

ABSTRACT
Sterile alpha motif and HD domain-containing protein 1 (SAMHD1) is a mammalian dNTP hydrolase that acts as a negative regulator in the efficacy of cytarabine treatment against acute myeloid leukemia (AML). However, the role of SAMHD1 in AML development and progression remains unknown. We have reported that SAMHD1 knockout (KO) in the AML-derived THP-1 cells results in enhanced proliferation and reduced apoptosis, but the underlying mechanisms are unclear. Here we show that SAMHD1 KO in THP-1 cells increased PI3K activity and reduced expression of the tumor suppressor PTEN. Pharmacological inhibition of PI3K activity reduced cell proliferation specifically in SAMHD1 KO cells, suggesting that SAMHD1 KO-induced cell proliferation is mediated via enhanced PI3K signaling. However, PI3K inhibition did not significantly affect SAMHD1 KO-reduced apoptosis, implicating the involvement of additional mechanisms. SAMHD1 KO also led to enhanced phosphorylation of p27 at residue T157 and its mis-localization to the cytoplasm. Inhibition of PI3K activity reversed these effects, indicating that SAMHD1 KO-induced changes in p27 phosphorylation and localization is mediated via PI3K-Akt signaling. While SAMHD1 KO significantly enhanced THP-1 cell migration in vitro, SAMHD1 KO attenuated the ability of THP-1 cells to form subcutaneous tumors in xenografted immunodeficient mice. This effect correlated with significantly increased expression of tumor necrosis factor α (TNF-α) in tumors, which may suggest that TNF-α-mediated inflammation could account for the decreased tumorigenicity in vivo. Our findings implicate that SAMHD1 can regulate AML cell proliferation via modulation of the PI3K-Akt-p27 signaling axis, and that SAMHD1 may affect tumorigenicity by downregulating inflammation.
KEYWORDS: SAMHD1, cell proliferation, PI3K, PTEN, Akt, p27
Introduction
AML is the most common type of leukemia in adults over the age of 45 [1,2]. PI3K-Akt signaling plays a major role in maintaining cell proliferation and survival under normal physiological conditions. Dysregulation of this pathway occurs via overexpression or hyper-activation of the signaling mediators PI3K, Akt, PDK1 (3-phosphoinositide-dependent kinase 1), and/or loss-of-function mutations of the inhibitory molecule PTEN (phosphatase and tensin homolog) [3–5]. These alterations have been implicated in several malignancies, including AML [3–7]. Mediators of this pathway have been key drug targets in pre-clinical and clinical studies of AML [6–10]. In particular, specific inhibitors targeting PI3K, PDK1 and Akt have been reported to potentiate the cytotoxicity of the anti-cancer drug deoxycytidine analog cytarabine (Ara-C) in AML and other cancers [4,8,11,12], indicating that targeting the PI3K-Akt pathway may synergistically enhance the efficiency of Ara-C and other therapeutics against AML. Among other targets, the PI3K-Akt signaling regulates the activity of p27 (also known as Kip1), a cyclin-dependent kinase (CDK) inhibitor that induces cell cycle arrest in G1 and has been reported to be dysregulated in several malignancies [13–18]. Phosphorylation on serine, tyrosine or threonine residues modulates p27 activity and stability [13]. Akt phosphorylates p27 on threonine 157 (T157), which inhibits p27 nuclear import, thus impairing CDK inhibition by p27 and resulting in cell cycle progression [15].
SAMHD1 is the first identified mammalian dNTP hydrolase (dNTPase) that regulates intracellular dNTP homeostasis [19–23]. SAMHD1 inhibits infections of retroviruses and certain DNA viruses by reducing the dNTP levels in the host cells required for viral genome replication [19,21,22]. Besides its role in viral infections, SAMHD1 is emerging as a key player in regulating cancer development or treatment. Somatic mutations of SAMHD1 have been identified in chronic lymphocytic leukemia (CLL), glioblastoma, myeloma, medulloblastoma, lung adenocarcinoma, colorectal, breast, and pancreatic cancers [24–34]. In CLL patients, increased SAMHD1 somatic mutation rate (~ 11%) is associated with increased chemo-resistance [25]. In colorectal cancers, SAMHD1 is highly mutated leading to loss of its dNTPase activity [34]. Additionally, these mutations are associated with inactivated mismatch repair and increased mutation rates, suggesting that SAMHD1 function is required to maintain genomic integrity [34]. SAMHD1 has also been shown to facilitate DNA double-stranded break repair and to promote genomic integrity through mechanisms independent of its dNTPase function [35]. We initially reported that SAMHD1 expression is significantly downregulated in cell lines or primary cells derived from T-cell leukemia and lymphoma patients compared to those from healthy donors [36–38]. SAMHD1 protein and mRNA is also downregulated in CLL, lung adenocarcinoma, and breast cancers [25,39]. Collectively, these studies suggest an anti-tumor role of SAMHD1.
We have reported that SAMHD1 knockout (KO) in THP-1 cells via the CRISPR/Cas-9 approach increases cell proliferation and reduces apoptosis relative to control cells with endogenous SAMHD1 expression [40]. However, the underlying mechanisms of these effects remain unknown. Furthermore, we have shown that, in cutaneous T-cell lymphoma derived-cells, exogenous SAMHD1 expression inhibits cell growth and induces the extrinsic apoptotic pathway by activating caspase-8, −3 and −7 and by downregulating the expression of the anti-apoptotic molecule cFLIPS (short form of cellular FLICE-like inhibitory protein) [41]. These results implicate that SAMHD1 may affect tumor cell growth by regulating cell proliferation and apoptosis.
Herein, we report that SAMHD1 controls THP-1 cell proliferation via regulation of the PI3K-Akt-p27 signaling axis. SAMHD1 KO leads to increased PI3K activity, which mediates enhanced cell proliferation in THP-1 cells. Additionally, we show that SAMHD1 KO reduces PTEN expression, and modulates phosphorylation and intracellular localization of the cell cycle regulator p27 via the PI3K-Akt pathway, thereby affecting cell growth and migration. Our data suggest that SAMHD1 may modulate AML cell proliferation by regulating the key PI3K-Akt-p27 signaling axis.
Results
SAMHD1 expression negatively correlates with myeloblast percentage in AML patient-derived peripheral blood mononuclear cells (PBMCs)
We analyzed levels of SAMHD1 expression in PBMCs isolated from 22 AML patients (Figure 1(a)). SAMHD1 protein levels were variable among different donors, with cells expressing higher or lower levels of SAMHD1 compared to normal PBMCs or THP-1 cells (Figure 1(a)). These variations are likely due to genetic heterogeneity that is common in AML patients [42]. Interestingly, SAMHD1 protein levels negatively correlated with the percentage of myeloblasts in PBMCs, one of the factors required for AML diagnosis, in these samples (Figure 1(b)). This result indicates that SAMHD1 may have a key role in controlling myeloid cancer cell proliferation.
Figure 1.
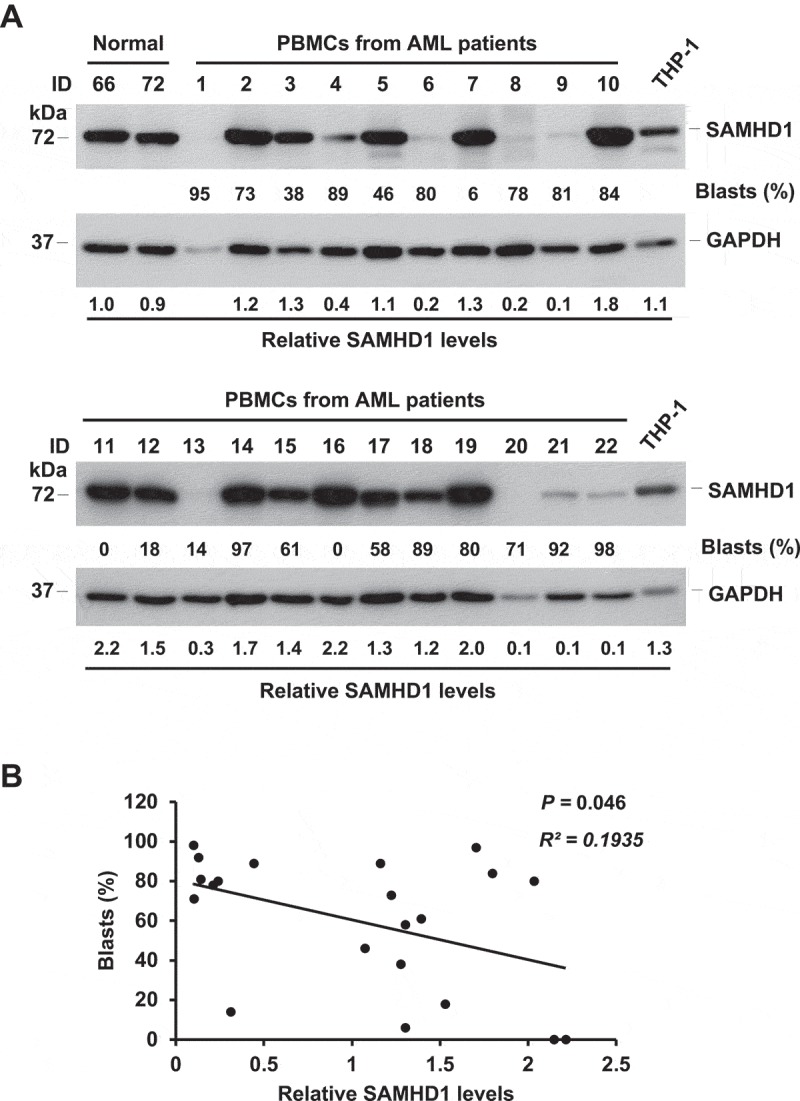
SAMHD1 expression negatively correlates with myeloblast levels in AML patient-derived PBMCs. (a) Five million PBMCs from 22 AML patients were collected and analyzed via immunoblotting for endogenous SAMHD1 protein. PBMCs from 2 ‘Normal’ (healthy) donors and THP-1 cells were used as a reference. GAPDH was a loading control. The percentages of blasts (%) in patient PBMCs are shown. Relative SAMHD1 levels were calculated by densitometry analysis. (b) Non-parametric correlation (Spearman) analysis between relative SAMHD1 levels and blast percentages (P = 0.046, R2 = 0.1935). Patient #1 sample was excluded from analysis due to low protein concentration of the cell lysate.
SAMHD1 KO leads to increased PI3K activity in THP-1 cells
We have reported that SAMHD1 KO in AML-derived THP-1 cells leads to a significant increase in cell growth and proliferation and reduces apoptosis levels compared to control cells [40]. However, the mechanisms underlying the anti-proliferative function of SAMHD1 in THP-1 cells remain unknown. To identify potential mechanisms, we performed a microarray analysis in THP-1 control and SAMHD1 KO (THP-1/KO) cells using the Affymetrix system. Complete microarray data have been deposited in NCBI’s Gene Expression Omnibus [43] and are accessible through GEO Series accession number GSE113610 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113610). We identified 1181 genes whose expression was significantly affected by SAMHD1. Pathway enrichment analysis revealed that the AML signaling is among the most up-regulated pathways in THP-1/KO cells compared to control cells (Figure 2(a)). SAMHD1 silencing resulted in significant downregulation of mRNA levels of the tyrosine kinase receptors CSF1R and FLT3 as well as the proto-oncogene KRAS, while the transcripts of other key mediators of the AML signaling, such as PI3K and Akt, were highly expressed in THP-1/KO cells relative to control cells (Figure 2(b)).
Figure 2.
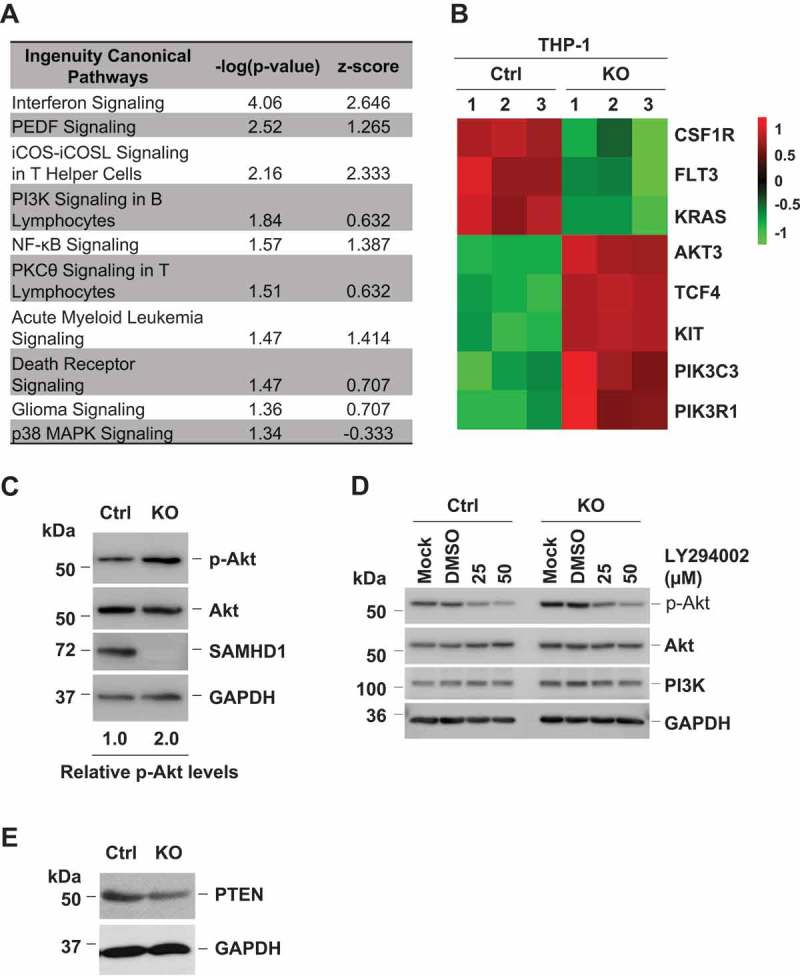
SAMHD1 KO enhances PI3K activity and reduces PTEN levels. (a) Significantly enriched pathways in THP-1/KO cells versus control cells were identified by ingenuity pathway analysis (IPA) of the microarray data. Statistically significant pathways are shown with their respective p values and z-scores. Pathway up-regulation is indicated by a positive z-score, while down-regulation is indicated by a negative z-score. PEDF, serpin family F member 1; iCOS, inducible T cell costimulator; iCOSL, inducible T cell costimulator ligand; PKCθ, protein kinase C theta; MAPK, mitogen-activated protein kinase. (b) Activation of the AML signaling in THP-1/KO cells relative to control (Ctrl) cells by microarray analysis. Each cell line was analyzed in three biological replicates as indicated by sample numbers 1–3. The heatmap shows mRNA levels of differentially expressed key regulators of the AML signaling pathway. Up-regulated genes are shown in red, down-regulated genes are shown in green (the numbers next to the legend represent raw Z-score). CSF1R, colony stimulating factor 1 receptor; FLT3, Fms related tyrosine kinase 3; KRAS, Kirsten ras oncogene homolog; AKT3, AKT serine/threonine kinase 3; TCF4, transcription factor 4; KIT, KIT proto-oncogene receptor tyrosine kinase; PIK3C3, phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R1, phosphoinositide-3-kinase regulatory subunit 1. (c) THP-1 control (Ctrl) and SAMHD1 KO cells were analyzed for the indicated protein expression by immunoblotting. The levels of phosphorylated AKT (p-AKT) expression were quantified by densitometry after normalization for total AKT and GAPDH. (d) THP-1 control and SAMHD1 KO cells were treated with LY294002 or DMSO (vehicle control of LY) or left untreated (Mock), and then analyzed for the indicated protein expression by immunoblotting. GAPDH was a loading control. (e) PTEN protein levels are lower in THP-1/KO cells compared to control (Ctrl) cells. Panels show representative results from three independent experiments.
Since PI3K-Akt signaling is a critical regulator of cell proliferation and is also one of the most frequently dysregulated pathways in AML, we hypothesized that the increased proliferation in THP-1/KO cells was mediated by altered PI3K-Akt signaling. To assess PI3K activity levels in THP-1 control or THP-1/KO cells and validate the microarray data, we performed immunoblotting and measured the phosphorylation of Akt at residue S473 (p-Akt), which is a downstream substrate of PI3K [5]. In line with a recent study [44], we observed that p-Akt level in THP-1/KO cells was 2-fold higher compared to control cells (Figure 2(c)), indicating that PI3K activity was elevated in THP-1 cells upon SAMHD1 KO. Notably, the total Akt levels were unaltered in these cells (Figure 2(c)), suggesting that the absence of SAMHD1 affects the activity but not the expression of Akt. Enhanced phosphorylation of Akt upon SAMHD1 KO was confirmed using additional two THP-1/KO and one control cell clones (Supplementary Figure 1). To validate the increased PI3K activity upon SAMHD1 KO, we treated THP-1 control and KO cells with LY294002 (LY), a specific inhibitor of the PI3K catalytic subunit [45]. As expected, p-Akt levels were significantly reduced upon LY treatment in a dose-dependent manner (Figure 2(d)), confirming that the increased p-Akt levels in THP-1/KO cells was due to enhanced PI3K activity. Since PTEN is a crucial negative regulator of the PI3K signaling [5], we examined whether increased PI3K activity could be mediated via altered PTEN expression. Indeed, we observed that PTEN expression was significantly reduced in THP-1/KO cells relative to control cells (Figure 2(e)), suggesting that increased PI3K activity may be partially due to reduced PTEN protein levels. Thus, SAMHD1 negatively regulates PI3K signaling activity in THP-1 cells.
SAMHD1 KO-induced proliferation in THP-1 cells is mediated via PI3K signaling
To determine whether increased cell proliferation in THP-1 cells upon SAMHD1 KO is due to increased PI3K activity, we assessed cell proliferation in control and THP-1/KO cells via an MTT-based assay after treatment with LY. Consistent with our previous observations [40], untreated THP-1/KO cells demonstrated significantly increased cell proliferation rate than control cells in the presence of DMSO as a vehicle control of LY (Figure 3(a)). Compared to cells treated with DMSO, treatment with LY significantly reduced the proliferation rate in both control and THP-1/KO cells (Figure 3(a)). However, the reduction of cell proliferation upon LY treatment was more pronounced in THP-1/KO cells than in control cells (at day 5, LY decreased proliferation of 1.2-fold in control cells and of 1.5-fold in THP-1/KO cells), suggesting that THP-1/KO cells are more sensitive to PI3K inhibitor treatment due to elevated PI3K activation. We have reported that SAMHD1 KO also reduces apoptosis and caspase activity in THP-1 cells [40]. To determine whether PI3K inhibition can also reverse the SAMHD1 KO-induced changes in apoptosis, we treated control and THP-1/KO cells with DMSO or LY and performed apoptosis and caspase-3/7 activity assays. Consistent with our previous observations [40], apoptosis and caspase activity were significantly lower in THP-1/KO cells than those in control cells in the presence of DMSO (Figure 3(b,c), respectively). Interestingly, PI3K inhibition via LY treatment did not significantly alter the apoptosis levels and caspase-3/7 activity in control or THP-1/KO cells (Figure 3(b,c) and Supplementary Fig. 2), suggesting that additional mechanisms may be involved in SAMHD1-regulation of apoptosis in THP-1 cells. These results also indicate that SAMHD1 may regulate cell proliferation and apoptosis via distinct mechanisms.
Figure 3.
