

ABSTRACT
Small molecule inhibitors of the checkpoint proteins CHK1 and WEE1 are currently in clinical development in combination with the antimetabolite gemcitabine. It is unclear, however, if there is a therapeutic advantage to CHK1 vs. WEE1 inhibition for chemosensitization. The goals of this study were to directly compare the relative efficacies of the CHK1 inhibitor MK8776 and the WEE1 inhibitor AZD1775 to sensitize pancreatic cancer cell lines to gemcitabine and to identify pharmacodynamic biomarkers predictive of chemosensitization. Cells treated with gemcitabine and either MK8776 or AZD1775 were first assessed for clonogenic survival. With the exception of the homologous recombination-defective Capan1 cells, which were relatively insensitive to MK8776, we found that these cell lines were similarly sensitized to gemcitabine by CHK1 or WEE1 inhibition. The abilities of either the CDK1/2 inhibitor roscovitine or exogenous nucleosides to prevent MK8776 or AZD1775-mediated chemosensitization, however, were both inhibitor-dependent and variable among cell lines. Given the importance of DNA replication stress to gemcitabine chemosensitization, we next assessed high-intensity, pan-nuclear γH2AX staining as a pharmacodynamic marker for sensitization. In contrast to total γH2AX, aberrant mitotic entry or sub-G1 DNA content, high-intensity γH2AX staining correlated with chemosensitization by either MK8776 or AZD1775 (R2 0.83 – 0.53). In summary, we found that MK8776 and AZD1775 sensitize to gemcitabine with similar efficacy. Furthermore, our results suggest that the effects of CHK1 and WEE1 inhibition on gemcitabine-mediated replication stress best predict chemosensitization and support the use of high-intensity or pan-nuclear γH2AX staining as a marker for therapeutic response.
Keywords: MK8776, AZD1775, CHK1, WEE1, gemcitabine, pancreatic cancer
Introduction
The cell cycle checkpoint kinases CHK1 and WEE1 are integral to the intra-S-phase [1] and G2 checkpoints [2] activated as part of the cellular DNA damage response (DDR). Both proteins affect cell cycle arrest through persistent inhibitory phosphorylation of CDK1 and CDK2 at T14/Y15: CHK1 through phosphorylation of the phosphatase CDC25A, targeting that protein for degradation, and WEE1 through the direct phosphorylation of CDK1/2 at these sites [3]. In addition to halting cell cycle progression, negative regulation of CDK1/2 activity by CHK1 or WEE1 promotes homologous recombination repair (HR) [4,5] and prevents untimely DNA replication origin firing, subsequent nucleotide shortage and the accumulation of stalled replication forks [6–8]. CHK1 and WEE1 inhibitors have each been developed as targeted therapies predicted to enhance existing treatments by magnifying chemotherapy-induced DNA damage, either by directly inhibiting DNA repair pathways, or indirectly, by permitting cell cycle progression in the presence of unrepaired lesions or exacerbating chemotherapy-induced replication stress [9].
While initial studies focused on abrogation of cell cycle checkpoints and aberrant mitotic entry as the major mechanisms underlying chemosensitization by CHK1 or WEE1 inhibitors [10–12], subsequent studies have demonstrated that aberrant mitotic entry is not required for chemosensitization and have alternatively suggested a role for DNA replication stress resulting from CDK2-mediated aberrant origin firing and nucleotide shortage [6,7,13]. The synergistic cytotoxicity of combined CHK1 and WEE1 inhibitors has also been attributed to their respective effects on DNA replication [14].
While the therapeutic mechanisms of both WEE1 and CHK1 inhibitors have been associated with regulation of CDK1/2 activity [6,8], the further ability of CHK1 to stabilize and facilitate restart of stalled replication forks may also contribute to its value as a therapeutic target [15]. Among the multiple functions of CHK1 at stalled forks are recruitment of RAD51 for HR [4], inhibition of MUS81 endonuclease [16], facilitation of translesion synthesis [17], and replication fork restart [18]. The broader role of CHK1 in the cellular response to stalled replication suggests that CHK1 and WEE1 inhibitors may have different mechanisms of action and may not be uniformly therapeutic across different model systems, a hypothesis supported by the synergistic cytotoxicity of combined CHK1 and WEE1 inhibition [19].
These observations led us to consider that either CHK1 inhibition or WEE1 inhibition may be superior in terms of gemcitabine chemosensitization in different pancreatic cancer cell lines and underlie the importance of characterizing their specific mechanisms of sensitization in order to develop markers that predict the efficacy of chemosensitization by either CHK1 or WEE1 inhibition. While we previously identified phospho-CHK1 (Ser345), a marker of DNA damage signaling, as a marker for sensitization to gemcitabine by CHK1 inhibitor [20], other studies have highlighted the importance of either high-intensity or pan-nuclear γH2AX staining as an important marker for replication stress caused by either CHK1 or WEE1 inhibition [7,21,22].
Since both CHK1 and WEE1 inhibitors are in clinical development both as single agents and in combination with gemcitabine [23–26], we initiated this study to evaluate the relative efficacies of CHK1 and WEE1 inhibitors for sensitizing pancreatic cancer cells to gemcitabine. Furthermore, we sought to establish a marker of response that could be incorporated into future clinical trials. We found that, with the exception of the HR-defective Capan1 cells which were relatively insensitive to MK8776, the pancreatic cancer cell lines in our panel were similarly sensitized to gemcitabine by either CHK1 or WEE1 inhibition by MK8776 or AZD1775, respectively. The abilities of either the CDK1/2 inhibitor roscovitine or exogenous nucleosides to prevent MK8776 or AZD1775-mediated chemosensitization, however, were both inhibitor-dependent and highly variable among cell lines, suggesting the importance of CDK-dependent aberrant origin firing and the resulting nucleotide exhaustion to gemcitabine chemosensitization is not universal. Despite these mechanistic differences, we found that the high-intensity, pan-nuclear γH2AX staining associated with replication stress best correlated with gemcitabine chemosensitization by either MK8776 or AZD1775.
