

Abstract
The identification of anaplastic lymphoma kinase (ALK), an oncogenetic driver mutation, in lung cancer has paved the way for a new era in the treatment of non-small cell lung cancer (NSCLC). Targeting ALK using tyrosine kinase inhibitors (TKI) has dramatically improved the prognosis of patients with ALK-rearranged NSCLC. However, most patients relapse on ALK-TKI therapy within a few years because of acquired resistance. One mechanism of acquiring resistance is a second mutation on the ALK gene, and the representative mutation is L1996M in the gatekeeper residue. In particular, the solvent-front ALK G1202R mutation is the common cause of resistance against first- and second-generation ALK-TKIs. Another major concern regarding ALK-TKI is metastasis to the central nervous system, commonly observed in patients relapsing after ALK-TKI therapy. The next-generation ALK inhibitor lorlatinib (PF-06463922) has therefore been developed to inhibit resistant ALK mutations, including ALK G1202R, and to penetrate the blood–brain barrier. In a Phase I/II trial, the safety and efficacy of lorlatinib were demonstrated in patients with advanced ALK-positive NSCLC, most of whom had central nervous system metastases and had previous ALK-TKI treatment. In this review, we discuss the structure, pharmacodynamics, and pharmacokinetics of lorlatinib and compare its characteristics with those of other ALK inhibitors. Furthermore, clinical trials for lorlatinib are summarized, and future perspectives in the management of patients with ALK-rearranged NSCLC are discussed.
Keywords: non-small cell lung cancer, anaplastic lymphoma kinase, ALK inhibitor, lorlatinib
Introduction
The identification of oncogenetic driver mutations in lung cancer has heralded a new era in the treatment of non-small cell lung cancer (NSCLC). In particular, oncogenic driver mutations in EGFR, anaplastic lymphoma kinase (ALK), ret proto-oncogene (RET), c-ros oncogene 1, and receptor tyrosine kinase (ROS1) have recently been identified in basic and clinical studies.1,2 These oncogenetic mutation profiles subclassify NSCLC, especially lung adenocarcinomas, and patients with these oncogenes can be successfully treated with specific kinase inhibitors.
The ALK rearrangement is a potent oncogene and was first identified in NSCLC by Soda et al in 2007.3 There are several partners that fuse with ALK, including echinoderm microtubule-associated protein-like 4 (EML4), Huntingtin interacting protein 1 (HIP1), and Translocated Promoter Region (TPR), resulting in a potent transforming activity in NSCLC.3–5 For instance, the EML4–ALK fusion gene was first identified by Soda et al, and it is constitutively oligomerized via the coiled coil domain within the EML4 region, leading to the activation of downstream signaling via the Ras/MAPK, PI3K/AKT, and JAK/STAT pathways, among others.3,6
Approximately 5% of patients with NSCLC harbor the ALK fusion gene, and the characteristics of this patient cohort are as follows: younger age, ever or never smoker, adenocarcinoma histology, no definite racial differences in frequency of ALK rearrangement, and mutual exclusion of other driver oncogenes.7–9
Importantly, ALK inhibition showed remarkable anti-tumor efficacy in mouse xenograft models transduced with NIH3T3 cells expressing the EML4–ALK fusion gene. In clinical settings, targeting of ALK using tyrosine kinase inhibitors (TKI), such as crizotinib, alectinib, and ceritinib, demonstrated remarkable antitumor efficacy and improvement of prognosis in patients with ALK-rearranged NSCLC.10–15 Unfortunately, despite these promising results, most patients relapsed on TKI therapy within a few years because of acquired resistance.13,15–18 There are two main resistance mechanisms to ALK inhibitors: ALK dominant or ALK nondominant.18,19 ALK dominant resistance mechanisms include secondary mutations and copy number gain in the ALK gene, while ALK nondominant resistance mechanisms include the activation of bypass downstream signaling via, for example, EGFR, Kirsten rat sarcoma viral oncogene homolog (KRAS), v-kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog (KIT), met proto-oncogene (MET), and insulin-like growth factor 1 receptor. With respect to the former type of mechanism, the solvent-front mutation ALK G1202R is established as the common cause of resistance against first- and second-generation ALK-TKI therapy.20
Another major concern is metastasis to the central nervous system (CNS), considered a sanctuary site owing to the blood–brain barrier (BBB).21 A limitation of the first-generation ALK inhibitor crizotinib was that relapse in the brain after treatment was commonly reported.21,22 Although second-generation ALK inhibitors ceritinib and alectinib have demonstrated effectiveness against brain metastasis in crizotinib-relapsed patients, these patients frequently relapse with CNS progression.21 In a Phase I/II trial of alectinib (AF-002JG), the CNS response rate was 52%.23 This effect may partly be associated with poor BBB permeability, which is attributed to the expression of p-glycoprotein (P-gp) at the luminal side of the BBB endothelium.24–27
Therefore, next-generation ALK inhibitors, such as brigatinib (AP26113) and lorlatinib (PF-06463922), were designed to inhibit resistant ALK mutants and to penetrate the BBB.28,29 Lorlatinib, was developed by Pfizer to specifically inhibit TKI-resistant ALK mutants with optimal brain penetration.29 A novel oral ATP-competitive macrocyclic TKI, targeting ALK as well as ROS1, lorlatinib received Breakthrough Therapy Designation from the US Food and Drug Administration (FDA) in April 2017. According to data presented at the 18th World Conference on Lung Cancer (WCLC) in October 2017, the systemic and intracranial overall response rates (ORR) were as high as 62.4% and 54.9%, respectively, in ALK-positive patients who had previously received ALK inhibitors.30 Thus, lorlatinib has attracted much attention because of its antitumor efficacy against both systemic and intracranial lesions.
