

Abstract
Background
Heterozygous gain-of-function mutations in PI3K110 lead to lymphadenopathy, lymphoid hyperplasia, Epstein-Barr virus (EBV) and cytomegalovirus (CMV) viremia, and sinopulmonary infections.
Objective
The known role of NK cell function in the control of EBV and CMV prompted us to investigate the functional and phenotypic effect of PI3K110δ mutations on NK cell subsets and cytotoxic function.
Methods
Patient mutations were identified by whole exome or targeted sequencing. We performed NK cell phenotyping and functional analysis of patient cells by flow cytometry, standard Cr51 cytotoxicity assays, and quantitative confocal microscopy.
Results
PI3K110δ mutations led to an altered NK cell developmental phenotype and cytotoxic dysfunction. Impaired NK cell cytotoxicity was due to decreased conjugate formation with susceptible target cells and abrogated activation of cell machinery required for target cell killing. These defects were partially restored following the initiation of treatment with rapamycin in three patients.
Conclusion
We describe novel NK cell functional deficiency due to PI3K110δ mutation, which is a likely contributor to the severe viremia observed in these patients. Rapamycin treatment partially restores NK cell function, providing further rationale for its use in this disease.
Keywords: NK cell deficiency, combined immunodeficiency, cytotoxicity, APDS, PI3K signaling
Capsule Summary
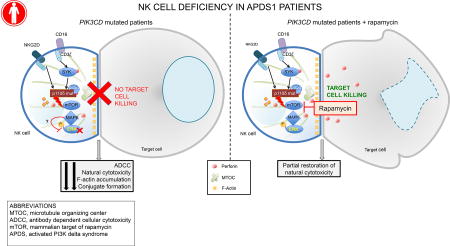
Gain-of-function mutations in PI3K110δ lead to impaired NK cell cytolytic function that can be partially restored with rapamycin treatment.
Introduction
NK cells play an important role in host defense, and NK cell deficiency (NKD) leads to severe and often fatal viral infection and malignancy1–4. While primary immunodeficiencies (PID) leading to isolated NKD are relatively rare, currently greater than 40 congenital deficiencies affect NK cell function in a broader immunological context5. Human NK cells play a critical role in the control of viral infections through the secretion of cytokines and the direct, contact dependent killing of virally infected cells. As such, PID that include an effect on NK cell function are frequently caused by mutations in genes that affect the cytolytic machinery of NK cells. These conditions include the familial hemophagocytic lymphohistiocytosis diseases, mutations affecting the cytoskeleton such as DOCK86, 7 and Wiskott-Aldrich syndrome8, 9, defects in the NFκB pathway10, 11, and others5.
NK cell killing of virally infected targets is mediated by the directed secretion of lytic granules following the formation of a lytic immunological synapse (IS). NK cell IS formation and function includes multiple tightly regulated steps, and the perturbation of these checkpoints leads to a blunting of the cytotoxic response. NK cell cytotoxicity is controlled through a balance of activating and inhibitory receptor signaling. Overcoming a critical threshold of activating receptor signaling leads to commitment to formation of an IS, which includes F-actin remodeling and de novo polymerization, polarization of the microtubule organizing center (MTOC) and lytic granules to the IS, and exocytosis of perforin- and granzyme-containing lytic granules. An early priming step in IS formation is the convergence of lytic granules to the MTOC, which occurs in response to diverse activating signals, including cytokine stimulation, integrin ligation and activating receptor ligation12–14. This step, which is independent of actin remodeling, precedes integrin-mediated firm adhesion and de novo F-actin polymerization, which occurs downstream of activating receptor signaling. Convergence is followed by polarization of the MTOC, which delivers lytic granules to the IS. Lytic granules then traverse the F-actin network at the IS and undergo exocytosis, leading to target cell apoptosis.
The phosphoinositide 3-kinase (PI3K) pathway is a key axis for NK cell cytotoxicity. Signaling downstream from NK cell activating receptors, such as CD16 and NKG2D, leads to PI3K recruitment and activation through adaptors including DAP10 and CD3ζ15–20. Downstream signalling to cytotoxicity is mediated by Rac1, p21 activated kinase-1 (PAK1), mitogen-activated protein kinase kinase (MEK) and extracellular regulated kinases (ERK) 1/221. The requirement for PI3K in NK cell cytotoxic function has been shown using pharmacologic inhibitors, with varying effects dependent upon the target and killing not being entirely abrogated in all cases16, 19. Further insight has been gained using isoform-specific mouse models to dissect the role of individual subunits in the cytotoxic process. Class 1A PI3K in lymphocytes are comprised of a p110 catalytic subunit, encoded by the PIK3CA, PIK3CB or PIK3CD gene, paired with a p85, p55 or p50 regulatory subunit. Catalytic inactivation specifically of the PI3K110δ subunit in mice leads to reduced NK cell number, impaired maturation, and decreased cytotoxic function22. The effect of gain-of-function Pik3cd mutations has not been directly tested in mice, however their effect can be predicted based on loss-of-function mutations in Pten, which have a similar effect of elevating PtdIns(3,4,5)P3 levels.
Mutations in PI3K isoforms lead to distinct PID; loss of p85 in both mice and humans leads to loss of B cell function and subsets, whereas gain-of-function mutations in p110δ patients lead to “p110δ-activating mutation causing senescent T cells, lymphadenopathy and immunodeficiency” (PASLI) disease23 or “activating PI3Kδ syndrome, class 1” (APDS1)24, characterized by EBV and CMV viremia in conjunction with T cell lymphoproliferation and CD8+ T cell metabolism defects, lymphopenia, increased IgM and circulating transitional B cells, and recurrent respiratory infections23–27. In these patients, hyperactive PI3K pathway signalling through protein kinase B (AKT) drives premature senescence of CD8+ T cells and subsequent loss of CD8+ T cell mediated IL-2 secretion23, as reviewed in Lucas et al.28. The hyperactivation of the PI3K pathway leads to increased mammalian target of rapamycin (mTOR) activity and glycolytic function. These effects are partially counteracted by treatment with the mTOR inhibitor rapamycin. Rapamycin treatment of APDS1 patients results in decreased CD8+ cellularity and increased IL-2 secretion, which is accompanied by observed clinical improvement of the disease23.
