

ABSTRACT
Double-strand breaks (DSBs) are repaired through two major pathways, homology-directed recombination (HDR) and non-homologous end joining (NHEJ). The choice between these two pathways is largely influenced by cell cycle phases. HDR can occur only in S/G2 when sister chromatid can provide homologous templates, whereas NHEJ can take place in all phases of the cell cycle except mitosis. Central to NHEJ repair is the Ku70/80 heterodimer which forms a ring structure that binds DSB ends and serves as a platform to recruit factors involved in NHEJ. Upon completion of NHEJ repair, DNA double strand-encircling Ku dimers have to be removed. The removal depends on ubiquitylation and proteasomal degradation of Ku80 by the ubiquitin E3 ligases RNF8. Here we report that RNF8 is a substrate of APCCdh1 and the latter keeps RNF8 level in check at DSBs to prevent premature turnover of Ku80.
KEYWORDS: Anaphase promoting complex, Cdh1, double-strand breaks, RNF8, NHEJ repair
Introduction
Double-strand breaks (DSBs) are the most dangerous form of DNA damage. They are repaired through two pathways, homology-directed recombination (HDR) and non-homologous end joining (NHEJ) [1–3]. The choice between these two pathways is largely influenced by cell cycle phases, with NHEJ primarily occurring in G1 and HDR in S/G2 when homologous sequences are available from sister chromosomes [3,4]. Central to NHEJ repair is the Ku70/80 heterodimer which forms a ring structure that binds DSB ends and serves as a platform to recruit factors involved in NHEJ [5,6]. Upon completion of NHEJ repair, DNA double strand-encircling Ku dimers have to be removed [5,6]. Two removal mechanisms are believed at work. One involves nicking of DNA to allow Ku escape as suggested by findings in yeasts [7–9]. Another involves ubiquitylation and proteasomal degradation of Ku80. The ubiquitin E3 ligase RNF8 was shown to ubiquitinate Ku80 and promote removal of Ku complex from DNA in mammalian cells [10].
The anaphase promoting complex/cyclosome (APC/C) is an E3 ubiquitin ligase critical for mitotic progression [11]. It utilizes two adaptor proteins, Cdc20 and Cdh1 (Fzr1) to bring in substrates for ubiquitylation (K11-linked) [12]. Besides the function in cell cycle regulation, APCCdh1 has been implicated in DNA damage response [13–16]. We showed recently that this E3 ligase helps HDR repair by protecting K-63 linked ubiquitin chains essential for the recruitment of BRCA1 at DSBs [17]. In addition, our work also implicated APCCdh1 in NHEJ repair [17,18], but how exactly the E3 ligase is involved in NHEJ repair remained unclear. Here we report that APCCdh1 regulates the expression of RNF8 in G1 and keeps RNF8 level in check at DSBs to prevent premature turnover of Ku80.
Results
Cdh1 is required for efficient NHEJ repair
We reported previously that Cdh1 plays an important role in HDR repair of DSBs by protecting ubiquitin signaling and promoting BRCA1 recruitment [17]. Interestingly, depletion of Cdh1 also resulted in a strong suppression of NHEJ repair efficiency (Figure 1(a)), although the suppression was not as strong as that caused by depletion of either RNF8 or Ku80, two known NHEJ factors. The knockdown efficiency was shown in supplemental Figure 1(a and b). On the other hand, the depletion of RNF138, an ubiquitin E3 ligase implicated in ubiquitylation of Ku80 in S/G2 cells [19], had little effect on NHEJ repair in G1. Consequently, the cells depleted of these factors were sensitized to ionizing radiation (Figure 1(b)). In agreement with above results, the depletion of Cdh1 showed a weaker sensitization than that of RNF8 or Ku80. To minimize cell cycle effect, the cells were synchronized to G1 (Figure 1(c)) with lovastatin treatment prior to radiation.
Figure 1.