Results
Comparison of gemcitabine chemosensitization by CHK1 and WEE1 inhibitors
To begin to compare the relative abilities of CHK1 and WEE1 inhibitors to sensitize cells to gemcitabine, we first determined the sensitivities of a panel of pancreatic cancer cell lines to either MK8776 or AZD1775-induced cytotoxicity (Suppl. Figure 1). Consistent with previous reports, 24 h MK8776 was relatively non-toxic in each of the cell lines examined [27]. There was, however, variability in sensitivity to AZD1775, with IC50 values ranging from 134 ± 24 nM in Capan1 cells to 890 ± 77 nM in MiaPaCa2 cells. The sensitivity of Capan1 cells to AZD1775 may be attributable to the presence of a BRCA2 mutation resulting in HR deficiency [21,28]. We next assessed the abilities of MK8776 and AZD1775 to sensitize cells to moderately toxic concentrations of gemcitabine. Using a previously optimized schedule of gemcitabine followed by CHK1 or WEE1 inhibitor (Figure 1(a)) [20], we found that MK8776 and AZD1775 each produced similar levels of gemcitabine chemosensitization in MiaPaCa2, BxPC3, AsPC1 and Panc1 cells (Figure 1(b-e)), although in MiaPaCa2 and Panc1 cells, AZD1775 demonstrated greater potency than MK8776. In contrast, Capan1 cells were only minimally sensitized by MK8776, while sensitization by AZD1775 was comparable to that achieved in other cell lines (Figure 1(f)). Western blot analysis of phospho-CHK1 (Ser345), a biomarker for CHK1 inhibition and the DNA damage response [20], confirmed that MK8776 inhibited CHK1 in Capan1 cells (Suppl. Figure 2). Of note, sensitivity to single agent cytotoxicity did not predict gemcitabine chemosensitization. For example, while MiaPaCa2 cells were relatively resistant to AZD1775-induced cytotoxicity, they were sensitized to gemcitabine by as little as 10 – 25 nM AZD1775. With the exception of Capan1 cells, in which MK8776 had minimal effects on survival, similarly chemosensitizing concentrations of each inhibitor were used in subsequent experiments.
Figure 1.
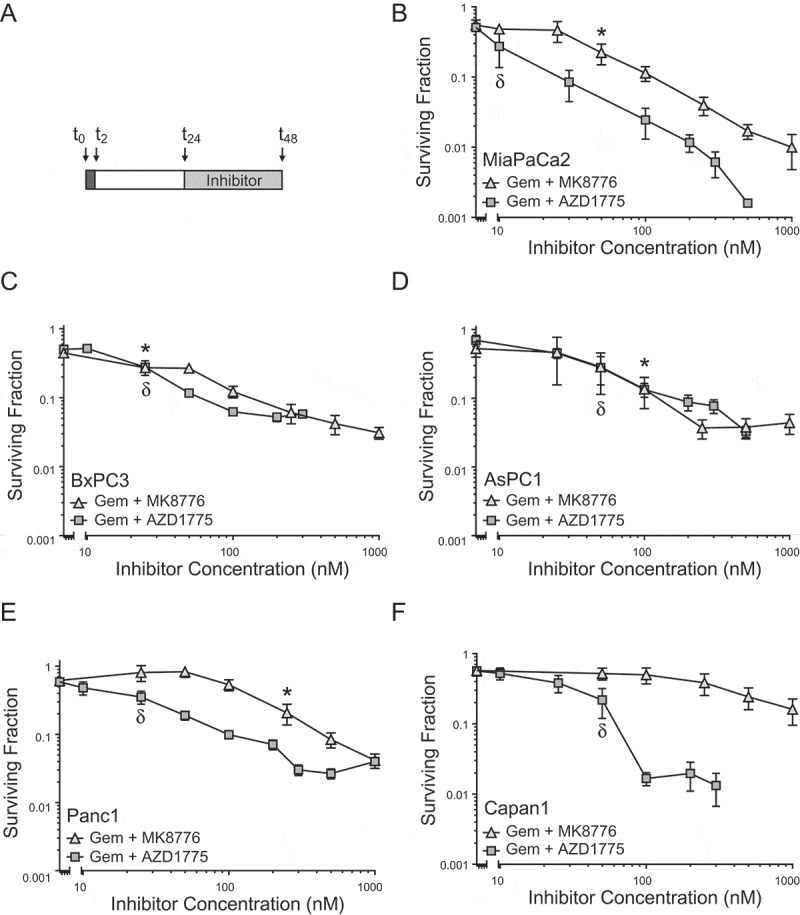
Comparison of gemcitabine chemosensitization by either MK8776 or AZD1775 in pancreatic cancer cells. Cells treated with equitoxic concentrations of gemcitabine from t0 – t2 (250 nM, MiaPaCa-2, BxPC3 and Panc1; 500 nM, AsPC1 or 50 nM, Capan1) and either MK8776 or AZD1775 from t24 – t48 as illustrated (A) were assayed for clonogenic survival (B-E). Data presented are the mean ± SEM of n = 2–6 independent experiments and are normalized to the plating efficiency of cells treated with inhibitor alone. The minimum concentrations of either MK8776* or AZD1775δ required to produce statistically significant gemcitabine chemosensitization are indicated (P < 0.05, one-way ANOVA).
Figure 2.