In this study, we identified a family of three siblings with E525K mutations in the PIK3CD gene29, 30. The 3 affected brothers had clinical and immunologic features consistent with PIK3CD gain-of-function disease, including persistently elevated EBV PCR titers, diffuse lymphadenopathy, hepatosplenomegaly, markedly elevated percentages of transitional B cells, and poor responses to polysaccharide antigens. The identification of this mutation in these patients afforded us the opportunity to evaluate NK cell function and phenotype prior to and following the initiation of rapamycin treatment. Further, we expanded our cohort to describe NK cell deficiency in 5 previously described patients23 and 2 additional previously unreported patients. We show that both E1021K and E525K gain-of-function mutations in PI3K110δ lead to functional NK cell deficiency because of decreased frequency of conjugate formation with susceptible target cells and impaired signalling leading to the execution of cytotoxicity. Further, skewing of the NK cell phenotype occurs, notably with decreased expression of the Fc receptor (CD16) and IL-2 receptor β (CD122) on patient cells. Following stabilization while on rapamycin therapy a significant improvement in NK cell function was observed, reflected by partially corrected immune synapse formation. Therefore, we define gain-of-function mutations in PI3K p110δ as a novel cause of NK cell functional and developmental impairment and a likely contributor to disease in these patients.
Materials and Methods
Cell isolation and cell lines
All human samples were obtained using written informed donor consent and were used with the approval of the National Institutes of Health (NIH) and Baylor College of Medicine Institutional Review Boards for the Protection of Human Subjects. All samples were obtained in compliance with the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMC) were isolated by density centrifugation over Ficoll-Paque according to manufacturer’s instructions. For Cr51 release assays, Raji and K562 cell lines were used as target cells for NK cell cytotoxicity and were maintained in supplemented RPMI media with 10% fetal calf serum. Target cells were routinely tested and confirmed to be mycoplasma free.
Genetic studies
Whole exome sequencing for research at Baylor College of Medicine was performed as previously described31. Clinical sequencing was performed by Baylor Genetics32, 33. Variants were confirmed by Sanger sequencing by the Baylor DNA Sequencing Core Facility. Patients from the NIH were identified genetically by targeted sequencing or whole exome sequencing as previously described23. For deep sequencing, the PIK3CD c.G1573A variant was validated by amplicon deep sequencing on MiSeq sequencer (Illumina). Briefly, an amplicon of 234bp defined by forward primer of 5’-ACCGAGGAGGAGGTGAGTG-3’ and reverse primer of 5’-ACTTGGTGACCAGCAGCAG-3’ was generated through polymerase chain reaction (PCR) from 6 genomic DNA samples of the proband and family members using PrimeSTAR GXL DNA Polymerase (Takara). PCR products were purified with AMPure beads (Beckman Coulter) and further end-repaired and ligated with TruSeq indexed adaptors. Equal amounts of ligated amplicons were pooled and sequenced by MiSeq Nano Kit v2 300 cycles with PhiX spike-in. The raw sequencing data were demultiplexed and converted to Fastq files. The reads were then aligned to human genome assembly 37/hg19 reference sequence by BWA-MEM. Variants were called and number of variants read were counted from each de-multiplexed sample.
Cr51 release assays
Chromium release assays for the analysis of NK cell activity were performed as previously described34. Freshly isolated PBMC were incubated at varying ratios (50:1, 25:1, 12.5:1, 6.25:1, 3.13:1) with 104 K562 or Raji target cells that had been labeled with 50 µCi of Cr51 then washed. Where indicated, assays were performed in the presence of 1000 U/ml IL-2 (Roche) or, as a stimulus for antibody-mediated cytotoxicity, 20 µg/ml Rituximab. Effector:target conjugates were incubated in 200 µl in round-bottomed 96 well plates (Corning). Following 4 hours of incubation, positive controls for maximal lysis were produced by lysing labeled target cells with 1% octylphenoxypolyethoxyethanol. Supernatant was harvested and transferred to LumaPlates (PerkinElmer). The supernatant was dried and plates were read on a TopCount gamma counter (PerkinElmer). Percent specific lysis was calculated as [(Experimental release – spontaneous release)/maximum release*100]. Lytic units were calculated as the number of effectors required to lyse 10% of K562 target cells using the slope of the curves generated by cytotoxicity assay data over the range of effector to target ratios8.
Flow cytometry
Multi-parametric flow cytometry was done using the antibodies described in Supplemental Table 1. For cell surface receptors, all incubations were performed for 30 minutes at 4°C. For intracellular staining of cytok ines and effector molecules, cells were fixed and permeabilized with Cytofix/Cytoperm (BD Bioscience) followed by incubation with antibodies of interest. For activating and functional assays, cells were pre-incubated with phorbol myristate acetate (PMA) and ionomycin (Sigma Aldrich) or vehicle control for 6 hours. Brefeldin A (10 µg/mL-Sigma Aldrich) was added 4 hours before antibody staining. Flow cytometric data were acquired on a BD Fortessa with 18-color configuration. Data were exported as FCS files. SPADE analysis was performed with Cytobank Premium35 following gating on lymphocytes then CD56+CD3− cells. Clustering analysis was based upon the following parameters: CD94, CD117, CD57, CD62L, CD16, CD27 and CD127. Pairwise comparison of single markers and routine flow cytometric analyses were performed using FlowJo X (TreeStar Inc.).
For detection of downstream signaling, NK cells were activated for 0, 2, 4, 8 or 16 minutes with PMA and ionomycin and then harvested, fixed, permeabilized and stained for intracellular phospho-p44/42 MAPK (ERK1/2; Thr202/Tyr2014, D13.14.4E, Cell Signaling Technology). Samples were acquired and analyzed as above.
For flow cytometric analysis of conjugation frequency, PBMC were pre-incubated with anti-CD56 (clone HCD56, Biolegend) and anti-CD3 (clone SK7, Biolegend) to identify NK cells as CD56+CD3−. Susceptible K562 target cells labeled with vital dye PKH26 were mixed with PBMC and incubated for 0, 20, 40 or 60 minutes prior to fixation. Data were acquired on a BD Fortessa and analyzed using FlowJo X. NK cells were gated and the frequency of cells found in conjugates was calculated as [PKH26+CD56+CD3−]/[CD56+CD3−].
