E and I–K). Further, increasing the concentration of anti-IgM failed to rescue the loss of Ca2+ oscillations mediated by R2 I-III+IV-V (tryp), R2 I-IV+V (calpain), R3 I-III+IV-V (tryp), or R3 I-IV+V (tryp) (Figs. 7L and 8F). These data strongly suggest that region-specific receptor fragmentation regulates R2 and R3 activities. These results were consistent with those obtained from the ex vivo pancreatic acini Ca2+ imaging assays (Fig. 1, A–D) and provide a possible explanation for the reduction of Ca2+ oscillations in PAC isolated from mice subjected to caerulein treatment. Based on these data, it can be envisioned that prematurely activated digestive enzymes cleave R2 and R3 at the third and fourth solvent-exposed regions and, as a consequence, significantly reduce the frequency of oscillatory Ca2+ signals of IP3R in PAC during the development of pancreatitis.
Figure 8.

Region-specific fragmentation alters the temporal Ca2+ release profile of R2 and R3. A–L, a panel of DT40–3KO cells, each stably expressing a pair of complementary polypeptides representing fragmented R2 or R3 at particular sites, were loaded with Fura-2/AM, followed by stimulation of B-cell receptor activation with anti-IgM. Single-cell Ca2+ imaging recording showed that fragmentation at the proximal N-terminal solvent-exposed region had no impact on the frequency of Ca2+ oscillations mediated by R2 (A, B, and E) and R3 (G, H, and K). However, receptor fragmentation more toward the C terminus resulted in a significant decrease in Ca2+ oscillations mediated by fragmented R2 (C–E) and fragmented R3 (I–K). This decrease in the frequency of Ca2+ oscillations could not be rescued by increasing the concentrations of stimuli (F and L). *, statistical significance determined by one-way ANOVA followed by Tukey's multiple comparisons test.
One caveat of studying the functional consequences of receptor fragmentation by using complementary receptor fragments is that fragmented IP3R are assembled from polypeptides but not generated in situ. In addition, measurements of global [Ca2+]i are an indirect measurement of channel activity. Therefore, we next performed single-channel patch clamp recording with the “on-nucleus” configuration to study the potential biophysical alteration of the R2 activity resulting from proteolytic fragmentation. An initial attempt was made to induce fragmentation of R2 with trypsin. However, no channel activity was recorded in the presence of trypsin, even at picomolar levels of enzyme, presumably because of the receptor destruction resulting from failure to control the amount of receptor proteolysis under these conditions. To circumvent this difficulty and fragment the receptor specifically at the fourth solvent-exposed region in situ in a controlled manner, we constructed a R2 with a tobacco etch virus (TEV) protease cleavage sequence inserted immediately after the tryptic site (Arg-1884) in this region. This construct, termed R2 (TEV), was stably expressed in DT40–3KO cells. TEV protease was capable of specifically cleaving purified R2 (TEV) at the fourth solvent-exposed region to result in fragments corresponding to R2 I-IV+V (TEV) (Fig. 9C). In contrast, incubation of purified full-length R2 with TEV protease failed to result in any receptor fragment (Fig. 9A), confirming the specificity of TEV protease–mediated receptor fragmentation. Patch clamp recording in the on-nucleus configuration demonstrated no difference in the single-channel open probability (Po) or mode of gating between R2 (TEV) and full-length R2 WT in the absence of TEV protease (Fig. 9, B and D–G). However, when R2 (TEV) was exposed to TEV, a significant decrease in channel Po, accompanied by a significant increase in interburst interval but no change in burst length (Fig. 9, D–G), was observed. TEV protease had no effect on full-length R2 (Fig. 9, B and E–G), strongly indicating that the alteration in R2 (TEV) channel biophysical properties resulted from receptor fragmentation. Taken together, these single-channel data are consistent with the results of the single-cell Ca2+ imaging assays (Fig. 9) and provide an underlying biophysical mechanism to explain the decrease in oscillatory Ca2+ signals in PAC during experimental acute pancreatitis (Fig. 1).
Figure 9.

Receptor fragmentation alters R2 activity at the single-channel level. A–G, a TEV protease cleavage sequence was inserted into R2 to achieve a specific receptor cleavage at the fourth solvent-exposed region. TEV protease neither cleaved the R2 WT nor had an impact on its channel activity (A, B, and E–G). In contrast, TEV protease specifically cleaved R2 (TEV) at the predicted site and significantly decreased the channel open probability and increased interburst interval with no impact on burst length (C–G). The NT2 antibody was used to detect R2 in the Western blot assays. Each experimental condition was repeated five times. *, statistical significance determined by one-way ANOVA followed by Tukey's multiple comparisons test. CTRL, control.
