

Abstract
Four porphyrinic ionic liquids and four higher melting salts (>100 °C) were synthesized as potential photosensitizers from highly symmetric porphyrins by introducing alkyl chains and exchanging anions to tune their solubility and singlet oxygen generation capability. Among the synthesized compounds was 5,10,15,20‐tetra(4‐dodecylpyridinum)porphyrin tetrakis‐bis(trifluoromethylsulfonyl)‐amide, a room‐temperature ionic liquid that could be crystallized as a solvate with nitrobenzene.
Keywords: photosensitizers, porphyrinic ionic liquids, porphyrins, singlet oxygen, solubility
Singlet oxygen, a very reactive molecular oxygen species to elicit cell deaths in photodynamic therapy, has attracted much recent attention.1 Singlet oxygen is generally produced by photosensitizers,2 such as porphyrins, phthalocyanines, or chlorin, by light irradiation. Of these, porphyrin derivatives, which are well known for their extensive π‐conjugated structures, have also been widely used in coordination chemistry,3 liquid crystals,4 catalysis,5 photodynamic therapy,6 and more. However, the tendency to form stacks and aggregates due to their planar aromatic nature, results in their high crystallinity, high melting points, and low solubility in organic solvents.7 The low solubility in organic solvents in particular is a major drawback for practical applications, as solution‐based processes,8 such as spin‐coating, are widely used for material and device fabrication.
In our recent efforts to overcome the limitations of porphyrins as photosensitizers, 4,4′,4′′,4′′′‐(porphine‐5,10,15,20‐tetrayl)tetrakis(benzoic acid) was attached to a biocompatible chitin support and used as singlet oxygen generator due to the increasing need of biocompatible supports to immobilize photosensitizers;9 however, the efficiency was low, owing to its heterogeneous nature. Therefore, we sought to prepare a porphyrin singlet oxygen generator in pure liquid form, which we believe will enhance its sorption into various membranes or solubility in solvents resulting in a better ease of use.
The conversion of a molecule with low solubility into an ionic liquid (IL, defined as a salt with melting point below 100 °C)10 has been reported as a suitable strategy to increase the solubility of active pharmaceutical ingredients (APIs).11 Incorporation of porphyrin molecules into IL form should also be a good strategy to increase the solubility of porphyrins, and thus expand their potential applications. Along these lines, Xu et al. combined cationic porphyrin derivative 5‐(4‐N‐dodecanoylpyridinium‐4‐yl)‐10,15,20‐tris[3,4,5‐tris(dodecyloxy)‐phenyl]porphyrin with the tetrakis(pentafluorophenyl)borate anion, resulting in porphyrinic ILs.12 Owing to the high molecular weight and relatively rigid structure of porphyrins, this approach was reported to be limited to porphyrin structures of low symmetry combined with large anions.12 Zagami et al.13 tried a different strategy by combining the anionic meso‐tetrakis(4‐sulfonatophenyl)porphyrin and cationic trihexyl(tetradecyl)‐phosphonium cation, but this approach yielded a salt with a melting point of 176.53 °C.
In the present work, we chose a design strategy that should be applicable to making IL porphryins for a host of applications, utilizing highly symmetric porphyrin (D 4h) precursors (which are easier to synthesize and cheaper in comparison with lower symmetry porphyrins), adding long alkyl chains (C12) into their meso positions,14 and pairing them with well‐known IL‐forming anions. The porphyrin derivatives 5,10,15,20‐tetrakis(4‐hydroxyphenyl)‐21 H,23H‐porphine (TOHPP) and 5,10,15,20‐tetra(4‐pyridyl)porphyrin (T4PyP) (Scheme 1) were used as starting materials, owing to their ease of functionalization, relatively high melting points, and low solubilities. In addition, these two different porphyrins also allow the investigation of changes in the electronic environment of the porphyrin core after functionalization, where modification of TOHPP by etherification at the OH groups would not be expected to affect the electronic environment of the porphyrin core, whereas modification of the pyridyl groups in T4PyP would change the electronic environment.
Scheme 1.
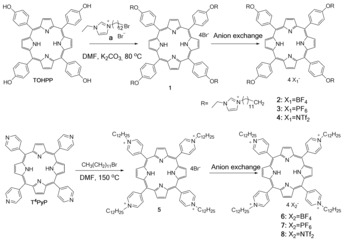
Synthetic procedures for the compounds made in this study.