Confocal microscopy
Confocal microscopy was performed as previously described36. Briefly, patient PBMC were incubated with susceptible K562 target cells for 45 minutes to facilitate conjugate formation. Following incubation, cells were fixed and permeabilized, then stained intracellularly with anti-tubulin biotin (Life Technologies) followed by streptavidin AlexaFluor 488 (Life Technologies). These steps were followed by staining with anti-perforin AlexaFluor 647 (clone δG9; Biolegend) and phalloidin AlexaFluor 568 (Life Technologies). Slides were mounted with ProLong Gold anti-fade media (Life Technologies). Images were acquired on a Zeiss Axio Observer Z1 microscope with Yokogowa CSU-10 spinning disk, Hamamatsu ORCA-AG camera, and Zeiss 63X 1.43 NA oil immersion lens. Excitation lasers (405 nm, 488 nm, 568 nm) were managed through an LMM5 laser merge unit (Spectral Research). Images were taken in a single Z-plane. Images were acquired and analyzed with Volocity 6.0 software (PerkinElmer). Image analysis was performed as described for F-actin accumulation, lytic granule convergence and MTOC polarization36–38. Briefly, F-actin accumulation at the synapse was calculated with Volocity software (PerkinElmer) by measuring the intensity and area of F-actin above a uniform threshold. Cortical actin from both effector and target cells was subtracted from F-actin at the synapse so that only specific F-actin accumulation was measured37. For measurement of granule convergence, centroids of lytic granules were detected by perforin intensity and the MTOC was detected by intensity of tubulin immunostaining. Distance of granules to the MTOC was measured for each granule using Volocity software; each data point represents the mean of all granules within a given cell38.
Graphing and statistical analysis
Pairwise comparisons were made by Student’s two-tailed t test. For cytotoxicity assays, conditions were compared by ordinary one-way ANOVA with post hoc analysis by Tukey’s multiple comparisons test. For conjugation assays, statistical outliers were identified using ROUT analysis (1%) and excluded from analysis. All graphing and statistical analyses was performed with Prism 6.0 (GraphPad Software).
Results
Clinical history and genetic confirmation of PI3K110δ mutation
The index family consists of 3 brothers from Qatar who presented at 9 years, 5 years, and 1 year of age, respectively, due to diffuse lymphadenopathy, hepatosplenomegaly, and persistently elevated EBV PCR titers. X-linked lymphoproliferative disease, autoimmune lymphoproliferative syndrome, and hemophagocytic lymphohistiocytosis were excluded, and the family returned to Qatar without a formal diagnosis. They returned 4 years later for further evaluation. The older brother had developed bronchiectasis and mild pulmonary fibrosis in the interim due to recurrent pneumonias. They otherwise denied any other recurrent infections. The parents denied consanguinity, and the 3 siblings had 2 other older twin brothers who were healthy. Immunologically, the 3 affected boys had markedly elevated transitional B cell percentages and poor antibody responses to polysaccharide antigens. Whole exome sequencing was performed and revealed the presence of a heterozygous missense PIK3CD E525K mutation in the 3 affected brothers; the 2 twin siblings carried wild-type PIK3CD sequences (see Supp. Table 2 for other variants found at low frequency within the EXaC database). The father was noted to have 6 variant reads out of 38 total reads, suggestive of mosaicism29, 30 or heterozygosity. As the resolution of Sanger sequencing did not permit us to determine whether the father was truly heterozygous (Supp. Fig 1A), we performed deep sequencing of the region of interest, generating greater than 200,000 reads (Supp. Fig 1B). This analysis confirmed the presence of the heterozygous mutation in the 3 affected siblings and homozygosity of the wild-type allele in the unaffected brothers. The father was also found to have full heterozygosity at this locus, with 49% of reads having the mutation present in the affected children. Unfortunately, the father was not available for further immunologic evaluation. It is regrettable that further clinical information about the father is not available as this would additionally demonstrate the pathogenicity of the mutation shared by the father and the patients, however the previous reports of the PIK3CD E525K mutation as disease-causing with high penetrance (23) underscore the likelihood that there is clinical impact of this mutation on affected family members.
We identified further individuals with PIK3CD mutations. First, we were introduced to a 5-year-old asymptomatic boy who had clinical whole exome sequencing performed due to mild developmental delay. The results incidentally revealed the presence of a single heterozygous E1021K mutation in PIK3CD, which was confirmed by clinical Sanger sequencing. This subject was also noted to have an elevated transitional B cell percentage (over 20% of CD19+ B cells) and lack of response to polysaccharide antigens. Serologic and PCR testing showed that he had never been previously exposed to EBV. Next, we obtained samples from 5 previously reported subjects and one unreported individual who had PIK3CD mutations identified by whole exome or targeted sequencing23. Three of the 6 subjects carried the E525K mutation, 2 had the E1021K mutation, and 1 patient had the N334K mutation (summarized in Table 1). As previously described23, 24, the E1021K mutation is located within the kinase domain of PI3K110δ and the E525K mutation is found within the helical domain (Supp. Fig 1C).
TABLE I.
Clinical and immunologic phenotype of PI3K110δ patients
| Patient | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Family* | NA | NA | NA | F.II.1* | E.1* | D.I.1* | B.III.1* | NA | NA | G.1* |
| Mutation | E525K | E525K | E525K | E525K | E525K | E525K | E1021K | E1021K | E1021K | N334K |
| Clinical/immunologic phenotype | ||||||||||
| Transitional B cells | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ND | ↑ | NL |
| Specific antibody response | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ND | ↓ | NL |
| IgA | NL | NL | NL | ↓ | NL | NL | NL | ND | NL | ↓ |
| IgM | ↑ | NL | NL | NL | NL | NL | ↑ | ND | NL | ↑ |
| IgG | ↑ | ↑ | ↑ | NL | NL | ↑ | NL | ND | NL | ↓ |
| Switched B memory | ↑ | ↑ | ↑ | ↓ | ↓ | NL | ↓ | ND | Low/NL | ↓ |
| CD4+ | NL | Low/NL | NL | NL | ↓ | ↓ | ↓ | ND | ↓ | ↓ |
| CD8+ effector | ND | ND | ND | ↑ | ↑ | ↑ | ↑ | ND | ND | ↑ |
| CD8+ central memory | ND | ND | ND | ↑ | NL | NL | NL | ND | ND | NL |
| EBV | Yes | Yes | Yes | Yes | Yes | Yes | Yes | ND | No | Yes |
| Lymphadenopathy | Yes | Yes | Yes | Yes | Yes | No | Yes | ND | No | Yes |
| Hepatosplenomegaly | Yes | Yes | Yes | No | No | No | No | ND | No | ND |
| Other | EBV+ lymphoma | CMV | CMV | Sinopulmonary bacterial infections | ||||||
CMV, Cytomegalovirus; NA, not applicable; ND, not determined; NL, normal.