Discussion
This study represents a comprehensive investigation of a novel modification of R2 and R3 and its consequences in PAC in the context of two rodent models of acute pancreatitis. A previous study, in which PAC were isolated by enzymatic digestion following induction of experimental pancreatitis, reported a significant decrease in the ability of PAC from the experimental pancreatitis animals to display sustained oscillatory Ca2+ signals. These data are established as strong evidence that the characteristics of the cytosolic Ca2+ signal, important for driving physiological secretion, is disrupted in experimental pancreatitis (24). However, the underlying mechanism for this striking transition in Ca2+ signaling was not elucidated. In this study, we first confirmed this alteration of Ca2+ signals in PAC in situ in excised pancreatic lobules prepared from control mice or from mice subjected to the experimental acute pancreatitis paradigm. This preparation has the significant advantage that the tissue is not exposed to prolonged enzymatic isolation and better reflects the heterogeneity of the impact on different regions of the tissue in the pancreatitis model. Notably, Western blot analysis demonstrated that, concurrent with a significant reduction in the ability of PAC from experimental pancreatitis animals to support sustained Ca2+ oscillations, these mice showed a marked decrease in the abundance of full-length R2 and R3 with the concomitant appearance of various species of fragments of R2 and R3. These observations, coupled with data showing that proteolytic cleavage of R1 alters the receptor activity without disabling the channel, led us to hypothesize that, during pancreatitis, proteases are prematurely activated, resulting in fragmentation of R2 and R3 and, in turn, altered [Ca2+]i signals.
To explore this hypothesis, experiments were designed to answer four major questions. Do R2 and R3 retain their tetrameric architectures after proteolytic fragmentation? Do fragmented R2 and R3 remain associated with ER membranes? Do fragmented R2 and R3 retain the ability to be gated by IP3 binding? Does receptor fragmentation alter the temporal profile of agonist-evoked Ca2+ signals? First, nondenaturing native gel analysis combined with co-immunoprecipitation assays strongly suggested that, although peptide continuity was lost, R2 and R3 retain a tetrameric structure after receptor fragmentation. This is likely due to significant noncovalent interactions between tightly folded globular domains in the proteolytically cleaved protein. Further, membrane fractionation followed by Western blot analysis indicated that all fragments of R2 and R3 were still located on ER membranes. To study the function of fragmented receptors, we generated a library of DT40–3KO cells stably expressing R2 or R3 assembled from complementary peptides representing one type of fragmented IP3R. Consistent with our previous findings for R1, all fragmented IP3R could support Ca2+ release in response to IP3 binding. However, when receptors were fragmented more toward the C terminus, the temporal characteristics of Ca2+ signals were greatly altered. Indeed, compared with full-length receptors, R2 I-III+IV-V, R2 I-IV+V, R3 I-III+IV-V, and R3 I-IV+V exhibited a significantly lower frequency of Ca2+ oscillations. At a biophysical level, this observation was supported mechanistically by single-channel patch clamp data, which revealed that receptor fragmentation dramatically decreased the single-channel open probability.
This study and our previous report have thoroughly investigated the functional consequences of IP3R fragmentation (12, 45, 46). Early studies showed that exposure of purified R1 to a low concentration of trypsin in vitro resulted in five receptor fragments (16, 21, 33). This result was interpreted as the overall structure of R1 consisting of five compact globular domains linked by four solvent exposed regions. Based on the sequence homology among all three isoforms of IP3R, R2 and R3 were also predicted to have the same overall structure (16, 19). It is worth noting that all putative fragmentation sites, including those reported in this study, are located in these solvent-exposed regions and may be explained by the relatively ease of accessibility of these regions to protease activity (16, 21, 33, 47, 48). The coupling domain (amino acids 586–2276) of IP3R generates interfaces where Ca2+, regulatory proteins, and nucleotides bind to mediate channel activities (1, 2, 49). The distinct regulatory events among the three isoforms of IP3R confer subtype-specific Ca2+ signals. We postulate that some of the regulatory events rely on peptide continuity for an appropriate communication between regulatory inputs and the channel domain. As a result, disruption of receptor continuity by proteases in specific regions of the coupling domain would potentially impact such communication and, subsequently, alter the single-channel Po and temporal characteristics of Ca2+ signals evoked by agonist stimulation.
Consistent with the idea that overall regulation of IP3R activity is altered following loss of peptide continuity, we reported previously that fragmented R1 at either the third or fourth solvent-exposed regions in the coupling domain, corresponding to the activity of caspase or calpain, significantly increased the ability of R1 to induce Ca2+ oscillations in cells (12, 41, 46). Notably, we have also reported that caspase-cleaved R1 exhibited a significantly augmented single-channel open probability (12). Here we extend this finding by showing that region-specific receptor fragmentation is a general regulatory event for all isoforms of IP3R, including R2 and R3. Interestingly, in marked contrast to R1, proteolytic fragmentation significantly decreased the single-channel open probability of R2 and the ability of R2 and R3 to induce oscillatory Ca2+ signals in cells. How does proteolysis result in subtype-specific effects on single channel activity and the temporal pattern of stimulated Ca2+ signals? Although extrapolating the effects on single channel activity to the global pattern of cellular Ca2+ signals is challenging, a current mathematical model for the generation of Ca2+ oscillations suggests that the oscillation frequency is dictated by the rate at which Ca2+ activates and subsequently inactivates the particular IP3R (46). We speculate that this form of regulation differs between IP3R subtypes in the native state and is altered by proteolysis. An increase in R1 Po following cleavage might reflect an increase in susceptibility to be activated (or, equally possible, a decreased sensitivity to be inactivated) by the fixed [Ca2+] in the patch pipette, with the opposite occurring for R2. In this scenario, differentially altering the relationship between IP3R activation and deactivation could result in changes to the oscillation period.