In this review, we discuss the structure, pharmacodynamics, and pharmacokinetics of lorlatinib, and compare its characteristics with those of other ALK inhibitors. Furthermore, clinical trials for lorlatinib are summarized and future perspectives in the management of patients with ALK-rearranged NSCLC are discussed.
Structural characteristics of lorlatinib
Lorlatinib (product name PF-06463922) is a small-molecule macrocyclic ALK-TKI with the molecular formula C21H19 FN6O2 and chemical structure (10R)–7-Amino-12-fluoro-2, 10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno) pyrazolo[4,3-hour][2,5,11]-benzoxadiazacyclotet-radecine-3-carbonitrile (Figure 1).31 It is an ATP-competitive inhibitor of recombinant ALK and ROS1 kinases, resulting in the deactivation of ALK tyrosine kinase in the cytoplasm. The macrocycle of lorlatinib (Figure 1) is its main structural difference from other ALK inhibitors. Lorlatinib was developed from crizotinib using a structure-based drug design approach to overcome ALK mutant resistance and high P-gp efflux (Figure 1).31,32 During the development process, the macrocyclic structure was associated with improved metabolic stability and low propensity for P-gp efflux than the acyclic analog.
Figure 1.
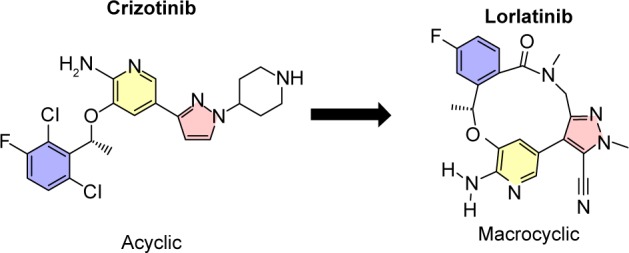
The structure of crizotinib and lorlatinib.
Pharmacodynamic properties
In biochemical assays, lorlatinib showed a mean Ki of <0.07 nM against wild-type ALK. In addition, mean Ki values of lorlatinib against crizotinib-resistant mutants such as L1196M, G1269A, 1151Tins, and F1174L were as low as <0.1–0.9 nM.29
In a cell viability assay comparing lorlatinib with crizotinib, ceritinib, and alectinib, lorlatinib was shown to be the most potent inhibitor against wild-type ALK as well as TKI-resistant ALK mutants including G1202R, which confers resistant to first- and second-generation ALK inhibitors.29 Furthermore, the mean inhibitory concentration 50 values of lorlatinib, crizotinib, ceritinib, and alectinib against G1202R mutant BaF3 cell-line proliferation were 80, 560, 309, and 595 nM, respectively, implying that only lorlatinib could inhibit the ALK G1202R mutant.29
Pharmacokinetics
Lorlatinib is orally bioavailable, with a time to maximum plasma concentration of 1–2 hours after repeated once-daily dosing of 10–200 mg. The elimination half-time of lorlatinib ranges from 19.0 to 28.8 hours across doses of 10, 50, 75, 100, and 200 mg.33 In vitro and in vivo metabolite assay results have shown that lorlatinib can potentially alter the pharmacokinetics of other coadministered drugs eliminated by the CYP/CYP450 (CYP) pathways. Therefore, concomitant use of CYP3A inhibitors was not permitted from 12 days prior to the first dose of lorlatinib in a Phase III trial.
Preclinical studies
During in vitro experiments, lorlatinib was found to potently suppress ALK-dependent signaling and inhibit cell growth in crizotinib- or alectinib-resistant ALK mutant lung cancer cell lines as well as in wildtype ALK cell lines.29 Lorlatinib also showed significantly greater cell growth inhibition compared with crizotinib in cell lines derived from patients with acquired resistance to crizotinib, ceritinib, or alectinib.29 Mouse models also demonstrated the systemic and intracranial efficacy of lorlatinib, leading to prolonged survival. In addition to its antitumor effect, BBB penetration by lorlatinib was confirmed by Collier et al using carbon11- and fluorine18-labeled lorlatinib and initial positron emission tomography imaging in a nonhuman primate model.34
Clinical trials
The efficacy of lorlatinib in patients with advanced ALK-positive or ROS1-positive NSCLC has been investigated, and the findings are summarized below.
Phase I/II studies
Phase I and II trials (NCT01970865) of lorlatinib were initiated to analyze the safety, pharmacokinetic, efficacy, and outcomes of lorlatinib in patients with advanced ALK-positive or ROS1-positive NSCLC.