Refers to Lucas et al.24
NK cells from PI3K110δ patients have decreased natural and antibody-mediated cytotoxicity
NK cells from PI3K110δ mutant mice have decreased natural cytotoxicity22, 39 and PASLI/APDS1 patients show susceptibility to viral infection. As NK cells participate in antiviral defense, we tested the function of patient NK cells using Cr51 release assays against susceptible K562 target cells in the presence or absence of IL-2. Patients showed decreased cytotoxic function, although interestingly stratification based on mutation was observed for both unstimulated and IL-2 stimulated cells (Fig 1A). Patients with E525K and N334K mutations had a greater reduction in NK cell lytic function than those with E1021K mutation relative to grouped healthy controls. While response to IL-2 was varied, a relative increase in cytolytic function in response to IL-2 was noted (Fig 1A, B). We further evaluated the cytolytic capacity of patient NK cells by calculating lytic units required to lyse 10% of K562 target cells following adjustment for the frequency of NK cells within the PBMC population (Fig. 1B). With the exception of Patient 5, patient NK cells had reduced lytic capacity when directly compared to the healthy donor control used for their respective assays.
Figure 1. PI3K110δ mutation results in impaired NK cell cytotoxic function.

PBMC isolated from healthy donors (circles, n=18), PI3K110δ E525K patients (squares, n=6), PI3K110δ E1021K patients (triangles, n=3) or PI3K110δ N334K (diamonds, n=1) were incubated with A) susceptible K562 target cells in the absence (left) or presence of 1000 U/ml IL-2 (right). Cytotoxicity was measured by Cr51 release assay. B) Results from (A) are shown as the lytic units (LU10) required to lyse 10% of target cells when adjusted for the frequency of NK cells within PBMC. Patient LU10 is normalized to the healthy donor control for each assay. C) PBMC isolated from healthy donors (circles, n=10), PI3K110δ E525K patients (squares, n=4) or PI3K110δ E1021K patients (triangles, n=2) were incubated with Patient Raji B cells in the presence of 20 µg/ml Rituximab. Cytotoxicity was measured by Cr51 release assay.
To determine the effect of PI3K110δ mutation on antibody-mediated cellular cytotoxicity (ADCC), Raji target cells were coated with rituximab, and cytotoxicity assays were performed as above. While Raji cells in the absence of antibody were not lysed (not shown), the presence of antibody led to effective killing of targets by controls. However, NK cells from patients with E525K and E1021K mutations had significantly decreased function (Fig. 1C). Taken together, these results show impaired NK cell function in patients with gain-of-function PI3K110δ mutations, producing significantly reduced cytolytic function towards both susceptible tumor cells and antibody-coated targets.
PI3K110δ mutation leads to skewed NK cell developmental phenotypes in human NK cells
Patients with PI3K110δ gain-of-function mutations have some variability in NK cell numbers and altered T cell subsets due to premature senescence25,23. In addition, alteration of PI3K function in mice is known to lead to reduced NK cell numbers with abnormal NK cell developmental subsets22, 39. To fully explore the NK cell phenotype in these patients, we performed quantitative flow cytometry analysis with 5 panels assessing 40 parameters including NK cell surface receptors, intracellular cytokines, and markers of NK cell activation (Supplemental Table 1). To visually compare the distribution of phenotypic subsets between healthy donor and patient samples and to identify novel populations, we performed visual spanning tree progression of density normalized events (SPADE)40. Analysis was performed on flow cytometry data of 8 key NK cell maturation markers following gating for CD56+CD3− NK cells from 2 patients with E525K mutations and 5 healthy donors from 3 independent experiments. Consistently, Patients 1 and 3 (E525K mutation) had unique subsets preserved between the two patients that were not seen in healthy donors (Fig 2). Healthy donor populations from 5 individuals were remarkably similar when visualized by SPADE given the known intra-individual heterogeneity in NK cell subsets (Fig 2, not shown)41, 42.
Figure 2. PI3K110δ mutation leads to altered NK cell subset distribution.

Flow cytometry data from 8-color analysis of NK cell surface receptors associated with developmental subsets on healthy donor (top) and two E525K PI3K110δ patients (bottom) and was visualized by spanning analysis of density events (SPADE). Gating strategy for each patient or control is shown on the left with the frequency of NK cells within the lymphocyte gate. Clustering parameters are shown in the center. SPADE plot node sizes and intensities are based upon cell counts as shown in the heat map legend.
Guided in part by our SPADE analysis that identified visually distinct subsets within patient NK cell populations, we defined markers from our panels that were differentially expressed on patient cells compared to healthy controls. To quantitatively compare these, we performed pairwise analyses of frequency of receptor expression (percent positive) on NK cells. Since CD56bright and CD56dim NK cells represent phenotypic and functional subsets, and CD56bright NK cells are thought to be the developmental precursors of CD56dim NK cells, we quantified expression on these subsets individually. As previously described, we saw significant reduction in NK cell numbers in many PI3K110δ patients23 (Fig 3A). While the frequency of patient CD56bright cells was normal (Fig 3B), skewing of the NK cell phenotype towards a generally immature one was present, with a decrease in CD16 and increase in NKG2A expression on CD56dim NK cells (Fig 3C). Further, CD62L and CD127 expression were decreased on CD56bright NK cells, and CD122 expression was diminished on both subsets (Fig 3C). In contrast to the phenotype observed in the mouse model, no detectable difference in CD27 expression was observed on patient NK cells (not shown)22. Granzyme B and CD57 were also unchanged between patient and healthy donor cells, in contrast to the phenotype of effector T cells in these patients23. Intracellular IFNγ expression in response to PMA/ionomycin stimulation was lower in patient NK cells, although not significantly. While TNFα expression was also not affected, a significant decrease in GM-CSF expression by patient NK cells was observed (Supp. Fig. 2). Taken together, these results indicate an alteration in NK cell phenotype or activation state. These effects are independent of global NK cell development when taken in light of the normal distribution of CD56bright and CD56dim subsets. Furthermore, they are distinct from effects on both murine NK cells and human CD8+ effector cells, which have unique markers affected by PI3K110δ mutation. Collectively, however, these significant alterations in NK cell phenotype provide interesting insight into the loss of cytotoxic function in patient NK cells.