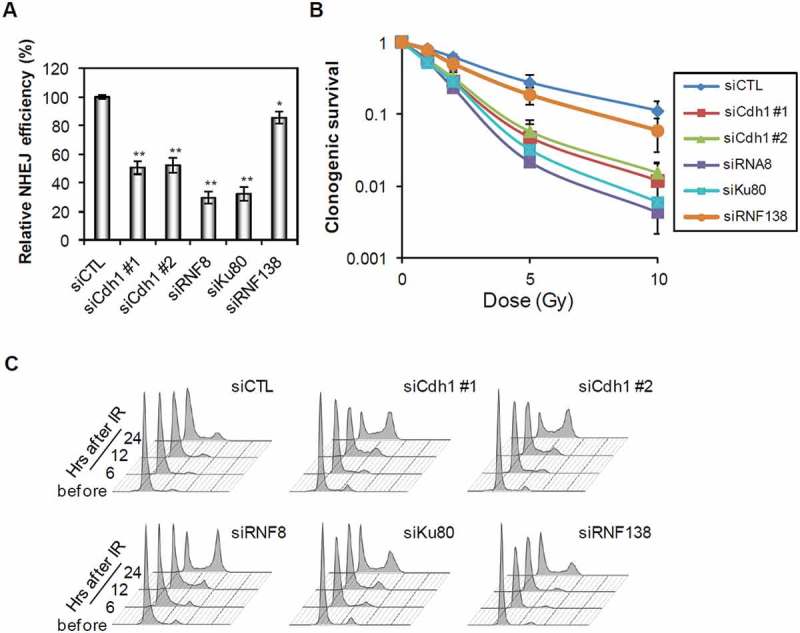
Inhibition of Cdh1 suppresses NHEJ in G1 cells. (A) Quantification of NHEJ assay in cells transfected with indicated siRNAs. U2OS cells were transfected with indicated siRNAs and treated with lovastatin. The NHEJ efficiency was normalized to that in control (siCTL) cells. (B) Survival of U2OS cells transfected with the indicated siRNAs after irradiation. Twenty-four hours after transfection of the siRNAs, the cells were treated with lovastatin for 36 h and then cultured in fresh media for 2 weeks after indicated doses of irradiation. (C) Cell cycle analysis of the cells transfected with indicated siRNAs. U2OS cells were transfected with indicated siRNAs and next day cells were treated with lovastatin for 36 h and then irradiated. Cell cycle distribution was measured at the indicated time points after release from lovastatin arrest and IR. Error bars indicate SEM from three independent experiments. ** (in A) indicates P < 0.001 (student’s t-test).
Cdh1 targets RNF8 in G1
Since APCCdh1 functions as an ubiquitin E3 ligase that promotes degradation of a large number of proteins, we reasoned it might target a factor important for NHEJ repair. As shown above, RNF8 as a key regulator of DNA damage response is critical for NHEJ repair. It was also reported to be involved in the regulation of mitotic exit as its expression reached the highest point in mitosis and declined upon mitotic exit [20]. Indeed, we observed a similar behavior of RNF8 protein expression during the cell cycle (Figure 2(a and b)), while its mRNA levels (and Cdh1 mRNA levels) remained steady throughout cell cycle (supplemental Figure 1C and D). RNF8 protein levels declined upon mitotic exit but re-accumulated upon entry into S phase, whereas Ku80 did not show such an expression pattern. Notably, RNF8 downregulation followed a pattern similar to SKP2, Cyclin A and Cyclin B1, which are known APCCdh1 substrates. Thus, it is highly likely that RNF8 might also be subjected to APCCdh1 regulation. To that end, we found that RNF8 protein levels as well as those of cyclin A and cyclin B1, increased after Cdh1 or APC3 depletion in HeLa cells (Figure 2(c)), while there were no changes in the mRNA expression levels of RNF8 (supplemental Figure 2A). On the other hand, depletion of RNF8, an ubiquitin ligase by itself, didn’t change Cdh1 levels (supplemental Figure 2B, C and D), indicating that RNF8 does not regulate Cdh1.
Figure 2.
