

Abstract
Background:
Neonatal candidiasis causes significant morbidity and mortality in high risk infants. The micafungin dosage regimen of 10 mg/kg established for the treatment of neonatal candidiasis is based on a laboratory animal model of neonatal hematogenous Candida meningoencephalitis and pharmacokinetic (PK)–pharmacodynamic (PD) bridging studies. However, little is known about the how these PK–PD data translate clinically.
Methods:
Micafungin plasma concentrations from infants were used to construct a population PK model using Pmetrics software. Bayesian posterior estimates for infants with invasive candidiasis were used to evaluate the relationship between drug exposure and mycologic response using logistic regression.
Results:
Sixty-four infants 3–119 days of age were included, of which 29 (45%) infants had invasive candidiasis. A 2-compartment PK model fits the data well. Allometric scaling was applied to clearance and volume normalized to the mean population weight (kg). The mean (standard deviation) estimates for clearance and volume in the central compartment were 0.07 (0.05) L/h/1.8 kg and 0.61 (0.53) L/1.8 kg, respectively. No relationship between average daily area under concentration–time curve or average daily area under concentration–time curve:minimum inhibitory concentration ratio and mycologic response was demonstrated (P > 0.05). Although not statistically significant, mycologic response was numerically higher when area under concentration–time curves were at or above the PD target.
Conclusions:
While a significant exposure–response relationship was not found, PK–PD experiments support higher exposures of micafungin in infants with invasive candidiasis. More patients would clarify this relationship; however, low incidence deters the feasibility of these studies.
Keywords: micafungin, neonates, infants, invasive candidiasis, pharmacokinetics-pharmacodynamics
Disseminated candidiasis results in significant morbidity and mortality in premature infants.1–3 The mortality associated with invasive candidiasis (IC) is 3 times higher than that of uninfected infants of similar gestational age and birth weight.4 The involvement of the central nervous system (CNS) is especially detrimental for subsequent neurodevelopmental outcomes and mandates meticulous attention to the selection of safe and effective antifungal regimens.5–7
There is a large safety and efficacy database that supports the role of micafungin for the treatment of IC in both adults and children.8–11 Micafungin has broad-spectrum anti-Candida coverage and a favorable safety profile.12,13 Micafungin demonstrates linear pharmacokinetics (PK); dose adjustment is not required in the setting of renal and/or hepatic impairment; and therapeutic drug monitoring is not required in any patient populations.14
The pharmacodynamics (PD) of micafungin for the treatment of hematogenous Candida meningoencephalitis has been explored in a well-characterized experimental model.15 PK–PD bridging studies suggest that a neonatal regimen of 10 mg/kg is required for effective treatment of Candida infections.15,16 These studies were used to justify the choice of this high-dose regimen for treatment of infants with IC. Unfortunately, however, inherent difficulties in conducting neonatal trials and the general decrease in the incidence of neonatal candidiasis resulted in early termination of one of the studies after the enrollment of only 30 infants with the primary and safety results reported elsewhere.17
Ideally, predictions from preclinical models should be tested clinically to prospectively validate the PK–PD relationships. Establishing preclinical-to-clinical linkages are increasingly viewed as fundamental for the establishment of effective antimicrobial therapies. In this study, we examined the clinical relevance of reaching the PD target established in preclinical models. We combined micafungin plasma concentrations from 4 neonatal clinical trials to construct a population PK (PPK) model and then explored whether increasing micafungin drug exposures resulted in improved clinical outcomes in infants with IC.
METHODS
Study Design
Micafungin plasma concentrations from 4 pediatric clinical trials were available for PPK modeling. Each study has been described in detail elsewhere.16–20 Briefly, the dataset consists of 2 PK and safety studies with dosages ranging from 0.75 to 10 mg/kg and 2 efficacy, safety and PK studies with dosages ranging from 2 to 10 mg/kg/d. The local Institutional Review Board/Ethics Committee at each site approved the studies, and parental consent was obtained for each infant before initiation of study procedures within each study. Plasma sampling is described in the respective publications. The micafungin concentrations were analyzed using high performance liquid chromatography, as detailed in the individual publications.16,18,19 Estimated gestational age was not available for all clinical trials so we were unable to incorporate this variable in the analysis or to use it to calculate other age descriptions such as post-menstrual age.