Figure 3. Pairwise analysis of differentially expressed receptors on NK cells from patients and healthy donors.

PBMC from 5 healthy donors (black circles) and PI3K110δ patients with E525K (red), E1021K (blue) or N334K (green) mutations were analyzed by flow cytometry as in Figure 2. A) NK cell frequency and B) CD56bright subset distribution. C) NK cell receptor expression on CD56bright and CD56dim subsets as indicated. *p<0.05 by Student’s two-tailed T test.
Impaired conjugate formation, ERK phosphorylation and lytic machinery polarization in PI3K110δ NK cells
To further probe the mechanism of cytotoxic dysfunction, we sought to identify the affected stage(s) of PI3K110δ patient NK cell-mediated killing of target cells. The initial stage of NK cell cytotoxicity is recognition and conjugate formation leading to firm adhesion (reviewed in43). Given the requirement for F-actin rearrangement in conjugate formation and the potential impact of deregulated PI3K activity on this pathway, we analyzed the frequency of NK- target cell conjugate formation by FACS. Differentially labeled NK cells and targets were mixed and allowed to form conjugates for 0, 10, 20, 30 and 60 minutes. Despite comparable expression of integrins and activating receptors between healthy donors and patient cells (not shown), NK cells from PI3K110δ patients showed a trend towards lower conjugate formation that was significant in patients with E1021K mutations at later time points (60 minutes: HD 7.9±1.9%; E525K 4.4±0.7%; E1021K 1.9±0.4%) (p<0.05 by one-way ANOVA, Fig 4A).
Figure 4. Impaired conjugate formation, ERK phosphorylation and effector cell polarization in NK cells from PI3K110δ patients.

A) Differentially labeled NK cells within PBMC and K562 targets were incubated together for times indicated, then fixed, and conjugate frequency was determined by flow cytometry. Healthy donor, n=7; E525K, n=5; E1021K, n=4. B) Intracellular phospho-ERK was analyzed by flow cytometry following activation by plate bound anti-NKG2D. *p<0.05 by ordinary one-way ANOVA with post hoc comparison, n=5. C) NK cells were conjugated to K562 targets then fixed, permeabilized and stained with anti-perforin, anti-α-tubulin and phalloidin. Scale bar=5 µm. D) F-actin accumulation, lytic granule convergence and MTOC polarization were measured for >25 conjugates. *p<0.05 by Student’s two-tailed T test.
PI3K110δ catalytically inactive mutant mice show impaired signaling in the MAPK pathways, specifically phosphorylation of JNK1/222, and Pik3cd−/− Pik3cg−/− mice have defective ERK1/2 signaling39. To test this pathway in patient cells, we performed intracellular FACS for phosphorylated ERK1/2, JNK1/2 and p38 following NK cell activation by PMA and ionomycin. While phosphorylation of JNK1/2 and p38 were intact in patient cells, the percentage of NK cells with ERK1/2 phosphorylation was significantly reduced in patients with both E525K and E1021K mutations 15 minutes following activation (HD 43.3±9.2%; E525K 7.2±2.2%; E1021K 9.2±1.4%) (p<0.05 by one-way ANOVA, Fig 4B).
Upon conjugate formation and activating signaling, NK cells rapidly converge lytic granules to the MTOC, which is followed by the effector stage of cytotoxicity, marked by accumulation of F-actin and polarization of lytic granules and the MTOC to the immunological synapse38, 43. To further investigate the impact of impaired PI3K110δ signaling on NK cell lytic function, we performed quantitative analysis of the polarization of lytic machinery in NK cells conjugated to susceptible target cells and imaged by confocal microscopy (Fig. 4C). While healthy donor NK cells had accumulation of Factin following conjugation to target cells, aggregation of F-actin at the immunological synapse was significantly decreased in all patient cells tested, regardless of mutation (Fig 4D). Furthermore, significant defects in the convergence of lytic granules to the MTOC and MTOC polarization were present in the E525K cohort (Fig 4D). Interestingly, these parameters were not significantly affected in the E1021K patients.
Partial increase in NK cell function following initiation of treatment with rapamycin
Rapamycin treatment has proven relevant for the amelioration of symptoms in PI3K110δ patients23. To determine the specific effect of treatment on NK cell function, we evaluated NK cell cytotoxicity in Patients 1–3 both prior to initiation of rapamycin and following the establishment of treatment. While NK cell number increased in one patient only (Fig 5A) and cell surface receptor phenotype was not significantly changed (not shown), improvement in the specific lysis of K562 target cells was seen in all three patients (Fig 5B). When normalized to healthy donor controls, the mean specific lysis at 50:1 effector to target for all three patients was raised from 0.14±0.01 to 0.49±0.1 (p<0.01, Fig 5B). Finally, to ascertain if this increase in cytotoxic function correlated with a restoration of previously impaired effector mechanism, we re-evaluated NK-target conjugates by confocal microscopy. We found that following the establishment of rapamycin therapy in the patients, who had all failed to accumulate F-actin at the synapse previously, we now observed that F-actin rearrangement was comparable to that of healthy donor controls (Fig 5C). Therefore, the increase in NK cell function following mTOR inhibition in PI3K110δ patients occurs in concert with restoration of cytolytic machinery.
Figure 5. Partial restoration of NK cell function following initiation of rapamycin treatment in PI3K110δ patients.