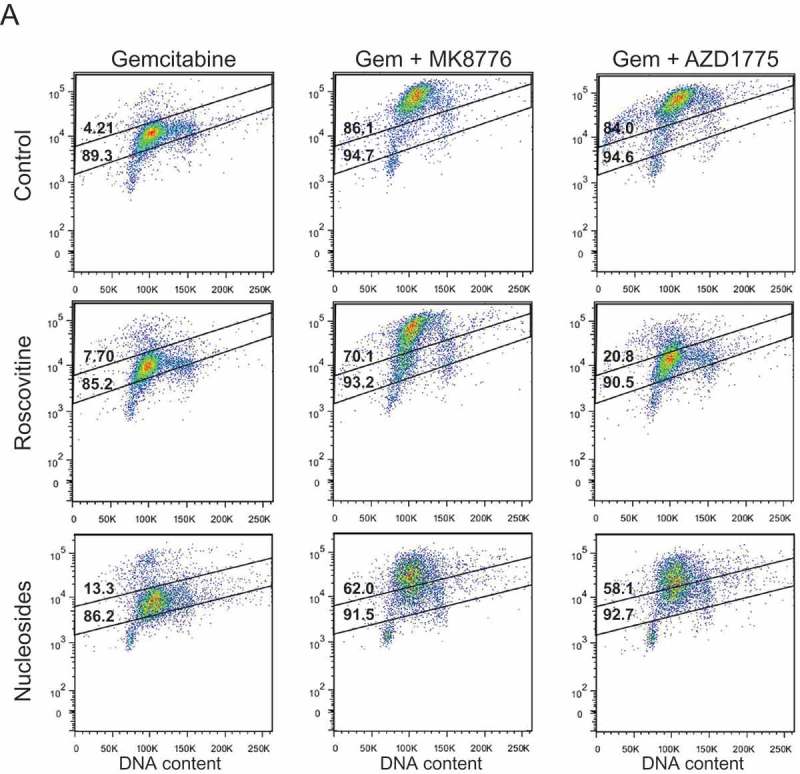
The effects of roscovitine and exogenous nucleosides on MK8776 or AZD1775-mediated high-intensity γH2AX-staining in gemcitabine-treated pancreatic cancer cells. MiaPaCa2 cells were treated as illustrated in Figure 1(a) with the addition of either 20 μM roscovitine or exogenous nucleosides during inhibitor treatment, collected 30 h post-gemcitabine and assayed for γH2AX by flow cytometry (A). In each representative dot plot, the lower number is the total percentage of cells in the population considered γH2AX-positive, as defined by the larger gate, while the upper number is the percentage of cells with a high-intensity γH2AX-staining pattern, as defined by the upper gate. Cells treated with gemcitabine ± MK8776 or AZD1775 ± roscovitine or nucleosides were collected 30 or 48 h post-gemcitabine and assayed for γH2AX by flow cytometry (B – F). Data presented are the mean ± SEM from 3–8 independent experiments. Conditions that significantly attenuated high-intensity (symbols in bars) or total (symbols above bars) γH2AX staining compared to gemcitabine + either MK8776 or AZD1775 alone are indicated (*p < 0.05, one-way ANOVA). Significant differences between the effects of roscovitine, compared to the effects of nucleosides, on γH2AX staining are also indicated (δp<0.05, one-way ANOVA).
Contribution of DNA replication stress to gemcitabine chemosensitization by CHK1 and WEE1 inhibitors
The therapeutic activities of gemcitabine, MK8776 and AZD1775 have each been associated (in part) with replication stress; gemcitabine through its mis-incorporation into DNA [29], and MK8776 and AZD1775 through inhibition of CHK1 or WEE1, respectively, resulting in CDK2 hyperactivation, aberrant origin firing and subsequent nucleotide depletion [6,7,27]. To evaluate the relative contribution of replication stress resulting from CDK2 hyperactivation to gemcitabine chemosensitization by either MK8776 or AZD1775, we first assessed the effects of the CDK1/2 inhibitor roscovitine on the abilities of these inhibitors to sensitize cells to gemcitabine. While roscovitine alone had no effect on gemcitabine cytotoxicity (Suppl. Figure 3), concurrent roscovitine prevented MK8776-mediated gemcitabine chemosensitization in three of five pancreatic cancer cell lines (BxPC3, AsPC1 and Capan1; Table 1) and partially protected Panc1 cells from MK8776-mediated sensitization. In addition, with the exception of a partial effect in MiaPaCa2 cells, roscovitine prevented AZD1775-mediated gemcitabine chemosensitization. These results are consistent with previous studies suggesting CDK1/2 hyperactivity is a significant factor in gemcitabine chemosensitization by either CHK1 [30] or WEE1 inhibitors [31] in some, but not all, cell lines.
Figure 3.
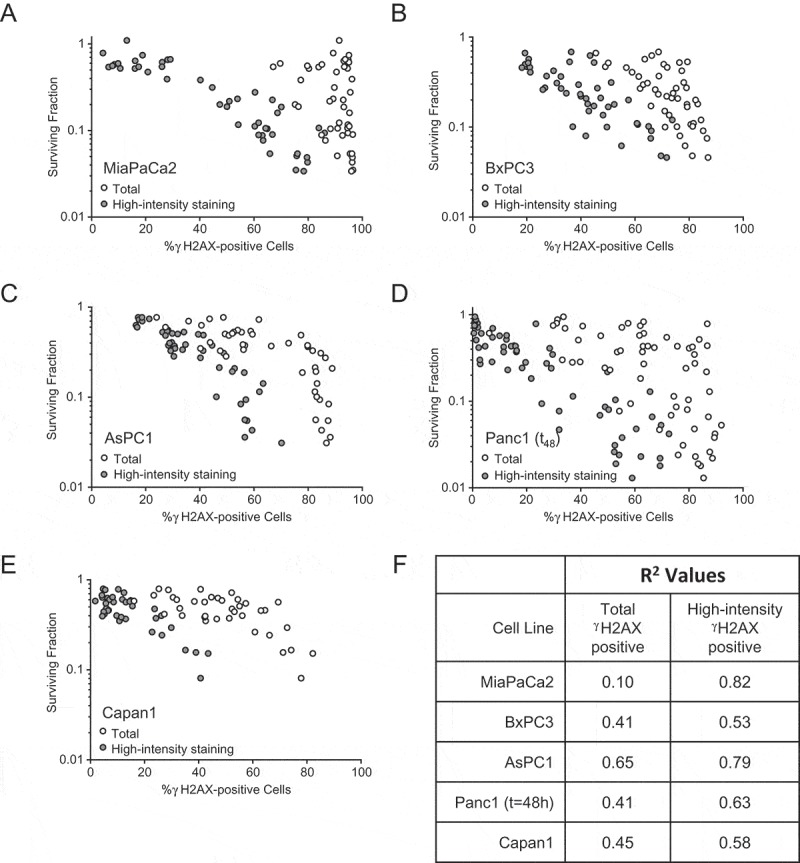
Correlation between high-intensity γH2AX staining and gemcitabine-sensitization by MK8776 or AZD1775. Cells treated as described in Figure 1(a) were collected either 30 h or 48 h post-gemcitabine and assayed both for clonogenic survival and for γH2AX staining intensity by flow cytometry. Circles represent a single experimental sample for each data set (total or high-intensity γH2AX staining) from one of at least 8 independent experiments (A-E). Sample conditions include gemcitabine alone, gemcitabine + MK8776 ± roscovitine or nucleosides, and gemcitabine + AZD1775 ± roscovitine or nucleosides. R2 values calculated for survival vs. total γH2AX staining and survival vs. high-intensity γH2AX staining are tabulated in (F).