PPK Modeling
A PPK model was constructed using the PPK program Pmetrics (v1.5.1; University of Southern California, Los Angeles, CA).21 The observations were weighted by the inverse of the estimated assay variance. Initial parameter estimates were anchored on 2 prior PPK models. The first study included 47 infants <4 months which used prior knowledge from adult PPK modeling and standardized estimates of clearance and volume in the central compartment to a 70-kg adult weight.22 The second study utilized 293 pediatric patients including the 64 infants in the present analysis.23 This PPK model normalized the clearance and volume to the mean weight of the 293 pediatric patients based on prior knowledge from previous pediatric PPK analysis.23 The structural model included allometric scaling of clearance, as previously described.21 Clearance and volume were also standardized to the mean body weight of the population (1.8 kg). The differential equations for the final structural model with allometric scaling are as follows:
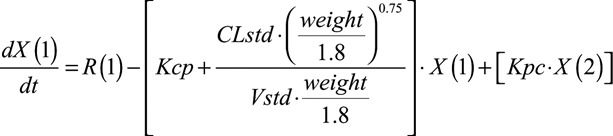 |
(1) |
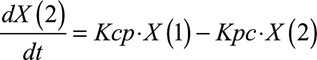 |
(2) |
where CLstd and Vstd represent the normalized clearance and volume values using the mean body weight of the population, R represents the infusion of micafungin into compartment 1, and Kcp and Kpc represent the rate of drug transfer to and from the central (compartment 1) and peripheral (compartment 2) compartments, respectively.
The fit of the model to the data was evaluated by visual inspection of the observed versus predicted concentrations before and after the Bayesian step and the coefficient of determination (r2) from the linear regression of the observed versus predicted values. In addition, the estimates for bias (mean weighted error), imprecision (adjusted mean weighted squared error), objective function and log likelihood were assessed. Non-compartmental analyses (NCA) were conducted for the infants with 4 or more samples in a 24-hour period to generate non-compartmental analysis area under concentration–time curve (AUC0–24) and compared with the population model-predicted AUC0–24.
Bayesian posterior estimates from the final model were used to estimate AUC for each patient for the entire dosing interval. The average daily AUC was then determined by dividing the total AUC for the treatment course by the number of days of micafungin therapy. Using daily average AUC avoids the issue of having to define what time in the course of therapy that AUC is important for efficacy (eg, AUC at the end of dosing or on a specific day during therapy).
Exposure–Response Analysis
A subset of 29 infants who received micafungin for the treatment of proven IC was used for the exposure–response analysis. Mycologic response was used as an outcome measure. Successful mycologic response was defined as eradication (documented by negative fungal cultures) through 1 week after the receipt of the last dose of micafungin. In most patients, after documentation of eradication during therapy, follow-up cultures were sparse or not performed because of lack of clinical need (ie, a successful response). Therefore, an additional part of the definition of “successful response” was that no new antifungal therapy was required after completion of micafungin. Failure (persistence of infection) was defined as continued positive cultures, or in the absence of repeat cultures, there was a requirement to switch to an alternative antifungal therapy for further treatment. Survival reported anytime during the course of the study was used in the analysis.
The relationship of average daily AUC (AUCave) and AUCave:minimum inhibitory concentration (MIC) to mycologic response (binary data) was analyzed using logistic regression in SAS (version 9.3; SAS Institute Inc., Cary, NC). The logit function was used without covariates illustrated by the following equation:
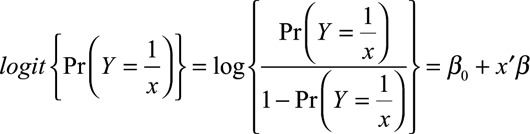 |
where Y is response, β0 is the intercept parameter, and β is the vector of slope parameters.
The exposure parameters were added in an automated stepwise approach with α = 0.3 for model inclusion and α = 0.05 for model retention. Additional statistical comparisons were performed in MYSTAT 12 (version 12.02, http://www.systat.com).
Attainment of Pharmacodynamic Targets
Drug exposures from the 29 infants with IC were used to verify if the 10 mg/kg dosage ensured attainment of the PD target (AUC and AUC:MIC ratio) representing the near-maximal effect determined in the in vivo rabbit model of Candida meningoencephalitis15,22 and to assess if achieving this target improved survival and/or mycologic response.