What is the role of IP3R fragmentation in acute pancreatitis? We used both in vitro and in vivo rodent models with different toxic stimuli to characterize the modification of IP3R. Consistently, IP3R were fragmented in all tested models, indicating that IP3R fragmentation may be a general event occurring in acute pancreatitis. Our studies show that IP3R fragmentation likely occurs at an early stage in models of acute pancreatitis. Repetitive injections of supramaximal concentrations of caerulein are widely used to induce acute pancreatitis in mice, and commonly seven to 12 injections are utilized (24, 35, 50, 51). Our data demonstrate that fragmentation of IP3R is already initiated after the third injection. Based on these data, we speculate that IP3R fragmentation occurs at an early stage of acute pancreatitis and may represent a protective strategy employed by the cell to limit Ca2+ signaling. The signaling cascade linking pathological Ca2+ signals to acute pancreatitis has been well characterized. Specifically, a globally sustained increase of [Ca2+]i in PAC is thought to contribute to secretory inhibition, premature intracellular digestive enzyme activation, cell death, and, eventually, acute pancreatitis (52–54). Based on these ideas, experimental strategies aimed at inhibiting pathological Ca2+ signals, such as attenuating Ca2+ channel activity (55–57), chelating intracellular Ca2+ (58–60), or promoting Ca2+ clearance (61), have been shown to be protective in acute pancreatitis. Notably, we demonstrated that fragmented R2 and R3 may play a similar functional role by reducing the frequency of Ca2+ oscillations and essentially decreasing the overall [Ca2+]i in PAC in models of acute pancreatitis. Specifically, R2 and R3 fragmentation transformed sustained Ca2+ responses or robust elevated Ca2+ oscillations into lower-frequency Ca2+ transients and, thereby, would be predicted to decrease the total amount of Ca2+ release into the cytosol by both internal Ca2+ release and, subsequently, store-dependent Ca2+ influx. This process may either delay or suppress the premature intracellular digestive enzyme activation and, thereby, be protective at the early stage of acute pancreatitis. In conclusion, combined with our previous reports (12, 45), our data are the first to systemically investigate and characterize the functional consequences of proteolytic fragmentation of all three isoforms of IP3R. Further, to our knowledge, this is the first report to show the modification of IP3R in acute pancreatitis and provide a possible explanation for the alteration of the spatial and temporal properties of Ca2+ signals observed in the early stages of acute experimental pancreatitis.
Materials and methods
Reagents
All restriction enzymes and T4-DNA ligase were from New England Biolabs. RPMI 1640 medium, penicillin/streptomycin, G418 sulfate, β-mercaptoethanol, and chicken serum were purchased from Invitrogen. Fetal bovine serum was from Gemini Bio-products. Fura-2/AM was from TEFLabs. Enhanced chemiluminescent substrate and 800CWTM secondary antibodies were from Thermo Scientific. The Dc protein assay kit, Tris base, glycine, horseradish peroxidase–conjugated secondary antibodies, and all reagents used for SDS-PAGE were from Bio-Rad. Mouse anti-chicken IgM was from SouthernBiotech. The antibody against the N terminus of R2 (NT2) was generated by Pocono Rabbit Farms and Laboratories. The mouse mAb against the N terminus of R3 (NT3) was from BD Transduction Laboratories. Both NT2 and NT3 were diluted (1:1000) for Western blotting. CT2 and CT3 were gifts from the Richard Wojcikiewicz laboratory. CT2 and CT3 were raised against the extreme carboxyl aa 2686–2701 of R2 and aa 2658–2670 of R3, respectively (28). Both CT2 and CT3 were diluted (1:200) for Western blotting. All IP3R antibodies used in this study have been shown to be subtype-specific based on appropriate recognition of a single isoform by Western blotting cell lysates containing only a specific isoform (10, 18, 28, 42). Caerulein and taurolithocholic acid 3-sulfate disodium salt were from Sigma-Aldrich. Gabexate mesylate and camostat mesilate were from Selleckchem.
Animal husbandry
Experiments with animals were conducted in accordance with protocol 100783/UCAR-2001-214R, approved by the University of Rochester. The mice used for this work were 12-week-old C57/BL6NJ mice. The rats used for this work were adult male Wistar rats.
Cell culture and plasmid transfection
DT40–3KO cells were grown in RPMI 1640 medium supplemented with 1% chicken serum, 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 39 °C with 5% CO2. DT40–3KO cell transfection and generation of stable cell lines were performed as described previously using the Amaxa nucleofector (Lonza Laboratories). DT40–3KO were generated and authenticated in the Riken Institute (Japan) and originally obtained directly from this source (62).