Table 1.
Clonogenic survival of pancreatic cancer cells treated with Gem + MK8776 or Gem + AZD1775 concurrent with roscovitine or exogenous nucleosides. Cells treated under equitoxic conditions with gemcitabine followed by either MK8776 or AZD1775 ± nucleosides or 20 μM roscovitine (as illustrated in Figure 1A) were trypsinized and collected at 30 h (MiaPaCa2, BxPC3, AsPC1, Capan1) or 48 h post-Gem (Panc1) (6 or 24 h MK8776 or AZD1775 ± nucleosides or roscovitine, respectively) and assayed for drug-induced loss of clonogenicity. Non-toxic, similarly sensitizing concentrations of each inhibitor were used: 500 nM MK8776 (MiaPaCa2, BxPC3, Panc1, Capan1), 250 nM MK8776 (AsPC1), 200nM AZD1775 (MiaPaCa2, AsPC1, Panc1) or 100nM AZD1775 (BxPC3, Capan1). Data are the mean clonogenic survival ± SEM (n = 3–7; p < 0.05, one-way ANOVA) and are normalized to the plating efficiency for either MK8776 alone, AZD1775 alone or the combinations of MK8776 or AZD1775 with roscovitine or exogenous nucleosides. Two-way analysis of variance was used to test for statistically significant differences in nucleoside or roscovitine protection from chemosensitization by AZD1775 compared to MK8776 (p < 0.05¥). Under these treatment conditions, roscovitine was not toxic, and did not significantly affect Gem-induced loss of clonogenicity (Suppl. Figure 3).
| Cell Line | MiaPaCa2 | BxPC3 | AsPC1 | Panc1 (t = 48h) |
Capan1 |
|---|---|---|---|---|---|
| Gemcitabine | 0.65 ± 0.04 | 0.44 ± 0.06 | 0.53 ± 0.03 | 0.49 ± 0.06 | 0.61 ± 0.02 |
| Gem + MK8776 | 0.07 ± 0.01* | 0.08 ± 0.01* | 0.13 ± 0.03* | 0.05 ± 0.01* | 0.38 ± 0.03* |
| Gem + MK8776 + roscovitine | 0.20 ± 0.01*¥ | 0.36 ± 0.07ɸ | 0.56 ± 0.08ɸ | 0.28 ± 0.05*ɸ | 0.66 ± 0.06ɸ |
| Gem + MK8776 + nucleosides | 0.19 ± 0.02*¥ | 0.24 ± 0.04 | 0.29 ± 0.05* | 0.34 ± 0.03ɸ | 0.66 ± 0.06ɸ |
| Gem + AZD1775 | 0.08 ± 0.01* | 0.10 ± 0.01* | 0.07 ± 0.01* | 0.03 ± 0.01* | 0.17 ± 0.03* |
| Gem + AZD1775 + roscovitine | 0.48 ± 0.05*δ¥ | 0.45 ± 0.08δ | 0.56 ± 0.07δ | 0.41 ± 0.06δ | 0.60 ± 0.05δ |
| Gem + AZD1775 + nucleosides | 0.32 ± 0.03*δ¥ | 0.33 ± 0.07δ | 0.31 ± 0.01*δ | 0.10 ± 0.01* | 0.55 ± 0.01δ |
Versus Control*, Gem+MK8776ɸ, Gem+AZD1775δ or Gem+MK8776 vs Gem+AZD1775¥ (P < 0.05)
One limitation of the previous experiments is that roscovitine has multiple targets throughout the cell cycle, including both CDK7 and CDK9 [32], in addition to CDK1 and CDK2. We found, however, that purvalanol-A, a CDK1/2 inhibitor that does not target CDK9, largely replicated the effects of roscovitine on gemcitabine chemosensitization by either MK8776 or AZD1775 (Suppl. Figure 4A) and siRNA-mediated depletion of CDK7 had no effect on either MK8776 or AZD1775-mediated chemosensitization in Panc1 cells (Suppl. Figure 4B). Furthermore, we previously found that depletion of Cyclin B1 with siRNA and subsequent loss of Cyclin B-CDK1 activity (as indicated by inhibition of aberrant mitotic entry) did not affect gemcitabine sensitization by the CHK inhibitor AZD7762, suggesting that inhibition of CDK2, rather than CDK1, is responsible for this effect[30]. These results, combined with the results from multiple studies documenting the effects of roscovitine on either the CHK1-mediated intra-S-phase checkpoint or CHK1 and WEE1-mediated replication stress [6,7,27,33], are consistent with the hypothesis that roscovitine-mediated CDK2 inhibition is responsible for the effects of roscovitine on gemcitabine chemosensitization.
Figure 4.

Confocal immunofluorescent γH2AX staining patterns in cells treated with gemcitabine and AZD1775. A) BxPC3 cells treated as described in Figure 1(a) were collected either 2 h post-radiation (7.5 Gy) or 30 h post-gemcitabine (6 h AZD1775) and sorted by γH2AX staining intensity with flow cytometry: R1, negative; R2, positive, low-intensity; or R3, positive, high-intensity. B) The percentages of cells within each gate are given for the samples shown in (A). C) Confocal immunofluorescent images of representative focal, ring and pan-nuclear γH2AX staining patterns, labeled in green. Nuclei were co-stained with propidium iodide, shown in red. D) Sorted cells were spotted on slides and scored for focal (0–10 or >10), ring, or pan-nuclear γH2AX staining. Data are from either a single control experiment (7.5 Gy condition) or are the mean ± SD of the percentage of cells with the indicated γH2AX staining pattern (n = 2 independent experiments). The numbers of cells scored for each experimental sample are given in parentheses. No cells were recovered from the 7.5 Gy, γH2AX-positive, high-intensity gate (R3).