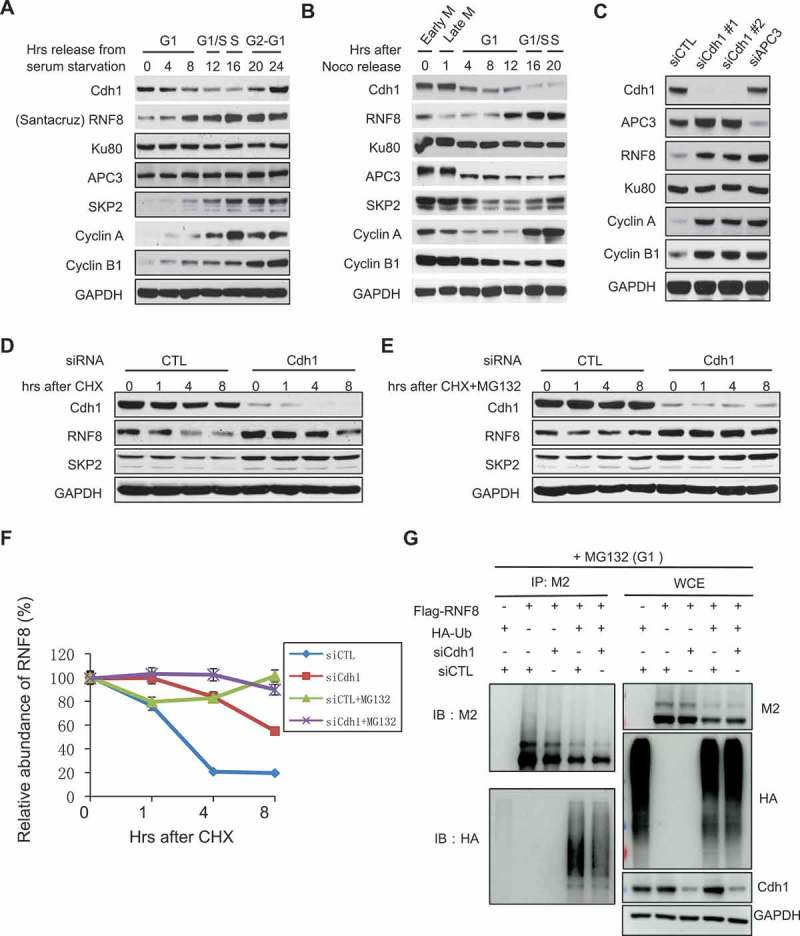
Cdh1 targets RNF8 in G1. (A, B) Differential expression of RNF8 during the cell cycle. (A) HeLa cells were cultured in media containing 0.5% of FBS for 48 h and released from serum starvation. Cells were harvested at indicated time points and western blot analysis was performed to probe the indicated proteins. (B) U2OS cells were treated with nocodazole for 16 h and released from the G2/M arrest. Then cells were harvested at the indicated time points for western blotting. (C) Regulation of RNF8 stability by Cdh1. U2OS cells were transfected with indicated siRNAs and cultured for 72 h before harvest. (D, E, F) Stability of RNF8 in Cdh1-depleted cells. U2OS cells were treated with indicated siRNAs for 72 h and then treated with either cycloheximide (D) or cycloheximide and MG132 (E) for indicated times. (F) Quantification of RNF8 levels shown in (E, F). (G) Ubiquitination assay of RNF8 in the presence or absence of Cdh1 in G1 cells. HEK293 cells were transfected with indicated siRNA or constructs and treated with lovastatin. Cell lysates were prepared as described in methods for the analysis of RNF8 ubiquitination.
To further demonstrate the regulation of RNF8 by Cdh1, we measured the half-life of RNF8 in control and Cdh1-depleted cells. RNF8 was degraded much faster in cells transfected with control siRNA than in Cdh1 siRNA-transfected cells (Figure 2(d and f)). More importantly, the degradation of RNF8 was inhibited by the 26S proteasome inhibitor MG132 (Figure 2(a and f)). In addition, we measured the levels of ubiquitylation on RNF8 in cells. As shown in Figure 2(g), RNF8 was ubiquitylated in cells and the ubiquitylation was severely compromised in Cdh1-depleted cells. These data strongly suggest that Cdh1 targets RNF8 for ubiquitylation-mediated degradation.
Cdh1 regulates RNF8 accumulation at DBSs
We showed previously that APCCdh1 was recruited to DNA damage sites and catalyzed the formation of K11-linked ubiquitin chains on its substrates such as USP1 [17]. As shown in Figure 3(a), K11-linked Ub chains could be detected at micro-irradiation induced DNA damage sites. The intensity of K11-linked Ub signal diminished in cells transfected with Cdh1, RNF8 or ATM siRNAs. However, as expected, the remaining K-11 Ub chain signal was more intense in RNF8-depleted cells than in Cdh1-depleted cells (Figure 3(b)). This is likely because there are substrates other than RNF8 such as USP1 at DSB sites [13,17]. More significantly, Cdh1 knockdown markedly increased the amount of RNF8 recruited to damage sites in both G1 and S/G2 cells (Figure 3(c and d), supplemental Figure 3A and B).
Figure 3.
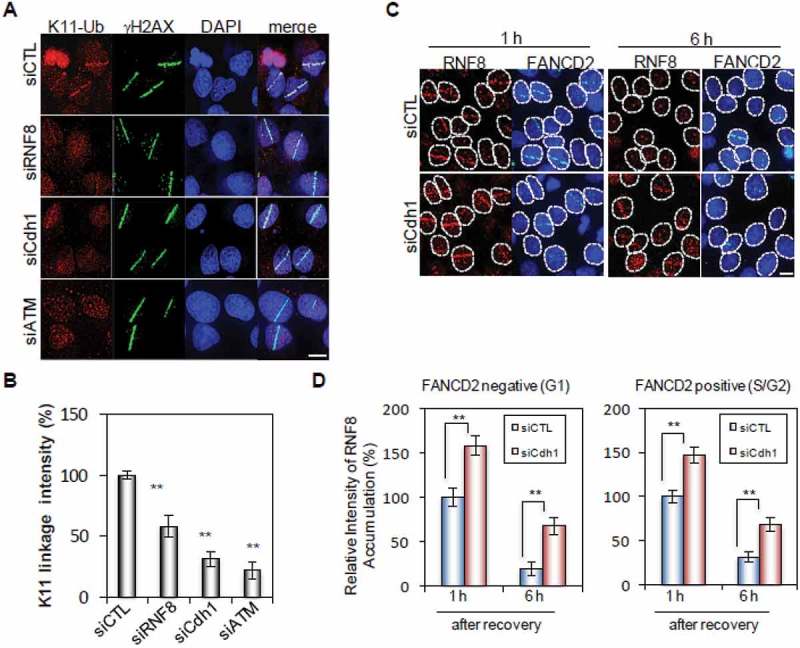
Cdh1 regulates RNF8 accumulation at DSBs. (A, B) The accumulation of K11-linked poly-ubiquitin at sites of DNA damage. (A) U2OS cells were transfected with indicated siRNAs for 72 h, microirradiated and processed 30 min later for immunostaining of K11-Ub and γH2AX. (B) Quantification of K11 linkage intensity at microirradiated regions (>110 cells quantificated). (C) The accumulation of RNF8 at sites of DNA damage. U2OS cells were transfected with indicated siRNAs and immunostaining of RNF8 and FANCD2 was performed 1 h and 6 h after microirradiation. (D) Quantification of RNF8 intensity at microirradiated regions in G1 and S/G cells (>100 cells quantificated). Error bars indicate SEM from three independent experiments. ** (in B) indicates P < 0.001 (student’s t-test). Scale bars represent 10 μm.
Cdh1 protects Ku80 at DSBs
The dynamics of Ku80 at DSB sites is important for its function in promoting NHEJ. Previous reports suggest that RNF8 mediates Ku80 ubiquitylation and removal from DSBs to facilitate NHEJ repair [10]. Consistent with that, we observed that Ku80 accumulation at damage sites was stronger and longer in RNF8-depleted cells than in control cells (Figure 4(a and b)). Strikingly, depletion of Cdh1 dramatically shortened the retention time of Ku80 at damage sites, which could be extended again with simultaneous silencing of RNF8 (Figure 4(a and b)). The cells in the experiment was synchronized to G1 with lovastatin treatment (Figure 4(c)) and the knockdown efficiency was shown in Figure 4(d).
Figure 4.
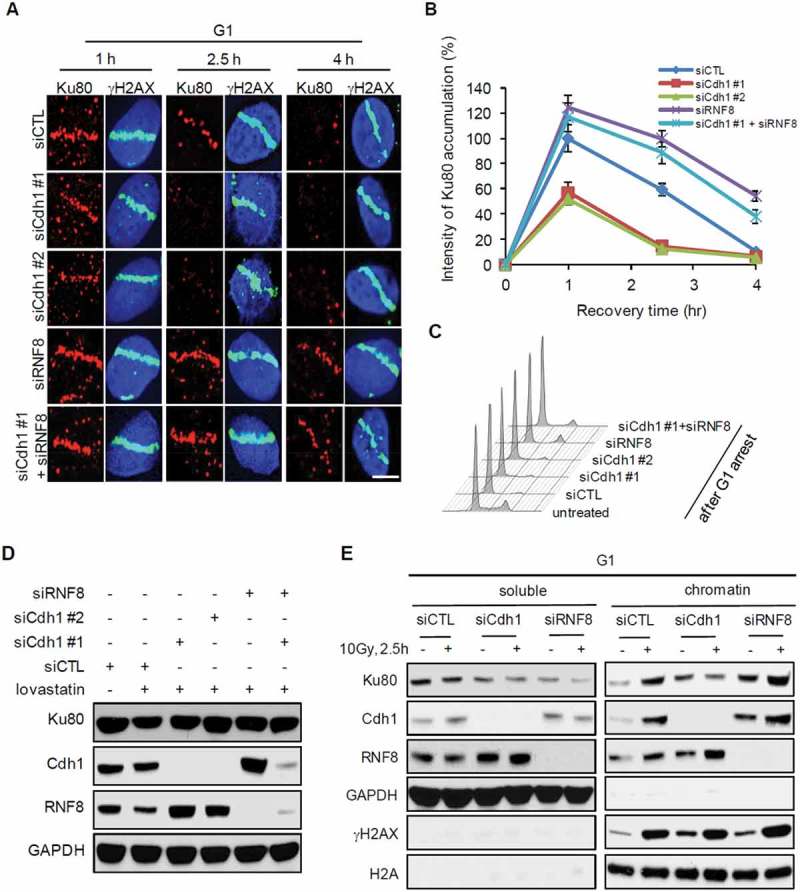
Cdh1 protects Ku80 at DSBs in G1 phase. (A, B) The accumulation of Ku80 at sites of DNA damage. (A) U2OS cells were transfected with indicated siRNA and next day treated with lovastatin for 2 days. Then, cells were subjected to microirradiation and immunostaining of Ku80 and γH2AX was performed at 1 h, 2.5 h and 4 h after microirradiation. (B) Quantification of Ku80 intensity at microirradiated regions shown in (A) (n > 80). (C) Analysis of cell cycle arrest in cells treated with lovastatin. U2OS cells were treated as indicated in (A) and then cell cycle analysis was performed. (D) The efficiency of siRNA-mediated target gene knockdown was shown by Western Blot analysis. (E) Regulation of Ku80 dissociation from chromatin after DNA damage. U2OS cells were transfected with indicated siRNAs and treated with lovastatin. After isolation of soluble and chromatin fractions, the expression of indicated proteins was measured by Western blot analysis. Error bars indicate SEM from three independent experiments. Scale bars represent 10 μm.
To extend above microscopic observation, we measured the levels of Ku80 bound to chromatin before and after DNA damage (IR). In G1 cells, DNA damage induced enrichment of Ku80 at chromatin (Figure 4(e)). The enrichment diminished in Cdh1-depleted cells but was enhanced by RNF8 depletion (Figure 4(e)). Taken together, these results suggest that APCCdh1 keeps RNF8 levels in check to protect Ku80 at damage sites, and therefore indirectly helps NHEJ repair (Figure 5).
Figure 5.
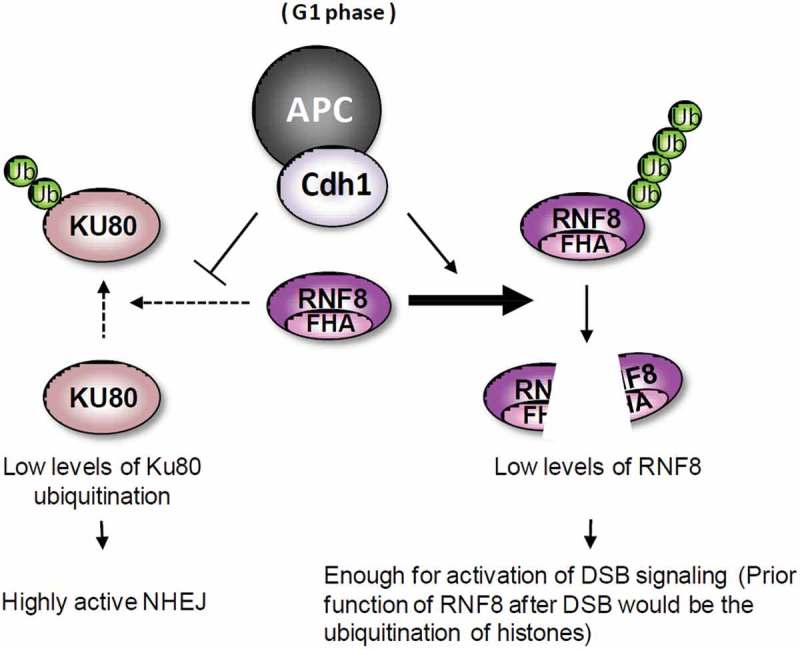
A model for the regulation of NHEJ by APCCdh1 in G1 cells. The DSBs in G1 cells are mainly repaired by Ku heterodimer-mediated NHEJ. RNF8-mediated polyubiquitination of histones activates the DNA damage signaling. However, RNF8 also induces polyubiquitination of Ku80, promoting dissociation of Ku80 from DSBs. In G1 phase, the destruction of RNF8 by active APCCdh1 complex suppresses the dissociation of Ku80 from DSBs, maintaining NHEJ in a highly active state.
Discussion
We present evidence here suggesting a requirement for APCCdh1 in efficient NHEJ repair of DSBs by direct regulation of RNF8, a key regulator of DNA damage response during NHEJ repair. Mechanistically, our data supports a model whereby APCCdh1 restrain RNF8 levels at damage sites in G1 and indirectly protects Ku80 from precocious removal by RNF8 (Figure 5). APCCdh1 contributes to a diverse array of cellular and organismal functions [21–28] including DNA damage response and damage repair. Here we added yet another role of APCCdh1, regulating NHEJ repair, to the growing list of roles played by APCCdh1. Our results strongly suggest that RNF8 is a substrate of APCCdh1 both in G1 and at DSB sites. However, we were unable to locate either a KEN box or a RXXL motif in RNF8 that could mediate its ubiquitylation by APCCdh1. The E3 complex might act on yet an uncharacterized motif of RNF8 or indirectly on RNF8.
Apparently, the level of RNF8 has to be fine regulated at DNA damage sites in order to properly control Ku80 dynamics. It is thought that Ku80 ubiquitylation by RNF8 is critical for Ku80 removal from the re-ligated broken ends to finish NHEJ repair. It is unknown what triggers the ubiquitylation process. Perhaps Ku80 is modified in some way once the ends are joined together. Or, the ubiquitylation could happen constantly at a steady rate, regardless the status of Ku complex. The latter would require precise control over RNF8 level and is supported by our findings here. In addition, we showed previously that Cdh1 was absent from DSB sites destined for NHEJ repair in S/G2 cells [17]. It is unclear what restrains RNF8’s action on Ku80 in non-G1 cells.
Experimental procedures
Cell culture
HeLa, HEK293 and U2OS cells were obtained from ATCC and maintained as recommended. HeLa and HEK293 cells were cultured in DMEM with 10% fetal bovine serum (FBS). U2OS cells were cultured in McCoy’s 5A medium with 10% FBS. All culture media were supplemented with 100 units/ml of penicillin and 100 μg/ml of streptomycin.
RNA interference
All siRNA oligonucleotides used in this study were from Sigma. The sequences of siRNAs are as follows: RNF8 #1 5ʹ-CAGAGAAGCUUACAGAUGUUUdTdT-3ʹ, RNF8 #2 5ʹ-GGACAAUUAUGGACAACAAdTdT-3ʹ, Cdh1 #1 5ʹ-UGAGAAGUCUCCCAGUCAGdTdT-3ʹ, Cdh1 #2 5ʹ-GGAUUAACGAGAAUGAGAAdTdT-3ʹ, APC3 5ʹ- CAAAAGAGCCUUAGUUUAAdTdT-3ʹ, ATM 5ʹ-CGCAUGUGAUUAAAGCAACdTdT-3ʹ, Ku80 5ʹ-AACCAGGUUCUCAACAGGCUGdTdT-3ʹ, RNF138 5ʹ-CCUGUGUCAAGAAUCAAdTdT-3ʹ. Cells were transfected with these siRNAs using Lipofectamine 2000 (Life technology) according to the manufacturer’s protocol. Cells were used for further analyses after transfection for 48–72 hrs.
Antibodies and reagents
The antibodies used in this study include: anti-Cdh1 (sc-56,312, 1:1000 for WB, Santa Cruz), anti-RNF8 (sc-271,462, 1:1000 for WB, 1:200 for IF, Santa Cruz), SKP2 (4313s, 1:1000 for WB, Cell signaling Technology), Ku80 (A302-627A-M, 1:1000 for WB, 1:200 for IF, Bethyl), APC3 (sc-13,154, 1:1000 for WB), Cyclin A (sc-751, 1:1000 for WB), GAPDH (MAB374, 1:5000 for WB, Millipore), anti-Ub K11 linkage (MABS107-I, 1:200 for IF, Millipore), anti-γH2AX (05–636, 1:250 for IF, Millipore), anti-Flag (M2, F3165, 1:10,000 for WB, Sigma), anti-HA (901,503, 1:5000 for WB, Biolegand), Cyclin B1 (4138s, 1:1000 for WB, Cell signaling Technology). Nocodazole was purchased from Sigma, Lovastatin and MG132 were purchased from Selleckchem.
Western blotting
The cells were harvested after the indicated treatments and lysed with lysis buffer (50mM Tris-HCl, 150mM NaCl, 1% Triton X-100 and 0.1% SDS), supplemented with proteintase inhibitors (Roche). The protein was loaded on SDS-PAGE gel and transferred to a PVDF membrane. Then detected with the indicated antibodies.
Ubiquitylation assay
HEK293 cells were transfected with indicated siRNA or expression constructs and treated with lovastatin. Cells were treated with MG132 (10 μM) for 6 hrs before harvested. The harvested cells were lysed with NETN buffer containing protease inhibitors (Roche). After sonication, the supernatants were incubated with anti-Flag M2 affinity beads (Sigma) for 6 hr at 4°C. The beads were then spun down, washed with the NETN lysis buffer, and prepared for Western blot analysis.
Quantitative RT-PCR
Total RNA was isolated from cells using Trizol reagent (Invitrogen). Then one microgram RNA extract was reverse transcribed using the RT reagent kit (Takara). FastStart Universal SYBR Green master mix (Roche) was used for quantitative PCR. The relative expression levels of target gene mRNA were normalized to the expression levels of GAPDH.
Laser microirradiation
Laser microirradiation was conducted as described previously [17]. Briefly, U2OS cells transfected with the indicated siRNA were seeded in a 35mm glass bottom dishes (MatTek). Cells were treated with Hoechst 33,258 (1ug ml-1, Sigma) for 2h, then exposed to the 405nm laser beam for micro-irradiation through a 60× (NA 1.4) oil microscope objective using Nikon A1R confocal microscope (Nikon). After micro-irradiation, cells were returned to culture in 37°C for the indicated time then fixed for immunofluorescence staining.
NHEJ assays
The NHEJ assay was performed as described in our previous reports [17]. Briefly, U2OS cells were transfected with the siRNAs indicated for 72h. Then each 106 cells were transfected with 5 μg of linearized pCSCMV:tdTomato (digested with BamHI) and 5μg of pEGFP-N1 plasmids. The percentage of GFP positive and Tomato positive cells was analysis by fluorescence activated cell sorter (FACS).
Immunofluorescence microscopy
The cells treated as indicated on the 35mm glass bottom dishes were washed with pre-cold PBS twice. The cells were then fixed with 4% paraformaldehyde (PFA) solution for 15 min at room temperature, permeabilized in 0.5% Triton X-100 solution for 15 min. They were blocked in 5% goat serum in PBST, and incubated with primary antibodies for one hour at 37°C. Subsequently, the cells were washed 2–3 times with PBS and incubated with secondary antibodies for one hour at 37°C. DAPI staining was performed to visualize nuclear DNA, and images were acquired on the Nikon A1R confocal microscope (Nikon).
Clonogenic survival
U2OS cells were transfected with indicated siRNAs for 48 hours. Then, the cells were exposed to the indicated doses of IR, and 300 cells from each condition were plated in triplicate and incubated at 37°C with 5% CO2 for 10 days. At end of the incubation, colonies consisting of 50 or more cells in each well were counted and the surviving fraction was calculated by the formula: (mean number of colonies)/(number of cells seeded × plating efficiency). Plating efficiency was defined as the mean number of colonies divided by the number of cells seeded for unirradiated control cells.
Funding Statement
This work was supported in part by a Chinese National Natural Science Foundation grant (# 81773032) and in part by an international collaboration grant (# 2013DFB30210) and a 973 Project grant (# 2013CB910300) from Chinese Minister of Science and Technology. PZ is supported by grants from NIH (CA116097 and CA122623). S.H. is supported by grants (# 81272488, 81472795, 81602802 and 81602680) from Chinese National Natural Science Foundation. Y.N. and M.K. are supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF-2016R1A6A3A04011000). We also thank other members of the Zhang lab for helpful discussion and support.
Acknowledgments
We thank S.-Y. Lin (MD Anderson Cancer Center) for cell lines; J. Rosen (Baylor College of Medicine) for reagents; Hisao Masai (Tokyo Metropolitan Institute of Medical Science) for U2OS-Fucci cell line; Daniel Durocher (University of Toronto) for HeLa-Fucci cell line. This work was performed with facilities and instruments in the Imaging Core of National Center for Protein Science (Beijing), the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574), the Integrated Microscopy Core at Baylor College of Medicine with funding from the NIH (HD007495, DK56338, and CA125123).
Author contributions
C.M. and K.H. performed most of the experiments; M. K., Y.N., H.L., L.T. and D.Z. contributed to experimental work; Q.Z., H.C. and S.H. contributed to experimental design and data analysis; P.Z. designed and supervised the study, secured funding, analyzed the data. All authors discussed the results and commented on the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Jackson SP, Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. [DOI] [PubMed] [Google Scholar]
- [4].Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. [DOI] [PubMed] [Google Scholar]
- [5].Fell VL, Schild-Poulter C. The Ku heterodimer: function in DNA repair and beyond. Mutat Res Rev Mutat Res. 2015;763:15–29. [DOI] [PubMed] [Google Scholar]
- [6].Kragelund BB, Weterings E, Hartmann-Petersen R, et al. The Ku70/80 ring in non-homologous end-joining: easy to slip on, hard to remove. Front Bioscience. 2016;21:514–527. [DOI] [PubMed] [Google Scholar]
- [7].Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philosophical Transactions Royal Society London Series B, Biological Sciences. 2011;366:3562–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Neale MJ, Pan J, Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu D, Topper LM, Wilson TE. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics. 2008;178:1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Feng L, Chen J. The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat Struct Mol Biol. 2012;19:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li M, York JP, Zhang P. Loss of Cdc20 causes a securin-dependent metaphase arrest in two-cell mouse embryos. Mol Cell Biol. 2007;27:3481–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jin L, Williamson A, Banerjee S, et al. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cotto-Rios XM, Jones MJ, Busino L, et al. APC/CCdh1-dependent proteolysis of USP1 regulates the response to UV-mediated DNA damage. J Cell Biol. 2011;194:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang L, Park CH, Wu J, et al. Proteolysis of Rad17 by Cdh1/APC regulates checkpoint termination and recovery from genotoxic stress. EMBO J. 2010;29:1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bassermann F, Frescas D, Guardavaccaro D, et al. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134:256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lafranchi L, De Boer HR, De Vries EG, et al. APC/C(Cdh1) controls CtIP stability during the cell cycle and in response to DNA damage. EMBO J. 2014;33:2860–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ha K, Ma C, Lin H, et al. The anaphase promoting complex impacts repair choice by protecting ubiquitin signalling at DNA damage sites. Nat Commun. 2017;8:15751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lin H, Ha K, Lu G, et al. Cdc14A and Cdc14B redundantly regulate DNA double-strand break repair. Mol Cell Biol. 2015;35:3657–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ismail IH, Gagne JP, Genois MM, et al. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat Cell Biol. 2015;17:1446–1457. [DOI] [PubMed] [Google Scholar]
- [20].Plans V, Guerra-Rebollo M, Thomson TM. Regulation of mitotic exit by the RNF8 ubiquitin ligase. Oncogene. 2008;27:1355–1365. [DOI] [PubMed] [Google Scholar]
- [21].Almeida A. Regulation of APC/C-Cdh1 and its function in neuronal survival. Mol Neurobiol. 2012;46:547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cooper KF, Strich R. Meiotic control of the APC/C: similarities & differences from mitosis. Cell Div. 2011;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Eguren M, Manchado E, Malumbres M. Non-mitotic functions of the anaphase-promoting complex. Semin Cell Dev Biol. 2011;22:572–578. [DOI] [PubMed] [Google Scholar]
- [24].Fuchsberger T, Lloret A, Vina J. New functions of APC/C ubiquitin ligase in the nervous system and its role in alzheimer’s disease. Int J Mol Sci. 2017;18:1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hu D, Qiao X, Wu G, et al. The emerging role of APC/CCdh1 in development. Semin Cell Dev Biol. 2011;22:579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li M, Zhang P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div. 2009;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Qiao X, Zhang L, Gamper AM, et al. APC/C-Cdh1: from cell cycle to cellular differentiation and genomic integrity. Cell Cycle. 2010;9:3904–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhou Z, He M, Shah AA, et al. Insights into APC/C: from cellular function to diseases and therapeutics. Cell Div. 2016;11:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.