Construct preparation
The method for generation of fragmented IP3R was first described elsewhere in details (41). cDNAs encoding R2 I-II+III-V (trypsin), R2 I-III+IV-V (trypsin), R2 I-IV+V (calpain), R3 I-II+III-V (trypsin), R3 I-III+IV-V (trypsin), and R3 I-IV+V (trypsin) were constructed using the corresponding primers: forward, 5′-GATGGAAGCAACAATGTCATGAGGTAGAATTCGCGGCCGCGCTAGCATGACCATCCACGGAGTGGGAGAGATGA-3′; forward, 5′-ATGGGCTGGAGACTCTCAGCTCGCTAGAATTCGCGGCGCGCTAGCATGTCTGGACCTCGCTTCAAGGAAG-3′; forward, 5′-GGAATGAAAGGGCAGTTAACAGAATAGAATTCGCGGCCGCGCTAGCATGGCGTCTTCAGCCACATCC-3′; forward, 5′-CCTGGTGGCAAGAATGTGCGGAGGTAGAATTCGCGGCCGCGCTAGCATGTCCATCCAGGGGGTGGGGCACA-3′; forward, 5′-GCCAACTACAAGACGGCCACCAGGTAGAATTCGCGGCCGCGCTAGCATGACCTTCCCTCGGGTCATCCCC-3′; and 5′-CACCGGGGGCACGACGTGAGCGAGTAGAATTCGCGGCCGCGCTAGCATGCGTGCGCAGAACAACGAGATGGGG-3′, respectively. To introduce a TEV protease cleavage site after the Arg-1884 in R2, primer 5′-AGCATACTGTGTGTACAGAGAAAACCTGTACTTCCAATCTAGAGAGATGGACCCGGAAAT-3′ was used.
Native gel analysis
Cells were harvested by centrifugation and lysed in CHAPS lysis buffer (40 mm NaCl, 25 mm HEPES, 10 mm CHAPS, and 1 mm EDTA (pH 7.4)) supplemented with protease inhibitors. After 20 min on ice at 4 °C, lysates were cleared by centrifugation at 16,000 × g for 10 min at 4 °C. Cleared lysates were mixed with of 4× sample buffer, 5% G-250 sample additive, and fractionated 3–12% native PAGE Novex gels. Separated proteins were transferred to polyvinylidene difluoride membranes and probed using the indicated primary antibodies and the appropriate horseradish peroxidase–conjugated secondary antibodies. Protein bands were detected using enhanced chemiluminescent substrate.
Subcellular fractionation
Cells were harvested and washed with ice-cold PBS and then resuspended in homogenization buffer containing 20 mm HEPES, 5 mm NaN3, 0.5 mm EGTA, and 320 mm sucrose (pH 7.4) supplemented with protease inhibitors. Cells were homogenized using a Teflon glass homogenizer. Homogenates were cleared by centrifugation at 1000 × g for 10 min at 4 °C. The resulting supernatants were centrifuged at 100,000 × g at 4 °C for 1 h. The supernatants designated as the cytosolic fraction were removed, and the microsomal pellet was resuspended in lysis buffer. Equivalent amounts of proteins were fractionated and processed for immunoblot analyses with the indicated antibodies.
Fluorescence imaging
DT40 cells expressing defined IP3R constructs were loaded with 2 μm Fura-2/AM on a glass coverslip mounted onto a Warner chamber at room temperature for 20–30 min. Loaded cells were perfused with HEPES imaging buffer (137 mm NaCl, 4.7 mm KCl, 1.26 mMCaCl2,1 mm Na2HPO4, 0.56 mm MgCl2, 10 mm HEPES, and 5.5 mm glucose (pH 7.4)) and stimulated with the indicated agonist. Ca2+ imaging was performed using an inverted epifluorescence Nikon microscope with a ×40 oil immersion objective (numerical aperture = 1.3). Cells were alternately excited at 340 and 380 nm, and emission was monitored at 505 nm. Images were captured every second with an exposure of 10 ms and 4 × 4 binning using a digital camera (Cooke Sensicam QE) driven by TILL Photonics software.
In vivo acute pancreatitis model
Twelve-week-old C57/BL6NJ mice were starved overnight and then given three consecutive i.p. injections of caerulein (50 μg/kg/injection) hourly. Mice in the control group received the same amount of 0.9% saline instead. Mice were euthanized 1 h after the third injection, and pancreata were removed for further analysis. All animal procedures were approved by the University of Rochester University Committee on Animal Resources.
Pancreatic lobule imaging using multiphoton microscopy
Mice were euthanized, and pancreata were removed and placed into oxygen-bubbled ice-cold imaging buffer (137 mm NaCl, 4.7 mm KCl, 1.26 mMCaCl2,1 mm Na2HPO4, 0.56 mm MgCl2, 10 mm HEPES, and 5.5 mm glucose (pH 7.4)). Pancreatic lobules were excised and exposed to liberase for 10 min, followed by loading with Fluo-2/AM. Fluo2-loaded lobules were excited at 810 nm using a Spectra Physics tunable fS pulsed Ti-Sapphire laser controlled by Fluoview software on an Olympus FV1000MP microscope using a ×25 water immersion objective (1.03 numerical aperture). The clusters were stimulated with various concentrations of CCh. Images were acquired at a resolution of 512 × 512 pixels. Regions of interest were selected, and fluorescence intensity in that region was determined as a function of time and expressed relative to the initial fluorescence.