A) NK cell number in three patients prior to (open squares) and following initiation of (open circles) rapamycin treatment. B) Evaluation of NK cell cytotoxic function without IL-2 stimulation in the same three patients relative to normal controls (dashed line). C) NK cell-target conjugates were analyzed as in Figure 4. **p<0.01 by Student’s two-tailed T test at 50:1 ratio. *p<0.05 by ordinary one-way ANOVA with post hoc comparison.
Discussion
The PI3K signaling axis is an important one in NK cell biology, as demonstrated by studies in both mouse22, 39 and humans23, 24, 44. In particular, the PI3K signaling pathway is required for the development of NK cells in mouse22, 39 and the polarization of lytic machinery during human NK cell-mediated killing of target cells45. The recent description of patients with gain-of-function mutations in PIK3CD highlights the importance of balanced signaling through this pathway in human health, as these patients suffer severe lymphoproliferative disease and immunodeficiency. The prevalence of EBV disease, a hallmark of NK cell dysfunction, and the documented role for this signaling pathway in NK cell function led us to investigate further the functional and developmental phenotype of NK cells in these patients. Here we show that in addition to the reduced NK cell number seen in some PI3K110δ patients23, significant impairments in NK cell phenotype and function exist in patients with PIK3CD gain-of-function mutations.
Previous studies of murine models have revealed both developmental and functional abnormalities in NK cells following disruption of PI3K110δ. Loss of PI3K110δ function leads to reduced NK cell number in the periphery and an immature phenotype when combined with loss of the PI3K110γ subunit39. Catalytically inactive PI3K110δ results in a similar phenotype, with a block in NK cell development leading to decreased NK cell numbers and specific decrease in the CD27hi mature population22. In both previous studies, developmental defects were accompanied by specific loss of NK cell function, as impairment in JNK1/2 and ERK1/2 activation leads to defective in vitro and in vivo tumor clearance22, 39. We have now demonstrated that hyperactive PI3K110δ function in humans leads to NK cell derangements.
The careful examination of the NK cell phenotype in PI3K110δ patients shows select developmental defects. Interesting conserved defects were observed between the gain-of function mutations in our patients and murine loss-of-function models. This semblance includes the immature NK cell phenotype seen in patient cells, although this defect is not as absolute as the halt in NK cell development seen in the mouse model. The presence of CD27 on patient cells at comparable levels to healthy donors is a notable difference between gain- and loss-of-function mutations, although the significant divergence between mouse and man in terms of NK cell development may also account for this discrepancy. Whereas human NK cells are defined largely phenotypically and functionally by CD56 density, with CD56bright being the cytokine producing subset and CD56dim the predominantly cytotoxic subset, murine NK cells do not express a CD56 homologue. As human NK cells are thought to pass first through the CD56bright stage and then mature to the CD56dim stage, it has been difficult to completely correlate these stages in the mouse. However, a surrogate scheme based on CD27 and CD11b (Mac-1) density has been designed in terms of function46, and thus the lack of CD27hi NK cells in the mouse model is reflective of a block in maturation. That said, the relative frequency of CD56bright and CD56dim cells was not directly affected as a result of gain-of-function mutations in the patients we examined. Instead, other very consistent differences were observed that indicated a less mature NK cell functional phenotype as a result of gain-of-function mutations in patient NK cells.
These phenotypic changes were initially illuminated by SPADE analysis that showed unique NK cell subsets in patient cells relative to healthy donors. Further analysis showed a generally immature CD56dim population, with decreased CD16 and increased NKG2A expression. Interestingly, both CD56bright and CD56dim subsets had decreased CD122 expression, which could indicate that at least some of the functional hyporesponsiveness shown by patient NK cells is due to decreased IL-2 mediated activation. While CD56dim NK cells appeared less mature, the CD56bright NK cells in our patients had decreased CD62L and CD127 receptors, and it would be of interest to determine whether patient T cells were similarly affected and whether this decreased expression contributes to the T cell phenotype seen in these patients23, 24, 44. Of note, CD56bright NK cells appeared to have normal cytokine production, including IFNγ, upon stimulation, and levels of granzyme B were not affected unlike in cytotoxic T lymphocytes23.
The significance of the differences between the mutations is not entirely clear. While both E525K and E1021K result in impaired NK cell cytotoxic function and seemingly similarly affect NK cell number and phenotype, the E1021K mutation had a significant effect on conjugate formation, whereas the E525K mutation had a greater effect on polarization of lytic machinery following the formation of conjugates. The E1021K mutation, located in the kinase domain, has been shown to increase membrane association and kinase activity24, 47. In contrast, both N334K and E525K mutations interrupt inhibitory interactions between the regulatory p85α subunit and p110δ catalytic subunit, leading to a different mechanism of PI3K hyperactivation. While both mutations have the effect of promoting PtdIns(4,5)P2 phosphorylation, subtle differences may exist as a result of the mislocalization of protein due to E1021K mutations that have yet to bet determined.
Despite hyperphosphorylation of AKT as a result of both E525K and E1021K mutations23, 24, the effect of gain-of-function mutations on both T cells and NK cells appears to be one of cellular hyporesponsiveness. This conclusion is supported by our results obtained on a single cell level by confocal microscopy image analysis. It is interesting that rarer loss-of-function mutations in PI3K also lead to immunodeficiency and impaired immune responses48, 49, however the cellular phenotype of NK cells was not specifically studied in these instances. Gain-of-function mutations in PTEN, which would be predicted to have an effect counter to that of PI3K gain-of-function, also lead to a very similar NK cell phenotype. Human NK cells overexpressing PTEN have impaired F-actin accumulation, granule convergence and MTOC polarization as seen in APDS1 patients50. Interestingly, PI3K inhibitors also impair conjugate formation, cytotoxic function and MTOC polarization51. This effect underscores the importance of carefully regulated PtdIns(4,5)P2 and PtdIns(3,4,5)P3 signaling in NK cell cytotoxic function and suggests that deregulation of this signalling globally leads to hyporesponsiveness. In T cells, this hyporesponsiveness is likely due to premature senescence of the effector population. However, as NK cell senescence is poorly defined it is difficult to draw a parallel. The similarities between loss- and gain-of-function mutations in terms of NK cell phenotype suggests that overactivation of this signaling pathway leads to an inability to tune its responsiveness and may be as detrimental as inactivation through loss of function. This signaling can be partially restored through dampening hyperactive signaling, and inhibition of mTOR signaling is effective in partially restoring NK cell function in these patients. Treatment with rapamycin in our cohort led to increased cytotoxic function with increased F-actin accumulation at the NK cell synapse, a critical step in NK cell cytotoxicity. Interestingly, this was independent of reversal of the phenotypic changes that we detected in patient NK cells, suggesting that it may represent a partial restoration of intracellular activation that has been rendered hyporesponsive in APDS patient NK cells. While the exact mechanism of action is not known, the known importance of the PI3K signaling pathway through AKT and mTOR in NK cells suggests that re-tuning responsiveness to cytokine or activating receptor signaling may enable restoration of function in patient cells. mTOR plays a role in both the generation and activation of NK cells, and primary human cells from healthy donors treated with rapamycin have impaired cytotoxic function, suggesting a direct role for mTOR signaling in the regulation of lytic function52.