Initially, TOHPP and T4PyP were reacted with 1‐(12‐bromododecyl)‐3‐ethylimidazolium bromide and 1‐bromododecane, respectively, in dimethylformamide (DMF) (Scheme 1) to obtain 1 and 5. (See the Supporting Information for detailed synthetic procedures.) After synthesis and isolation of the bromide (Br−) salts, they were used to prepare salts of hexafluorophosphate ([PF6]−), tetrafluoroborate ([BF4]−), and bis(trifluoromethylsulfonyl)amide ([NTf2]−) by anion exchange. All of the synthesized TOHPP‐ and T4PyP‐based compounds were prepared at yields higher than 70 % (Table 1). Attenuated total reflection Fourier transform infrared spectroscopy (ATR‐FTIR), nuclear magnetic resonance (NMR), and mass spectrometry (MS) were used to confirm the structures and purity of the resulting compounds.
Table 1.
Thermal parameters.
| Salts | Physical state[a] | Yield [%] |
T
5 %dec
[b]
[°C] |
T
m
[c]
[°C] |
T
c
[c]
[°C][ |
|---|---|---|---|---|---|
| TOHPP | purple solid | – | 496.015 | 422.816 | – |
| 1 | dark purple solid | 71 | 254.1 | 139.4 | – |
| 2 | dark chocolate solid | 80 | 348.3 | 144.2 | – |
| 3 | dark chocolate solid | 73 | 334.6 | 137.9 | – |
| 4 | dark chocolate solid | 85 | 353.5 | 113.2 | – |
| T4PyP | purple solid | – | >50117 | >30018 | – |
| 5 | dark brown solid | 75 | 239.6 | 93.4 | – |
| 6 | dark brown solid | 90 | 296.8 | 87.0 | – |
| 7 | dark brown solid | 95 | 272.1 | 67.6 | – |
| 8 | dark brown highly viscous liquid | 95 | 360.9 | 15.6 | 10.8 |
[a] The state at room temperature. [b] T 5 %dec: 5 % mass loss temperature (determined by TGA). [c] T m: melting point; T c: crystallization temperature (determined by DSC).
As shown in Table 1, most of the synthesized compounds are solids at room temperature, except 8, which is a highly viscous room‐temperature liquid. Differential scanning calorimetry (DSC) was conducted by three cooling and heating cycles (Figures S27‐S28) between −90 to 50 °C below their T 5 %dec (from TGA). As expected, the melting points decreased with increasing anion size,19 except in the case of 1.
All derivatives of TOHPP would not be considered as ILs, owing to their melting points exceeding 100 °C; nonetheless, their melting points were much lower than that of TOHPP (422.8 °C). On the other hand, all compounds synthesized from T4PyP have melting points below 100 °C, including one room‐temperature IL (8), which melts at 15.56 °C and crystallizes at 10.81 °C. These salts exhibited both glass transitions and melting temperatures, but only in the first cycle, after which they remained in a supercooled state20 (Figures S27 and S28). The three solids, 5, 6, and 7, exhibited broad melting peaks, suggesting that these compounds behaved like waxy solids.21 These results confirm that the introduction of long alkyl chains into the porphyrin molecule and the correct selection of the counter ions to interfere in the packing can be used to lower the melting point of even symmetric porphyrins.14
Thermal gravimetric analysis (TGA) indicated that all of the prepared compounds decomposed above 230 °C (T 5 %dec, Table 1), suggesting good thermal stability, although less thermally stable than the starting materials (TOHPP=496.0 °C; T4PyP >501 °C). A comparison between compounds with a common cation indicated that the decomposition temperature followed the trend of Br− < [PF6]− < [BF4]− < [NTf2]−. A similar decomposition trend has been previously reported for ILs of these anions with imidazolium‐ and ammonium‐based cations.22
Crystals of the room‐temperature IL 8 as a nitrobenzene solvate were obtained from a mixture of nitrobenzene (50 μL) and dichloromethane (DCM; 1 mL), as dark reddish needle‐like crystals, which were analyzed by single crystal X‐ray diffraction (SCXRD) (Figure 1). The compound, 8⋅4C6H5NO2 crystallizes in the triclinic centrosymmetric space group P‐1, with one half of a porphyrin cation, two [NTf2]− anions, and two nitrobenzene molecules in the asymmetric unit. One of the dodecyl chains is disordered.