Shaw et al reported the results of the open-label, single-arm, first-in-human Phase I trial.33 Fifty-four patients with advanced ALK-positive or ROS1-positive NSCLC received lorlatinib orally at doses ranging from 10 to 200 mg once daily or 35 to 100 mg twice daily. The most common adverse events (AEs) included hypercholesterolemia (39 [72%] of 54 patients), hypertriglyceridemia (21 [39%] of 54 patients), peripheral edema (21 [39%] of 54 patients), and peripheral neuropathy (21 [39%] of 54 patients). One dose-limiting toxicity, a grade 2 CNS effect (slowed speech, mentation, and word-finding difficulty), occurred at 200 mg, and no maximum tolerated dose was identified. A dose of 100 mg once daily was well tolerated, and none of the patients required dose reduction. In addition, pharmacokinetics data showed that this dose was the lowest dose that exceeded the efficacious concentration of 150 ng/mL. Therefore, 100 mg once daily was adopted as the recommended dose in a subsequent Phase II study.30 Among ALK-positive patients, the proportion who achieved an objective response was 19 (46%; 95% CI 31–63) of 41 ALK-positive patients and 11 (42%; 95% CI 23–63) of 26 patients who had previously received two or more ALK inhibitors. Among 12 ROS1-positive patients, 6 (50%; 95% CI 21–79) achieved an objective response. Of the 24 patients who had measurable CNS target lesions, 11 (46%; 95% CI 26–67) had an intracranial objective response.
At the 18th WCLC in October 2017, the results of a Phase II trial (NCT01970865) of lorlatinib were reported in six expansion cohorts according to prior treatment.30 A total of 275 ALK- or ROS1-positive patients received lorlatinib at the recommended Phase II dose of 100 mg once daily. In four cohorts encompassing 197 patients who had previously received ALK inhibitors, ORR was 62.4% (ranging from 33% to 74%) and intracranial ORR was 54.9% (ranging from 39% to 75%). In addition, 90% (27/30) of patients who received lorlatinib as a first-line therapy had a confirmed ORR. Lorlatinib subsequently received breakthrough therapy designation for patients with advanced ALK-positive NSCLC previously treated with one or more ALK inhibitors as well as for first-line treatment of ALK-positive NSCLC. The clinical outcomes from Phase II studies of ceritinib, alectinib, brigatinib, and lorlatinib used after crizotinib with or without chemotherapy are briefly summarized in Table 1.15,16,30,39
Table 1.
Clinical outcomes of Phase II studies of ALK inhibitors used after crizotinib with or without chemotherapy
| Clinical trials and cohorts | Ceritinib39 | Alectinib16 | Brigatinib15 | Lorlatinib30 | |
|---|---|---|---|---|---|
|
| |||||
| Phase II study | Phase II global study | Phase II study | Phase II study | ||
| (ASCEND-2) | 180 mg cohort | ALK-positive cohorts | |||
| Previous treatment | Crizotinib + chemo | Crizotinib ± chemo | Crizotinib ± chemo | Crizotinib ± chemo | Treatment naïve |
| Number of patients | n=140 | n=138 | n=110 | n=59 | n=30 |
| Systemic response | |||||
| ORR (%) | 39 | 50 | 54 | 69 | 90 |
| 95% CI | (31%–47%) | (41%–59%) | (43%–65%) | (NA) | (NA) |
| Median PFS (months) | 5.7 | 8.9 | 12.9 | (NA) | (NA) |
| 95% CI | (5.4–7.6) | (5.6–11.3) | (11.1–NR) | (NA) | (NA) |
| Intracranial efficacy (measurable baseline CNS metastases) | |||||
| Number of patients | n=20 | n=35 | n=18 | n=37 | n=8 |
| Intracranial ORR (%) | 45 | 57 | 67 | 68a | 75a |
| 95% CI | (23%–69%) | (39%–74%) | (41%–87%) | (NA) | (NA) |
Note:
The information whether ORR was calculated from measurable baseline CNS metastases was not available.
Abbreviations: ALK, anaplastic lymphoma kinase; CNS, central nervous system; NA, not available; NR, not reached; ORR, overall response rates; PFS, progression-free survival.
Ongoing Phase III study
The ongoing Phase III CROWN study (NCT03052608) began in April 2017 and the estimated primary completion date of study is August 31, 2018. This study is an open-label, randomized, double-blind, two-arm trial with an estimated enrollment of 280 patients. The aim of the trial is to compare the efficacy of lorlatinib with crizotinib as a first-line treatment in patients with advanced ALK-positive NSCLC. The primary endpoint of this study is progression-free survival (PFS), and the main secondary outcomes are objective response, intracranial objective response, clinical benefit response, and AEs.