We next tested the hypothesis that MK8776 or AZD1775-mediated gemcitabine chemosensitization more specifically results from the nucleotide depletion and subsequent replication stress caused by aberrant CDK2 activity. We found that in some cell lines (MiaPaCa2, Panc1 and Capan1; Table 1) the magnitude of protection afforded by exogenous nucleosides concurrent with MK8776 was similar to that of roscovitine, while in others (BxPC3 and AsPC1) roscovitine was more effective than nucleoside repletion. These differences suggest that not only does the magnitude of the CDK-dependent component of gemcitabine chemosensitization after CHK1 inhibition vary between cell lines, as reflected by the range of chemoprotection afforded by roscovitine, but also the extent which that component results from nucleotide-depletion. In contrast, with the exception of Panc1 cells, nucleoside repletion significantly protected cells from AZD1775-mediated chemosensitization, and, in MiaPaCa2 cells, nucleoside repletion resulted in significantly greater protection from AZD1775 compared to MK8776-mediated chemosensitization (P < 0.05, 2-way ANOVA). Western blot analysis confirmed that both roscovitine and nucleosides rescued MK8776 and AZD1775-induced replication stress, as indicated by a substantial reduction in the levels of both phospho-RPA2(S4/S8) and phospho-RPA2(S33) (Suppl. Fig. 5). Thus, while nucleotide depletion contributes to gemcitabine chemosensitization by both CHK1 and WEE1 inhibitors, in some cell lines the relative contribution of this replication stress to overall sensitization is more significant following WEE1 inhibition.
Correlation between high intensity γH2AX staining and gemcitabine chemosensitization by CHK1 and WEE1 inhibitors
As we and others have shown, persistent replication stress resulting from either gemcitabine [34], CHK1 [7,22] or WEE1 inhibitor [21] is associated with a high-intensity, pan-nuclear staining pattern that has been proposed as a potential biomarker for therapeutic response to these drugs [30]. To further test this hypothesis, we next assessed the contribution of CDK2 hyperactivity and nucleotide depletion to high-intensity γH2AX staining in pancreatic cancer cells treated with gemcitabine and either MK8776 or AZD1775. We first established that the non-toxic concentrations of MK8776 and AZD1775 used in these experiments did not induce significant high-intensity γH2AX staining when given as single agents (Suppl. Fig 6A-B) and that neither roscovitine nor nucleosides, when given 24 h post-gemcitabine, inhibited the high-intensity γH2AX staining resulting from treatment with gemcitabine alone (Suppl. Fig. 6C). With the exception of AsPC1 cells, we found that neither MK8776 nor AZD1775 consistently increased the total number of γH2AX-positive cells after treatment with gemcitabine, in part because in cell lines such as MiaPaCa2, even a moderately toxic concentration of gemcitabine resulted in >80% of cells staining positive for γH2AX (Figure 2(a-b)). Both inhibitors did, however, significantly increase the number of S-phase cells with a high-intensity γH2AX staining pattern (Figure 2(b-f)). We also found that the ability of roscovitine or nucleosides to prevent high-intensity γH2AX staining varied across cell lines and with treatment conditions. In MiaPaCa2 cells for example, roscovitine and nucleosides had only a minor effect on high-intensity γH2AX staining in response to gemcitabine and MK8776 (Figure. 2(a,b)). This result is consistent with the minimal protection from CHK1 inhibitor-mediated sensitization conferred by either roscovitine or nucleosides in MiaPaCa2 cells. In contrast, both gemcitabine chemosensitization and high-intensity γH2AX staining induced by AZD1775 were significantly reduced in MiaPaCa2 cells treated with nucleosides or roscovitine. In other cell lines (BxPC3, AsPC1, Panc1), nucleosides and roscovitine attenuated high-intensity γH2AX staining caused by either MK8776 or AZD1775, (Figure 2(c-e)) a finding consistent with the protection from gemcitabine-chemosensitization observed across most of these treatment conditions (Table 1). Taken together, these data suggest an association between high-intensity γH2AX staining and the magnitude of gemcitabine chemosensitization by CHK1 or WEE1 inhibitor.
Figure 2.
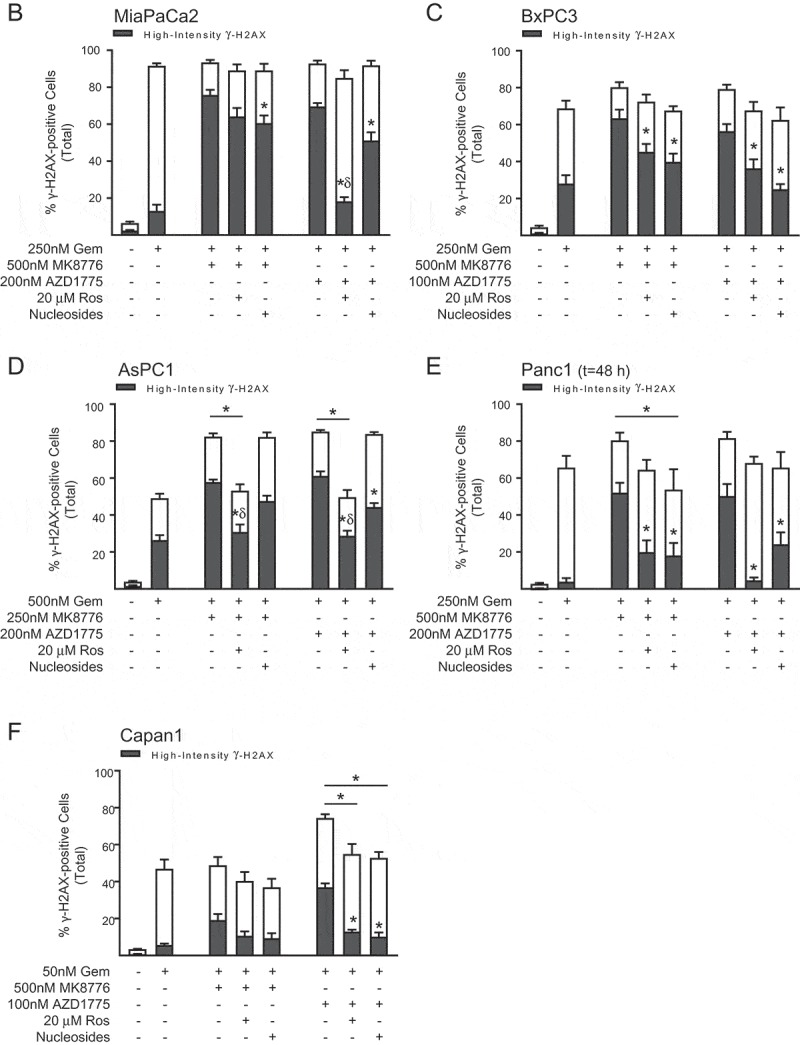
(Continued).