RESULTS
Study Population for PPK
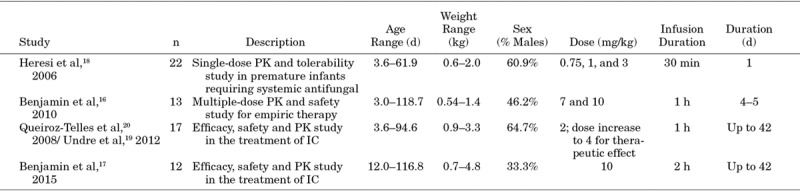
A summary of the demographics of patients enrolled included from the 4 studies is provided in Table 1. Four micafungin clinical trials with a combined total of 64 infants 3–119 days of age were available. There were slightly more males than females (n = 35 males, 55%), the mean (standard deviation [SD]) age was 35 days (27 days), and the mean (SD; range) weight was 1.8 kg (1.1 kg; 0.5–4.8 kg). The treatment duration ranged from 1 to 34 days, with a mean (SD) of 6.8 days (7.9 days).
TABLE 1.
Description of Micafungin Pediatric Studies Included in the PPK Model
PPK Model
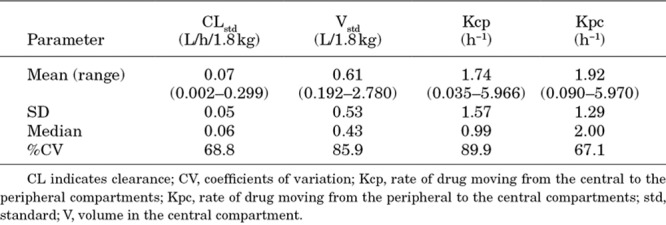
A total of 287 micafungin concentrations from 64 infants were retrieved for the PPK analysis. Forty infants had multiple samples over a 24-hour dosing interval with an average of 5 samples each (range 3–9). A description of the 4 studies is included in Table 1. A 2-compartment model with allometric scaling fits the data well. A visual inspection of the observed versus predicted concentrations after the Bayesian step was acceptable with a coefficient of determination (r2) of 0.945 (Fig. 1) using the median parameter values. Similar results were observed for the mean posterior predicted values (r2 = 0.941 for the linear regression of the observed versus predicted values). Estimates of bias and imprecision were also acceptable (−0.126 and 0.902, respectively). The mean parameter estimates are included in Table 2. The mean (SD) clearance and volume in the central compartment were 0.07 (0.05) L/h/1.8 kg and 0.61 (0.53) L/1.8 kg, respectively.
FIGURE 1.
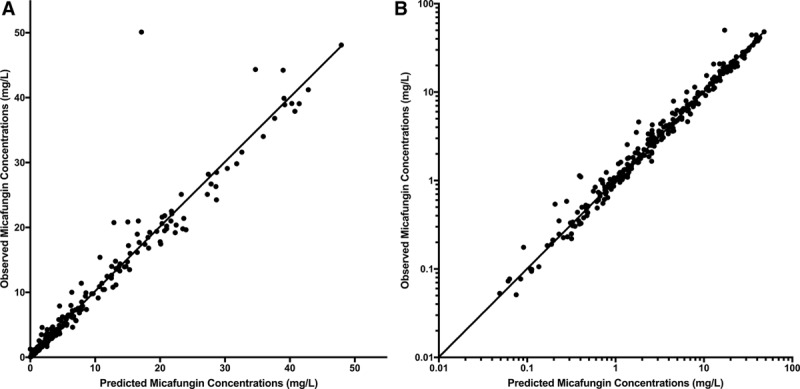
Observed versus posterior predicted concentrations (mg/L) from the final model after the Bayesian step on a linear scale (A; r2 = 0.945; slope = 0.995; 95% confidence interval [CI]: 0.967–1.02; intercept = 0.24; 95% CI: −0.104 to 0.584) and on a log scale (B; r2 = 0.947; slope = 0.946 95% CI: 0.92–0.972; intercept = 0.0496; 95% CI: 0.0284–0.0709). Dotted line is line of unity where observed concentrations equal predicted concentrations.
TABLE 2.
Mean, Medians, SD and %CV for the Parameter Estimates
Thirty-seven infants had 4 or more samples in the 24-hour period. AUC0–24 calculated using non-compartmental analysis analyses for these 37 subjects were compared with AUC0–24 calculated from the model predictions. Spearman correlation rank order test demonstrated that the results were not significantly different (S = 140; P value < 2.2e-16; ρ = 0.983).