Safety, tolerability, and adverse events
In Phase I study, only one dose-limiting toxicity (grade 2 CNS effect: slowed speech, mentation, and word-finding difficulty) occurred in cohorts of those who were treated with 200 mg of lorlatinib once daily, and a maximum tolerated dose was not defined.33,35 Among the combined cohorts of 54 patients, the common AEs were hypercholesterolemia (72%), hypertriglyceridemia (39%), peripheral neuropathy (39%), and peripheral edema (39%). These AEs differ from those reported for other ALK inhibitors. The AEs associated with ALK inhibitors are summarized in Table 2, which shows that hepatotoxicity (elevated aspartate aminotransferase or alanine aminotransferase) and gastrointestinal disorders (eg, nausea, diarrhea, or vomiting) were mainly associated with other ALK inhibitors.13,14,16,23,36–40 In the Phase II cohort of 17 patients receiving 100 mg of lorlatinib once daily, no patient required dose reduction or permanently discontinued treatment because of treatment-related AEs. The most common reasons for temporary treatment discontinuation in this cohort were hypercholesterolemia (12%) and increased lipase (12%). No cases of interstitial lung disease or pneumonitis were reported in clinical studies of lorlatinib.
Table 2.
Characteristics of AEs observed with crizotinib, ceritinib, alectinib, brigatinib, and lorlatinib use in clinical trials
| Crizotinib | Ceritinib | Alectinib | Brigatinib | Lorlatinib | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phase I36 | Phase II37 | Phase I38 | Phase II39 | Phase I/II23 | Phase II16 | Phase I/II40 | Phase I/II33 | |||||||||
| Patients (n) | 144 | 1,066 | 246 | 140 | 47 | 138 | 137 | 54 | ||||||||
| Major AEsa | All grades (%) | Grade 3–4 (%) | All grades (%) | Grade 3–4 (%) | All grades (%) | Grade3–4 (%) | All grades (%) | Grade 3–4 (%) | All grades (%) | Grade 3–4 (%) | All grades (%) | Grade 3–4 (%) | All grades (%) | Grade 3–4 (%) | All grades (%) | Grade 3–4 (%) |
| Gastrointestinal disorders | ||||||||||||||||
| Nausea | 56 | – | 51 | – | 83 | 6 | 81 | 6 | 15 | – | – | – | 53 | – | 11 | – |
| Diarrhea | 50 | – | 47 | – | 86 | 6 | 80 | 6 | – | – | – | – | 41 | – | – | – |
| Constipation | 28 | – | 35 | – | 30 | – | 29 | – | 11 | – | 33 | – | 23 | – | 22 | – |
| Vomiting | 39 | – | 47 | – | 61 | 4 | 63 | 4 | – | – | – | – | 21 | – | – | – |
| General disorders | ||||||||||||||||
| Fatigue | 24 | – | 21 | 3 | 43 | 5 | 36 | 6 | 30 | – | 26 | – | 43 | 4 | 15 | – |
| Peripheral edema | 30 | – | – | – | – | – | 17 | – | 25 | – | 13 | – | 39 | – | ||
| Nervous system disorders | ||||||||||||||||
| Cognitive disturbance | –b | – | – | – | – | – | – | – | – | – | – | – | – | – | 24 | – |
| Peripheral neuropathy | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 39 | – |
| Headache | – | – | – | – | 19 | – | – | – | – | – | 16 | – | 33 | – | – | – |
| Respiratory disorders | ||||||||||||||||
| Cough | – | – | – | – | 29 | – | 21 | – | – | – | 14 | – | 31 | – | – | – |
| Dyspnea | – | – | – | – | 21 | 4 | 21 | 6 | – | – | – | 3 | 23 | 6 | – | – |
| Others | ||||||||||||||||
| Hypertension | – | – | – | – | – | – | – | – | – | – | – | – | 15 | 5 | – | – |
| Myalgia | – | – | – | – | 15 | – | – | – | 17 | – | 23 | – | – | – | – | – |
| Weight increase | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 11 | 6 |
| Pneumonitis/ILDc | 3 | 2 | 2 | <1 | 4 | 3 | 1 | <1 | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 |
| Eye disorders | 64 | – | 58 | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Representative abnormal laboratory values | ||||||||||||||||
| Neutropenia | – | – | 21 | 6 | – | – | – | – | – | 4 | – | – | – | – | – | – |
| Increased AST | 10 | 4 | 30 | 7 | 33 | 10 | 32 | 5 | – | – | 12 | – | 13 | – | 13 | – |
| Increased ALT | 12 | 3 | 44 | 27 | 44 | 17 | 15 | 4 | 10 | – | 11 | – | – | – | ||
| Increased γ-GTP | – | – | – | – | – | – | 18 | 12 | – | – | – | – | – | – | – | – |
| Increased CPK | – | – | – | – | – | – | – | – | 15 | – | – | – | – | – | – | – |
| Increased creatine | – | – | – | – | 17 | – | – | – | – | – | – | – | – | – | – | – |
| Increased amylase | – | – | – | – | – | 3 | – | – | – | – | – | – | 20 | 4 | 13 | – |
| Increased lipase | – | – | – | – | – | 5 | – | – | – | – | – | – | 17 | 9 | 17 | 4 |
| Hypercholesterolemia | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 72 | 13 |
| Hypertriglyceridemia | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 39 | 6 |
Notes:
Major AEs were selected from common AEs ($10% of frequency).
“–” in All grades: <10% or not available, “–” in Grade 3–4: <3% or not available.