Isolation of rat pancreatic acinar cells
Rat pancreata were obtained from male adult Wistar rats. Pancreata were enzymatically digested with type II collagenase (Sigma) in Dulbecco's modified Eagle's medium (Invitrogen) with 0.1% BSA and 1 mg/ml soybean trypsin inhibitor for 30 min, followed by gentle trituration. Acini were then filtered through 350-μm nylon mesh, centrifuged at 75 × g through 1% BSA in Dulbecco's modified Eagle's medium, and resuspended in 1% BSA in Dulbecco's modified Eagle's medium.
Single-channel patch clamp with the on-nucleus configuration
Isolated DT40 nuclei were prepared by homogenization as described previously (63). Single IP3R channel potassium currents were measured in the on-nucleus patch clamp configuration using pCLAMP 9 and an Axopatch 200B amplifier (Molecular Devices) as described previously (63). TEV protease was included in the pipette solution for the corresponding experiments. Gigaohm seals were attained, and channel activity was verified at −100 mV. Patches were then depolarized to 0 mV for 15 min to allow TEV protease to cleave the receptor. The patches were again repolarized to −100 mV, and channel activity was measured. Traces were consecutive 3-s sweeps sampled at 20 kHz and filtered at 5 kHz. Pipette resistances were typically 20 megaohm and seal resistances were >5 gigaohm.
Author contributions
L. W. and L. E. W. data curation; L. W. and L. E. W. formal analysis; L. W. and L. E. W. investigation; L. W. and L. E. W. methodology; L. W. writing-original draft; K. J. A. and D. I. Y. conceptualization; K. J. A. and D. I. Y. writing-review and editing; D. I. Y. supervision; D. I. Y. funding acquisition; D. I. Y. project administration.
Acknowledgment
We thank Dr. Ashok Saluja (University of Miami Medical School) for providing the trypsinogen 7–null animals.
This work was supported by NIDCR, National Institutes of Health Grants R01DE014756 and R01DE019245. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IP3R
- inositol 1,4,5-trisphosphate receptor(s)
- ER
- endoplasmic reticulum
- i.p.
- intraperionteal(ly)
- PAC
- pancreatic acinar cell(s)
- CCh
- carbachol
- TLCS
- taurolithocholic acid 3-sulfate disodium salt
- cDNA
- complementary DNA
- TEV
- tobacco etch virus
- Po
- open probability
- aa
- amino acid(s).
References
- 1. Foskett J. K., White C., Cheung K. H., and Mak D. O. (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658 10.1152/physrev.00035.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yule D. I., Betzenhauser M. J., and Joseph S. K. (2010) Linking structure to function: recent lessons from inositol 1,4,5-trisphosphate receptor mutagenesis. Cell Calcium 47, 469–479 10.1016/j.ceca.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alzayady K. J., Sebé-Pedrós A., Chandrasekhar R., Wang L., Ruiz-Trillo I., and Yule D. I. (2015) Tracing the evolutionary history of inositol 1,4,5-trisphosphate receptor: insights from analyses of Capsaspora owczarzaki Ca2+ release channel orthologs. Mol. Biol. Evol. 32, 2236–2253 10.1093/molbev/msv098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chandrasekhar R., Yule D. I., and Wang L. (2017) Inositol 1,4,5-trisphosphate receptors (InsP3R). Pancreapedia: Exocrine Pancreas Knowledge Base 10.3998/panc.2017.07 [DOI] [Google Scholar]
- 5. Berridge M. J. (2009) Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 1793, 933–940 10.1016/j.bbamcr.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 6. Berridge M. J. (2016) The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296 10.1152/physrev.00006.2016 [DOI] [PubMed] [Google Scholar]
- 7. Luyten T., Welkenhuyzen K., Roest G., Kania E., Wang L., Bittremieux M., Yule D. I., Parys J. B., and Bultynck G. (2017) Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca2. Biochim. Biophys. Acta 1864, 947–956 10.1016/j.bbamcr.2017.02.013 [DOI] [PubMed] [Google Scholar]
- 8. Chandrasekhar R., Alzayady K. J., Wagner L. E. 2nd, and Yule D. I. (2016) Unique regulatory properties of heterotetrameric inositol 1,4,5-trisphosphate receptors revealed by studying concatenated receptor constructs. J. Biol. Chem. 291, 4846–4860 10.1074/jbc.M115.705301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patterson R. L., Boehning D., and Snyder S. H. (2004) Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu. Rev. Biochem. 73, 437–465 10.1146/annurev.biochem.73.071403.161303 [DOI] [PubMed] [Google Scholar]
- 10. Alzayady K. J., Wang L., Chandrasekhar R., Wagner L. E. 2nd, Van Petegem F., and Yule D. I. (2016) Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal. 9, ra35 10.1126/scisignal.aad6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wright F. A., and Wojcikiewicz R. J. (2016) Chapter 4: inositol 1,4,5-Trisphosphate Receptor Ubiquitination. Prog. Mol. Biol. Transl. Sci. 141, 141–159 10.1016/bs.pmbts.2016.02.004 [DOI] [PubMed] [Google Scholar]