Exposure–Response Population and Analysis
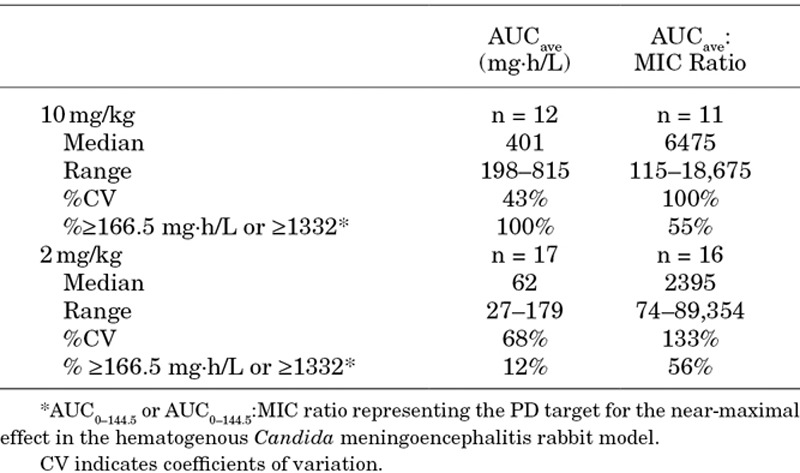
Twenty-nine infants ranging in age from 4 to 117 days received micafungin for the treatment of proven IC or candidemia; 17 infants received a dose of 2 mg/kg in a trial comparing micafungin to liposomal amphotericin B20 and 12 infants received a dose of 10 mg/kg in a trial comparing micafungin to conventional amphotericin B.17 The median (range) body weight of the 29 infants was 1.95 kg (0.68–4.85 kg). The median (range) treatment duration was 14 days (1–34 days). MIC values from the 2 mg/kg and 10 mg/kg studies were available for all but 2 infants with values ranging from 0.004 to 2 mg/L and 0.03 to 2 mg/L, respectively. All but 3 patients had candidemia. The other 3 patients had proven infections in the urinary tract (n = 2) and disseminated disease (n = 1; eye, cerebrospinal fluid, blood). Seventy-six percent and 92% of infants receiving 2 and 10 mg/kg survived, respectively. Of the 5 patients who died, 3 deaths occurred in the first week of starting micafungin (2 at 2 mg/kg and 1 at 10 mg/kg), 1 infant (2 mg/kg) died during the first week after the last dose of micafungin and 2 infants (2 mg/kg) died within the 30 days after the last dose of study drug. Successful mycologic response was achieved in 76% and 83% of patients receiving 2 and 10 mg/kg, respectively. Both infants with urinary tract infections administered 10 mg/kg/d had a successful mycologic response. The infant with a disseminated infection (2 mg/kg/d) failed mycologically; however, all 3 survived. Table 3 summarizes the exposure estimates (AUCave and AUCave:MIC) for the 2 and 10 mg/kg dosages from each study.
TABLE 3.
Estimates of AUCave and AUCave:MIC for Infants Less Than 4 Months of Age with IC Treated with Micafungin at doses of 2 mg/kg (n = 17) and 10 mg/kg (n = 12)
The CNS PD target AUC for near-maximal effect that was demonstrated in the rabbit model of hematogenous Candida meningoencephalitis was approximately 166.5 mg·h/L.15,22 All infants treated at a dose of 10 mg/kg achieved the CNS PD target, while only 2 (12%) of the infants receiving 2 mg/kg achieved the CNS PD target (Table 4). Successful mycologic response was achieved by 86% of patients who reached the PD target when compared with 73% of patients who did not meet the PD target, but this difference was not statistically significant (P = 0.396). Of those infants reaching the AUC:MIC ratio PD target for near-maximal effect of 1332, successful mycologic response was achieved by 73% and 83% of those who reached and did not reach the target, respectively. There was no clear relationship between mycologic response (success or failure) and either AUCave and AUCave:MIC when examined using logistic regression (P > 0.3). Figure 2A and 2B illustrates the similarity in the drug exposure measures for those patients with successful and failure of mycologic response.
TABLE 4.