Taken together, our data confirm speculation28 that extreme susceptibility to herpesviral infection in APDS patients is due in part to impaired NK cell function and provides additional rationale for the use of rapamycin in patients, particularly those with viral illness.
Supplementary Material
Key Messages.
Patients with PIK3CD gain-of-function mutations have impaired NK cell cytotoxic function due to decreased conjugate formation and NK cell polarization
Patients with PIK3CD gain-of-function mutations have decreased NK cell frequency which is accompanied by decreased expression of CD16, CD122 and CD127 and increased expression of NKG2A
Functional, but not phenotypic, defects are partially restored by rapamycin treatment
Acknowledgments
We thank the patients and their families for their participation in this study. This work was supported by NIH-NIAID Grants R01AI067946 and R01 AI120989 to JSO, the Jeffrey Modell Foundation, the American Society of Hematology Junior Scholar Award to EMM, and NIH-NHGRFI/NHHLBI Grant U54HG006542 to the Baylor Hopkins Center for Mendelian Genomics.
Baylor College of Medicine (BCM) and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of the Baylor Genetics (BG), which performs clinical exome sequencing. JRL is an employee of BCM and derives support through a professional services agreement with the BG. JRL serves on the Scientific Advisory Board of the BG. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, has stock options in Lasergen, Inc., and is a coinventor of US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting.
Abbreviations
- NKD
NK cell deficiency
- PID
primary immunodeficiency
- MTOC
microtubule organizing center
- EBV
Epstein-Barr virus
- CMV
cytomegalovirus
- PASLI disease
p110δ-activating mutation causing senescent T cells, lymphadenopathy and immunodeficiency
- APDS
activated PI3K delta syndrome
- PI3K
phosphoinositide 3 OH-kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The remaining authors declare that they have no competing interests.
References
- 1.Ham H, Billadeau DD. Human immunodeficiency syndromes affecting human natural killer cell cytolytic activity. Front Immunol. 2014;5:2. doi: 10.3389/fimmu.2014.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orange JS. Human natural killer cell deficiencies. Curr Opin Allergy Clin Immunol. 2006;6:399–409. doi: 10.1097/ACI.0b013e3280106b65. [DOI] [PubMed] [Google Scholar]
- 3.Orange JS, Ballas ZK. Natural killer cells in human health and disease. Clin Immunol. 2006;118:1–10. doi: 10.1016/j.clim.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Voss M, Bryceson YT. Natural killer cell biology illuminated by primary immunodeficiency syndromes in humans. Clin Immunol. 2015;177:29–42. doi: 10.1016/j.clim.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol. 2013;132:515–25. doi: 10.1016/j.jaci.2013.07.020. quiz 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ham H, Guerrier S, Kim J, Schoon RA, Anderson EL, Hamann MJ, et al. Dedicator of cytokinesis 8 interacts with talin and Wiskott-Aldrich syndrome protein to regulate NK cell cytotoxicity. J Immunol. 2013;190:3661–9. doi: 10.4049/jimmunol.1202792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizesko MC, Banerjee PP, Monaco-Shawver L, Mace EM, Bernal WE, Sawalle-Belohradsky J, et al. Defective actin accumulation impairs human natural killer cell function in patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2013;131:840–8. doi: 10.1016/j.jaci.2012.12.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orange JS, Ramesh N, Remold-O'Donnell E, Sasahara Y, Koopman L, Byrne M, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci U S A. 2002;99:11351–6. doi: 10.1073/pnas.162376099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orange JS, Roy-Ghanta S, Mace EM, Maru S, Rak GD, Sanborn KB, et al. IL-2 induces a WAVE2-dependent pathway for actin reorganization that enables WASp-independent human NK cell function. J Clin Invest. 2011;121:1535–48. doi: 10.1172/JCI44862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orange JS, Brodeur SR, Jain A, Bonilla FA, Schneider LC, Kretschmer R, et al. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. J Clin Invest. 2002;109:1501–9. doi: 10.1172/JCI14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willmann KL, Klaver S, Dogu F, Santos-Valente E, Garncarz W, Bilic I, et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun. 2014;5:5360. doi: 10.1038/ncomms6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu HT, Mace EM, Carisey AF, Viswanath DI, Christakou AE, Wiklund M, et al. NK cells converge lytic granules to promote cytotoxicity and prevent bystander killing. J Cell Biol. 2016;215:875–89. doi: 10.1083/jcb.201604136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James AM, Hsu HT, Dongre P, Uzel G, Mace EM, Banerjee PP, et al. Rapid activation receptor- or IL-2-induced lytic granule convergence in human natural killer cells requires Src, but not downstream signaling. Blood. 2013;121:2627–37. doi: 10.1182/blood-2012-06-437012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang M, March ME, Lane WS, Long EO. A signaling network stimulated by beta2 integrin promotes the polarization of lytic granules in cytotoxic cells. Sci Signal. 2014;7:ra96. doi: 10.1126/scisignal.2005629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat Immunol. 2003;4:557–64. doi: 10.1038/ni929. [DOI] [PubMed] [Google Scholar]
- 16.Bonnema JD, Karnitz LM, Schoon RA, Abraham RT, Leibson PJ. Fc receptor stimulation of phosphatidylinositol 3-kinase in natural killer cells is associated with protein kinase C-independent granule release and cell-mediated cytotoxicity. J Exp Med. 1994;180:1427–35. doi: 10.1084/jem.180.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerboni C, Gismondi A, Palmieri G, Piccoli M, Frati L, Santoni A. CD16-mediated activation of phosphatidylinositol-3 kinase (PI-3K) in human NK cells involves tyrosine phosphorylation of Cbl and its association with Grb2, Shc, pp36 and p85 PI-3K subunit. Eur J Immunol. 1998;28:1005–15. doi: 10.1002/(SICI)1521-4141(199803)28:03<1005::AID-IMMU1005>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 18.Jiang K, Zhong B, Gilvary DL, Corliss BC, Vivier E, Hong-Geller E, et al. Syk regulation of phosphoinositide 3-kinase-dependent NK cell function. J Immunol. 2002;168:3155–64. doi: 10.4049/jimmunol.168.7.3155. [DOI] [PubMed] [Google Scholar]