Figure 1.
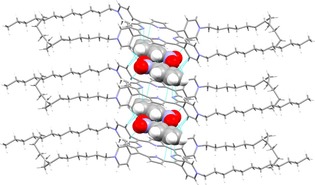
π–π stacking interactions between the porphyrin and nitrobenzene in 8⋅4C6H5NO2; nitrobenzene is depicted in space‐fill form, whereas the porphyrin is shown as capped sticks. Disorder was omitted for clarity.
The cations are aligned on top of each other along the a axis, with one of the nitrobenzene molecules π‐stacked with one of the porphyrin aromatic rings in a sandwich‐like fashion (Figure 1). The shortest π–π stacking distance between nitrobenzene and one of the porphyrin's pyrrole rings is a short 3.178(3) Å, which is even shorter than the π–π stacking reported by Goldberg and co‐workers in 5,10,15,20‐tetra(4‐hydroxyphenyl)‐porphyrin⋅3(pyrene), 3.393(5) Å.23 The second unique nitrobenzene is located between the hydrophobic dodecyl chains of adjacent porphyrins. The trapped nature of the nitrobenzenes in the 3D network of the porphyrinic IL, is similar to the liquid clathrate behavior reported by Holbrey et al.,24 where the low‐temperature‐melting 1‐methyl‐3‐methylimidazolium hexafluorophosphate ([C1mim][PF6]) traps benzene in a crystalline inclusion complex [C1mim][PF6]⋅0.5C6H6.
The anions are located above and below the four cationic pyridinium functionalities, exhibiting typical ion arrangements for salts (Figure S29 a). The cation–anion interactions are depicted by series of C−Hcation⋅⋅⋅Oanion contacts. Interestingly, one of the anions forms self‐complementary 1D chains parallel to the a axis through F⋅⋅⋅F bonds [2.849(5) Å], which is slightly shorter than the sum of the van der Waals (vdW) radii.25 The second unique anion is disordered and is locked between the hydrophobic dodecyl chains. The overall supramolecular arrangement of the ionic porphyrin crystal suggests the formation of two separate domains (apolar and polar) throughout the crystal lattice as shown in Figure S29 b.
To determine the effect of the anions on the solubility of the porphyrin‐based salts, their solubilities were determined in protic (water and methanol) and aprotic solvents [DCM, tetrahydrofuran (THF), and DMF] by dissolving a certain amount of sample (ca. 4 mg) in various solvents till saturation. The supernatants were further diluted 10–1000 times with the same solvent and analyzed by using a UV/Vis spectrometer. The solubilities (Table 2) were calculated according to the UV/Vis absorbance by using calibration curves.
Table 2.
Solubilities of porphyrin‐based compounds in solvents.
| Water[a]
[mm] |
Methanol [mm] |
DCM [mm] |
DMF [mm] |
THF [mm] |
|
|---|---|---|---|---|---|
| TOHPP | NS | 2.61 | 1.39 | 1.67 | 22.43 |
| 1 | 5.91 | 8.64 | 1.52 | 19.8 | NS |
| 2 | NS | 0.55 | 1.99 | 7.81 | NS |
| 3 | NS | 0.63 | 245.00 | 5.35 | NS |
| 4 | NS | 3.08 | 4.37 | 2.20 | 4.72 |
| T4PyP | NS | 0.02 | 3.70 | 0.04 | 0.04 |
| 5 | NS | 2.41 | 37.15 | 0.69 | NS |
| 6 | NS | 0.69 | 150.77 | 3.65 | 1.13 |
| 7 | NS | 0.78 | 109.13 | 8.69 | 10.49 |
| 8 | NS | 3.64 | 5.50 | 12.67 | 50.56 |
[a] NS=not soluble.
As shown in Table 2, all the porphyrin salts, except 1, were insoluble in water, as were the unmodified porphyrins, which is in line with the hydrophobic nature of the [BF4]−, [PF6]−, and [NTf2]− anions and the long alkyl chains on the cations. All of the porphyrin salts were soluble in methanol; however, the T4PyP‐based salts have higher solubilities in methanol than T4PyP.
The solubilities of the T4PyP‐based ILs in aprotic solvents such as DCM, DMF, and THF were higher as compared with T4PyP (Table 2). TOHPP‐based salts have higher solubilities in DCM and DMF, whereas a poorer solubility in THF with respect to TOHPP. These results suggest that the IL approach allows one to tune solubilities of the porphyrins either higher or lower.