MIC Values and Treatment Response by Dose Groups and by Patients Who Did or Did not Achieved the CNS PD Target
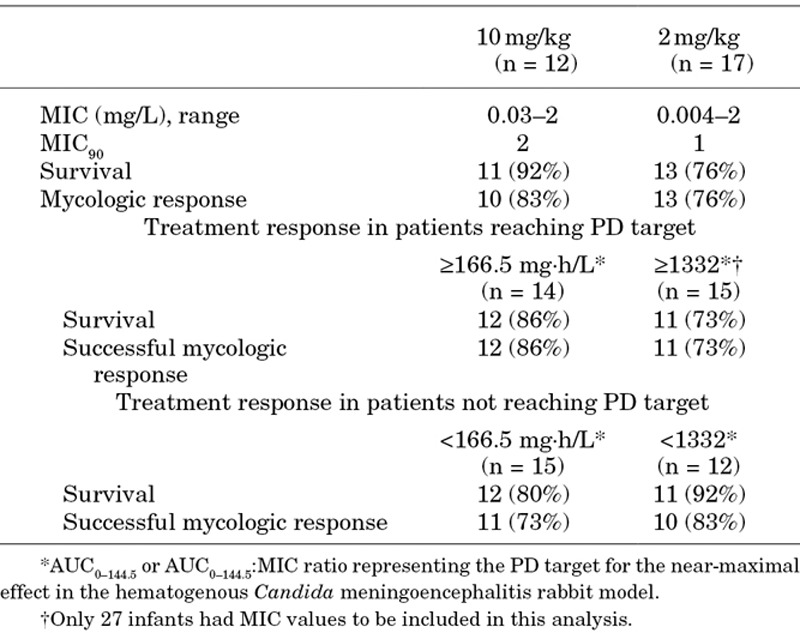
FIGURE 2.
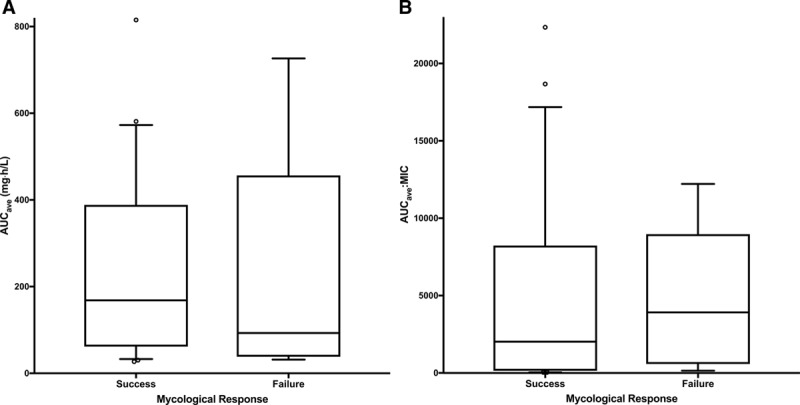
Boxplot illustrating the relationship between mycologic response and AUCave (A) and AUCave:MIC (B). (The box represents the interquartile range of AUCave and AUCave:MIC, respectively, with the median displayed as the band inside the box, the lines extending from the boxes represent the overall range of values.)
DISCUSSION
In the current PPK analysis, robust estimates of drug exposures for individual patients were obtained. However, no statistically significant relationship between drug exposure and treatment outcomes was demonstrated. There are several potential reasons for this observation. First, the number of infants (n = 29) was small, and therefore, the study lacked adequate power to detect a difference despite the numerically better response rate in the group with higher exposures (86% versus 73%; P = 0.396). Assuming the mycologic responses rates of 86% and 73% from the 2 groups, the study would require a sample size of approximately 142 to yield at least 80% power (2-sided, 5% significance level). Second, there is extreme heterogeneity in the clinical characteristics of neonates and young infants with many factors that are extraneous to the infection that potentially confound outcome measures (eg, gestational age, birth weight and other concomitant comorbidities). Third, there is significant heterogeneity in the clinical presentation and prognosis of Candida infections in this population. Disease may range from simple colonization to dissemination and devastating involvement of the brain. It may be possible to stratify patients to more effectively account for the heterogeneity; however, there simply are not adequate laboratory and clinical tools to do this accurately.