Pneumonitis/ILD were included as major AEs as critical side effects in using ALK inhibitors.
Frequency of AEs:
 ,
,
 ,
,
 ,
,
 .
.
Abbreviations: AE, adverse event; ALK, anaplastic lymphoma kinase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; ILD, interstitial lung disease; γ-GTP, γ-glutamyltransferase.
Acquired resistance to lorlatinib
As described above, lorlatinib has shown great efficacy in patients who were resistant to first- and second-generation ALK-TKIs. However, acquired resistance to lorlatinib can be expected, as for other ALK-TKIs. Shaw et al demonstrated a novel mechanism of lorlatinib resistance by sequencing the DNA of lorlatinib-resistant patient and detecting a double mutation (ALK C1156Y/L1198F) that unexpectedly restored sensitivity to crizotinib.41 Although the patient had previously relapsed with crizotinib and ceritinib therapies, the patient subsequently again responded to crizotinib. Furthermore, activation of the MET pathway might be associated with the acquisition of resistance to lorlatinib, as for alectinib, and it is possible that this resistance mechanism might be overcome using crizotinib.42 These findings are consistent with the observation that gefitinib (a first-generation EGFR-TKI) can overcome osimertinib (a third-generation EGFR-TKI) resistance via the C797S mutation in the EGFR gene.43,44
Recently, the mechanisms of lorlatinib resistance have been described by screening accelerated mutagenesis in vitro and sequencing 20 lorlatinib-resistant biopsy specimens from patients.45 Interestingly, the ALK-dominant lorlatinib-resistance mechanism was primarily caused by multiple different compound ALK mutations. In other words, lorlatinib alerts the single ALK gene to a lorlatinib-resistant compound ALK mutation, a so-called double mutation. For instance, patients harboring ALK C1156Y become lorlatinib resistant by acquiring ALK C1156Y/L1198F. Figure 2 shows the sensitivity of Ba/F3 cells expressing EML4-ALK variant 1, either wild type or mutant, to ALK inhibitors.45
Figure 2.

Sensitivity of Ba/F3 cells expressing EML4-ALK variant 1, either wild type or mutant, to ALK inhibitors.
Abbreviation: ALK, anaplastic lymphoma kinase.
Future directions
Lorlatinib shows potent activity against acquired ALK mutations and high brain permeability in targeting CNS metastasis.29 Based on Phase I/II trial data, lorlatinib may represent an effective therapeutic strategy for ALK-TKI-resistant mutant NSCLC as well as progressive CNS metastases after ALK-TKI treatment.33 A Phase III trial (NCT03052608) comparing the efficacy of lorlatinib with crizotinib as a first-line treatment for patients with advanced ALK-positive NSCLC is currently ongoing. As with alectinib and ceritinib, lorlatinib is expected to show superior efficacy to crizotinib in treating ALK-positive NSCLC.46,47 If so, the question is which ALK inhibitor should be used for the first-line treatment of ALK-positive NSCLC: lorlatinib, alectinib, or ceritinib.
Recently, osimertinib, a third-generation EGFR-TKI, has shown superior efficacy to standard EGFR-TKI therapy (gefitinib) in treating EGFR-mutant NSCLC.48 In April 2018, the FDA approved osimertinib as a first-line treatment for EGFR-mutant NSCLC. Like osimertinib, lorlatinib may become standard-of-care first-line treatment for ALK-positive NSCLC. Then, can we say that lorlatinib is the best for first-line therapy which prolongs PFS and OS?
At this point, the optimal treatment strategy using lorlatinib remains unclear. First-line use of lorlatinib, the effect of which on PFS remains unknown, can be followed by crizotinib treatment if lorlatinib resistance is mediated by the L1198F-containing double mutation conferring sensitivity to crizotinib.41,45 What, then, about the sequential use of alectinib or ceritinib followed by lorlatinib? Although the significance of alectinib or ceritinib treatment after lorlatinib remains unclear, preclinical studies demonstrated that ceritinib and alectinib were also resistant to a lorlatinib-resistant mutation.45 Considering that most ALK secondary mutations for alectinib and ceritinib can be overcome using lorlatinib, sequential therapy of lorlatinib after alectinib or ceritinib might prolong PFS and OS compared with using lorlatinib as a first-line therapy. The outcome of the Phase III CROWN study (NCT03052608) and a retrospective study on the sequential use of lorlatinib followed by alectinib or ceritinib may verify the value of this strategy. Brigatinib also joins this complex sequential therapy, contributing to a median OS of 5 years among ALK-rearranged patients, which is a landmark in the treatment of advanced NSCLC.
A recent meta-analysis of alectinib showed that ALK inhibitor-naïve patients tended to have a higher ORR than crizotinib-pretreated patients (87%; 95% CI 81%–92% vs 52%; 95% CI 46%–58%).49 Furthermore, the recent Phase III ALUR study (NCT02604342; alectinib vs chemotherapy in crizotinib-pretreated patients) has reported a median PFS of 9.6 months (95% CI 6.9–12.2) and 1.4 months (95% CI 1.3–1.6) with alectinib and chemotherapy, respectively.50 In the J-ALEX trial, the median PFS of ALK inhibitor-naïve patients treated with alectinib as a second-line therapy was 20.3 months (95% CI 20.3–not estimable), while that of crizotinib-pretreated patients was 8.2 months (95% CI 6.4–15.7).51 Therefore, the efficacy of alectinib seemed to decrease when administered after crizotinib.