To check the effects of modification on porphyrin electronic properties, UV/Vis spectra were used to study their characteristic Soret and Q bands26 (Figure S31). As anticipated, for the TOHPP‐based compounds (Figure S31 a), the Soret band (423 nm) and Q bands (518, 556, 594, and 548 nm) were the same as TOHPP, indicating that the electronic structure of the porphyrin did not change after introducing the long alkyl chains. This is likely due to the oxygen atom between the porphyrin ring and the long alkyl chains (C12), which should not alter the conjugated structure of the porphyrin ring.
On the other hand, the Soret band (424 nm) and Q bands (517, 550, 588, and 617 nm) of the T4PyP‐based porphyrin salts (Figure S31 b), where the C12 groups are directly appended to the porphyrinic aromatic rings, were shifted with respect to T4PyP (Soret band: 415 nm; Q bands: 512, 544, 575, and 607 nm) after quaternization. This confirms that the electronic structure of the porphyrin ring can be changed by direct connection of a long alkyl chain to the porphyrin ring, which is likely caused by the electron‐donating nature of the alkyl groups. The anion exchange did not affect the positions of the absorption bands. Taken together, the results suggest that the absorption properties of porphyrin salts can be tuned by introducing different functional groups.
Having achieved the low‐melting and liquid porphyrinic salts that we had sought, we next investigated their abilities to generate singlet oxygen (1O2), which is key to their performance as a photosensitizer in photodynamic therapy. The 1O2 production capacity can be investigated through the photo‐oxidation of 9,10‐dimethylanthracene (DMA) in DMF.9, 27 Any generated 1O2 is captured by DMA through a photo‐oxidation reaction (Scheme S1), which can be detected by measuring the change of absorbance at 379 nm in the UV/Vis spectrum of DMA solution. In our studies, the performance of the synthesized porphyrin‐based compounds was compared with that of meso‐tetraphenylporphyrin (TPP, a standard dye used to study 1O2 generation)9, 27 and DMA solution without photosensitizer.
The 1 cm cuvettes containing DMA solution (8.4×10−5 m) without photosensitizer or DMA solution (8.4×10−5 m) with the porphyrins or TPP (0.75×10−6 m) were irradiated by using a halogen lamp (150 W). The peak intensity for DMA solution without photosensitizer remained the same, indicating no 1O2 production. For DMA solutions containing the porphyrin salts and TPP, the peak intensity at 379 nm decreased with increasing irradiation time (up to 10 min), suggesting that the concentration of DMA in the solution decreased, owing to the reaction of DMA with 1O2.27 These results confirmed that the porphyrin salts can generate 1O2 upon light irradiation.
The DMA photo‐oxidation kinetic behavior in the presence of the porphyrin compounds was studied by linear least‐squares fit of the semilogarithmic plot of ln A 0/A versus time, where A 0 is the absorbance at 379 nm before irradiation and A is the absorbance at 379 nm after a certain irradiation time. The observed rate constants (k) were calculated based on the fitting curves from Figure 2 (slope, Table 3). For DMA without photosensitizer, the k value was negligible. For the TOHPP‐based porphyrin salts, their k values were comparable with the reference TPP and TOHPP, indicating that the TOHPP‐based compounds had similar 1O2 generation rate with TPP and TOHPP. Accordingly, TOHPP‐based salts had similar Φ Δ values (calculated based on k) compared to TPP.
Figure 2.
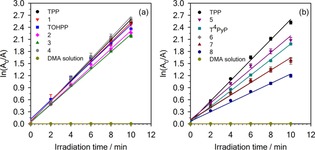
First‐order plots of DMA photo‐oxidation by a) TOHPP‐based compounds and b) T4PyP‐based compounds with TPP reference, and DMA solution.
Table 3.