These results led us to evaluate whether or not high-intensity γH2AX staining may predict gemcitabine chemosensitization by CHK1 and WEE1 inhibitors. To test this hypothesis, we assessed the relationship between survival and either high-intensity or total γH2AX staining in response to gemcitabine alone or gemcitabine in combination with MK8776 or AZD1775. While the total levels of γH2AX staining showed a relatively poor correlation with survival in most cell lines (r2 0.10–0.65), high-intensity γH2AX staining better correlated with survival (r2 0.53–0.82) (Figure 3). In MiaPaCa2 cells, for example, total γH2AX staining did not correlate with survival (r2 0.10) while high-intensity γH2AX staining strongly correlated with survival (r2 0.83). These data are in the context of poor correlations between either premature mitosis (r2 0.01–0.59) or apoptosis (sub-G1 DNA content; r2 0.01–0.55) and survival (Suppl. Fig. 7). The lack of correlation between protection from checkpoint abrogation and protection from either MK8776 or AZD1775-mediated gemcitabine chemosensitization is consistent with previous studies demonstrating that the ability of either roscovitine or purvalanol-A to prevent aberrant mitotic entry does not correlate with protection from AZD7762-mediated gemcitabine chemosensitization [30]. Taken together, these data demonstrate that high-intensity γH2AX staining is a marker of DNA replication stress that predicts for gemcitabine chemosensitization by either CHK1 or WEE1 inhibition.
Characterization of pan-nuclear γH2AX staining in high intensity γH2AX cells
While flow cytometry has the advantage of being a high-throughput system with good reproducibility, it does not allow for the characterization of staining patterns within individual cells, only staining intensity. Therefore the previous data did not address part of our hypothesis; that the high-intensity γH2AX staining we measured by flow reflects the pan-nuclear γH2AX staining pattern previously associated with DNA replication stress [21,22]. In order to test this hypothesis, we conducted a flow sorting experiment to determine the pattern of γH2AX staining in cells with either low or high γH2AX staining intensity. Since there are several possible mechanisms for high-intensity γH2AX staining, including focal staining associated with DNA double strand breaks, ring staining of pre-apoptotic cells [35] or pan-nuclear staining resulting from DNA replication stress [21,22], each of these staining patterns was assessed. BxPC3 cells were treated with either radiation (as a positive control for γH2AX staining in response to DNA double strand breaks), or gemcitabine ± AZD1775 and sorted by γH2AX staining intensity as negative, low, or high-intensity (Figure 4(a)). As anticipated, under control conditions the majority of cells were negative for γH2AX staining (R1 gate) while in response to radiation there was an increase in low-intensity γH2AX staining (R2; Figure 4(b)). In response to gemcitabine alone, both low and high-intensity γH2AX staining increased with the high-intensity staining further enhanced by the combination with AZD1775. These low and high-staining populations were then microscopically analyzed for γH2AX staining patterns and classified as having a focal, ring, or pan-nuclear γH2AX staining pattern (Figure 4(c)). Consistent with radiation-induced γH2AX staining occurring as a function of DNA double strand breaks, the majority of irradiated cells within the low-intensity γH2AX staining gate were composed of cells with >10 γH2AX foci with little pan-nuclear or ring staining (Figure 4(d)). In response to gemcitabine alone, cells with >10 γH2AX foci accounted for the majority of the low-intensity stained cells, while cells with either focal (36%) and pan-nuclear γH2AX (54%) staining accounted for the majority of cells in the high-intensity γH2AX staining gate. In contrast, after treatment with gemcitabine in combination with AZD1775, the majority of cells in the high intensity population displayed a pan-nuclear γH2AX staining pattern (76%). These staining patterns are consistent with both direct immunofluorescence staining of γH2AX in gemcitabine-treated BxPC3 or MiaPaCa2 cells (Suppl. Fig. 8) and the flow data presented in Figure 2. Taken together, these data support the hypothesis that high-intensity γH2AX staining assessed by flow cytometry in response to gemcitabine and CHK1 or WEE1 inhibition is largely attributable to a pan-nuclear γH2AX staining pattern that may serve as a marker for sensitization by CHK1 and WEE1 inhibitors.
Discussion
Recent studies suggest that while CHK1 and WEE1 inhibitors have similar potential for abrogating the DNA damage response, their distinct mechanisms for inhibiting Cyclin-CDK activities, either indirectly through CDC25A (CHK1), or by direct phosphorylation of CDK1/2 (WEE1), can significantly influence therapeutic response [27]. In this study, we found that despite these mechanistic differences, both the CHK1 inhibitor MK8776 and the WEE1 inhibitor AZD1775 sensitize many pancreatic cancer cell lines to gemcitabine with similar efficacy. Furthermore, we found that gemcitabine chemosensitization by either MK8776 or AZD1775 correlates with a high-intensity, pan-nuclear γH2AX staining pattern previously associated with DNA replication stress. These results validate the further clinical development of CHK1 and WEE1 inhibitors as chemosensitizing agents, underscore the importance of DNA replication stress to their mechanisms of action, and support the use of high-intensity or pan-nuclear γH2AX staining as a pharmacodynamic marker for tumor cell sensitization by CHK1 and WEE1 inhibitors.
The finding that replication stress is a critical mechanism of sensitization to gemcitabine by CHK1 or WEE1 inhibitor is consistent with our prior study which demonstrated a dissociation between cell cycle checkpoint abrogation mediated by the CHK inhibitor AZD7762 and gemcitabine chemosensitization. In that study, we found that while either the CDK1/2 inhibitor roscovitine or depletion of Cyclin B1 with siRNA prevented AZD7762-mediated aberrant mitotic entry, these effects did not correlate with protection from chemosensitization [30]. In both studies, we have instead found that sensitization correlates with a high-intensity, pan-nuclear γH2AX staining pattern associated with DNA replication stress [7,21,22]. While previous studies have found that MK8776 or AZD1775-mediated cytotoxicity depends on their relative abilities to promote aberrant CDK2 activity, the inability of roscovitine to uniformly prevent gemcitabine chemosensitization in our studies suggests CDK-independent mechanisms may also contribute.