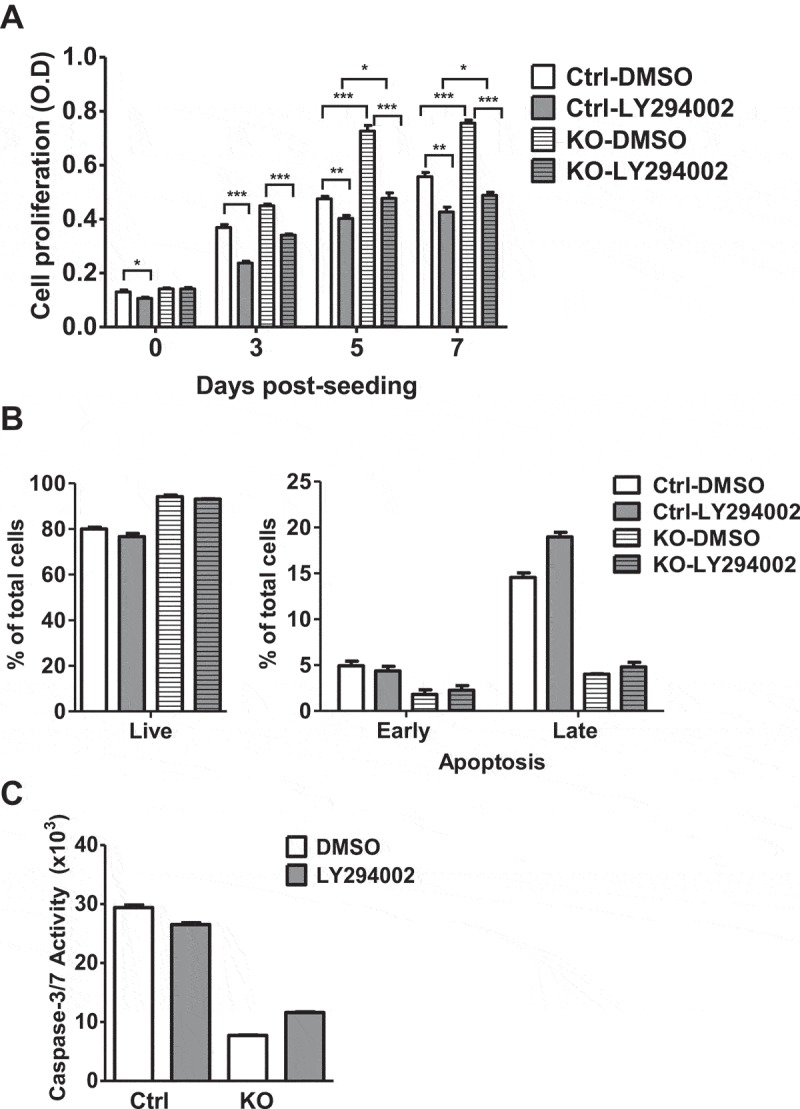
PI3K inhibition reduces SAMHD1 KO-induced proliferation in THP-1 cells but does not affect apoptosis. (a) THP-1 control (Ctrl) and SAMHD1 KO cells were treated with DMSO or the PI3K inhibitor LY294002 at 5 µM. Cells were analyzed by an MTT-based cell proliferation assay at the indicated time points. One representative experiment performed in three replicates is shown. OD, optical density at 490 nm. *, P < 0.01; **, P < 0.001; ***, P ≤ 0.0001. (b-c) THP-1 control (Ctrl) or SAMHD1 KO cells were treated with DMSO or 25 µM LY294002 for 18 h and were analyzed via (b) annexin-V/7-AAD apoptosis assay and (c) via caspase-glo 3/7 activity assay to determine the apoptosis levels and caspase activities, respectively. Each experiment was performed in three biological replicates.
SAMHD1 KO inhibits nuclear localization of p27
In several cancers including AML, enhanced and constitutive PI3K-Akt activation may lead to mis-localization of p27 to the cytoplasm, thus preventing cell cycle arrest [18,46,47]. To further understand the mechanism by which PI3K signaling regulates cell proliferation in THP-1/KO cells, we determined expression and localization of p27 in these cells. Interestingly, subcellular fractionation analysis revealed that p27 in THP-1/KO cells was predominantly localized to the cytoplasm (94% in the cytosol and 6% in the nucleus). In contrast, p27 in THP-1 control cells was predominantly localized to nucleus (39% in the cytosol and 61% in the nucleus). The purity of the cytosolic and nuclear fractions was validated by immunoblotting of GAPDH and Lamin A/C, respectively (Figure 4(a)). Consistent with a previous report [48], SAMHD1 localized in both cytoplasm and nucleus in THP-1 control cells. To further confirm the reduced nuclear localization of p27 in THP-1/KO cells, we performed immunofluorescence (IF) microscopy of SAMHD1 and p27. Consistently, IF analysis indicated that p27 was mainly localized out of nucleus in THP-1/KO cells relative to control cells (Figure 4(b)). Together, these results indicate that SAMHD1 KO reduces nuclear translocation of p27 in THP-1 cells. As p27 nuclear import is required for inhibition of cyclin-CDK complexes [49], cytosolic accumulation of p27 in THP-1/KO cells may result in increased cell proliferation.
Figure 4.
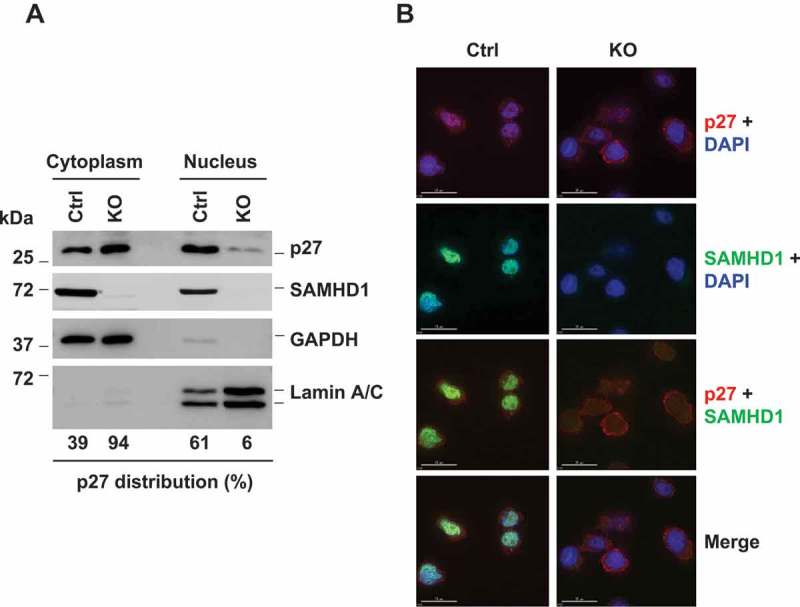
SAMHD1 KO inhibits nuclear localization of p27 in THP-1 cells. (a) THP-1 control and SAMHD1 KO cells were analyzed via subcellular fractionation followed by immunoblotting. GAPDH and Lamin A/C were used as markers for cytosolic and nuclear fraction, respectively. Results are representative of three independent experiments. (b) Cells were analyzed via indirect immunofluorescence for p27 (red) and SAMHD1 (green) expression. DAPI (blue) was used to stain the nuclei. Scale bars, 15 µm. Results are from one representative experiment of two independent assays.
SAMHD1 KO induces mis-localization of p27 to the cytoplasm via enhanced PI3K signaling
Since activated PI3K-Akt signaling has been identified to regulate p27 expression and localization [50], we hypothesize that p27 mis-localization in THP-1/KO cells is due to enhanced PI3K activity in these cells. To test this, we treated control and THP-1/KO cells with DMSO or LY, and then assessed the localization of p27 via subcellular fractionation. As shown in Figure 5(a), PI3K inhibition via LY treatment in THP-1/KO cells resulted in a significant increase (from 9% to 39%) in p27 nuclear localization relative to DMSO-treated cells in a dose dependent manner. In contrast, there was only a modest increase (from 42% to 47%) in nuclear p27 levels upon LY treatment relative to DMSO treatment in control cells, suggesting that SAMHD1 KO-induced mis-localization of p27 to cytoplasm is in part due to enhanced PI3K activity. We further validated the role of PI3K signaling in SAMHD1 KO-induced changes in p27 localization by IF microscopy and quantification. Consistent with our previous data (Figure 4(a,b)), nuclear levels of p27 were significantly lower in DMSO-treated THP-1/KO cells compared to control cells (Figure 5(b,c)). However, PI3K inhibition in SAMHD1 KO cells resulted in a significant increase in nuclear localization of p27 compared to DMSO-treated cells (Figure 5(b,c)). These results indicate that SAMHD1 KO-mediated p27 mis-localization to cytoplasm is due to enhanced PI3K activity in THP-1 cells.
Figure 5.
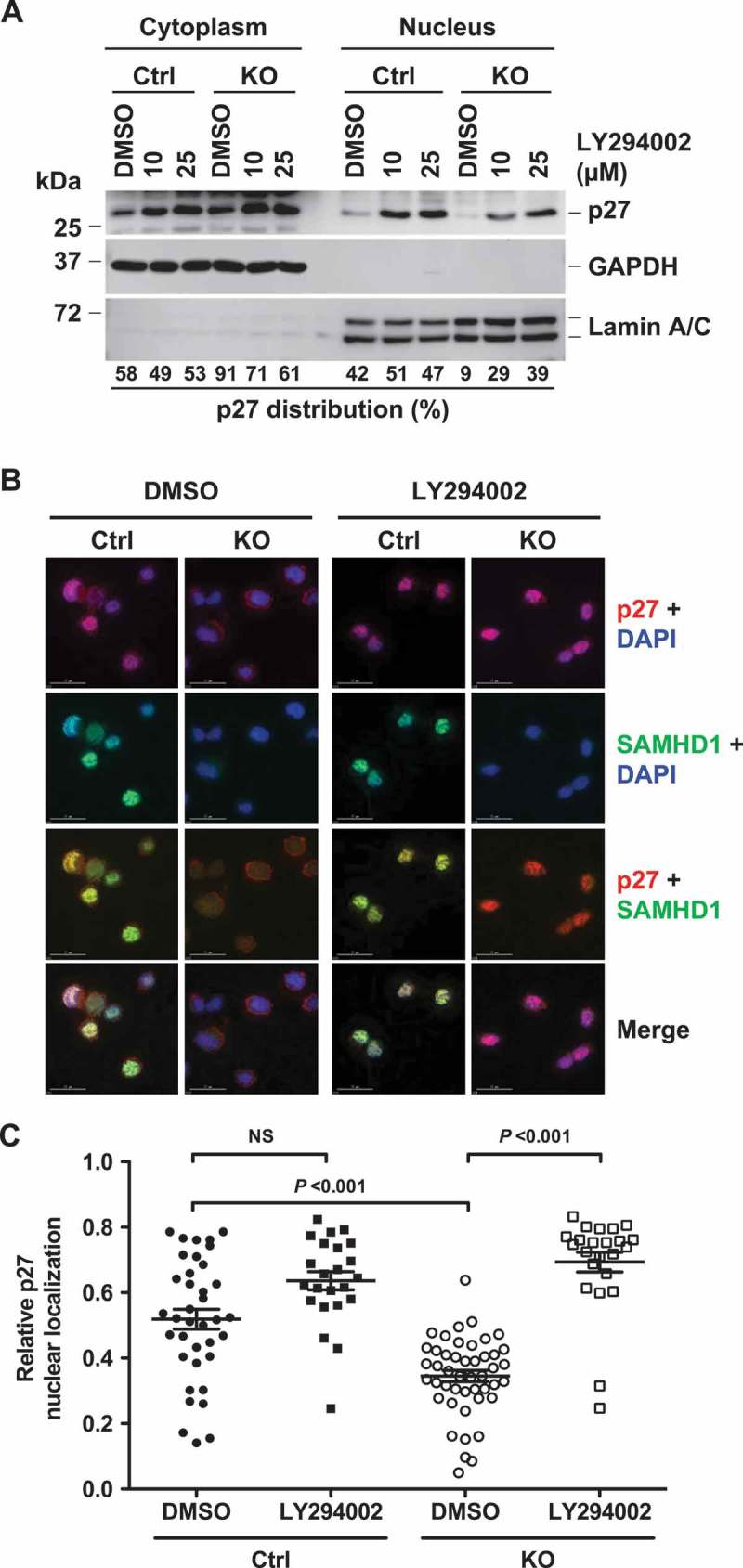
PI3K inhibition reverses SAMHD1 KO-induced inhibition of p27 nuclear localization in THP-1 cells. (a) THP-1 control and SAMHD1 KO cells treated with DMSO or LY294002 (LY) for 18 h were analyzed via subcellular fractionation followed by immunoblotting for the indicated proteins. GAPDH and Lamin A/C were used as markers for cytosolic and nuclear fraction, respectively. Results are from one representative experiment of two independent assays. (b) THP-1 control and SAMHD1 KO cells were treated with DMSO or 25 µM of LY and indirect immunofluorescence was performed against p27 and SAMHD1 using specific antibodies. DAPI was used for nuclear staining. Representative images are shown. Scale bars, 15 µm. (c) Pearson’s co-localization coefficient between p27 and DAPI was quantified to determine the nuclear localization of p27 in cells. Each dot in the plot represents a cell analyzed. NS, not significant (P > 0.05). One representative experiment of three independent assays is shown.
SAMHD1 KO leads to increased T157 phosphorylation of p27 via PI3K signaling and enhanced cell migration
Upon activation by PI3K, Akt leads to phosphorylation of p27 at residue T157 located in its nuclear localization signal, thus preventing p27 nuclear translocation and leading to uncontrolled cell proliferation [15–17]. To further understand the mechanisms of SAMHD1 KO-induced effect on p27 localization, we assessed the T157 phosphorylation of p27 (p-p27) using a specific antibody. The p-p27 level in THP-1/KO cells was significantly increased (~1.8 fold) compared to control cells (Figure 6(a,b)). Increased p-p27 upon SAMHD1 KO was confirmed using additional THP-1/KO and control clones (Supplementary Fig. 1). Additionally, p-p27 was observed only in the cytoplasmic fractions of control and KO cells (Figure 6(c)), possibly because phosphorylation at T157 blocks p27 nuclear translocation [15]. Consistent with total cell lysates (Figure 6(a)), cytoplasmic p-p27 levels were higher in THP-1/KO cells compared to control cells. Inhibition of PI3K activity using LY specifically reduced the p-p27 levels in THP-1/KO cells, but not in control cells (Figure 6(d)). These results indicate that SAMHD1 silencing, through enhanced PI3K signaling, leads to increased phosphorylation of p27 at T157, thus preventing its translocation to the nucleus.
Figure 6.
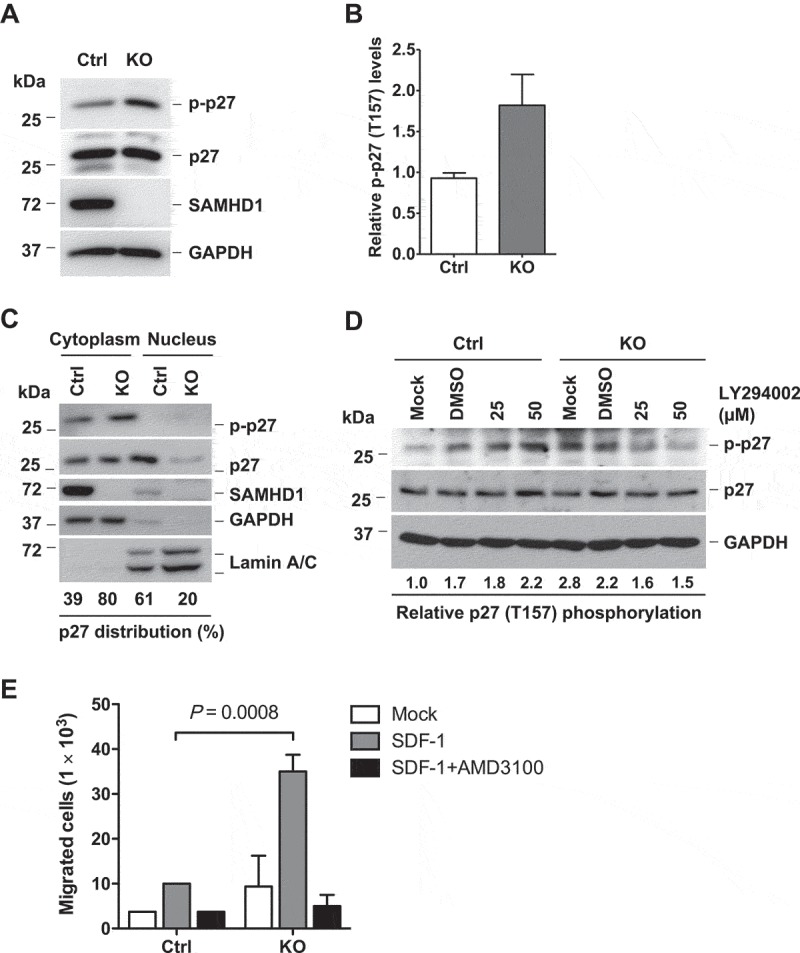
SAMHD1 KO leads to increased T157 phosphorylation of p27 via PI3K signaling and enhanced cell migration. (a) THP-1 control or SAMHD1 KO cells were analyzed by immunoblotting for the indicated protein. GAPDH was used as a loading control. (b) Densitometric analysis was performed to determine the relative levels of T157 phosphorylation of p27 (p-p27) based on the immunoblotting results of (a). (c) Subcellular fractionation in THP-1 control and SAMHD1 KO cells was performed as described in Figure 4a. (d) THP-1 control and SAMHD1 KO cells were left untreated (Mock) or treated with DMSO or LY294002 for 18 h, and p27 (total and phosphorylated form) levels were determined by immunoblotting. Densitometric analysis was performed to quantify the relative levels of T157 phosphorylation of p27 (p-p27). GAPDH was used as a loading control. (e) THP-1 control and SAMHD1 KO cells were grown in 0.1% FBS containing media with or without AMD3100 (10 µg/ml, DMSO as the vehicle control) for 90 min. Cells were then seeded in the upper chamber of a transwell plate containing media only (Mock) or media with SDF-1 in the lower chamber, and further incubated for 3 h to allow cell migration. The graph shows data from one representative experiment performed in biological duplicates. The SDF-1-induced cell migration assay represents one of three independent experiments.
As cytoplasmic p27 has been reported to promote cell migration in various cancer cell types [51–53], we performed a migration assay in cells treated with the stromal-derived factor 1 (SDF-1), the specific ligand of the CXC motif chemokine receptor 4 (CXCR4), which is involved in control of migration and metastasis of tumor cells including AML cells [54–56]. Flow cytometry analysis revealed similar low levels of surface CXCR4 expression on THP-1 control and THP-1/KO cells (10.7% and 10.4% CXCR4-positive population, respectively) (Supplementary Fig. 3A). Compared to control cells, THP-1/KO cells showed a ~ 4-fold increase (P = 0.0008) in migration ability (Figure 6(e)). Treatment with the CXCR4 antagonist AMD3100 efficiently inhibited SDF-1-induced migration in both cell lines, indicating that the effect observed was specifically due to CXCR4 activation by SDF-1 (Figure 6(e)). These data suggest that SAMHD1 can negatively regulate THP-1 cell migration in vitro. Interestingly, LY treatment did not affect SDF-1-induced migration in both cell lines (Supplementary Fig. 3B), suggesting that the effect of SAMHD1 on the migration of THP-1 may be mediated by a PI3K-independent mechanism.
SAMHD1 KO attenuates THP-1 cell tumorigenicity in xenograft mice
In order to validate the growth inhibitory function of SAMHD1 in AML cells, we assessed the effect of SAMHD1 on THP-1 cell tumorigenicity using a xenograft mouse model [57]. We obtained NSG (non-obese diabetic/severe combined immune deficient-gamma) mice (n = 7 per group), subcutaneously injected control or THP-1/KO cells and monitored the rate of tumor growth. Unexpectedly, THP-1/KO cells did not demonstrate higher rate of subcutaneous tumor formation in mice relative to control cells (Figure 7(a)). Furthermore, the THP-1/KO cells formed significantly smaller tumors relative to control cells at 24 days post-injection (Figure 7(b)), indicating that the absence of SAMHD1 expression (Figure 7(c)) attenuated the ability of THP-1 cells to form subcutaneous tumors in these mice.
Figure 7.
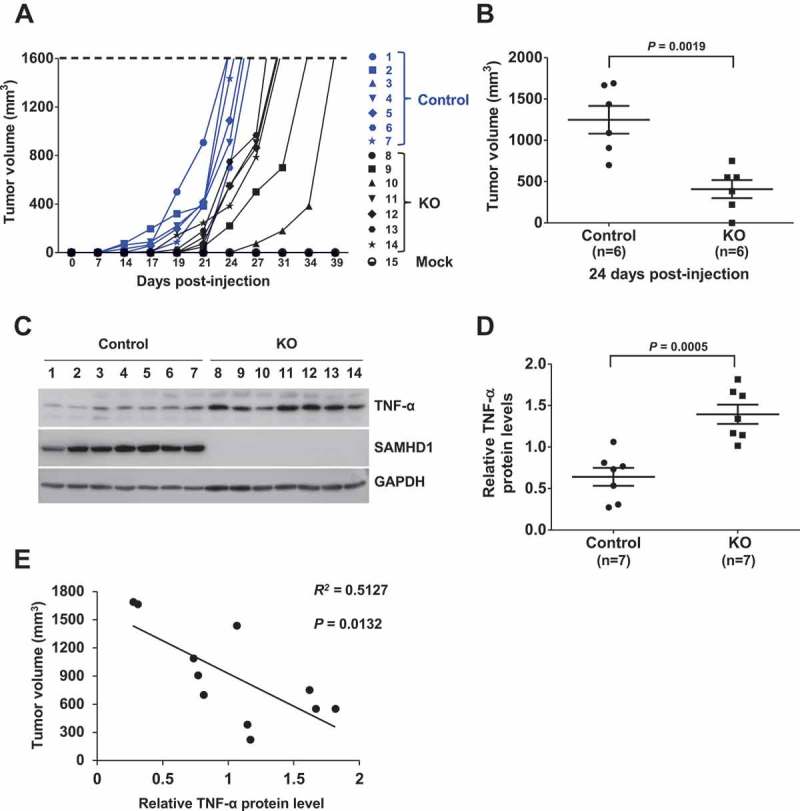
SAMHD1 KO attenuates THP-1 cell tumorigenicity in xenograft mice. Female, 4–6-week old NSG mice (n = 7 per group) were subcutaneously injected with THP-1 control or THP-1/KO cells (5 × 106 per mouse). (a) The rate of tumor growth and tumor volume were assessed at the time indicated. Mice were sacrificed when the tumors reached a volume of 1,600 mm3. (b) Average volume of subcutaneous tumors from the mice of both groups was calculated at 24 days post-injection (6 out of 7 mice developed measurable tumors). (c) Upon sacrificing the mice, tumor tissues were collected and harvested proteins were analyzed via immunoblotting for TNF-α and SAMHD1. GAPDH was a loading control. (d) Densitometry analysis was performed to quantify the relative TNF-α protein levels in control and THP-1/KO cell-derived tumors. The results were based on the normalization to GAPDH levels. (e) Pearson’s correlation analysis was performed to determine correlation between subcutaneous tumor volume and the TNF-α protein levels. All results are representative of two independent experiments. (b, d and e) Statistical significance was determined by unpaired Students t-test and the P values are shown on the figures.
Of note, upstream regulator analysis of our microarray data predicted tumor necrosis factor α (TNF-α), one of the major cytokines involved in inflammation, to be significantly activated in THP-1/KO cells relative to control cells (Supplementary Table 1). This may indicate that THP-1/KO cells are characterized by spontaneously increased inflammation status. Moreover, TNF-α has been described to regulate cancer progression and development with antineoplastic activity in hematological malignancies [58]. These studies prompted us to analyze TNF-α expression levels in THP-1 cell-derived tumors in mice. Interestingly, human TNF-α protein levels in the tumors derived from THP-1/KO cells were significantly higher than in control cells (Figure 7(c,d)). Furthermore, we found a significant inverse correlation between the tumor volumes and TNF-α protein levels in the tumors (Figure 7(e)). These results show that the loss of SAMHD1 in THP-1/KO cells may lead to significantly increased TNF-α expression in subcutaneous tumors, suggesting that TNF-α-mediated inflammation responses may possibly contribute to decreased tumor growth in vivo.
Discussion
In this study, we identify a novel mechanism involving the PI3K-Akt-p27 signaling axis through which SAMHD1 regulates THP-1 cell proliferation, and thus may perform an anti-proliferative function in AML-derived cells. However, our results of mouse xenograft study suggest that the role of SAMHD1 in tumorigenicity is complex, likely involving negative regulation of TNF-α-mediated inflammation responses (Figure 8).
Figure 8.
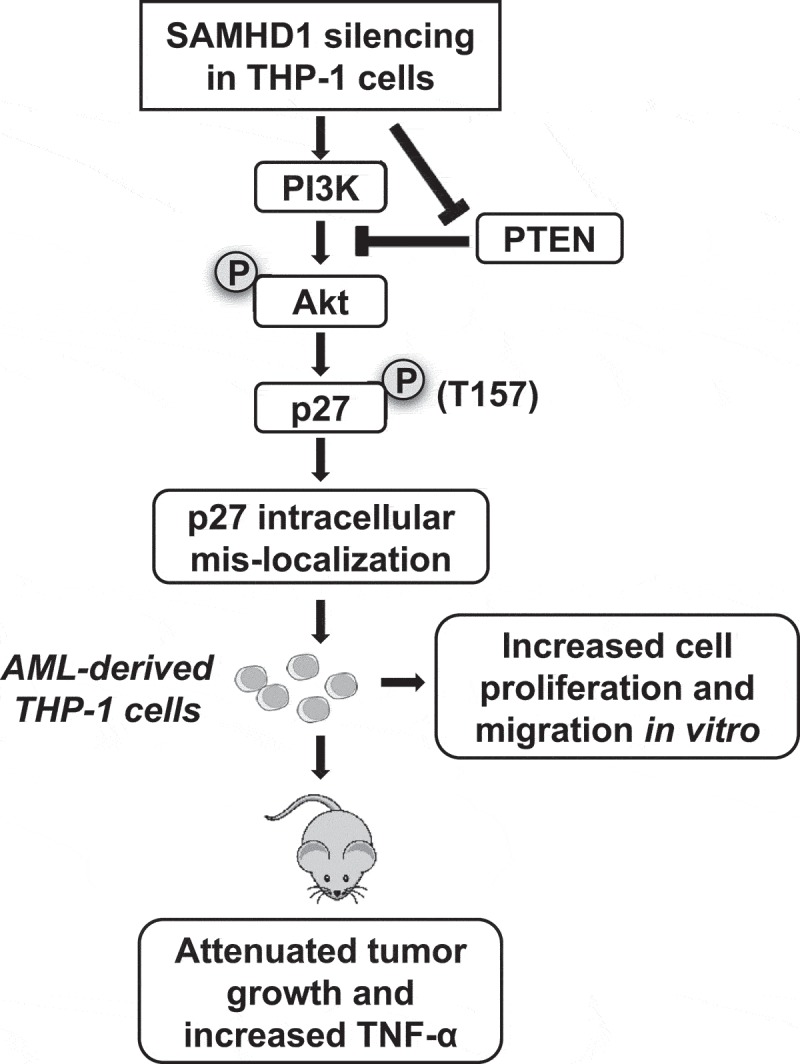
Proposed mechanisms by which SAMHD1 regulates AML cell proliferation in vitro and affects tumorigenicity in xenografted mice. Silencing of SAMHD1 in AML-derived THP-1 cells causes down-regulation of PTEN and increase of PI3K activity, which in turn induces phosphorylation of Akt and of its substrate p27. Phosphorylation of p27 at residue T157 results in its mis-localization to the cytoplasm and impairment of its CDK inhibitory function, leading to increased cell proliferation and enhanced cell migration. The letters P with a circle indicates phosphorylation of Akt and p27. In contrast, SAMHD1 KO attenuates THP-1 cell tumorigenicity in xenografted immunodeficient mice, possibly due to increased TNF-α expression and inflammation responses in tumors.