Kinetic parameters for the photo‐oxidation reaction of DMA.
| Sample | Observed rate constant (k) [min−1] |
Φ Δ |
|---|---|---|
| TPP | 0.25 | 0.62 |
| TOHPP | 0.23 | 0.57 |
| 1 | 0.24 | 0.60 |
| 2 | 0.23 | 0.56 |
| 3 | 0.23 | 0.53 |
| 4 | 0.26 | 0.64 |
| T4PyP | 0.20 | 0.48 |
| 5 | 0.21 | 0.51 |
| 6 | 0.16 | 0.38 |
| 7 | 0.16 | 0.38 |
| 8 | 0.12 | 0.29 |
| DMA solution | 0.00 | 0.00 |
Unlike the results with the TOHPP derivatives, the k and Φ Δ values of the T4PyP‐based porphyrinic ILs (Table 3) were smaller compared to TPP, suggesting the 1O2 generation rate of the T4PyP‐based ILs was lower than TPP. Having noticed (Figure S31) that the modification of the porphyrin by direct attachment of the alkyl groups to the aromatic porphyrin core in the T4PyP salts changed the spectral responses, we expected the changes in these electronic properties to be responsible for the lower k value. As previously seen in the literature, the 1O2 generation is related to the electronic structure of the photosensitizer,28 which is also observed in this case. It is also noteworthy that the k values of the T4PyP‐based porphyrin ILs decreased with increasing anion size, something not observed for the TOHPP salts. Overall, these results suggest that low‐melting and IL porphyrinic salts are capable of generating 1O2, thus demonstrating their potential as photosensitizers.
In conclusion, our strategy to prepare low‐melting and IL porphyrinic salts using highly symmetric porphyrin precursors was successful. Four TOHPP‐based porphyrin salts with melting points >100 °C (although much lower melting than TOHPP) were obtained by introducing C12 alkyl chains without changing the electronic nature of the porphyrin. Four T4PyP‐based porphyrinc ILs were obtained in a similar fashion, including one room‐temperature IL; however, the electronic properties of the porphyrin were changed.
The synthesized porphyrinic salts showed higher solubilities in different solvents than their neutral precursors. More importantly, these porphyrinic salts generate 1O2 upon light irradiation, and thus exhibit a potential use as photosensitizers. The synthesis of liquid photosensitizers with tunable solubilities opens new research areas and numerous promising applications based on porphyrinic ILs.
Experimental Section
Detailed information on experimental (synthesis of porphyrin salts and characterization methods), NMR spectra, FT‐IRs, TGAs, DSCs, XRD,29 UV/Vis, and singlet oxygen quantum yield are available in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support. This research was undertaken, in part, thanks to funding from the Canada Excellence Research Chairs Program.
K. Li, H. M. Titi, P. Berton, R. D. Rogers, ChemistryOpen 2018, 7, 659.
References
- 1.
- 1a. Li X., Lee S., Yoon J., Chem. Soc. Rev. 2018, 47, 1174–1188; [DOI] [PubMed] [Google Scholar]
- 1b. Ethirajan M., Chen Y., Joshi P., Pandey R. K., Chem. Soc. Rev. 2011, 40, 340–362. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Ferreira D. P., Conceicao D. S., Calhelha R. C., Sousa T., Socoteanu R., Ferreira I. C. F. R., Ferreira L. F. V., Carbohydr. Polym. 2016, 151, 160–171; [DOI] [PubMed] [Google Scholar]
- 2b. DeRosa M. C., Crutchley R. J., Coord. Chem. Rev. 2002, 233, 351–371; [Google Scholar]
- 2c. Bonnett R., Buckley D. G., Burrow T., Galia A. B. B., Saville B., Songca S. P., J. Mater. Chem. 1993, 3, 323–324. [Google Scholar]
- 3. Redman J. E., Feeder N., Teat S. J., Sanders J. K., Inorg. Chem. 2001, 40, 2486–2499. [DOI] [PubMed] [Google Scholar]
- 4. Concellón A., Marcos M., Romero P., Serrano J. L., Termine R., Golemme A., Angew. Chem. Int. Ed. 2016, 56, 1259–1263; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 129, 1279–1283. [Google Scholar]
- 5. Li G., Dilger A. K., Cheng P. T., Ewing W. R., Groves J. T., Angew. Chem. Int. Ed. 2017, 57, 1251–1255; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 130, 1265–1269. [Google Scholar]
- 6. Schmitt J., Heitz V., Sour A., Bolze F., Ftouni H., Nicoud J. F., Flamigni L., Ventura B., Angew. Chem. Int. Ed. 2015, 54, 169–173; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 171–175. [Google Scholar]
- 7.K. M. Kadish, K. M. Smith, R. Guilard, The Porphyrin Handbook: Inorganic, organometallic and coordination chemistry, Vol. 3, Elsevier, 2000.