In contrast, alectinib showed roughly equal efficacy in chemotherapy-pretreated and chemotherapy-naïve patients in the same study.51 The Japanese AF-001JP trial also demonstrated the remarkable efficacy of alectinib in chemotherapy-pretreated ALK inhibitor-naïve patients, with an ORR of 93.5% (95% CI 82–99) and median PFS still not reached after a 3-year follow-up.12,52 Taken together, chemotherapy followed by alectinib may prolong PFS and OS to a greater extent than first-line use of alectinib. These findings related to alectinib may also apply to lorlatinib, highlighting the importance of investigating the treatment order of lorlatinib in future studies.
In recent years, immune checkpoint inhibitors (ICIs) have presented a new approach to the treatment of NSCLC.53 At present, several clinical trials of ICIs with ALK-TKI are ongoing.54 In a recent Phase I/II study of nivolumab plus crizotinib for the first-line treatment of NSCLC (CheckMate 370), the primary endpoints of safety and tolerability were not achieved.55 The results of other ongoing clinical studies are therefore required to evaluate the combinational use of ALK inhibitors with ICIs.
Another possible treatment strategy is the combination of ALK inhibitors with other inhibitors that bypass signaling pathways such as EGFR, MET, or KIT, which represent ALK nondominant resistant mechanisms. Among 20 lorlatinib-resistant biopsies, 12 (60%) did not show an ALK mutation, and likely harbored an ALK-nondominant mechanism.45
Recently, specific ALK fusion variants were shown to be associated with clinical outcome.56 EML4-ALK variant 3, in particular, was significantly associated with the development of ALK resistance mutations, particularly G1202R. Interestingly, an exploratory analysis of 29 patients who received lorlatinib showed that those harboring variant 3 had a significantly longer median PFS than those harboring variant 1 (11.0 vs 3.3 months; P=0.011). Therefore, this specific ALK fusion variant could represent a potential biomarker for response to lorlatinib.
The suggested treatment strategies using lorlatinib are summarized in Figure 3.
Figure 3.
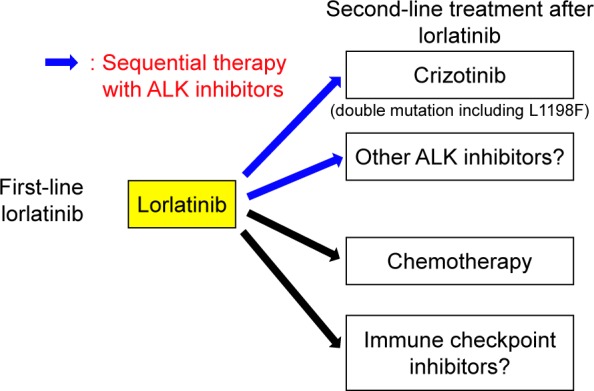
The proposed positioning of lorlatinib in the treatment of ALK-positive patients with NSCLC.
Abbreviations: ALK, anaplastic lymphoma kinase; NSCLC, non-small cell lung cancer.
Conclusion
Lorlatinib has attracted significant attention because of its potent antitumor effects against both systemic and intracranial lesions in preclinical and Phase I/II studies.29,33 A Phase III study to compare the efficacy of lorlatinib with crizotinib as a first-line treatment in patients with advanced ALK-positive NSCLC is currently underway. The outcome of the trial will likely influence the future treatment options for patients with ALK-positive NSCLC.
Acknowledgments
We thank Clare Cox, PhD, from Edanz Group (www.edan-zediting.com/ac) for editing a draft of this manuscript.
Footnotes
Author contributions
Takaki Akamine wrote the manuscript and designed figures with support from Gouji Toyokawa who was in charge of overall direction. Takashi Seto and Tetsuzo Tagawa supervised the revising of the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
Takashi Seto reports grants from Bayer Yakuhin, Eisai, Merck Serono, Novartis Pharma, and Verastem; personal fees from Bristol-Myers Squibb, Kyowa Hakko Kirin, Mochida Pharmaceutical, Nippon Kayaku, Ono Pharmaceutical, Roche Singapore, Sanofi, Showa Yakuhin, Taiho Pharmaceutical, and Takeda Pharmaceutical; grants and personal fees from Astellas Pharma, AstraZeneca, Chugai Pharmaceutical, Daiichi Sankyo, Eli Lilly Japan, Kissei Pharmaceutical, MSD, Nippon Boehringer Ingelheim, Pfizer Japan, Yakult Honsha, outside the submitted work. The other authors report no conflicts of interest in this work.