Outcome measures used in these studies have a number of inherent limitations. Previous studies have demonstrated that infants who survive IC have poorer neurodevelopment outcomes compared with those without IC regardless of the presence of candidemia or confirmed CNS infection.7 The extent of correlation between short-term outcome measures and longer-term neurodevelopment outcomes is not known. The demonstration of negative fungal cultures is central to definitions of disease resolution. However, the lack of sensitivity of fungal cultures (<50%)24 impairs the clinical utility of this metric. Clinical signs and symptoms are neither sensitive nor specific enough to assess therapeutic response.25 The strongest predictor of septicemia in 1 study was hypotension, present in less than 5% of the infants, which had only a 31% positive predictive value.26 All-cause mortality is obviously an important endpoint, but is invariably confounded by comorbidities that may overwhelm the signal coming from the drug–pathogen interaction. The use biomarkers such as (1→3)-β–D-glucan could aid in the objective assessment of the response to therapy.27,28 These data and others suggest that fungal biomarkers be included for the assessment of therapeutic response in future clinical trials.29
A further problem resides in substantial difficulties in conducting clinical trials in this patient population. Enrollment in the micafungin clinical trials was extremely slow. For example, a recently terminated study enrolled only 30 of the 225 planned patients in two and a half years.17 Reasons for such slow recruitment may be related to the decreased incidence of IC in this population.30 A recent study reported in 2014 that the annual US incidence of IC decreased dramatically.30 The terminated study provides a recent example of the difficulty of conducting these studies.17 This study had 70 sites from 23 countries available to screen patients. Only half of the sites found appropriate infants to screen and only 22% of them were able to enroll at least 1 infant.
Clinical trials may not be the most efficient way to identify safe and effective regimens for relatively rare fungal infections. Preclinical-to-clinical bridging studies represent one of the few ways regimens can be de-risked for clinical study. Importantly, however, there is relatively little experience with this approach and certainly no consensus on the type of studies that are required. We have recently reflected and summarized some of the necessary factors in a PK–PD package for the development of new antifungal agents in adults.31 The same exercise now needs to be performed for infants. This topic was discussed in detail at a recent Food and Drug Administration workshop (http://www.fda.gov/Drugs/NewsEvents/ucm507958.htm). Some important considerations include (1) using preclinical models that are a faithful mimic of human neonatal disease. Such an approach enables pharmacodynamic idiosyncrasies in infants to be examined (2) using neonatal strains and studying more than 1 strain; (3) cross-validating findings in several experimental model systems; (4) using compounds for which there is an established regimen and indication to enable the outcomes from new agents to be benchmarked; (5) setting up experimental models that produce “on scale” readouts. Model behavior is governed by the chosen experimental conditions, such as strain, inoculum, background immunosuppression, delay in initiation of treatment and treatment duration. The key idea is that clinically relevant exposures of a positive control should induce a response in the middle of the drug exposure–response relationship.
Clinical data remain central to establishing safe and effective anti-infective regimens for neonates. A toxicodynamic relationship was not established in the current dataset, which was too small to enable meaningful analyses. Clear relationships between drug exposure and toxicity have not been established for the echinocandins in any clinical context. The safety of neonatal dosages as high 15 mg/kg is published elsewhere.16,17,32,33
Regarding the PPK model employed for the current analysis, we used a previously described PPK model for micafungin, which applied an exponent of 0.75 on clearance estimates. A recent report suggests that allometric scaling for neonatal data may be closer to 1.34 To check our results, we performed a sensitivity analysis using 1 as the allometric scaling exponent. However, the resulting estimates for clearance and AUCave did not differ from the model described herein.
In conclusion, we could not establish a statistically significant relationship between micafungin exposure and the clinical outcome of neonatal candidiasis. However, prior PK–PD studies suggest that relatively high drug exposures are required for the effective treatment of CNS candidiasis. Without larger numbers of patients and more accurate clinical outcome measures, there will be persistent problems in establishing direct preclinical-to-clinical linkages. At the present time carefully designed experimental programs coupled with PK–PD bridging studies provide the best way to develop new drugs for premature infants.
Footnotes
This work was supported by Astellas Pharma Inc.
L.L.K., A.V.D., P.L.B. and A.K. were employed by Astellas Pharma Global Development, Inc. during the conduct of the study and outside the submitted work. T.J.W. reports participation on advisory boards for Astellas and grants from Astellas, outside the submitted work. D.K.B. reports personal fees from Allergan, Astellas, Cempra Pharmaceuticals, Shionogi, Inc., and The Medicines Company, outside the submitted work. A.A. reports grants from Astellas during the conduct of the study and grants from Merck and Pfizer, outside the submitted work. P.B.S. reports personal fees as a consultant for Astellas, outside the submitted work. P.M. reports personal fees as a consultant for Astellas during the conduct of the study. W.W.H. reports grants from Pfizer, personal fees from Basilea and grants and personal fees from Amplyx, Astellas and F2G, outside the submitted work. The other author has no conflicts of interest to disclose.
REFERENCES
- 1.Manzoni P, Mostert M, Castagnola E.Update on the management of Candida infections in preterm neonates. Arch Dis Child Fetal Neonatal Ed. 2015;100:F454–F459.. [DOI] [PubMed] [Google Scholar]
- 2.Arsenault AB, Bliss JM.Neonatal candidiasis: new insights into an old problem at a unique host-pathogen interface. Curr Fungal Infect Rep. 2015;9:246–252.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith PB, Steinbach WJ, Benjamin DK., JrNeonatal candidiasis. Infect Dis Clin North Am. 2005;19:603–615.. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin DK, DeLong E, Cotten CM, et al. Mortality following blood culture in premature infants: increased with Gram-negative bacteremia and candidemia, but not Gram-positive bacteremia. J Perinatol. 2004;24:175–180.. [DOI] [PubMed] [Google Scholar]
- 5.Watt KM, Cohen-Wolkowiez M, Ward RM, et al. Commentary: pediatric antifungal drug development: lessons learned and recommendations for the future. Pediatr Infect Dis J. 2012;31:635–637.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoll BJ, Hansen NI, Adams-Chapman I, et al. National Institute of Child Health and Human Development Neonatal Research Network. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA. 2004;292:2357–2365.. [DOI] [PubMed] [Google Scholar]
- 7.Benjamin DK, Jr, Stoll BJ, Fanaroff AA, et al. National Institute of Child Health and Human Development Neonatal Research Network. Neonatal candidiasis among extremely low birth weight infants: risk factors, mortality rates, and neurodevelopmental outcomes at 18 to 22 months. Pediatrics. 2006;117:84–92.. [DOI] [PubMed] [Google Scholar]
- 8.Pappas P, Kauffman C, Andes D, et al. Clinical practice guideline for the management of Candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. [published online ahead of print February 15, 2016]. doi: 10.1093/cid/civ933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ullmann AJ, Akova M, Herbrecht R, et al. ESCMID Fungal Infection Study Group. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: adults with haematological malignancies and after haematopoietic stem cell transplantation (HCT). Clin Microbiol Infect. 2012;18(suppl 7):53–67.. [DOI] [PubMed] [Google Scholar]
- 10.Cornely OA, Bassetti M, Calandra T, et al. ESCMID Fungal Infection Study Group. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: non-neutropenic adult patients. Clin Microbiol Infect. 2012;18(suppl 7):19–37.. [DOI] [PubMed] [Google Scholar]
- 11.Hope WW, Castagnola E, Groll AH, et al. ESCMID Fungal Infection Study Group. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: prevention and management of invasive infections in neonates and children caused by Candida spp. Clin Microbiol Infect. 2012;18(suppl 7):38–52.. [DOI] [PubMed] [Google Scholar]
- 12.Cornely OA, Pappas PG, Young JA, et al. Accumulated safety data of micafungin in therapy and prophylaxis in fungal diseases. Expert Opin Drug Saf. 2011;10:171–183.. [DOI] [PubMed] [Google Scholar]
- 13.Arrieta AC, Maddison P, Groll AH.Safety of micafungin in pediatric clinical trials. Pediatr Infect Dis J. 2011;30:e97–e102.. [DOI] [PubMed] [Google Scholar]
- 14.Astellas Pharma US Inc. MYCAMINE® (micafungin sodium) prescribing information. Available at: https://www.mycamine.com. August 2016.
- 15.Hope WW, Mickiene D, Petraitis V, et al. The pharmacokinetics and pharmacodynamics of micafungin in experimental hematogenous Candida meningoencephalitis: implications for echinocandin therapy in neonates. J Infect Dis. 2008;197:163–171.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benjamin DK, Jr, Smith PB, Arrieta A, et al. Safety and pharmacokinetics of repeat-dose micafungin in young infants. Clin Pharmacol Ther. 2010;87:93–99.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benjamin D, Jr, Kaufman D, Hope W, et al. Micafungin Versus Conventional Amphotericin B in the Treatment of Invasive Candidiasis in Infants, Abstr Interscience Conference on Antimicrobial Agents and Chemotherapy. 2015San Diego, CA; [Google Scholar]
- 18.Heresi GP, Gerstmann DR, Reed MD, et al. The pharmacokinetics and safety of micafungin, a novel echinocandin, in premature infants. Pediatr Infect Dis J. 2006;25:1110–1115.. [DOI] [PubMed] [Google Scholar]
- 19.Undre NA, Stevenson P, Freire A, et al. Pharmacokinetics of micafungin in pediatric patients with invasive candidiasis and candidemia. Pediatr Infect Dis J. 2012;31:630–632.. [DOI] [PubMed] [Google Scholar]
- 20.Queiroz-Telles F, Berezin E, Leverger G, et al. Micafungin Invasive Candidiasis Study Group. Micafungin versus liposomal amphotericin B for pediatric patients with invasive candidiasis: substudy of a randomized double-blind trial. Pediatr Infect Dis J. 2008;27:820–826.. [DOI] [PubMed] [Google Scholar]
- 21.Neely MN, van Guilder MG, Yamada WM, et al. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit. 2012;34:467–476.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hope WW, Smith PB, Arrieta A, et al. Population pharmacokinetics of micafungin in neonates and young infants. Antimicrob Agents Chemother. 2010;54:2633–2637.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hope WW, Kaibara A, Roy M, et al. Population pharmacokinetics of micafungin and its metabolites M1 and M5 in children and adolescents. Antimicrob Agents Chemother. 2015;59:905–913.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berenguer J, Buck M, Witebsky F, et al. Lysis-centrifugation blood cultures in the detection of tissue-proven invasive candidiasis. Disseminated versus single-organ infection. Diagn Microbiol Infect Dis. 1993;17:103–109.. [DOI] [PubMed] [Google Scholar]
- 25.Kelly MS, Benjamin DK, Jr, Smith PB.The epidemiology and diagnosis of invasive candidiasis among premature infants. Clin Perinatol. 2015;42:105–117, viii.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fanaroff AA, Korones SB, Wright LL, et al. Incidence, presenting features, risk factors and significance of late onset septicemia in very low birth weight infants. The National Institute of Child Health and Human Development Neonatal Research Network. Pediatr Infect Dis J. 1998;17:593–598.. [DOI] [PubMed] [Google Scholar]
- 27.Salvatore CM, Chen TK, Toussi SS, et al. (1→3)-β-d-Glucan in cerebrospinal fluid as a biomarker for Candida and Aspergillus infections of the central nervous system in pediatric patients. J Pediatric Infect Dis Soc. 2016;5:277–286.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salvatore C, Petraitiene R, Sitaras L, et al. Prospective study and analytical performance of serum (1->3)-β-D-glucan in pediatric patients (Abstract Poster #1570). 2016New Orleans, LA: IDWeek; [Google Scholar]
- 29.Benjamin DK, Jr, Stoll BJ, Gantz MG, et al. Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Neonatal candidiasis: epidemiology, risk factors, and clinical judgment. Pediatrics. 2010;126:e865–e873.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aliaga S, Clark RH, Laughon M, et al. Changes in the incidence of candidiasis in neonatal intensive care units. Pediatrics. 2014;133:236–242.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hope W, Drusano GL, Rex JH.Pharmacodynamics for antifungal drug development: an approach for acceleration, risk minimization and demonstration of causality. J Antimicrob Chemother. 2016;71:3008–3019.. [DOI] [PubMed] [Google Scholar]
- 32.Smith PB, Walsh TJ, Hope W, et al. Pharmacokinetics of an elevated dosage of micafungin in premature neonates. Pediatr Infect Dis J. 2009;28:412–415.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Auriti C, Falcone M, Ronchetti MP, et al. High-dose micafungin for preterm neonates and infants with invasive and central nervous system Candidiasis. Antimicrob Agents Chemother. 2016;60:7333–7339.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calvier EA, Krekels EH, Välitalo PA, et al. Allometric scaling of clearance in paediatric patients: when does the magic of 0.75 fade? Clin Pharmacokinet. 2017;56:273–285.. [DOI] [PMC free article] [PubMed] [Google Scholar]