- 19.Upshaw JL, Arneson LN, Schoon RA, Dick CJ, Billadeau DD, Leibson PJ. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol. 2006;7:524–32. doi: 10.1038/ni1325. [DOI] [PubMed] [Google Scholar]
- 20.Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science. 2004;306:1517–9. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 21.Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, et al. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat Immunol. 2000;1:419–25. doi: 10.1038/80859. [DOI] [PubMed] [Google Scholar]
- 22.Guo H, Samarakoon A, Vanhaesebroeck B, Malarkannan S. The p110 delta of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J Exp Med. 2008;205:2419–35. doi: 10.1084/jem.20072327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15:88–97. doi: 10.1038/ni.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342:866–71. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: A large patient cohort study. J Allergy Clin Immunol. 2017;139:597–606. e4. doi: 10.1016/j.jaci.2016.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: A cohort study. J Allergy Clin Immunol. 2016;138:210–8. e9. doi: 10.1016/j.jaci.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 27.Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hulsdunker J, Eskandarian Z, et al. Activating PI3Kdelta mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol. 2016;183:221–9. doi: 10.1111/cei.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kdelta and primary immunodeficiencies. Nat Rev Immunol. 2016;16:702–14. doi: 10.1038/nri.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eldomery MK, Coban-Akdemir Z, Harel T, Rosenfeld JA, Gambin T, Stray-Pedersen A, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26. doi: 10.1186/s13073-017-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139:232–45. doi: 10.1016/j.jaci.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, Buhay CJ, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med. 2013;5:57. doi: 10.1186/gm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–11. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–9. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanborn KB, Rak GD, Maru SY, Demers K, Difeo A, Martignetti JA, et al. Myosin IIA associates with NK cell lytic granules to enable their interaction with F-actin and function at the immunological synapse. J Immunol. 2009;182:6969–84. doi: 10.4049/jimmunol.0804337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom. 2010;(Unit10):7. doi: 10.1002/0471142956.cy1017s53. Chapter 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanborn KB, Rak GD, Mentlik AN, Banerjee PP, Orange JS. Analysis of the NK cell immunological synapse. Methods Mol Biol. 2010;612:127–48. doi: 10.1007/978-1-60761-362-6_9. [DOI] [PubMed] [Google Scholar]
- 37.Banerjee PP, Orange JS. Quantitative measurement of F-actin accumulation at the NK cell immunological synapse. J Immunol Methods. 2010;355:1–13. doi: 10.1016/j.jim.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mentlik AN, Sanborn KB, Holzbaur EL, Orange JS. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol Biol Cell. 2010;21:2241–56. doi: 10.1091/mbc.E09-11-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tassi I, Cella M, Gilfillan S, Turnbull I, Diacovo TG, Penninger JM, et al. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity. 2007;27:214–27. doi: 10.1016/j.immuni.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 40.Qiu P, Simonds EF, Bendall SC, Gibbs KD, Jr, Bruggner RV, Linderman MD, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–91. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angelo LS, Banerjee PP, Monaco-Shawver L, Rosen JB, Makedonas G, Forbes LR, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res. 2015;62:341–56. doi: 10.1007/s12026-015-8664-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med. 2013;5:208ra145. doi: 10.1126/scitranslmed.3006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mace EM, Dongre P, Hsu HT, Sinha P, James AM, Mann SS, et al. Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol Cell Biol. 2014;92:245–55. doi: 10.1038/icb.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hulsdunker J, Eskandarian Z, et al. Activating PI3Kdelta mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol. 2015;183:221–229. doi: 10.1111/cei.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X, Trivedi PP, Ge B, Krzewski K, Strominger JL. Many NK cell receptors activate ERK2 and JNK1 to trigger microtubule organizing center and granule polarization and cytotoxicity. Proc Natl Acad Sci U S A. 2007;104:6329–34. doi: 10.1073/pnas.0611655104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. 2006;176:1517–24. doi: 10.4049/jimmunol.176.3.1517. [DOI] [PubMed] [Google Scholar]
- 47.Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, Burke JE. Conformational disruption of PI3Kdelta regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci U S A. 2017;114:1982–7. doi: 10.1073/pnas.1617244114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85alpha subunit of PI3K. J Exp Med. 2012;209:463–70. doi: 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang KJ, Husami A, Marsh R, Jordan MB. Identification of a phosphoinositide 3-kinase (PI-3K) p110δ (PIK3CD) deficient individual. Journal of Clinical Immunology. 2013;33:673–4. [Google Scholar]
- 50.Briercheck EL, Trotta R, Chen L, Hartlage AS, Cole JP, Cole TD, et al. PTEN is a negative regulator of NK cell cytolytic function. J Immunol. 2015;194:1832–40. doi: 10.4049/jimmunol.1401224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Segovis CM, Schoon RA, Dick CJ, Nacusi LP, Leibson PJ, Billadeau DD. PI3K links NKG2D signaling to a CrkL pathway involved in natural killer cell adhesion, polarity, and granule secretion. J Immunol. 2009;182:6933–42. doi: 10.4049/jimmunol.0803840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marcais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15:749–57. doi: 10.1038/ni.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.