CHK1 and WEE1 inhibitors are each in clinical development in combination with gemcitabine for patients with advanced solid tumors. Initial Phase 1 clinical trials of either MK8776 or AZD1775 in combination with gemcitabine have demonstrated tolerability at doses with pharmacokinetic/pharmacodynamic activity [23,36]. Furthermore, our ongoing clinical trial using gemcitabine and AZD1775 alone and combined with radiation therapy has shown promising results [37]. Although it is difficult to make comparisons across studies, the response rates (partial responses and stable disease) for WEE1 inhibitor (69%) are comparable with those for CHK1 inhibitor (50%). In the context of the present study, while we initially sought to identify cell line-dependent differences in the efficacy of CHK1 and WEE1 inhibitors, we found that the overall effects on gemcitabine chemosensitization were similar, consistent with both CHK1 and WEE1 inhibitors being clinically promising agents.
Staining for γH2AX has been used as a pharmacodynamic biomarker in many clinical trials of agents such as MK8776 and AZD1775 that target the DNA damage response [26,36,38]. These trials have assessed either γH2AX foci in surrogate or tumor tissues by immunofluorescent staining, or γH2AX staining intensity by flow cytometry in ex vivo assays. While the high-intensity and pan-nuclear γH2AX staining patterns highlighted in this study have not specifically been evaluated in clinical specimens, the establishment and broad application of validated γH2AX immunofluorescence assays [39] facilitates the use of high-intensity and/or pan-nuclear γH2AX staining as a potential marker of therapeutic response in future clinical trials of CHK1 or WEE1 inhibitors. It will be important in future studies to evaluate pan-nuclear γH2AX staining, distinct from total or focal γH2AX, in clinical specimens.
In conclusion, this study demonstrates the promise of agents targeting CHK1 and WEE1 for improved therapy of pancreatic cancers and supports the continued development of agents targeting DNA replication and the DNA replication stress response as a therapeutic strategy. Furthermore, this study highlights the importance of mechanistic studies to inform the optimal biomarkers for predicting therapeutic efficacy.
Materials and methods
Cell culture and drug solutions
AsPC1, BxPC3, MiaPaCa2 and Panc1 cells were obtained from and authenticated by the American Type Culture Collection. Capan1.NEO is a clonal cell line expressing the neomycin resistance gene obtained from S. Powell (Memorial Sloan Kettering Cancer Center, New York, NY) [40]. Cells were grown in either RPMI (AsPC1 and BxPC3; Invitrogen), DMEM (MiaPaCa2 and Panc1; Invitrogen), or IMDM medium (Capan1.NEO; Invitrogen) supplemented with 10% fetal bovine serum (Premium Select; Atlanta Biologicals). Gemcitabine (Gemzar, Eli Lilly) was dissolved in PBS and stored in aliquots at −20°C. MK8776 (Merck), AZD1775 (AstraZeneca), and roscovitine (Cell Signaling) were each dissolved in DMSO and stored in aliquots at −20°C. EmbryoMAX nucleoside solution (Millipore) was used at a 1:12.5-fold dilution (8x) concurrently with either MK8776 or AZD1775.
Clonogenic survival assay
Cells were processed for clonogenic survival as previously described [41]. All experiments were carried out as illustrated in Figure 1(a): cells were treated with gemcitabine for 2 h, followed 24 h later by either MK8776 or AZD1775. MiaPaCa2, BxPC3 AsPC1 and Capan1 cells were assayed 30 h post-gemcitabine (6 h MK8776 or AZD1775) and Panc1 cells were assayed 48 h post-gemcitabine (24 h MK8776 or AZD1775). Surviving fractions represent the plating efficiency for a given drug-treated sample divided by the plating efficiency for the corresponding non-gemcitabine condition. Sensitization or protection was defined by statistically significant differences in normalized surviving fractions between drug-treated groups.
Flow cytometry
For γH2AX flow cytometry, treated cells were trypsinized, washed with PBS, and fixed in ice-cold 70% ethanol. Samples were incubated overnight at 4°C with a mouse monoclonal anti-γH2AX antibody (JBW301, EMD Millipore) diluted 1:500 in PBS buffer containing 1% fetal bovine serum (Premium Select; Atlanta Biologicals) and 0.2% Triton X-100 (Sigma), followed by incubation with a FITC-conjugated anti-mouse secondary antibody (Sigma). Samples were then stained with propidium iodide to assess total DNA content and analyzed on a FACScan flow cytometer (BD Biosciences) with FlowJo software (Tree Star). For flow sorting experiments, cells were sorted and collected according to negative (R1), low-intensity (R2) or high-intensity γH2AX staining (R3; Figure 4(a)) on a FACSAria IIU flow cytometer (BD Biosciences). Sorted cells were washed with a 0.2% Triton-X100/0.5% BSA solution in ddH2O and spotted on to positively-charged glass slides as previously described [42]. After drying, slides were washed with ice-cold methanol for 5 minutes, dried, mounted with Prolong Gold (Molecular Probes) and visualized with an Olympus IX81 FluoView confocal microscope (Olympus America) with a 60x oil objective. Fields to score were chosen at random based on propidium iodide staining.
Statistical analyses
One- or two- way analysis of variance was performed in GraphPad Prism with the Dunnett’s or Sidak’s multiple comparison test, respectively. Calculations of R2 values for correlation analyses were also performed in GraphPad Prism using data from unique samples assayed for two or more different variates (ie survival and γH2AX staining or survival and premature mitotic entry).
Funding Statement
This work was supported by AstraZeneca and the National Institutes of Health [R01CA138723, U01CA216449, R01CA163895, P50CA130810].
Acknowledgments
This work was funded by National Institute of Health grants R01CA163895, R01CA138723, P50CA130810, U01CA216449, and an AstraZeneca research grant. Research reported in this publication was supported by the National Cancer Institutes of Health under Award Number P30CA046592 by the use of the following Cancer Center Shared Resource(s): Flow Cytometry.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data can be accessed here.
References
- [1].Sorensen CS, Syljuasen RG, Falck J, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–258. [DOI] [PubMed] [Google Scholar]
- [2].O’Connell MJ, Raleigh JM, Verkade HM, et al. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997;16:545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ashwell S. Checkpoint kinase and Wee1 inhibitors as anticancer therapeutics. DNA Repair in Cancer Therapy 2012; Chapter 10:211–234.
- [4].Sorensen CS, Hansen LT, Dziegielewski J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. [DOI] [PubMed] [Google Scholar]
- [5].Krajewska M, Heijink AM, Bisselink YJ, et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2013;32:3001–3008. [DOI] [PubMed] [Google Scholar]
- [6].Beck H, Nahse-Kumpf V, Larsen MS, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Syljuasen RG, Sorensen CS, Hansen LT, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40:477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Carrassa L, Damia G. DNA damage response inhibitors: mechanisms and potential applications in cancer therapy. Cancer Treat Rev. 2017;60:139–151. [DOI] [PubMed] [Google Scholar]
- [10].Hirai H, Iwasawa Y, Okada M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. [DOI] [PubMed] [Google Scholar]
- [11].Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Morgan MA, Parsels LA, Parsels JD, et al. The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle. 2006;5:1983–1988. [DOI] [PubMed] [Google Scholar]
- [13].Koh SB, Courtin A, Boyce RJ, et al. CHK1 inhibition synergizes with gemcitabine initially by destabilizing the DNA replication apparatus. Cancer Res. 2015;75:3583–3595. [DOI] [PubMed] [Google Scholar]
- [14].Hauge S, Naucke C, Hasvold G, et al. Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage in S-phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget. 2017;8:10966–10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gonzalez Besteiro MA, Gottifredi V. The fork and the kinase: a DNA replication tale from a CHK1 perspective. Mutat Res Rev Mutat Res. 2015;763:168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Techer H, Koundrioukoff S, Carignon S, et al. Signaling from Mus81-Eme2-dependent DNA damage elicited by Chk1 deficiency modulates replication fork speed and origin usage. Cell Rep. 2016;14:1114–1127. [DOI] [PubMed] [Google Scholar]
- [17].Speroni J, Federico MB, Mansilla SF, et al. Kinase-independent function of checkpoint kinase 1 (Chk1) in the replication of damaged DNA. Proc Natl Acad Sci U S A. 2012;109:7344–7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Petermann E, Woodcock M, Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci U S A. 2010;107:16090–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Carrassa L, Chila R, Lupi M, et al. Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012;11:2507–2517. [DOI] [PubMed] [Google Scholar]
- [20].Parsels LA, Qian Y, Tanska DM, et al. Assessment of chk1 phosphorylation as a pharmacodynamic biomarker of chk1 inhibition. Clin Cancer Res. 2011;17:3706–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Aarts M, Bajrami I, Herrera-Abreu MT, et al. functional genetic screen identifies increased sensitivity to WEE1 inhibition in cells with defects in fanconi anemia and HR pathways. Mol Cancer Ther. 2015;14:865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gagou ME, Zuazua-Villar P, Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell. 2010;21:739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leijen S, Van Geel RM, Pavlick AC, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34:4371–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Calvo E, Braiteh F, Von Hoff D, et al. Phase I study of CHK1 inhibitor LY2603618 in combination with gemcitabine in patients with solid tumors. Oncology. 2016;91:251–260. [DOI] [PubMed] [Google Scholar]
- [25].Daud AI, Ashworth MT, Strosberg J, et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol. 2015;33:1060–1066. [DOI] [PubMed] [Google Scholar]
- [26].Do K, Wilsker D, Ji J, et al. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol. 2015;33:3409–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sakurikar N, Thompson R, Montano R, et al. A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK-8776 as monotherapy due to CDK2 activation in S phase. Oncotarget. 2016;7:1380–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hattori H, Skoulidis F, Russell P, et al. Context dependence of checkpoint kinase 1 as a therapeutic target for pancreatic cancers deficient in the BRCA2 tumor suppressor. Mol Cancer Ther. 2011;10:670–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Plunkett W, Huang P, Searcy CE, et al. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol. 1996;23:3–15. [PubMed] [Google Scholar]
- [30].Parsels LA, Tanska DM, Parsels JD, et al. Dissociation of gemcitabine chemosensitization by CHK1 inhibition from cell cycle checkpoint abrogation and aberrant mitotic entry. Cell Cycle. 2016;15:730–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kreahling JM, Foroutan P, Reed D, et al. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS One. 2013;8:e57523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cicenas J, Kalyan K, Sorokinas A, et al. Roscovitine in cancer and other diseases. Ann Transl Med. 2015;3:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hughes BT, Sidorova J, Swanger J, et al. Essential role for Cdk2 inhibitory phosphorylation during replication stress revealed by a human Cdk2 knockin mutation. Proc Natl Acad Sci U S A. 2013;110:8954–8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007;6:1239–1248. [DOI] [PubMed] [Google Scholar]
- [35].Solier S, Pommier Y. The nuclear gamma-H2AX apoptotic ring: implications for cancers and autoimmune diseases. Cell Mol Life Sci. 2014;71:2289–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Daud A, Springett GM, Mendelson DS, et al. A phase I dose-escalation study of SCH 900776, a selective inhibitor of checkpoint kinase 1 (CHK1), in combination with gemcitabine (Gem) in subjects with advanced solid tumors. ASCO Meet Abstr. 2010;28:3064. [Google Scholar]
- [37].Cuneo KC, Morgan M, Schipper MJ, et al. Abstract CT035: A dose escalation trial of the WEE1 inhibitor AZD1775, gemcitabine, and concurrent radiation in locally advanced pancreatic cancer:. Proceedings of the American Association for Cancer Research Annual Meeting 2016 2016; Abstract no. CT035. [Google Scholar]
- [38].Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. [DOI] [PubMed] [Google Scholar]
- [39].Kinders RJ, Hollingshead M, Lawrence S, et al. Development of a validated immunofluorescence assay for gammaH2AX as a pharmacodynamic marker of topoisomerase I inhibitor activity. Clin Cancer Res. 2010;16:5447–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xia F, Taghian DG, DeFrank JS, et al. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc Natl Acad Sci U S A. 2001;98:8644–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lawrence TS. Ouabain sensitizes tumor cells but not normal cells to radiation. Int J Radiat Oncol Biol Phys. 1988;15:953–958. [DOI] [PubMed] [Google Scholar]
- [42].Johansson P, Muslimovic A, Hultborn R, et al. In-solution staining and arraying method for the immunofluorescence detection of gammaH2AX foci optimized for clinical applications. Biotechniques. 2011;51:185–189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.