References
- 1.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 3.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 4.Hong M, Kim RN, Song JY, et al. HIP1-ALK, a novel fusion protein identified in lung adenocarcinoma. J Thorac Oncol. 2014;9(3):419–422. doi: 10.1097/JTO.0000000000000061. [DOI] [PubMed] [Google Scholar]
- 5.Choi YL, Lira ME, Hong M, et al. A novel fusion of TPR and ALK in lung adenocarcinoma. J Thorac Oncol. 2014;9(4):563–566. doi: 10.1097/JTO.0000000000000093. [DOI] [PubMed] [Google Scholar]
- 6.Mano H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci. 2008;99(12):2349–2355. doi: 10.1111/j.1349-7006.2008.00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soda M, Isobe K, Inoue A, et al. A prospective PCR-based screening for the EML4-ALK oncogene in non-small cell lung cancer. Clin Cancer Res. 2012;18(20):5682–5689. doi: 10.1158/1078-0432.CCR-11-2947. [DOI] [PubMed] [Google Scholar]
- 8.Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19(15):4273–4281. doi: 10.1158/1078-0432.CCR-13-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camidge DR, Kono SA, Flacco A, et al. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res. 2010;16(22):5581–5590. doi: 10.1158/1078-0432.CCR-10-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 11.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 12.Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013;14(7):590–598. doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- 13.Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: a randomized, multicenter Phase II trial. J Clin Oncol. 2017;35(22):2490–2498. doi: 10.1200/JCO.2016.71.5904. [DOI] [PubMed] [Google Scholar]
- 16.Ou SH, Ahn JS, de Petris L, et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a Phase II Global Study. J Clin Oncol. 2016;34(7):661–668. doi: 10.1200/jco.2015.63.9443. [DOI] [PubMed] [Google Scholar]
- 17.Toyokawa G, Seto T. ALK inhibitors: what is the best way to treat patients with ALK+ non-small-cell lung cancer? Clin Lung Cancer. 2014;15(5):313–319. doi: 10.1016/j.cllc.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Toyokawa G, Seto T. Updated evidence on the mechanisms of resistance to ALK inhibitors and strategies to overcome such resistance: clinical and preclinical data. Oncol Res Treat. 2015;38(6):291–298. doi: 10.1159/000430852. [DOI] [PubMed] [Google Scholar]
- 19.Camidge DR, Doebele RC. Treating ALK-positive lung cancer-early successes and future challenges. Nat Rev Clin Oncol. 2012;9(5):268–277. doi: 10.1038/nrclinonc.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toyokawa G, Seto T, Takenoyama M, Ichinose Y. Insights into brain metastasis in patients with ALK+ lung cancer: is the brain truly a sanctuary? Cancer Metastasis Rev. 2015;34(4):797–805. doi: 10.1007/s10555-015-9592-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa DB, Shaw AT, Ou SH, et al. Clinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastases. J Clin Oncol. 2015;33(17):1881–1888. doi: 10.1200/JCO.2014.59.0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadgeel SM, Gandhi L, Riely GJ, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15(10):1119–1128. doi: 10.1016/S1470-2045(14)70362-6. [DOI] [PubMed] [Google Scholar]
- 24.Kodama T, Hasegawa M, Takanashi K, Sakurai Y, Kondoh O, Sakamoto H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother Pharmacol. 2014;74(5):1023–1028. doi: 10.1007/s00280-014-2578-6. [DOI] [PubMed] [Google Scholar]
- 25.Bartels AL, Kortekaas R, Bart J, et al. Blood-brain barrier P-glycoprotein function decreases in specific brain regions with aging: a possible role in progressive neurodegeneration. Neurobiol Aging. 2009;30(11):1818–1824. doi: 10.1016/j.neurobiolaging.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29(15):e443–e445. doi: 10.1200/JCO.2010.34.1313. [DOI] [PubMed] [Google Scholar]
- 27.Katayama R, Sakashita T, Yanagitani N, et al. P-glycoprotein mediates ceritinib resistance in anaplastic lymphoma kinase-rearranged non-small cell lung cancer. EBioMedicine. 2016;3:54–66. doi: 10.1016/j.ebiom.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S, Anjum R, Squillace R, et al. The potent ALK inhibitor brigatinib (AP26113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical models. Clin Cancer Res. 2016;22(22):5527–5538. doi: 10.1158/1078-0432.CCR-16-0569. [DOI] [PubMed] [Google Scholar]
- 29.Zou HY, Friboulet L, Kodack DP, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. 2015;28(1):70–81. doi: 10.1016/j.ccell.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solomon B, Shaw A, Ou S, et al. OA 05.06 Phase 2 study of lorlatinib in patients with advanced ALK +/ROS1 + non-small-cell lung cancer. J Thorac Oncol. 2017;12(11):S1756. [Google Scholar]
- 31.Johnson TW, Richardson PF, Bailey S, et al. Discovery of (10R)-7- amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57(11):4720–4744. doi: 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- 32.Huang Q, Johnson TW, Bailey S, et al. Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to crizotinib. J Med Chem. 2014;57(4):1170–1187. doi: 10.1021/jm401805h. [DOI] [PubMed] [Google Scholar]
- 33.Shaw AT, Felip E, Bauer TM, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017;18(12):1590–1599. doi: 10.1016/S1470-2045(17)30680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collier TL, Normandin MD, Stephenson NA, et al. Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib. Nat Commun. 2017;8:15761. doi: 10.1038/ncomms15761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw AT, Ou S-HI, Felip E. Efficacy and safety of lorlatinib in patients (pts) with ALK+ non-small cell lung cancer (NSCLC) with one or more prior ALK tyrosine kinase inhibitor (TKI): a phase I/II study. J Clin Oncol. 2017;35(15_suppl):9006. [Google Scholar]
- 36.Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blackhall F, Ross Camidge D, Shaw AT, et al. Final results of the large-scale multinational trial PROFILE 1005: efficacy and safety of crizotinib in previously treated patients with advanced/metastatic ALK-positive non-small-cell lung cancer. ESMO Open. 2017;2(3):e000219. doi: 10.1136/esmoopen-2017-000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim DW, Mehra R, Tan DSW, et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016;17(4):452–463. doi: 10.1016/S1470-2045(15)00614-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crinò L, Ahn MJ, de Marinis F, et al. Multicenter Phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: results from ASCEND-2. J Clin Oncol. 2016;34(24):2866–2873. doi: 10.1200/JCO.2015.65.5936. [DOI] [PubMed] [Google Scholar]
- 40.Sabari JK, Santini FC, Schram AM, et al. The activity, safety, and evolving role of brigatinib in patients with ALK-rearranged non-small cell lung cancers. Onco Targets Ther. 2017;10:1983–1992. doi: 10.2147/OTT.S109295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374(1):54–61. doi: 10.1056/NEJMoa1508887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gouji T, Takashi S, Mitsuhiro T, Yukito I. Crizotinib can overcome acquired resistance to CH5424802: is amplification of the MET gene a key factor? J Thorac Oncol. 2014;9(3):e27–e28. doi: 10.1097/JTO.0000000000000113. [DOI] [PubMed] [Google Scholar]
- 43.Chic N, Mayo-de-Las-Casas C, Reguart N. Successful treatment with gefitinib in advanced non-small cell lung cancer after acquired resistance to osimertinib. J Thorac Oncol. 2017;12(6):e78–e80. doi: 10.1016/j.jtho.2017.02.014. [DOI] [PubMed] [Google Scholar]
- 44.Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21(17):3924–3933. doi: 10.1158/1078-0432.CCR-15-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoda S, Lin JJ, Lawrence MS, et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 2018;8(6):714–729. doi: 10.1158/2159-8290.CD-17-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. 2017;377(9):829–838. doi: 10.1056/NEJMoa1704795. [DOI] [PubMed] [Google Scholar]
- 47.Tan DS, Araújo A, Zhang J, et al. Comparative efficacy of ceritinib and crizotinib as initial ALK-targeted therapies in previously treated advanced NSCLC: an adjusted comparison with external controls. J Thorac Oncol. 2016;11(9):1550–1557. doi: 10.1016/j.jtho.2016.05.029. [DOI] [PubMed] [Google Scholar]
- 48.Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 49.Fan J, Xia Z, Zhang X, et al. The efficacy and safety of alectinib in the treatment of ALK+ NSCLC: a systematic review and meta-analysis. Onco Targets Ther. 2018;11:1105–1115. doi: 10.2147/OTT.S156170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Novello S, Mazières J, Oh IJ, et al. Alectinib versus chemotherapy in crizotinib-pretreated anaplastic lymphoma kinase (ALK)-positive non-small-cell lung cancer: results from the phase III ALUR study. Ann Oncol. 2018;29(6):1409–1416. doi: 10.1093/annonc/mdy121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hida T, Nokihara H, Kondo M, et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial. Lancet. 2017;390(10089):29–39. doi: 10.1016/S0140-6736(17)30565-2. [DOI] [PubMed] [Google Scholar]
- 52.Tamura T, Kiura K, Seto T, et al. Three-year follow-up of an alectinib Phase I/II study in ALK-positive non-small-cell lung cancer: AF-001JP. J Clin Oncol. 2017;35(14):1515–1521. doi: 10.1200/JCO.2016.70.5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lambrechts D, Buysschaert I, Zanen P, et al. The 15q24/25 susceptibility variant for lung cancer and chronic obstructive pulmonary disease is associated with emphysema. Am J Respir Crit Care Med. 2010;181(5):486–493. doi: 10.1164/rccm.200909-1364OC. [DOI] [PubMed] [Google Scholar]
- 54.Moya-Horno I, Viteri S, Karachaliou N, Rosell R. Combination of immunotherapy with targeted therapies in advanced non-small cell lung cancer (NSCLC) Ther Adv Med Oncol. 2018;10 doi: 10.1177/1758834017745012. 1758834017745012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spigel DR, Reynolds C, Waterhouse D, et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation – positive advanced non-small cell lung cancer (CheckMate 370) J Thorac Oncol. 2018;13(5):682–688. doi: 10.1016/j.jtho.2018.02.022. [DOI] [PubMed] [Google Scholar]
- 56.Lin JJ, Zhu VW, Yoda S, et al. Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. J Clin Oncol. 2018;36(12):1199–1206. doi: 10.1200/JCO.2017.76.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]