- 8. Arias A. C., MacKenzie J. D., McCulloch I., Rivnay J., Salleo A., Chem. Rev. 2010, 110, 3–24. [DOI] [PubMed] [Google Scholar]
- 9. Li K., Berton P., Kelley S. P., Rogers R. D., Biomacromolecules 2018, 19, 3291–3300. [DOI] [PubMed] [Google Scholar]
- 10. Welton T., Chem. Rev. 1999, 99, 2071–2084. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Shadid M., Gurau G., Shamshina J. L., Chuang B. C., Hailu S., Guan E., Chowdhury S. K., Wu J. T., Rizvi S. A. A., Griffin R. J., Rogers R. D., MedChemComm 2015, 6, 1837–1841; [Google Scholar]
- 11b. Shekaari H., Zafarani-Moattar M. T., Mirheydari S. N., J. Solution Chem. 2016, 45, 624–663; [Google Scholar]
- 11c. Zavgorodnya O., Shamshina J. L., Mittenthal M., McCrary P. D., Rachiero G. P., Titi H. M., Rogers R. D., New J. Chem. 2017, 41, 1499–1508. [Google Scholar]
- 12. Xu H.-J., Gros C. P., Brandès S., Ge P.-Y., Girault H. H., Barbe J.-M., J. Porphyrins Phthalocyanines 2011, 15, 560–574. [Google Scholar]
- 13. Zagami R., Trapani M., Castriciano M. A., Romeo A., Mineo P. G., Scolaro L. M., J. Mol. Liq. 2017, 229, 51–57. [Google Scholar]
- 14. Nowak-Król A., Gryko D., Gryko D. T., Chem. Asian J. 2010, 5, 904–909. [DOI] [PubMed] [Google Scholar]
- 15. Smykalla L., Shukrynau P., Mende C., Rüffer T., Lang H., Hietschold M., Surf. Sci. 2014, 628, 92–97. [Google Scholar]
- 16. Jiang X., Du Y., Liu C., Geng Z., Huo P., Zhang S., Wang G., High Perform. Polym. 2013, 25, 843–853. [Google Scholar]
- 17. Berezina N., Minh D., Tikhonova Y., Tumanova N., Guseinov S., Bazanov M., Berezin M., Glazunov A., Semeikin A., Russ. J. Gen. Chem. 2016, 86, 835–839. [Google Scholar]
- 18. Henriques C. A., Gonçalves N. P. F., Abreu A. R., Calvete M. J. F., Pereira M. M., J. Porphyrins Phthalocyanines 2012, 16, 290–296. [Google Scholar]
- 19. Carmichael A. J., Seddon K. R., J. Phys. Org. Chem. 2000, 13, 591–595. [Google Scholar]
- 20. Weingärtner H., Angew. Chem. Int. Ed. 2008, 47, 654–670; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 664–682. [Google Scholar]
- 21. Brittain H. G., Bruce R. D., in Comprehensive Anal. Chem. Vol. 47 (Eds.: S. Ahuja, N. Jespersen), Elsevier, 2006, pp. 63–109. [Google Scholar]
- 22. Maton C., De Vos N., Stevens C. V., Chem. Soc. Rev. 2013, 42, 5963–5977. [DOI] [PubMed] [Google Scholar]
- 23. Roy S., Titi H. M., Goldberg I., CrystEngComm 2016, 18, 3372–3382. [Google Scholar]
- 24. Holbrey J. D., Reichert W. M., Nieuwenhuyzen M., Sheppard O., Hardacre C., Rogers R. D., Chem. Commun. 2003, 476–477. [DOI] [PubMed] [Google Scholar]
- 25. Gavezzotti A., J. Am. Chem. Soc. 1983, 105, 5220–5225. [Google Scholar]
- 26. Tanaka T., Osuka A., Chem. Soc. Rev. 2015, 44, 943–969. [DOI] [PubMed] [Google Scholar]
- 27. Scalise I., Durantini E. N., J. Photochem. Photobiol. A 2004, 162, 105–113. [Google Scholar]
- 28. Schweitzer C., Schmidt R., Chem. Rev. 2003, 103, 1685–1758. [DOI] [PubMed] [Google Scholar]
- 29. https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/open.201800166 1845858 and 1845857 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary