

Abstract
Human epidermal growth factor receptor 2 (HER2) is a validated breast cancer drug target for small molecule inhibitors that target the ATP-binding pocket of the kinase domain. In this work, a large-scale virtual screen was performed to a novel homology model of HER2, in a hypothesized “fully active” state, that considered water-mediated interactions during the prioritization of compounds for experimental testing. This screen led to the identification of a new inhibitor with micro molar affinity and potency (Kd = 7.0 μM, IC50 = 4.6 μM). Accompanying molecular dynamics simulations showed that inhibitor binding likely involves water coordination through an important water-mediated network previously identified in our laboratory. The predicted binding geometry also showed a remarkable overlap with the crystallographic poses for two previously reported inhibitors of the related Chk1 kinase. Concurrent with the HER2 studies, we developed formalized computational protocols that leverage solvated footprints (per-residue interaction maps that include bridging waters) to identify ligands that can “coordinate” or “displace” key binding site waters. Proof-of-concept screens targeting HIVPR and PARP1 demonstrate that molecules with high footprint overlap can be effectively identified in terms of their coordination or displacement patterns relative to a known reference. Overall, the procedures developed as a result of this study should be useful for researchers targeting HER2 and, more generally, for any protein in which the identification of compounds that exploit binding site waters is desirable.
Human epidermal growth factor receptor 2 (HER2)1,2 is an important transmembrane receptor tyrosine kinase in the epidermal growth factor receptor/ErbB family (EGFR/HER1/ErbB1, HER2/ErbB2, HER3/ErbB3, and HER4/ErbB4).3 ErbB receptors regulate processes such as cell proliferation, migration, angiogenesis, and apoptosis suppression.3−5 Deregulated activity of ErbB receptors is potentially oncogenic,4−6 which makes them promising drug targets. Several FDA-approved small-molecule inhibitors (e.g., gefitinib, erlotinib, lapatinib, and vandetanib)7−12 have been identified, which target HER2 and/or EGFR in the ATP-binding pocket of their intracellular kinase domains. However, despite the clinical utility of currently available HER2 inhibitors, considerable challenges for the field remain including loss of efficacy as a result of acquired drug resistance arising from continued use or, alternatively, the effects of somatic mutations, which can affect kinase activity or drug potency.13
The ErbB family is well-known for its conformational plasticity with regards to the kinase domain adopting different conformations or activation states.14,15 For example, based on crystallographic evidence, the approved drug erlotinib preferentially binds the “fully active” form of EGFR (PDB 1M17),16 while lapatinib targets the “CDK/Src-like inactive” form of EGFR (PDB 1XKK).17 An “active-like” state of HER2 has also been reported (PDB 3PP0).18 These different forms are typically defined based on the position of the αC helix, which leads to the presence or absence of a specific intramolecular salt bridge (K753-E770, equivalent to K745-E762 in EGFR), the side chain conformation of D863 (equivalent to D855 in EGFR) in the DFG-motif, and the conformation adopted by the A-loop (Figure 1).14,15 Readers should note that HER2 numbering is used through this manuscript.
Figure 1.

Ribbon representations based on crystallographic data showing EGFR in the fully active form (PDB 1M17, left), HER2 in the active-like form (PDB 3PP0, middle), and EGFR in the CDK/Src-like inactive form (PDB 1XKK, right). Key structural indicators that help define the three different states include the rotation of the αC helix, the A-loop, and D863 in the DFG-motif as highlighted. Residue labels based on HER2 numbering. K753, E770, and D863 are equivalent to K745, E762, and D855 in EGFR.
Importantly, previous studies19−22 have shown that different somatic point mutations (e.g., D769Y, G776V, G776C, and V777L) as well as insertion mutants (e.g., YVMA776–779 ins, VC777–778 ins, and GSP781–783 ins), although not directly in the ATP-binding pocket, can lead to constitutive or hyper-activation of the HER2 kinase domain. We hypothesize that since these mutants are activating, despite being distal from the ATP site, they would likely drive the kinase toward the “fully active” conformation. Accordingly, it would be reasonable to capture the underlying effects of the different mutants, without having to model each of them individually, by using a wild-type sequence but in the “fully active” state. By targeting this conformation, it may be possible to identify HER2 inhibitors that also inhibit some activating mutants. However, a crystal structure of HER2 in the fully active conformation has not yet been reported. A primary goal of this work was construction and refinement of a robust homology model of HER2 in the fully active form, based on the highly homologous EGFR protein (78% identity),23 for which crystallographic coordinates are available. The model was subsequently used to perform a large-scale virtual screen24−26 to identify and prioritize candidate inhibitors compatible with the fully active state for experimental testing.
Historically, one of the simplest scoring schemes used in virtual screens consists of intermolecular nonbonded steric (van der Waals, VDW) and Coulombic (electrostatic, ES) interaction energies. Alternatively, knowledge-based scoring functions can be used to identify compounds that make interaction patterns similar to that of a known ligand reference. Recent examples from our own work, which were also used here for the present project, include those implemented into the docking program DOCK6,27−29 based on footprint similarity,30,31 pharmacophore matching similarity,32 and Hungarian matching similarity,33 which can be used alone, or in combination.
In addition to direct interactions, interfacial water molecules often mediate (i.e., bridge)34−38 ligand binding through hydrogen bonds with the target, which affects affinity, specificity, and resistance.39−48 Water-mediated interaction appear to be particularly important for ligands binding to members of the ErbB family. For example, Balius et al.39 used molecular dynamics simulations, free energy calculations, and H-bond analysis to show that fold resistance trends for small-molecule inhibitors (erlotinib, gefitinib, and AEE788) that target the EGFR active form likely involve disruption of water-mediated interactions with nearby residues (T854, T790, and Q791). Similarly, for the inactive kinase form, Huang et al.47 used computer modeling to show that the specificity of lapatinib for EGFR > HER2 > ErbB4, and the effects of EGFR (C775F, T854A, and T790M) and HER2 (T790I) point mutations could be compellingly explained through water-pattern changes that were a direct result of the variations in protein sequence.
As outlined in this manuscript, the prioritization of the HER2 virtual screen results also included some rank-ordering procedures geared to capture the effects of bridging waters that were included during refinement of the kinase homology model. Concurrently, we explored different computational strategies to more systematically identify docked ligands that could incorporate the energetic contributions of water, which led to a more formalized docking protocol. From a general perspective, two strategies to exploit the effects of waters that mediate binding are to (1) “coordinate” and maintain an existing interaction that is enthalpically favorable36 or (2) “displace” and supplant an existing interaction, which may be entropically favorable due to water returning to bulk solvent.40,49−51 As an example, Figure 2 shows two different ligands binding to the protein scytalone dehydratase.50,51 In Figure 2a, a water is shown bridging interactions between the ligand BFS and two key residues Tyr22 and Tyr42. In Figure 2b, a rationally designed ligand called MQ0 displaces the same bridging water with a nitrile group that directly interacts with the same two residues.
Figure 2.

Examples of bridging water coordination (a, c) and displacement (b, d) in scytalone dehydratase (PDB 4STD and 3STD). In panels a and b, protein residues Y22 and Y42 are in gray, the coordinating ligand is in magenta, the displacing ligand is in cyan, and hydrogen bonding is shown as dashed lines. In panels c and d, the molecular footprints are shown where the y axis is the electrostatic (ES) interaction energy between the receptor and different species (coordinating ligand in magenta, coordinating ligand and bridging water in blue, displacing ligand in cyan). The x axis is the receptor sequence showing residues that make the most prominent ES interactions.
Importantly, molecular footprints,30,31 defined as per-residue interaction energy (VDW or ES) maps between two species, can be used to pinpoint which specific protein residues are involved in molecular recognition (qualitative information) and their approximate interaction magnitudes (quantitative information). We have previously leveraged footprint patterns to characterize and/or identify inhibitor binding to a variety of clinically relevant drug targets including HIVgp41,52−57 FABP,58,59 BoNT/A,60 and BoNT/E.61 As shown in Figure 2c, incorporating key waters into the footprints (termed solvated footprints) yields additional and potentially useful information. Here, the ES pattern made by the BFS ligand alone shows there are favorable interactions primarily with the receptor at position Asn123 and to a lesser extent Tyr42 (Figure 2c, magenta line). However, including the bridging water as part of the ligand when computing the footprint (Figure 2c, blue line) yields dramatically increased favorable interactions with Tyr22 and Tyr42 of ca. −2 to −2.5 kcal/mol, which, in this example, are important for binding.62 For the displacing case, a comparison of the footprint in Figure 2 (d, cyan; c, blue) shows that the residues previously engaged in the water bridge with ligand BFS now make direct favorable ES interactions with ligand MQ0 (blue vs cyan peaks at Tyr22 and Tyr42).
In the first part of this study, we present computational and experimental outcomes based on a large-scale virtual screen to a homology model of fully active HER2, which led to the successful identification of a compound with micro molar binding affinity and a predicted binding pose that coordinates a bridging water and resembles a previously identified inhibitor of the related Chk1 kinase. In the second part of this study, we outline and test a conceptually simple virtual screening protocol for the program DOCK6 that incorporates solvated molecular footprints. The protocol is based on the hypothesis that solvated footprint patterns can be used to identify compounds based on their footprint overlap30,31 to one of two references: (i) interaction patterns derived from ligands in a solvated binding site (coordination) or (ii) interaction patterns derived from ligands and water in a binding site without water (displacement). The protocol was tested and refined using two systems where binding site waters are known to be important: HIV-1 protease (HIVPR) and poly(ADP-ribose) polymerase 1 (PARP1). Given the key roles specific water molecules often play in molecular recognition, continued efforts to develop new computational methods that can effectively incorporate their effects into the structure-based design process is essential.
Methods Part 1: HER2 Virtual Screen
Virtual Screening Protocols
To date, no crystallographic structures of HER2 in the hypothesized fully active state14,15 have been reported. Therefore, we constructed a homology model of this form (see Supporting Information) for virtual screening based on EGFR, which is highly homologous to HER2 (78% identity).23 Structural analysis using the software package PROCHECK63 confirmed the overall quality of the model relative to the template (Table S1). A library of 1 929 663 commercially available organic compounds (ZINC12 database)64 was docked to the ATP-binding site on the HER2 model using our standard FLX docking protocol.27 The library was initially docked in grid space followed by energy-minimized in Cartesian space to enable footprint similarity scoring30,31 to be performed, as well as other properties (e.g., pharmacophore matching similarity,32 Hungarian matching similarity33) relative to the erlotinib reference (ligand + bridging waters). Following the screen, the 100 000 most favorable compounds (DOCK energy function) were clustered into families based on 2D structural similarity (Tanimoto cutoff 0.95, MOE program).65 Eight different scoring methods (energy-based and/or similarity-based), as implemented into DOCK6.8, were then used to uniquely rank-order the master list of the top 100 000 compounds in different ways to arrive at individually unique lists of 1000 “clusterheads” each (top-scoring family members). Prioritization of compounds for purchase and experimental testing was based on their scores within each of the seven rank-ordered lists, visualization of 3D binding geometries, and consideration of other drug-like properties (e.g., molecular weight, number of chiral centers, LogP).
Experimental Characterization of Binding Affinity
To assess the experimental affinity of compounds purchased based on the virtual screen to HER2, a competition binding assay was performed by the contract research organization DiscoverX (www.discoverx.com) using their scanELECT Kinase Selectivity and Profiling Assay Panel technology.66 Their HER2 assay67 quantifies the effect of candidate molecules on the amount of kinase in solution captured on an immobilized surface, compared to that of positive and negative (DMSO) controls, and reports a value termed “% control” for which zero indicates complete binding and 100 indicates no binding. The experiments evaluated candidate inhibitors at a concentration of 10 μM (n = 2). For comparison, the FDA-approved kinase inhibitors lapatinib and erlotinib were also tested but at a lower concentration of 1 μM.
For each of the test compounds with sufficiently low % controls values (<60.0), a dose–response curve was obtained by DiscoverX using their KdELECT Kinase Assay Panel technology to facilitate calculation of a binding constant (Kd). The experiments yielded an 11-point dose–response curve (n = 2 for each data point). For comparison, a dose–response curve for the FDA-approved inhibitor erlotinib was generated as a positive control.
Measurement of HER2 Inhibition
Compounds showing binding affinity for HER2 were also examined for their ability to inhibit kinase phosphorylation of the synthetic peptide substrate Poly(Glu, Tyr). The HER2 kinase assays were performed using radiolabeled [γ-32P] adenosine triphosphate (ATP) (Perkin-Elmer NEG002A100UC) with Poly(Glu, Tyr) (Sigma P0275) as a substrate as described by Sun and Budde.68 Purified HER2 kinase domain (see Supporting Information) was incubated with the compound of interest at various concentrations or dimethyl sulfoxide (DMSO) for 5 min at room temperature. Kinase reactions contained 50 nM HER2, 25 mM HEPES (pH 7.5), 0.2 μCi [γ-32P] ATP, 0.1 mM ATP, 1 mg/mL Poly(Glu, Tyr), 10 mM MgCl2, 10 mM MnCl2, 1 nM BSA, 0.5 mM DTT, and 0.5 mM activated sodium orthovanadate in 1% DMSO. After incubation at 30 °C for 20 min, 35 μL of each reaction was spotted onto a 2.5 cm × 2.5 cm square of Whatman 3MM paper. The paper squares were washed three times with 400 mL of warm (65 °C) 5% trichloroacetic acid (TCA), dried, and counted in a scintillation counter. IC50 values were calculated by nonlinear regression analysis of the percent activity.
Methods Part 2: Protocols to Coordinate or Displace Bridging Waters
Theoretical Overview
Concurrent with the HER2 studies, we evaluated more formalized virtual screening procedures to directly exploit the effects of bridging waters during compound prioritization. Two proteins (HIVPR and PARP1) with previously annotated bridging waters were used as test systems. As shown in Figure 3, from a virtual screening perspective, a bridging water can be either part of the receptor (termed here the COOR protocol for coordination) or part of the reference ligand (termed here the DISP protocol for displacement). In the COOR protocol, we hypothesize that using a molecular footprint of the cognate ligand in a solvated protein site (protein + bridging water) as a reference (termed the COOR reference) will enrich for compounds that make a similar interaction pattern to the reference and coordinate the bridging water (Figure 3, left). Alternatively, in the DISP protocol, we hypothesize that employing a molecular footprint of the solvated ligand (cognate ligand + bridging water) in an unsolvated protein site as a reference (termed the DISP reference) will enrich for compounds that mimic the combined interaction, which should therefore displace the bridging water (Figure 3, right).
Figure 3.

Workflows for two distinct virtual screen protocols to coordinate (COOR, left branch) or to displace (DISP, right branch) bridging water molecules.
Solvated Footprint Virtual Screening Details
We performed smaller-scale virtual screens of ∼500 000 molecules (equally sampled from the previous 2 M ZINC12 library) to the HIVPR and PARP1 test systems using the previously described docking and postprocessing protocol used for HER2, except that only molecular footprints (no other properties) of the candidate molecules were compared with references (COOR or DISP, see Supporting Information) using the footprint similarity score.30 For these calculations, scoring employed the ES footprint component (FPSES), which appeared to capture hydrogen bonding signatures of water more explicitly than did use of the VDW footprint components or the sum of both terms. The coordinating or displacing candidates were subsequently selected from among 1000 clusterheads only if their ES interaction values at the key residues are sufficiently close to those of the reference.
For these studies, we quantitatively define two interaction energy values E1 and E2 sufficiently close to each other as follows: E1/E2 ≥ 0.5. In the COOR context (eq 1), the ES interaction energy between a docked candidate and the water is labeled Ecan-wat and that for the reference (ligand alone) with the bridging water is labeled Eref-wat. A docked candidate is classified as “coordinating” if the ratio is greater than or equal to 0.5.
| 1 |
Similarly, in the DISP context (eq 2), the ES interaction energy between a docked candidate and a protein residue Rn is labeled Ecan-Rn and that for the reference (ligand and water) with Rn is labeled Eref-Rn.
| 2 |
Here, a docked candidate is classified as “displacing” if the relationship is satisfied for at least one Rn.
Results and Discussion
Results Part 1: Identification of HER2 Inhibitors Incorporating Bridging Waters
Virtual Screen Results
Following construction and refinement of the HER2 homology model (see Supporting Information), a library of ∼2 M purchasable molecules was docked to the kinase domain and reranked using eight different DOCK scoring methods and 3D visualization to prioritize compounds for experimental testing. Eight scoring functions were employed: (1) DCESUM (DOCK Cartesian energy, VDW + ES, kcal/mol), (2) FPSSUM (footprint similarity score, VDW + ES, Euclidian distance, kcal/mol), (3) FPSES (footprint similarity, ES pattern only, Euclidian distance, kcal/mol), (4) TS (total score = DCESUM + FPSSUM, kcal/mol), (5) FMS (pharmacophore matching similarity), (6) VOS (volume overlap similarity), (7) HMS (Hungarian matching similarity), and (8) FPSWGT (FPSES with four copies of WAT1 included in the reference to increase the contribution of water). Figure 4a shows the overall DOCK setup including protein (gray surface), DOCK anchor orientation sites (orange spheres), energy-grid bounding box (purple), and erlotinib reference (green surface). Figure 4b shows a close-up view of the MD frame selected from the refinement (see Methods section) highlighting the water network involving residues S783, T798, Q799, K860, T862, and the reference.
Figure 4.

(a) DOCK setup for HER2 showing protein in a gray surface, energy grid bounding box in purple, binding pocket spheres in yellow, and erlotinib in a green surface. (b) Close-up view of erlotinib, showing the water-mediated network. Atoms within H-bonding distance shown as dashed magenta lines.
Ultimately, 149 compounds were selected for experimental testing (ChemDiv vendor) as highlighted in Table 1 and Figure 5, and these were roughly evenly disrupted (N = 17–25) among the different functions except FPSWGT (N = 2). The use of unique functions helps to provide diversity and reduce overreliance on any one scoring method. Table 1 shows average scores for each of the eight groups along with molecular weight and number of ligand rotatable bonds. The diagonal of Table 1 highlights the self-consistency of the scoring approach. For example, the use of a given function yields the most favorable scores for that group relative to the other selection methods. Stated another way, the use of DCESUM yields the most favorable DCESUM scores (−70.3 kcal/mol) compared to any of the other seven methods. Similarly, the use of FPSSUM yields the lowest Euclidean distance for FPSSUM (6.2 kcal/mol) compared to the other groups (8.0–16.4 kcal/mol) and so forth (Table 1). As expected, the use of energy-dominated functions DCESUM and TS (DCESUM + FPSSUM) yields molecules with higher MWs (505–516 vs 408–463 g/mol) and greater numbers of ligand rotatable bonds (10.1–11.9 vs 7.2–8.4), which provides justification for using rank-ordering functions that reduce MW bias and increase diversity.
Table 1. Average Scores and Properties for 149 Small Molecules Selected from the Virtual Screen.
| lista | Nb | DCESUM | FPSSUM | FPSES | TS | FMS | VOS | HMS | MWc | RBd |
|---|---|---|---|---|---|---|---|---|---|---|
| DCESUM | 20 | –70.3 ± 1.1 | 16.4 ± 4.2 | 7.9 ± 3.0 | –53.9 ± 4.2 | 6.0 ± 3.3 | 0.3 ± 0.1 | 5.0 ± 1.8 | 516.1 ± 17.5 | 11.9 ± 1.5 |
| FPSSUM | 25 | –57.2 ± 1.2 | 6.2 ± 0.3 | 3.3 ± 0.4 | –50.9 ± 1.3 | 5.1 ± 0.4 | 0.5 ± 0.0 | 1.7 ± 1.2 | 419.2 ± 27.5 | 7.2 ± 2.0 |
| FPSES | 17 | –57.3 ± 1.3 | 8.0 ± 1.6 | 2.9 ± 0.2 | –49.3 ± 2.3 | 5.1 ± 0.3 | 0.4 ± 0.1 | 3.5 ± 1.5 | 459.4 ± 41.5 | 8.4 ± 2.3 |
| TS | 17 | –66.3 ± 1.3 | 9.4 ± 1.3 | 4.3 ± 0.7 | –57.0 ± 0.8 | 6.1 ± 3.6 | 0.4 ± 0.1 | 2.7 ± 1.0 | 504.5 ± 26.6 | 10.1 ± 1.8 |
| FMS | 23 | –58.4 ± 2.1 | 9.6 ± 2.7 | 4.9 ± 2.1 | –48.8 ± 2.4 | 4.1 ± 0.1 | 0.5 ± 0.1 | 2.8 ± 1.7 | 439.1 ± 34.9 | 8.1 ± 2.3 |
| VOS | 22 | –57.1 ± 1.1 | 8.3 ± 1.6 | 4.5 ± 1.4 | –48.8 ± 1.4 | 5.0 ± 0.4 | 0.6 ± 0.0 | 2.0 ± 1.2 | 408.0 ± 26.3 | 7.4 ± 1.3 |
| HMS | 23 | –57.3 ± 1.5 | 7.8 ± 1.5 | 4.0 ± 1.0 | –49.5 ± 2.2 | 5.7 ± 3.2 | 0.5 ± 0.0 | 0.0 ± 0.3 | 427.4 ± 30.9 | 7.5 ± 2.2 |
| FPSWGT | 2 | –56.6 ± 0.2 | 10.0 ± 0.9 | 4.2 ± 1.7 | –46.6 ± 0.7 | 4.5 ± 0.5 | 0.4 ± 0.1 | 3.0 ± 0.2 | 462.6 ± 48.2 | 8.0 ± 1.4 |
DCESUM (DOCK Cartesian energy, VDW + ES, kcal/mol), FPSSUM (VDW + ES footprint similarity, Euclidian distance, kcal/mol), FPSES (ES footprint similarity, Euclidian distance, kcal/mol), TS (total score = DCESUM + FPSSUM, kcal/mol), FMS (pharmacophore matching similarity), VOS (volume overlap similarity), HMS (Hungarian matching similarity), FPSWGT (FPSES with four copies of WAT1 in the reference to increase contribution of water).
N (number of molecules selected from each scoring method).
MW (molecular weight, g/mol).
RB (number of rotatable bonds).
Figure 5.

Overlap of the 149 purchased molecules with the erlotinib reference grouped by the primary scoring method used in prioritization. Values in parentheses indicate the number in each group. Erlotinib in a gray transparent surface, bridging water oxygens as red spheres. Functions employed include (1) DCESUM (DOCK Cartesian energy, VDW + ES, kcal/mol), (2) FPSSUM (VDW + ES footprint similarity, Euclidian distance, kcal/mol), (3) FPSES (ES footprint similarity, Euclidian distance, kcal/mol), (4) TS (total score = DCESUM + FPSSUM, kcal/mol), (5) FMS (pharmacophore matching similarity), (6) VOS (volume overlap similarity), (7) HMS (Hungarian matching similarity), and (8) FPSWGT (FPSES with four copies of WAT1 included in the reference to increase contribution of water).
In general, molecules selected using similarity-based functions (FPSSUM, FPSES, FMS, VOS, HMS, FPSWGT) tightly overlap with the reference volume (Figure 5). This is expected as similarity-based functions enrich for specific geometric features (FMS, VOS, HMS) or toward decomposed interaction features (FPSSUM, FPSES, FPSWGT) that are sensitive to the 3D geometry of the reference. Conversely, the two energy-dominated methods (DCESUM, total score) show less-tightly clustered poses that, in multiple instances, extend outside the volume occupied by the reference (Figure 5). The FPSSUM and FPSES groups also include examples of candidates that occlude water and thus could be considered displacing. The group labeled FPSWGT included four copies of WAT1 in the reference used with FPSES, which adds higher weighting to candidates that could displace water (possess functionality that overlaps water). In terms of coordination, visualization of the purchased candidates showed multiple DOCK poses in which ligand H-bond acceptors coordinated with WAT1 in a manner similar to the cognate ligand erlotinib as shown in Figure 6. The examples show two types of coordination involving aromatic ring nitrogens (left) or carbonyl oxygens (right).
Figure 6.
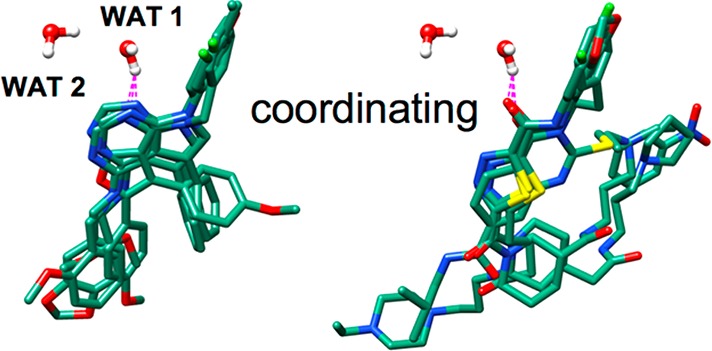
Representative examples of compounds from the virtual screen hypothesized to coordinate WAT1. Dashed magenta lines indicate atoms within H-bond distances.
Single-Concentration Binding Affinity and Inhibition
The initial experimental tests involved binding affinity and phosphorylation assays in which the 149 compounds were each tested at a single concentration of 10 μM (see Methods Part 1). For comparison, the known inhibitors lapatinib and erlotinib were tested at 1 μM. As shown in Figure 7a, encouragingly, seven of the tested compounds showed low % control scores (<60.0) in the scanELECT affinity assay (lower scores = higher affinity). Among the hits, ZINC01836093 had the best score of 30.5. The known inhibitors lapatinib and erlotinib showed 0 and 19 control scores, respectively. Importantly, in the companion kinase phosphorylation inhibition assay (Figure 7b), ZINC01836093 also inhibited 95.4% of HER2 kinase activity, which was close to the controls (lapatinib = 99.6%, erlotinib = 97.8%) and significantly stronger compared to the other six compounds, which confirms the results from scanELECT. The other six compounds similarly showed some inhibition (ranging from 14.6 to 55.8%) although the trend in the phosphorylation inhibition assay (Figure 7b) did not necessarily follow that of the binding affinity assay (Figure 7a).
Figure 7.

Activity for compounds with HER2 based on (a) the DiscoverX scanELECT affinity (n = 2) assay and the (b) phosphorylation inhibition assay (n = 3). Compounds tested at 10 μM; lapatinib and erlotinib tested at 1 μM. (c) Inhibition of Src by dasatinib and ZINC01836093. (d) 2D representations for compounds showing erlotinib in green box consensus scaffolds circled in orange or cyan.
To rule out the possibility that the activity of ZINC01836093 was due to the presence of PAINS (pan assay interference compounds)69 functionality or a result of colloidal aggregation,70 as reiterated in a recent editorial,71 a structural pattern search on the ZINC1572 Web site was performed. Importantly, ZINC01836093 does not appear to be similar to any previously reported PAINS or colloidal aggregators. As an additional test of specificity for the intended target, the activity of ZINC01836093 was tested using a similar enzyme activity assay against Src, a homologous nonreceptor tyrosine kinase. The activity of Src was almost completely inhibited by its selective inhibitor, dasatinib,73 but not by ZINC01836093 (Figure 7c), indicating selective inhibition of HER2. In terms of structure, Figure 7d shows 2D representations for the seven hits, relative to the erlotinib control (green box), along with the % control scores. Inspection reveals two groups of compounds with consensus scaffolds encompassing 5,6-diphenyl-furo[2,3-d]pyrimidin-4-amine (orange circle) or 8,9-diphenyl-furo[3,2-e][1,2,4]triazolo[1,5-c]pyrimidine (cyan circle) functionality. The seven compounds originated from four different rank-ordered lists: HMS = ZINC01836093, TS = ZINC13361691, VOS = ZINC02346332, and FPSSUM = ZINC02259460, ZINC02263039, ZINC02714631, and ZINC01833578.
Dose–Response Binding Affinity and Inhibition
The concentration-dependent activity of the seven hits for HER2 relative to erlotinib as a control were subsequently evaluated using the DiscoverX KdELECT assay (11-point dose–response curve). Among the compounds, only ZINC01836093 (the top-scoring hit) showed a clear dose–response behavior, which yielded a dissociation constant (Kd) = 7.0 μM (Figure 8a) within the range of concentrations tested. The control compound erlotinib similarly yielded a well-behaved dose–response curve with a Kd of 0.1 μM (Figure 8b). Importantly, the companion phosphorylation assays also yielded a clear dose–response inhibition for ZINC01836093 (Figure 8c) and erlotinib (Figure 8d) with IC50 values of 4.6 μM and 0.9 μM, respectively, which reassuringly confirms the binding affinity results. The lower Kd for erlotinib measured in the KdELECT assay (as compared to the kinase activity assay) is most likely due to the absence of ATP in the KdELECT measurement. HER2 inhibition experiments with varying concentrations of ATP and ZINC01836093 were consistent with a competitive mechanism of inhibition (Figure 9).
Figure 8.

Concentration-dependent activity for (a, b) ZINC01836093 and erlotinib with HER2 from the DiscoverX affinity assay (n = 2 each) and (c, d) the phosphorylation inhibition assay (erlotinib, n = 2; ZINC01836093, n = 7 for data points 0.5 μM and 1.0 μM, n = 4 for other points).
Figure 9.

HER2 radioactive kinase assays carried out with varying concentrations of ATP and ZINC01836093 and the results analyzed on a reciprocal plot.
In terms of hit rates, the present results for HER2 yield 4.7% (7/149) and 0.7% (1/149) for the single concentration and dose-dependent assays, respectively. For comparison, a recent study by Irwin and Shoichet74 showed widely varying hit rates of between 0.2% and 100% based on docking results taken from the literature across 53 systems. For kinases in particular, their analysis yielded DYRK1A (3.5%), CDK4 (5%), P38 MAPK (6%), MELK (19%), and PDK1 (20%). Finally, while the activity values for ZINC01836093 (Kd = 7.0 μM, IC50 = 4.6 μM) are less favorable than that of the approved drug erlotinib (Kd = 0.1 μM, IC50 = 0.9 μM) under the same conditions, they are reasonable for an early stage virtual screening hit. For comparison, the best hits obtained for the five kinases from the previous study74 ranged from 0.04 to 8 μM.
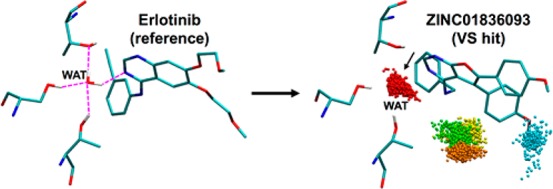
ZINC01836093 Binding Pose Suggests Water Coordination
From a computational perspective, the DOCK-predicted binding geometries for six of the seven hits appear within H-bonding distance of water molecule WAT1 that was incorporated into the footprint reference used during compound selection (Figure 4b) and thus binding could involve water coordination. Somewhat surprisingly, none of the active compounds appeared to displace WAT1. To examine in greater detail if the most promising hit, ZINC01836093, was likely to maintain its predicted coordination, the DOCK complex was subjected to short replicate MD simulations (three replicates with different random seeds), as shown in Figure 10, using the program AMBER16. The analysis confirmed stable coordination of the predicted water bridge as demonstrated by the tightly clustered ensemble of water oxygen atoms colored red (the most populated water cluster), positioned at the location of the expected site, from evenly spaced frames taken over the MD trajectories.
Figure 10.
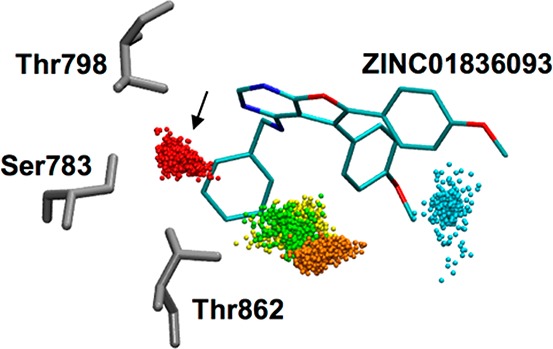
Solvent patterns for ZINC01836093 in HER2 derived from triplicate 20 ns MD ensembles (3 × 1000 frames each) color-coded by population (S1 red > S2 orange > S3 yellow > S4 green > S5 blue). ZINC01836093 in cyan with Ser783, Thr798, and Thr862 in gray. Water coordination prediction site indicated by black arrow.
At the suggestion of a reviewer, we also examined whether ZINC01836093 was compatible with the HER2 active-like conformation by redocking the compound to a model based on the X-ray structure of the HER2 active-like state originally complexed with ligand SYR127063 (PDB 3PP0).18 An overlay of the DOCK results, in their respective receptor conformations (fully active and active-like), showed predicted ligand poses that were highly similar (Hungarian RMSD 1.6 Å) suggesting the compound could bind to both forms. However, the DOCK interaction score (VDW + ES energies) in the active-like state was less favorable (−55.5 kcal/mol) than the fully active state (−60.8 kcal/mol), likely as a result of the more open binding site (outward rotation of the αC helix). Although the predicted poses suggest that both states would result in similar water coordination patterns, MD analysis of ZINC01836093 in the HER2 active-like state did not reveal a highly populated water site at the same spatial position as was observed in the fully active form (Figure 10, arrow). Taken together, the analysis suggests that ZINC01836093 is more compatible with, and thus would be expected to preferentially target, the fully active form.
Structural Similarity Search of ZINC01836093
To further investigate ZINC01836093 (Figure 11a), we used PubChem75 to perform a similarity search, which yielded 56 compounds with a Tanimoto coefficient of 95% or greater. Interestingly, ZINC01836093 has not been annotated as an inhibitor for any protein, although 10 of the 56 had been annotated as inhibitors for kinases other than HER2. One compound, in particular (ZINC02501415, Figure 11b), was reported by Foloppe et al.76 as an inhibitor of the Chk1 kinase, which has ∼24% kinase domain sequence identity23 with HER2. The same previous study also reported results for 14 analogues including one compound DF2 (Figure 11c) with a ca. 10-fold improvement in potency over ZINC02501415. Of particular interest for the present work is the fact that both ZINC02501415 and DF2 have been cocrystallized with Chk1 (PDB 2BR1 and 2BRO).76 Despite the relatively low sequence identity, an alignment of the Chk1 crystal structures with our HER2 homology model showed a good backbone overlap (Figure 11d) with Cα RMSDs of 1.070 and 1.034 Å, respectively (UCSF Chimera).77 Notably, as shown in Figure 11e,f, the X-ray poses for the two Chk1 inhibitors show a striking overlap (Hungarian RMSDs33 of 2.32 and 3.51 Å) with the DOCK-predicted binding pose for ZINC01836093 in HER2.
Figure 11.

Comparison of the newly identified HER2 inhibitor ZINC01836093 (a) with two Chk1 inhibitors (b, c) ZINC02501415 and DF2 from the PubChem similarity search. Structure overlay (d) of the HER2 (homology model, cyan) with the Chk1 kinase domain (crystal structure, gray) from PDB 2BR1. Binding pose comparison for ZINC01836093 with HER2 (cyan) vs ZINC02501415 (orange, e) or DF2 (pink, f). Water-mediated H-bonding for ZINC01836093 with HER2 (g) and a similar view for ZINC02501415 (h) or DF2 (i) with Chk1. H-bonding shown as dashed lines.
Intriguingly, although the two Chk1 inhibitors are similar with respect to 2D topology and the predicted binding geometry for ZINC01836093, they do not coordinate active-site waters in the same way as inhibitors of the ErbB family (in particular, EGFR and HER2)39,47 (Figures 11g–i). The primary reason appears to be a result of binding site residue differences between Chk1 (Val68, Leu84, and Ser147) and HER2 (Ser783, Thr798, and Thr862), which do not allow the same water coordination network to be formed, even when binding similarly structured ligands (Figures 11g–i). From a structural perspective, these observations demonstrate the importance of water, in terms of atomic level detail, in the classic lock-and-key model of ligand–protein binding.78 More generally, they provide support for developing a more systematic and direct way to exploit “solvated” molecular footprints in virtual screens as presented below in Results Part 2.
Computational Refinement of ZINC01836093
An examination of the binding site environment surrounding the predicted pose for ZINC01836093 (Figure 12a) revealed several nearby residues that could potentially be targeted in an attempt to improve HER2 affinity. As a proof-of-concept, we employed the de novo DOCK software package57 to automatically construct a series of computationally designed analogues, based on the parent compound, by sampling a series of functional groups (termed side chains, N = 217) to one of the five (R1–R5) attachment points shown in Figure 12a. In particular, we sought to identify analogues that could make new H-bonds with the receptor to residues K724, E770, T798, C805, R849, or D863, while retaining the predicted water-mediated interaction. Among these, E770 and D863, in particular, have been shown to be critical for kinase domain activation and catalysis.15
Figure 12.

(a) Predicted binding pose for parent compound ZINC01836093 (green) showing five refinement positions (R1–R5, magenta spheres) used in the construction of analogues to improve H-bonding with nearby residues (gray) in the HER2 kinase binding pocket. (b) Overlay showing 15 examples of analogues (orange) that maintain the original binding pose of the parent, but make new H-bond interactions with the intended group of residues (gray). Atoms within H-bonding distance shown as dashed magenta lines.
The refinement calculations yielded five different ensembles containing analogues and their associated energy-minimized conformations, for each of the side chains sampled. Figure 12b shows 15 examples (three from each of the five R-group positions), in which analogues retained the original 3D binding pose predicted for the parent (Figure 12a, green) and made the intended H-bond interaction (Figure 12b, dashed magenta lines) with at least one of the targeted residues. For comparison, the DOCK energy score (VDW + ES) for the parent was −74.56 kcal/mol, while the mean across the 15 analogues was −78.84 kcal/mol, indicating an overall improvement in the number of favorable interactions with HER2. Figure 13 shows 2D structures for the analogues along with their individual DOCK scores. Highlighted in green are the position and chemical makeup of the added functionality (shaded ovals). As expected, the added groups contain H-bond donors and acceptors that interact with their polar complements on K724, E770, T798, C805, R849, or D863. Overall, the refinement experiments have suggested several promising avenues for the development of analogues for future experimental testing. Additional computational studies to more thoroughly interrogate analogues using MD simulations are also planned.
Figure 13.

2D structures, code numbers, and energy scores (VDW + ES) for analogues constructed using de novo DOCK57 at one of the five refinement positions (R1–R5). Added functionalities, for each of the three representative examples at the five refinement sites, are highlighted in shaded ovals.
Results Part 2: Development of Virtual Screen Protocols to Incorporate Bridging Waters
Virtual Screen Results: Water Coordination
As noted above, concurrent with the HER2 studies, we sought to establish more well-defined protocols to include the effects of water in our DOCK virtual screening workflows based on the concepts of solvated footprints (Figure 3). The procedures, developed using HIVPR and PARP1 as test cases, rely on the construction of appropriate coordination (COOR protocol) or displacement (DISP protocol) references (Figure S1). Following the generation of the solvated footprint references, we docked a library of 490 235 small organic compounds to each of the two solvated protein binding sites and retained the top 1000 compounds based on their footprint similarity scores in terms of electrostatic overlap (FPSES scores) with the COOR references (Figure S1b,e). The procedure identified 263 and 77 out of 1000 candidates (eq 1) that make the expected interactions (i.e., coordinate water) in the HIVPR and PARP1 binding sites, respectively. Figure 14 shows the predicted binding geometries and ES footprint overlap for four representative compounds from each screen that coordinate the intended water. The specific molecules shown all meet the defined criteria for coordination (eq 1) and were chosen from among all possible candidates (HIVPR = 263) and (PARP1 = 77) after visualization of each binding site. Here, candidates coordinate water through different polar functionalities including sulfones, carbonyls, and amines. Some coordination is bifurcated.
Figure 14.

Representative coordinating compounds identified in the virtual screens to HIVPR (top) and PARP1 (bottom) with a significant solvated electrostatic (ES) footprint overlap. Coordinating compounds in cyan, water-mediated H-bonding in dashed lines, and binding pocket residues in gray. Bottom panels show the solvated ES footprints for each coordinating compound (blue line) vs the COOR reference (red line).
Consistent with the intent of the computational protocol, the docked candidates (Figure 14, blue footprints) show nearly complete overlap with the reference (Figure 14, red footprints). Specifically, for HIVPR, the magnitudes of the interaction between the candidates with the two catalytically important residues Asp25, Asp124, as well as the water (WAT) are essentially identical. Similarly, for PARP1, the candidates make the same three key interactions (and in general their magnitudes) as the reference at positions Gly202, Ser243, Glu327, and WAT. The fact that numerous compounds with the desired “coordinating” bridging interactions were identified in both systems using the solvated footprints is highly encouraging.
Interestingly, the number of candidates identified as coordinating is larger in HIVPR (N = 263) than in PARP1 (N = 77), although the bridging water appears to contribute somewhat less for HIVPR (0.16) than PARP1 (0.22) to the total ligand–receptor ES interaction defined here as the ratio [LIG–WAT/LIG–(REC+WAT)]. Thus, fewer coordinating molecules might have been expected for HIVPR than PARP1 under the same solvated screening conditions although we in fact see the opposite trend.
Virtual Screen Results: Water displacement
The use of solvated footprints with the complementary DISP protocol (Figure 3) yielded 154 and 848 docked molecules for HIVPR and PARP1, respectively, which met the criteria (eq 2) for displacing water. As before, four representative examples each were selected after visual inspection in the HIVPR and PARP1 binding sites and are shown in Figure 15. As expected, candidates that displace bridging water (Figure 15, red spheres) do so through direct interactions with the receptor that involve polar functionality positioned near where the water was originally located. As was observed in the bridging examples, different types of functionality were seen here for displacement, including sulfonamide, carbonyl, carboxylic acid, and amine groups, some in a bifurcated way. As expected, the accompanying solvated footprints (Figure 15) show nearly complete ES overlap between docked candidates from the screen (blue) and the reference (red) suggesting that the interactions made by the originally bridging water with Ile50/Ile149 in HIVPR, or with Glu327 in PARP1, were effectively mimicked.
Figure 15.

Representative displacing compounds identified in the virtual screens to HIVPR (top) and PARP1 (bottom) with a significant solvated electrostatic (ES) footprint overlap. Displacing compounds in cyan, H-bonding in dashed lines, and binding pocket residues in gray. Original locations for displaced waters represented as red spheres. Bottom panels show the solvated ES footprints for each displacing compound (blue) vs the DISP reference (red).
In contrast to the COOR screens in which HIVPR yielded a larger number of coordinating candidates than PARP1 (263 vs 77), the use of the DISP protocol led to the opposite trend (154 vs 848). A possible explanation is that the ES interaction contribution from the bridging water is smaller for HIVPR (0.11) than for PARP1 (0.52) compared to the total ES interaction defined here as the ratio [(LIG+WAT–I50+I49)/(LIG+WAT–REC)]. This suggests that for PARP1 the ES contribution from the bridging water in the DISP screens is more dominant than in HIVPR and thus more likely to be mimicked using ES footprint similarity alone.
MD-Stability: Water Coordination
To examine whether candidate compounds predicted to coordinate bridging waters (Figure 14) would remain geometrically and energetically stable under normal thermal fluctuations, MD simulations of each protein–ligand complex were performed using protocols outlined in the Supporting Information. For each candidate, three 20 ns simulations were performed and combined into one ensemble (3 × 1000 frames) from which the five most populated water sites were identified as described in the Methods section and color-coded for visualization (see Figure 16). In parallel, footprint similarity scores (i.e., overlap) were computed using Euclidean distance (Euc dist) from the average ES interaction patterns made by the waters at each site versus the water contribution from the COOR reference (Table 2). Water sites maintaining the intended bridge (ligand–water–protein) in a stable manner would be expected to have a low Euclidian distance (zero being perfect overlap). Such sites would also be expected to have a relatively high water population.
Figure 16.

Solvent patterns for predicted coordinating compounds in HIVPR (top) and PARP1 (bottom) derived from triplicate MD ensembles (3 × 1000 frames each) color-coded by population (S1 red > S2 orange > S3 yellow > S4 green > S5 blue). Key residues for HIVPR (Ile50, Ile149) and PARP1 (Glu327) in thick lines with ligands in thin lines. Arrows indicate the expected water sites (oxygen atoms only).
Table 2. Properties of Top-Populated Water Sites Derived from MD Simulations of Candidate Compounds Predicted to Coordinate the Bridging Water.
| HIVPR |
PARP1 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| KNI-272
(control) |
NU1098
(control) |
|||||||||
| property | S1 | S2 | S3 | S4 | S5 | S1 | S2 | S3 | S4 | S5 |
| Euc dista | 5.19 | 2.74 | 5.21 | 1.87b | 1.08 | 0.64b | 3.74 | 3.97 | 4.30 | 4.29 |
| population | 0.34 | 0.15 | 0.14 | 0.13b | 0.06 | 0.25b | 0.22 | 0.13 | 0.12 | 0.12 |
| ZINC02501244 | ZINC46021741 | |||||||||
| Euc dist | 4.61 | 5.04 | 3.83 | 3.77 | 1.48b | 4.05 | 5.02 | 4.13 | 4.14 | 4.91 |
| population | 0.52 | 0.27 | 0.08 | 0.07 | 0.03b | 0.54 | 0.30 | 0.06 | 0.02 | 0.02 |
| ZINC96361218 | ZINC15786589 | |||||||||
| Euc dist | 1.39b | 4.48 | 4.15 | 4.12 | 4.09 | 0.43b | 4.26 | 4.93 | 3.80 | 3.94 |
| population | 0.60b | 0.19 | 0.10 | 0.09 | 0.02 | 0.24b | 0.19 | 0.15 | 0.12 | 0.10 |
| ZINC95851204 | ZINC35446303 | |||||||||
| Euc dist | 0.55b | 1.64 | 2.22 | 4.15 | 0.91 | 1.10b | 5.01 | 4.16 | 4.19 | 4.43 |
| population | 0.57b | 0.24 | 0.06 | 0.05 | 0.03 | 0.25b | 0.23 | 0.12 | 0.10 | 0.06 |
| ZINC09716176 | ZINC07539129 | |||||||||
| Euc dist | 0.96b | 3.83 | 0.69 | 1.24 | 5.81 | 4.08 | 4.26 | 4.59 | 3.81 | 3.96 |
| population | 0.37b | 0.23 | 0.17 | 0.09 | 0.07 | 0.55 | 0.24 | 0.05 | 0.04 | 0.03 |
Euclidian distance between footprints in kcal/mol. Water sites ordered by highest to lowest population S1 > S2 > S3 > S4 > S5.
The intended water coordination sites.
On the basis of visual inspection of the five most populated sites (colored clusters in Figure 16) and their associated Euclidean distances (Table 2), the intended bridging waters (arrows in Figure 16) were observed in simulations of both positive controls (KNI-272, NU1098) and for six out of the eight candidate compounds. For six of the ten simulations, the S1 sites with the highest population also yielded low Euclidian distances, indicating highly stable coordination. For example in HIV protease, three of the four ligands from the virtual screen yield the lowest (ZINC96361218 = 1.39, ZINC95851204 = 0.55) or second lowest (ZINC09716176 = 0.96) Euclidian values at S1 (Table 2). Interestingly, ZINC09716176 showed a strong coordination with both S1 and S3 with each site bridging one of the two protease flaps (Ile149 and Ile50, respectively). For ZINC02501244, although the intended water bridge was observed at S5 (Figure 16 top, blue cluster, Table 2), the relative population was significantly lower (0.03), suggesting a less favorable interaction compared to the other three ligands. Surprisingly, the control ligand KNI-272 did not yield a low Euclidean distance for the S1 site, although the intended water was in fact observed at the less populated S4 site (Figure 16 top, green cluster, Table 2). Examination of the underlying footprints for KNI-272 (data not shown) revealed S4 waters interacting with the ligand and only one of the protease flaps (Ile50). Under these simulation conditions, the more stable water coordination by candidates ZINC96361218, ZINC95851204, and ZINC09716176 relative to the control ligand is notable and highlights the potential utility of the method.
For the PARP1 studies, two of the four ligands identified in the virtual screen (ZINC15786589 and ZINC35446303) maintained the intended water coordination during MD (Figure 16, bottom). The coordination was with the most populated S1 site that also yielded the lowest Euclidian distances relative to the footprint pattern derived from the water in the COOR reference (Table 2). Simulations of the control ligand NU1098 for PARP1 also showed coordination with the intended water at the S1 site that had the lowest Euclidean distance. To investigate the absence of coordination for the other two candidates, we examined the underlying MD trajectories. Although all four compounds initially coordinated the intended waters (Figure 14, bottom), conformational sampling during the simulations led to, in the case of ZINC46021741, coordination of a water site that bridged with two other residues (His201 and Tyr235), or, in the case of ZINC07539129, displacement of the intended water that resulted in a direct hydrogen bond to Glu327.
MD-Stability: Water Displacement
For ligands predicted to displace bridging waters (Figure 15), MD-based analysis (see Supporting Information) was similarly used to evaluate pose stability in the HIVPR and PARP1 binding pockets. Here, an absence of occupancy near the site of the original bridging water, as well as stable interactions between the ligand and key protein residues over time, would be expected for candidates that stably displace the intended water. Figure 17 shows solvent patterns for the five most populated water sites identified from the simulations, which, as before, were derived from triplicate MD runs of each candidate and known controls. To further characterize displacement, ES interactions between each ligand and key protein residues (HIVPR = Ile50, Ile149, PARP1 = Glu327) were plotted as a function of time (Figure 18).
Figure 17.

Solvent patterns for predicted displacing compounds in HIVPR (top) and PARP1 (bottom) derived from triplicate MD ensembles (3 × 1000 frames each) color-coded by population (S1 red > S2 orange > S3 yellow > S4 green > S5 blue). Key residues for HIVPR (Ile50, Ile149) and PARP1 (Glu327) in thick lines with ligands in thin lines. Key H-bonding between ligand and protein shown as dashed lines. Water sites intended to be displaced indicated by arrows.
Figure 18.

ES interaction energy (kcal/mol) between docked candidates and control ligands and key protein residues in HIVPR (Ile50, Ile149) or PARP1 (Glu327) versus time. Running block averages (50 frame blocks) plotted in colored lines with raw fluctuations in gray for three independent 20 ns MD simulations each.
Examination of the clusters in Figure 17 shows that the intended bridging waters remained successfully displaced (panels without arrows) in MD simulations for six out of the eight candidates from the DISP screens and for both positive controls (DMP323, 4AN). Specifically, for HIVPR, ZINC35481061, and ZINC19883914 showed no water occupancy at the intended displacement site (Figure 17) and plots of ligand–residue ES energy vs time showed strong stable favorable interactions with Ile50 and Ile149 of ca. −1 to −1.5 kcal/mol (Figure 18, left), confirming successful displacement. For ZINC35686312 and ZINC96361225, however, only partial displacement was observed. For ZINC35686312, favorable ES interactions were obtained for only one of the flaps (Figure 18, Ile 50 ca. −1.5 kcal/mol, Ile 149 ca. 0). For ZINC96361225, initially favorable ES interactions with the flaps weaken and then over time disappear in three out of three replicas for Ile149 and in one out of three replicas for Ile50. In both of these cases, the initially displaced water appears to come back and compete with the ligand resulting in partial displacement and partial coordination, suggesting that the two interaction types are similarly favorable.
For PARP1, the results show that all four candidates maintained successful displacement of the intended water (no occupancy) and in general direct favorable ES interactions with the intended residue Glu327 (Figure 18, right), although for some ligands different MD runs yielded different interaction strengths. For example, while ZINC65034286 maintained interaction strengths of ca. −6 kcal/mol across all three simulations (Figure 18, right), results for ZINC65304869 showed a significant regime switch (from ∼ −6 kcal/mol to ∼ −12 kcal/mol) at around 8 ns in one simulation. In contrast, for ZINC01107820, one of the simulations showed a significant ES loss. For ZINC35966985, all three MD runs eventually converged to the same ca. interaction strength between −1.5 and −3 kcal/mol.
A noteworthy outcome from the analysis is that several of the DISP screen candidates make significantly more favorable ES interactions with their intended protein residues compared to the positive controls. For example, for HIVPR (Figure 18, left), the ES interactions between Ile flap residues and the control DMP323 are between 0.5 and −1 kcal/mol, while simulations for at least two of the candidates increase to between −1 and −2 kcal/mol. Similarly, for PARP1 (Figure 18, right), the control ligand 4AN makes direct ES interactions with Glu327 of ca. −4.5 kcal/mol, and favorable increases of up to ca. −6 kcal/mol (on average) were observed for two of the four candidates.
Conclusions
The goals of this study were 2-fold: (1) identification of inhibitors with activity to the breast cancer target HER2 and (2) development of protocols to identify ligands that can coordinate or displace bridging water. In Results Part 1 (Table 1, Figures 4–13), computational and experimental results are presented from a large-scale virtual screen of ∼2 M compounds to a fully active homology model of the HER2 kinase domain (Figure 4a). Docked molecules were rescored using multiple scoring functions (Table 1, Figure 5), including those that considered a previously identified water-mediated interaction (Figure 4b) and prioritized by visual inspection and drug-likeness, which led to 149 candidates being purchased for experimental testing. Several candidates had DOCK-predicted poses that appeared to be capable of water coordination (Figure 6) or displacement (Figure 5).
Subsequent experimental testing led to seven compounds with a binding affinity for HER2 (Figure 7a) or inhibition of HER2 phosphorylation (Figure 7b). One of the hits, ZINC01836093, showed clear dose–response behavior in terms of affinity (Kd = 7.0 μM, Figure 8a) and inhibition (IC50 = 4.6 μM, Figure 8c) and appeared to be selective for HER2 over the related Src kinase (Figure 7c) compared to relevant controls. Importantly, 20 ns MD simulations of ZINC01836093 revealed stable coordination with a binding site water (Figure 10). A similarity search of the hit (Figure 11a) identified two structurally related inhibitors of the Chk1 kinase (Figure 11b,c). Notably, the DOCK6-predicted pose for ZINC01836093 with HER2 shows striking overlap with the crystallographic poses for the two Chk1 inhibitors (Figure 11e,f), although in the latter case structural differences between the HER2 and Chk1 binding site preclude the same water coordination network to be formed (Figure 11g–i). Efforts to refine ZINC01836093 (Figures 12 and 13) and improve affinity for HER2 are ongoing. Evaluating activity against a panel of HER2 activating mutants is also planned.
Results Part 2 (Table 2, Figure S1, Figures 14–18) outlines the development of new DOCK6 virtual screening protocols based on “solvated” footprints, which were tested through proof-of-concept virtual screens to HIVPR and PARP1. Specifically, molecular references derived from a ligand alone in a solvated binding site (COOR reference) or a solvated ligand in an apo binding site (DISP reference) were prepared (Figure S1) and ∼500 K ligands were screened to identify compounds with a high footprint similarity. Examination of top-scoring coordinating candidates (Figure 14) confirmed a high ES overlap with polar functionality interacting with the intended water, which in turn interacted favorably with the intended protein residues (HIVPR = Ile50/Ile149, PARP1 = Glu327). Examination of top-scoring displacing candidates (Figure 15) similarly showed a high ES overlap. In this case, compounds interacted with the intended residues through direct interactions involving polar functionality positioned at or near where the bridging water was located originally. Overall, the new virtual screening protocols were deemed successful in that the use of solvated footprints resulted in a significant amount of coordinating (HIVPR = 263, PARP1 = 77) or displacing ligands (HIVPR = 154, PARP1 = 848) being identified from among top candidates from each screen.
Candidates from the test screens (Figures 14 and 15) were also examined to see if they would maintain their expected water coordination or displacement during triplicate MD simulations of each complex. For the COOR protocols case, the five most populated water sites were computed (Figure 16). To determine if the sites maintained the expected interactions, footprint overlaps were also computed (Table 2) between the ES patterns derived from each of the five sites (water interacting with protein and ligand) with those made by the intended bridging water in the original COOR references used in the screens. Encouragingly, bridging waters were observed in simulations for six out of the eight candidates from the screens (four from HIVPR, two from PARP1, Figure 16) and two positive controls. In six of the ten simulations, the S1 site with the highest population also yielded a low Euclidian distance, which indicates stable coordination (Table 2).
For candidates selected using DISP protocols, water displacement was similarly observed for six of the eight compounds examined (two from HIVPR, four from PARP1, Figure 17) and both positive controls. For the remaining two cases, however, only partial displacement was observed. Notably, as shown in Figure 18, an examination of the ES interactions across individual MD trajectories revealed fluctuations and variability over time indicated that several compounds were observed to make more favorable ES interactions than the controls at specific residues involved in direct interaction with the ligands, which highlighted the benefits of using multiple simulations to help gauge if water displacement was successful.
Finally, although not pursued here, we envision that the new solvated footprint method will be useful to drive from-scratch ligand construction or refinement using de novo design57 and genetic algorithm approaches undergoing development in our laboratory. Directly designing molecules to have a good footprint overlap, instead of searching through existing chemical libraries, would be an orthogonal and potentially more effective approach. Given the increasing appreciation that molecular recognition can be tailored through strategic use of coordination or displacement of water, the development of new computational procedures such as those presented here are likely to become increasingly important.
Acknowledgments
The authors thank Dwight McGee, Yuchen Zhou, Courtney Singleton, Lauren Prentis, Stephen Telehany, William Allen, Brian Fochtman, He Huang, and Mengru Zhang for inspiring discussion. We would also like to thank Stony Brook Research Computing and Cyberinfrastructure and the Institute for Advanced Computational Science at Stony Brook University for access to the high-performance LIred and SeaWulf computing systems, the latter of which was made possible by a $1.4M National Science Foundation grant (#1531492).
Glossary
Abbreviations
- ATP
adenosine triphosphate
- DCESUM
DOCK Cartesian energy
- EGFR
human epidermal growth factor receptor 1
- ES
electrostatic
- FMS
pharmacophore matching similarity
- FPSES
electrostatic footprint similarity
- FPSSUM
footprint similarity
- HER2
human epidermal growth factor receptor 2
- HMS
Hungarian matching similarity
- MD
molecular dynamics
- PDB
protein data bank
- VDW
van der Waals
- VOS
volume overlap similarity
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00524.
Protocols for kinase domain homology modeling, expression and purification of the HER2 kinase domain, preparation of solvated footprint references, geometric and energetic stability of water coordination and displacement, and results for solvated footprint references (PDF)
Author Contributions
# J.G. and S.C. contributed equally to this work.
This work was funded in part by the Stony Brook University Office of the Vice President for Research and the U.S. Department of Defense under contracts W81XWH-14-1-0419 (to W.T.M) and W81XWH-14-1-0420 (to R.C.R) and by NIH grant R01 CA58530 (to W.T.M.). Gratitude is also expressed to the Stony Brook University Chemical Biology Training Program T32GM092714 (to S.C.).
The authors declare no competing financial interest.
Supplementary Material
References
- Coussens L.; Yang-Feng T. L.; Liao Y. C.; Chen E.; Gray A.; McGrath J.; Seeburg P. H.; Libermann T. A.; Schlessinger J.; Francke U.; Levinson A.; Ullrich A. (1985) Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 230, 1132–1139. 10.1126/science.2999974. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.; Ikawa S.; Akiyama T.; Semba K.; Nomura N.; Miyajima N.; Saito T.; Toyoshima K. (1986) Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature 319, 230–234. 10.1038/319230a0. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y. (2001) Biology of HER2 and its importance in breast cancer. Oncology 61, 1–13. 10.1159/000055396. [DOI] [PubMed] [Google Scholar]
- Miller T. W.; Rexer B. N.; Garrett J. T.; Arteaga C. L. (2011) Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 13, 224. 10.1186/bcr3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arman K.; Ergun S.; Temiz E.; Oztuzcu S. (2014) The interrelationship between HER2 and CASP3/8 with apoptosis in different cancer cell lines. Mol. Biol. Rep. 41, 8031–8036. 10.1007/s11033-014-3700-x. [DOI] [PubMed] [Google Scholar]
- Ciardiello F.; Caputo R.; Bianco R.; Damiano V.; Pomatico G.; De Placido S.; Bianco A. R.; Tortora G. (2000) Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin. Cancer Res. 6, 2053–2063. [PubMed] [Google Scholar]
- Hidalgo M.; Siu L. L.; Nemunaitis J.; Rizzo J.; Hammond L. A.; Takimoto C.; Eckhardt S. G.; Tolcher A.; Britten C. D.; Denis L.; Ferrante K.; Von Hoff D. D.; Silberman S.; Rowinsky E. K. (2001) Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J. Clin. Oncol. 19, 3267–3279. 10.1200/JCO.2001.19.13.3267. [DOI] [PubMed] [Google Scholar]
- Xia W.; Mullin R. J.; Keith B. R.; Liu L. H.; Ma H.; Rusnak D. W.; Owens G.; Alligood K. J.; Spector N. L. (2002) Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 21, 6255–6263. 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- Wakeling A. E.; Guy S. P.; Woodburn J. R.; Ashton S. E.; Curry B. J.; Barker A. J.; Gibson K. H. (2002) ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 62, 5749–5754. [PubMed] [Google Scholar]
- Sridhar S. S.; Seymour L.; Shepherd F. A. (2003) Inhibitors of epidermal-growth-factor receptors: a review of clinical research with a focus on non-small-cell lung cancer. Lancet Oncol. 4, 397–406. 10.1016/S1470-2045(03)01137-9. [DOI] [PubMed] [Google Scholar]
- Wells S. A. Jr.; Robinson B. G.; Gagel R. F.; Dralle H.; Fagin J. A.; Santoro M.; Baudin E.; Elisei R.; Jarzab B.; Vasselli J. R.; Read J.; Langmuir P.; Ryan A. J.; Schlumberger M. J. (2012) Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J. Clin. Oncol. 30, 134–141. 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson F. M.; Gray N. S. (2018) Kinase inhibitors: the road ahead. Nat. Rev. Drug Discovery 17, 353–377. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Gureasko J.; Shen K.; Cole P. A.; Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149. 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Jura N.; Zhang X.; Endres N. F.; Seeliger M. A.; Schindler T.; Kuriyan J. (2011) Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22. 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamos J.; Sliwkowski M. X.; Eigenbrot C. (2002) Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277, 46265–46272. 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- Wood E. R.; Truesdale A. T.; McDonald O. B.; Yuan D.; Hassell A.; Dickerson S. H.; Ellis B.; Pennisi C.; Horne E.; Lackey K.; Alligood K. J.; Rusnak D. W.; Gilmer T. M.; Shewchuk L. (2004) A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 64, 6652–6659. 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- Aertgeerts K.; Skene R.; Yano J.; Sang B. C.; Zou H.; Snell G.; Jennings A.; Iwamoto K.; Habuka N.; Hirokawa A.; Ishikawa T.; Tanaka T.; Miki H.; Ohta Y.; Sogabe S. (2011) Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 286, 18756–18765. 10.1074/jbc.M110.206193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu H.; Takahashi T.; Nomura M.; Majmudar K.; Suzuki M.; Lee H.; Wistuba II; Fong K. M.; Toyooka S.; Shimizu N.; Fujisawa T.; Minna J. D.; Gazdar A. F. (2005) Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 65, 1642–1646. 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- Wang S. E.; Narasanna A.; Perez-Torres M.; Xiang B.; Wu F. Y.; Yang S.; Carpenter G.; Gazdar A. F.; Muthuswamy S. K.; Arteaga C. L. (2006) HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 10, 25–38. 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Bose R.; Kavuri S. M.; Searleman A. C.; Shen W.; Shen D.; Koboldt D. C.; Monsey J.; Goel N.; Aronson A. B.; Li S.; Ma C. X.; Ding L.; Mardis E. R.; Ellis M. J. (2013) Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discovery 3, 224–237. 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazieres J.; Peters S.; Lepage B.; Cortot A. B.; Barlesi F.; Beau-Faller M.; Besse B.; Blons H.; Mansuet-Lupo A.; Urban T.; Moro-Sibilot D.; Dansin E.; Chouaid C.; Wislez M.; Diebold J.; Felip E.; Rouquette I.; Milia J. D.; Gautschi O. (2013) Lung Cancer That Harbors an HER2Mutation: Epidemiologic Characteristics and Therapeutic Perspectives. J. Clin. Oncol. 31, 1997–2003. 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W. Z.; Lopez R.; McWilliam H.; Remmert M.; Soding J.; Thompson J. D.; Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz I. D. (1992) Structure-Based Strategies for Drug Design and Discovery. Science 257, 1078–1082. 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. (2004) Virtual screening of chemical libraries. Nature 432, 862–865. 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebe G. (2006) Virtual ligand screening: strategies, perspectives and limitations. Drug Discovery Today 11, 580–594. 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Balius T. E.; Rizzo R. C. (2010) Docking Validation Resources: Protein Family and Ligand Flexibility Experiments. J. Chem. Inf. Model. 50, 1986–2000. 10.1021/ci1001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozell S. R.; Mukherjee S.; Balius T. E.; Roe D. R.; Case D. A.; Rizzo R. C. (2012) Evaluation of DOCK 6 as a pose generation and database enrichment tool. J. Comput.-Aided Mol. Des. 26, 749–773. 10.1007/s10822-012-9565-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen W. J.; Balius T. E.; Mukherjee S.; Brozell S. R.; Moustakas D. T.; Lang P. T.; Case D. A.; Kuntz I. D.; Rizzo R. C. (2015) DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem. 36, 1132–1156. 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balius T. E.; Mukherjee S.; Rizzo R. C. (2011) Implementation and Evaluation of a Docking-Rescoring Method Using Molecular Footprint Comparisons. J. Comput. Chem. 32, 2273–2289. 10.1002/jcc.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balius T. E.; Allen W. J.; Mukherjee S.; Rizzo R. C. (2013) Grid-based molecular footprint comparison method for docking and de novo design: Application to HIVgp41. J. Comput. Chem. 34, 1226–1240. 10.1002/jcc.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.; Rizzo R. C. (2015) Pharmacophore-Based Similarity Scoring for DOCK. J. Phys. Chem. B 119, 1083–1102. 10.1021/jp506555w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen W. J.; Rizzo R. C. (2014) Implementation of the Hungarian algorithm to account for ligand symmetry and similarity in structure-based design. J. Chem. Inf. Model. 54, 518–529. 10.1021/ci400534h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poornima C. S.; Dean P. M. (1995) Hydration in drug design. 1. Multiple hydrogen-bonding features of water molecules in mediating protein-ligand interactions. J. Comput.-Aided Mol. Des. 9, 500–512. 10.1007/BF00124321. [DOI] [PubMed] [Google Scholar]
- Poornima C. S.; Dean P. M. (1995) Hydration in drug design. 2. Influence of local site surface shape on water binding. J. Comput.-Aided Mol. Des. 9, 513–520. 10.1007/BF00124322. [DOI] [PubMed] [Google Scholar]
- Ladbury J. E. (1996) Just add water! The effect of water on the specificity of protein-ligand binding sites and its potential application to drug design. Chem. Biol. 3, 973–980. 10.1016/S1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
- Lu Y. P.; Wang R. X.; Yang C. Y.; Wang S. M. (2007) Analysis of ligand-bound water molecules in high-resolution crystal structures of protein-ligand complexes. J. Chem. Inf. Model. 47, 668–675. 10.1021/ci6003527. [DOI] [PubMed] [Google Scholar]
- Bodnarchuk M. S. (2016) Water, water, everywhere··· It’s time to stop and think. Drug Discovery Today 21, 1139–1146. 10.1016/j.drudis.2016.05.009. [DOI] [PubMed] [Google Scholar]
- Balius T. E.; Rizzo R. C. (2009) Quantitative Prediction of Fold Resistance for Inhibitors of EGFR. Biochemistry 48, 8435–8448. 10.1021/bi900729a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel J.; Tirado-Rives J.; Jorgensen W. L. (2009) Energetics of Displacing Water Molecules from Protein Binding Sites: Consequences for Ligand Optimization. J. Am. Chem. Soc. 131, 15403–15411. 10.1021/ja906058w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel J.; Tirado-Rives J.; Jorgensen W. L. (2009) Prediction of the Water Content in Protein Binding Sites. J. Phys. Chem. B 113, 13337–13346. 10.1021/jp9047456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. D.; Sherman W.; Farid R. (2010) Understanding Kinase Selectivity Through Energetic Analysis of Binding Site Waters. ChemMedChem 5, 618–627. 10.1002/cmdc.200900501. [DOI] [PubMed] [Google Scholar]
- Baron R.; Setny P.; McCammon J. A. (2010) Water in cavity-ligand recognition. J. Am. Chem. Soc. 132, 12091–12097. 10.1021/ja1050082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setny P.; Baron R.; McCammon J. A. (2010) How Can Hydrophobic Association Be Enthalpy Driven?. J. Chem. Theory Comput. 6, 2866–2871. 10.1021/ct1003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barillari C.; Duncan A. L.; Westwood I. M.; Blagg J.; van Montfort R. L. M. (2011) Analysis of water patterns in protein kinase binding sites. Proteins: Struct., Funct., Genet. 79, 2109–2121. 10.1002/prot.23032. [DOI] [PubMed] [Google Scholar]
- Huggins D. J.; Sherman W.; Tidor B. (2012) Rational approaches to improving selectivity in drug design. J. Med. Chem. 55, 1424–1444. 10.1021/jm2010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. L.; Rizzo R. C. (2012) A Water-Based Mechanism of Specificity and Resistance for Lapatinib with ErbB Family Kinases. Biochemistry 51, 2390–2406. 10.1021/bi2016553. [DOI] [PubMed] [Google Scholar]
- Levinson N. M.; Boxer S. G. (2014) A conserved water-mediated hydrogen bond network defines bosutinib’s kinase selectivity. Nat. Chem. Biol. 10, 127–132. 10.1038/nchembio.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunitz J. D. (1994) The Entropic Cost of Bound Water in Crystals and Biomolecules. Science 264, 670–670. 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- Chen J. M.; Xu S. L.; Wawrzak Z.; Basarab G. S.; Jordan D. B. (1998) Structure-based design of potent inhibitors of scytalone dehydratase: Displacement of a water molecule from the active site. Biochemistry 37, 17735–17744. 10.1021/bi981848r. [DOI] [PubMed] [Google Scholar]
- Wawrzak Z.; Sandalova T.; Steffens J. J.; Basarab G. S.; Lundqvist T.; Lindqvist Y.; Jordan D. B. (1999) High-resolution structures of scytalone dehydratase-inhibitor complexes crystallized at physiological pH. Proteins: Struct., Funct., Genet. 35, 425–439. . [DOI] [PubMed] [Google Scholar]
- McGillick B. E.; Balius T. E.; Mukherjee S.; Rizzo R. C. (2010) Origins of resistance to the HIVgp41 viral entry inhibitor T20. Biochemistry 49, 3575–3592. 10.1021/bi901915g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden P. M.; Kaur H.; Goyal R.; Gochin M.; Rizzo R. C. (2012) Footprint-based identification of viral entry inhibitors targeting HIVgp41. Bioorg. Med. Chem. Lett. 22, 3011–3016. 10.1016/j.bmcl.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden P. M.; Allen W. J.; Gochin M.; Rizzo R. C. (2014) Strategies for lead discovery: application of footprint similarity targeting HIVgp41. Bioorg. Med. Chem. 22, 651–661. 10.1016/j.bmc.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen W. J.; Yi H. A.; Gochin M.; Jacobs A.; Rizzo R. C. (2015) Small molecule inhibitors of HIVgp41 N-heptad repeat trimer formation. Bioorg. Med. Chem. Lett. 25, 2853–2859. 10.1016/j.bmcl.2015.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee T. D. Jr.; Yi H. A.; Allen W. J.; Jacobs A.; Rizzo R. C. (2017) Structure-based identification of inhibitors targeting obstruction of the HIVgp41 N-heptad repeat trimer. Bioorg. Med. Chem. Lett. 27, 3177–3184. 10.1016/j.bmcl.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen W. J.; Fochtman B. C.; Balius T. E.; Rizzo R. C. (2017) Customizable de novo design strategies for DOCK: Application to HIVgp41 and other therapeutic targets. J. Comput. Chem. 38, 2641–2663. 10.1002/jcc.25052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger W. T.; Ralph B. P.; Kaczocha M.; Sun J.; Balius T. E.; Rizzo R. C.; Haj-Dahmane S.; Ojima I.; Deutsch D. G. (2012) Targeting fatty acid binding protein (FABP) anandamide transporters - a novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS One 7, e50968. 10.1371/journal.pone.0050968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H. C.; Tong S. M.; Zhou Y. C.; Elmes M. W.; Yan S.; Kaczocha M.; Deutsch D. G.; Rizzo R. C.; Ojima I.; Li H. L. (2017) The Antinociceptive Agent SBFI-26 Binds to Anandamide Transporters FABP5 and FABP7 at Two Different Sites. Biochemistry 56, 3454–3462. 10.1021/acs.biochem.7b00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y. H.; Berger W. T.; Nesbitt N. M.; Kumar K.; Balius T. E.; Rizzo R. C.; Tonge P. J.; Ojima I.; Swaminathan S. (2015) Computer-aided identification, synthesis, and biological evaluation of novel inhibitors for botulinum neurotoxin serotype A. Bioorg. Med. Chem. 23, 5489–5495. 10.1016/j.bmc.2015.07.040. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; McGillick B. E.; Teng Y. G.; Haranahalli K.; Ojima I.; Swaminathan S.; Rizzo R. C. (2016) Identification of small molecule inhibitors of botulinum neurotoxin serotype E via footprint similarity. Bioorg. Med. Chem. 24, 4875–4889. 10.1016/j.bmc.2016.07.031. [DOI] [PubMed] [Google Scholar]
- Lundqvist T.; Rice J.; Hodge C. N.; Basarab G. S.; Pierce J.; Lindqvist Y. (1994) Crystal structure of scytalone dehydratase–a disease determinant of the rice pathogen, Magnaporthe grisea. Structure 2, 937–944. 10.1016/S0969-2126(94)00095-6. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Macarthur M. W.; Moss D. S.; Thornton J. M. (1993) Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 26, 283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Irwin J. J.; Shoichet B. K. (2005) ZINC–a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45, 177–182. 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2013) MOE, Version 2013.08, Chemical Computing Group Inc., Montreal, Canada.
- Discover X. https://www.discoverx.com/services/drug-discovery-development-services/kinase-profiling/kinomescan/scanelect (accessed June 13, 2018).
- Discover X. https://www.discoverx.com/kinase-data-sheets/erbb2 (accessed June 13, 2018).
- Sun G.; Budde R. J. (1999) Substitution studies of the second divalent metal cation requirement of protein tyrosine kinase CSK. Biochemistry 38, 5659–5665. 10.1021/bi982793w. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Duan D.; Torosyan H.; Doak A. K.; Ziebart K. T.; Sterling T.; Tumanian G.; Shoichet B. K. (2015) An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 58, 7076–7087. 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich C.; Bertozzi C.; Georg G. I.; Kiessling L.; Lindsley C.; Liotta D.; Merz K. M.; Schepartz A.; Wang S. M. (2017) The Ecstasy and Agony of Assay Interference Compounds. ACS Cent. Sci. 3, 143–147. 10.1021/acscentsci.7b00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T.; Irwin J. J. (2015) ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo L. J.; Lee F. Y.; Chen P.; Norris D.; Barrish J. C.; Behnia K.; Castaneda S.; Cornelius L. A.; Das J.; Doweyko A. M.; Fairchild C.; Hunt J. T.; Inigo I.; Johnston K.; Kamath A.; Kan D.; Klei H.; Marathe P.; Pang S.; Peterson R.; Pitt S.; Schieven G. L.; Schmidt R. J.; Tokarski J.; Wen M. L.; Wityak J.; Borzilleri R. M. (2004) Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 47, 6658–6661. 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. (2016) Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 59, 4103–4120. 10.1021/acs.jmedchem.5b02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Thiessen P. A.; Bolton E. E.; Chen J.; Fu G.; Gindulyte A.; Han L.; He J.; He S.; Shoemaker B. A.; Wang J.; Yu B.; Zhang J.; Bryant S. H. (2016) PubChem Substance and Compound databases. Nucleic Acids Res. 44, D1202–1213. 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foloppe N.; Fisher L. M.; Howes R.; Kierstan P.; Potter A.; Robertson A. G.; Surgenor A. E. (2005) Structure-based design of novel Chk1 inhibitors: insights into hydrogen bonding and protein-ligand affinity. J. Med. Chem. 48, 4332–4345. 10.1021/jm049022c. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler F. W. (1995) 100 Years “Schlussel-Schloss-Prinzip”: What Made Emil Fischer Use this Analogy?. Angew. Chem., Int. Ed. Engl. 33, 2364–2374. 10.1002/anie.199423641. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.