- 12. Wang L., Wagner L. E. 2nd, Alzayady K. J., and Yule D. I. (2017) Region-specific proteolysis differentially regulates type 1 inositol 1,4,5-trisphosphate receptor activity. J. Biol. Chem. 292, 11714–11726 10.1074/jbc.M117.789917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Futatsugi A., Nakamura T., Yamada M. K., Ebisui E., Nakamura K., Uchida K., Kitaguchi T., Takahashi-Iwanaga H., Noda T., Aruga J., and Mikoshiba K. (2005) IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309, 2232–2234 10.1126/science.1114110 [DOI] [PubMed] [Google Scholar]
- 14. Fujino I., Yamada N., Miyawaki A., Hasegawa M., Furuichi T., and Mikoshiba K. (1995) Differential expression of type 2 and type 3 inositol 1,4,5-trisphosphate receptor mRNAs in various mouse tissues: in situ hybridization study. Cell Tissue Res. 280, 201–210 10.1007/s004410050345 [DOI] [PubMed] [Google Scholar]
- 15. Teos L. Y., Zhang Y., Cotrim A. P., Swaim W., Won J. H., Ambrus J., Shen L., Bebris L., Grisius M., Jang S. I., Yule D. I., Ambudkar I. S., and Alevizos I. (2015) IP3R deficit underlies loss of salivary fluid secretion in Sjogren's syndrome. Sci. Rep. 5, 13953 10.1038/srep13953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bezprozvanny I. (2005) The inositol 1,4,5-trisphosphate receptors. Cell Calcium 38, 261–272 10.1016/j.ceca.2005.06.030 [DOI] [PubMed] [Google Scholar]
- 17. Diaz F., and Bourguignon L. Y. (2000) Selective down-regulation of IP3 receptor subtypes by caspases and calpain during TNFα-induced apoptosis of human T-lymphoma cells. Cell Calcium 27, 315–328 10.1054/ceca.2000.0126 [DOI] [PubMed] [Google Scholar]
- 18. Wojcikiewicz R. J., Ernst S. A., and Yule D. I. (1999) Secretagogues cause ubiquitination and down-regulation of inositol 1,4,5-trisphosphate receptors in rat pancreatic acinar cells. Gastroenterology 116, 1194–1201 10.1016/S0016-5085(99)70023-5 [DOI] [PubMed] [Google Scholar]
- 19. Maes K., Missiaen L., Parys J. B., De Smet P., Sienaert I., Waelkens E., Callewaert G., and De Smedt H. (2001) Mapping of the ATP-binding sites on inositol 1,4,5-trisphosphate receptor type 1 and type 3 homotetramers by controlled proteolysis and photoaffinity labeling. J. Biol. Chem. 276, 3492–3497 10.1074/jbc.M006082200 [DOI] [PubMed] [Google Scholar]
- 20. Soulsby M. D., and Wojcikiewicz R. J. (2005) The type III inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at three sites. Biochem. J. 392, 493–497 10.1042/BJ20051325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshikawa F., Iwasaki H., Michikawa T., Furuichi T., and Mikoshiba K. (1999) Trypsinized cerebellar inositol 1,4,5-trisphosphate receptor: structural and functional coupling of cleaved ligand binding and channel domains. J. Biol. Chem. 274, 316–327 10.1074/jbc.274.1.316 [DOI] [PubMed] [Google Scholar]
- 22. Adler G., Rohr G., and Kern H. F. (1982) Alteration of membrane fusion as a cause of acute pancreatitis in the rat. Dig. Dis. Sci. 27, 993–1002 10.1007/BF01391745 [DOI] [PubMed] [Google Scholar]
- 23. Lerch M. M., and Gorelick F. S. (2013) Models of acute and chronic pancreatitis. Gastroenterology 144, 1180–1193 10.1053/j.gastro.2012.12.043 [DOI] [PubMed] [Google Scholar]
- 24. Ward J. B., Sutton R., Jenkins S. A., and Petersen O. H. (1996) Progressive disruption of acinar cell calcium signaling is an early feature of cerulein-induced pancreatitis in mice. Gastroenterology 111, 481–491 10.1053/gast.1996.v111.pm8690215 [DOI] [PubMed] [Google Scholar]
- 25. Mooren FCh., Hlouschek V., Finkes T., Turi S., Weber I. A., Singh J., Domschke W., Schnekenburger J., Krüger B., and Lerch M. M. (2003) Early changes in pancreatic acinar cell calcium signaling after pancreatic duct obstruction. J. Biol. Chem. 278, 9361–9369 10.1074/jbc.M207454200 [DOI] [PubMed] [Google Scholar]
- 26. Voronina S., Collier D., Chvanov M., Middlehurst B., Beckett A. J., Prior I. A., Criddle D. N., Begg M., Mikoshiba K., Sutton R., and Tepikin A. V. (2015) The role of Ca2+ influx in endocytic vacuole formation in pancreatic acinar cells. Biochem. J. 465, 405–412 10.1042/BJ20140398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Smedt H., Missiaen L., Parys J. B., Henning R. H., Sienaert I., Vanlingen S., Gijsens A., Himpens B., and Casteels R. (1997) Isoform diversity of the inositol trisphosphate receptor in cell types of mouse origin. Biochem. J. 322, 575–583 10.1042/bj3220575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wojcikiewicz R. J. (1995) Type I, II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J. Biol. Chem. 270, 11678–11683 10.1074/jbc.270.19.11678 [DOI] [PubMed] [Google Scholar]
- 29. Sherwood M. W., Prior I. A., Voronina S. G., Barrow S. L., Woodsmith J. D., Gerasimenko O. V., Petersen O. H., and Tepikin A. V. (2007) Activation of trypsinogen in large endocytic vacuoles of pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 104, 5674–5679 10.1073/pnas.0700951104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saluja A. K., Donovan E. A., Yamanaka K., Yamaguchi Y., Hofbauer B., and Steer M. L. (1997) Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology 113, 304–310 10.1016/S0016-5085(97)70108-2 [DOI] [PubMed] [Google Scholar]
- 31. Hofbauer B., Saluja A. K., Lerch M. M., Bhagat L., Bhatia M., Lee H. S., Frossard J. L., Adler G., and Steer M. L. (1998) Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am. J. Physiol. 275, G352–G362 [DOI] [PubMed] [Google Scholar]
- 32. Balldin G., and Ohlsson K. (1979) Demonstration of pancreatic protease-antiprotease complexes in the peritoneal fluid of patients with acute pancreatitis. Surgery 85, 451–456 [PubMed] [Google Scholar]
- 33. Joseph S. K., Pierson S., and Samanta S. (1995) Trypsin digestion of the inositol trisphosphate receptor: implications for the conformation and domain organization of the protein. Biochem. J. 307, 859–865 10.1042/bj3070859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Betzenhauser M. J., Fike J. L., Wagner L. E. 2nd, and Yule D. I. (2009) Protein kinase A increases type-2 inositol 1,4,5-trisphosphate receptor activity by phosphorylation of serine 937. J. Biol. Chem. 284, 25116–25125 10.1074/jbc.M109.010132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dawra R., Sah R. P., Dudeja V., Rishi L., Talukdar R., Garg P., and Saluja A. K. (2011) Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 141, 2210–2217.e2 10.1053/j.gastro.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Williams J. A., Korc M., and Dormer R. L. (1978) Action of secretagogues on a new preparation of functionally intact, isolated pancreatic acini. Am. J. Physiol. 235, 517–524 [DOI] [PubMed] [Google Scholar]
- 37. Chaudhuri A., Kolodecik T. R., and Gorelick F. S. (2005) Effects of increased intracellular cAMP on carbachol-stimulated zymogen activation, secretion, and injury in the pancreatic acinar cell. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G235–243 10.1152/ajpgi.00334.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Voronina S., Longbottom R., Sutton R., Petersen O. H., and Tepikin A. (2002) Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J. Physiol. 540, 49–55 10.1113/jphysiol.2002.017525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Muili K. A., Wang D., Orabi A. I., Sarwar S., Luo Y., Javed T. A., Eisses J. F., Mahmood S. M., Jin S., Singh V. P., Ananthanaravanan M., Perides G., Williams J. A., Molkentin J. D., and Husain S. Z. (2013) Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J. Biol. Chem. 288, 570–580 10.1074/jbc.M112.428896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muili K. A., Jin S., Orabi A. I., Eisses J. F., Javed T. A., Le T., Bottino R., Jayaraman T., and Husain S. Z. (2013) Pancreatic acinar cell nuclear factor κB activation because of bile acid exposure is dependent on calcineurin. J. Biol. Chem. 288, 21065–21073 10.1074/jbc.M113.471425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alzayady K. J., Chandrasekhar R., and Yule D. I. (2013) Fragmented inositol 1,4,5-trisphosphate receptors retain tetrameric architecture and form functional Ca2+ release channels. J. Biol. Chem. 288, 11122–11134 10.1074/jbc.M113.453241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Betzenhauser M. J., Wagner L. E. 2nd, Iwai M., Michikawa T., Mikoshiba K., and Yule D. I. (2008) ATP modulation of Ca2+ release by type-2 and type-3 inositol (1,4,5)-triphosphate receptors: differing ATP sensitivities and molecular determinants of action. J. Biol. Chem. 283, 21579–21587 10.1074/jbc.M801680200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alzayady K. J., Wagner L. E. 2nd, Chandrasekhar R., Monteagudo A., Godiska R., Tall G. G., Joseph S. K., and Yule D. I. (2013) Functional inositol 1,4,5-trisphosphate receptors assembled from concatenated homo- and heteromeric subunits. J. Biol. Chem. 288, 29772–29784 10.1074/jbc.M113.502203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miyakawa T., Maeda A., Yamazawa T., Hirose K., Kurosaki T., and Iino M. (1999) Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 18, 1303–1308 10.1093/emboj/18.5.1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang L., Alzayady K. J., and Yule D. I. (2016) Proteolytic fragmentation of inositol 1,4,5-trisphosphate receptors: a novel mechanism regulating channel activity? J. Physiol. 594, 2867–2876 10.1113/JP271140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sneyd J., Han J. M., Wang L., Chen J., Yang X., Tanimura A., Sanderson M. J., Kirk V., and Yule D. I. (2017) On the dynamical structure of calcium oscillations. Proc. Natl. Acad. Sci. U.S.A. 114, 1456–1461 10.1073/pnas.1614613114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hirota J., Furuichi T., and Mikoshiba K. (1999) Inositol 1,4,5-trisphosphate receptor type 1 is a substrate for caspase-3 and is cleaved during apoptosis in a caspase-3-dependent manner. J. Biol. Chem. 274, 34433–34437 10.1074/jbc.274.48.34433 [DOI] [PubMed] [Google Scholar]
- 48. Kopil C. M., Vais H., Cheung K. H., Siebert A. P., Mak D. O., Foskett J. K., and Neumar R. W. (2011) Calpain-cleaved type 1 inositol 1,4,5-trisphosphate receptor (InsP3R1) has InsP3-independent gating and disrupts intracellular Ca2+ homeostasis. J. Biol. Chem. 286, 35998–36010 10.1074/jbc.M111.254177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fan G., Baker M. L., Wang Z., Baker M. R., Sinyagovskiy P. A., Chiu W., Ludtke S. J., and Serysheva I. I. (2015) Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527, 336–341 10.1038/nature15249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaiser A. M., Saluja A. K., Sengupta A., Saluja M., and Steer M. L. (1995) Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am. J. Physiol. 269, C1295–C1304 10.1152/ajpcell.1995.269.5.C1295 [DOI] [PubMed] [Google Scholar]
- 51. Gukovskaya A. S., Vaquero E., Zaninovic V., Gorelick F. S., Lusis A. J., Brennan M. L., Holland S., and Pandol S. J. (2002) Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 122, 974–984 10.1053/gast.2002.32409 [DOI] [PubMed] [Google Scholar]
- 52. Sah R. P., Garg P., and Saluja A. K. (2012) Pathogenic mechanisms of acute pancreatitis. Curr. Opin. Gastroenterol. 28, 507–515 10.1097/MOG.0b013e3283567f52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pandol S. J., Saluja A. K., Imrie C. W., and Banks P. A. (2007) Acute pancreatitis: bench to the bedside. Gastroenterology 132, 1127–1151 10.1053/j.gastro.2007.01.055 [DOI] [PubMed] [Google Scholar]
- 54. Saluja A. K., Lerch M. M., Phillips P. A., and Dudeja V. (2007) Why does pancreatic overstimulation cause pancreatitis? Annu. Rev. Physiol. 69, 249–269 10.1146/annurev.physiol.69.031905.161253 [DOI] [PubMed] [Google Scholar]
- 55. Wen L., Voronina S., Javed M. A., Awais M., Szatmary P., Latawiec D., Chvanov M., Collier D., Huang W., Barrett J., Begg M., Stauderman K., Roos J., Grigoryev S., Ramos S., et al. (2015) Inhibitors of ORAI1 prevent cytosolic calcium-associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology 149, 481–492.e7 10.1053/j.gastro.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang W., Cane M. C., Mukherjee R., Szatmary P., Zhang X., Elliott V., Ouyang Y., Chvanov M., Latawiec D., Wen L., Booth D. M., Haynes A. C., Petersen O. H., Tepikin A. V., Criddle D. N., and Sutton R. (2017) Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut 66, 301–313 10.1136/gutjnl-2015-309363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Husain S. Z., Prasad P., Grant W. M., Kolodecik T. R., Nathanson M. H., and Gorelick F. S. (2005) The ryanodine receptor mediates early zymogen activation in pancreatitis. Proc. Natl. Acad. Sci. U.S.A. 102, 14386–14391 10.1073/pnas.0503215102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saluja A. K., Bhagat L., Lee H. S., Bhatia M., Frossard J. L., and Steer M. L. (1999) Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am. J. Physiol. 276, G835–G842 [DOI] [PubMed] [Google Scholar]
- 59. Krüger B., Albrecht E., and Lerch M. M. (2000) The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am. J. Pathol. 157, 43–50 10.1016/S0002-9440(10)64515-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Raraty M., Ward J., Erdemli G., Vaillant C., Neoptolemos J. P., Sutton R., and Petersen O. H. (2000) Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc. Natl. Acad. Sci. U.S.A. 97, 13126–13131 10.1073/pnas.97.24.13126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Criddle D. N., Murphy J., Fistetto G., Barrow S., Tepikin A. V., Neoptolemos J. P., Sutton R., and Petersen O. H. (2006) Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 130, 781–793 10.1053/j.gastro.2005.12.031 [DOI] [PubMed] [Google Scholar]
- 62. Sugawara H., Kurosaki M., Takata M., and Kurosaki T. (1997) Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 16, 3078–3088 10.1093/emboj/16.11.3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wagner L. E. 2nd, and Yule D. I. (2012) Differential regulation of the InsP3 receptor type-1 and -2 single channel properties by InsP3, Ca2+ and ATP. J. Physiol. 590, 3245–3259 10.1113/jphysiol.2012.228320 [DOI] [PMC free article] [PubMed] [Google Scholar]