AML is a heterogeneous hematological malignancy characterized by uncontrolled proliferation of immature blood cells. Current AML treatment strategies predominantly consist of intensive chemotherapy with nucleoside analog drugs, including the cytidine analog Ara-C and anthracyclines, either alone or in combination with other drugs [4,5,59]. However, increased resistance to current treatment options has resulted in high incidence of tumor relapse and in an overall patient survival rate of just ~27%. Therefore, development of novel and more effective treatment strategies is urgently needed.
SAMHD1 has recently emerged as an important protein that may regulate the development and progression of solid and hematological malignancies, where SAMHD1 gene mutation or downregulation have been identified as primarily involved in cancer growth [60]. We have reported that SAMHD1 KO in AML-derived THP-1 cell lines increases cell proliferation and reduces apoptosis, suggesting a potential role of SAMHD1 in regulating AML pathogenesis [40]. Interestingly, recent studies discovered that SAMHD1, through its dNTPase activity, impairs therapeutic efficiency of Ara-C in AML patients, proposing SAMHD1 as a negative biomarker in AML treatment [61,62]. SAMHD1 gene silencing in AML-derived THP-1 cell line has been reported to increase the sensitivity to several other nucleoside-based antimetabolites [63], suggesting that SAMHD1 may impair treatment of solid cancers and hematological malignancies using these drugs. Despite the anti-therapeutic role of SAMHD1 in AML, increasing evidence suggests that SAMHD1 may be a potential tumor suppressor, as high expression of SAMHD1 is associated with more favorable AML progression [64]. These new studies implicate that SAMHD1 can be a double-edged sword for AML patients, and call for a better understanding of the function of SAMHD1 and mechanisms in AML pathophysiology.
Hyper-activation of the PI3K-Akt signaling drives uncontrolled cell proliferation and survival in many cancers, and constitutive PI3K activation has been reported in 50% of AML cases [4]. As a tumor suppressor, p27 acts downstream of the PI3K/Akt pathway. Gene mutations of p27, reduced p27 protein expression or its mis-localization to the cytoplasm have been identified as major mechanisms associated with malignant transformation in different cancer types [65]. Under normal physiological conditions, p27 is predominantly localized to the nucleus, where it binds to and inhibits cyclin-CDK complexes, thus controlling cell cycle progression and cell proliferation [14,15,50,66]. Impaired nuclear import of p27 as a consequence of PI3K/Akt-induced phosphorylation at residue T157 has been described as a negative prognostic factor and marker of poor survival in AML, breast and cervical cancer [15,17,50]. In addition to its cell cycle inhibitory activity, p27 possesses other important cellular functions [67]. In particular, cytoplasmic p27 has been described to sustain tumor migration and metastasis [51–53].
We found that SAMHD1 KO in THP-1 cells modulates the PI3K-Akt signaling by enhancing PI3K activity, reducing the expression of PTEN, and impairing the activity of the cyclin-CDK inhibitory protein p27 through block of its nuclear import. Importantly, this phenotype was confirmed in additional THP-1 control and THP-1/KO cell clones, excluding the possibility that these may be off-target effects of CRISPR-Cas9 expression, and confirming the important role of SAMHD1 in these processes.
Consistent with published studies [50], we found that mis-localization of p27 in the cytoplasm of AML-derived THP-1 cells is associated with sustained cell proliferation and migration. Notably, we provide first evidence that inhibition of p27 nuclear translocation in these cells is, at least in part, due to SAMHD1 silencing. Moreover, increased p27 cytoplasmic localization in SAMHD1-deficient cells was associated with enhanced migration potential, confirming the role of p27 in the control of cell migration and motility previously described [51,53]. These results suggest that the PI3K-Akt-p27 signaling axis is a key mediator of SAMHD1-mediated modulation of THP-1 cell proliferation. Interestingly, our data show that the PI3K/Akt pathway does not mediate SAMHD1-induced apoptosis, suggesting that additional pathways might be involved in SAMHD1-mediated regulation of apoptosis in these cells. Previous studies have shown that, when localized to the cytoplasm, p27 can have an anti-apoptotic function in certain cancer cells [68]. In our study, although PI3K inhibition rescued the nuclear p27 levels, p27 levels in the cytosol were not significantly reduced. Therefore, it would be interesting to determine whether cytoplasmic p27 plays a role in reducing apoptosis in the THP-1/KO cells. Additional studies are required to fully elucidate the molecular mechanisms associated with this effect.
Our in vivo study showing decreased tumor growth of THP-1/KO cells relative to control cells transplanted in immunodeficient NSG mice suggests that SAMHD1 may inhibit TNF-α-mediated inflammation responses in vivo. This observation is in line with our recent findings showing that, in SAMHD1 KO mice, SAMHD1 inhibits TNF-α responses [69]. Although NSG mice are severely immunodeficient and would not generate graft-versus-host diseases after the transplantation of human cells, subcutaneous tumors derived from the THP-1/KO or control cells could be affected by the microenvironment. Higher levels of human TNF-α protein or other proinflammatory cytokines expressed in THP-1/KO cell-derived tumor may lead to death of the cancer cells, thereby reducing the tumor size or limiting tumor growth rate. However, further studies are needed to determine whether there is a direct and causal effect of increased TNF-α on reduced tumorigenicity of THP-1/KO cells.
Although the mechanisms of AML development are well investigated and clinical significance of the PI3K-Akt-p27 signaling axis in AML treatment is well studied, there is an urgent need for novel and more effective strategies in AML therapy based on mechanistic studies. Our work provides new insights into AML pathology and highlights the role of SAMHD1 in this disease. Further studies to completely investigate the role of SAMHD1 and its interplay with PI3K-Akt-p27 axis may help to fully characterize the mechanisms by which SAMHD1 controls AML progression, and more importantly, will pave the way for the design of more effective therapeutic strategies for the treatment of this aggressive malignancy.
Materials and methods
PBMCs from AML patients and healthy donors
PBMCs derived from 22 de-identified AML patients of were obtained through the Leukemia Tissue Bank of the Ohio State University Comprehensive Cancer Center to evaluate the expression of SAMHD1 via immunoblotting as described [37]. Healthy blood donors’ leukocyte samples (buffy coats) were purchased from the Red Cross, Columbus, OH. PBMCs were isolated from the buffy coats using Ficoll gradient centrifugation (Sigma Aldrich) as described [37]. The Institutional Review Board at the Ohio State University has approved these in vitro experiments involving PBMCs from de-identified healthy blood donors and AML patients.
Cell culture and treatments
THP-1 cell lines were generated and cultured as previously described [40]. HEK293T cells were maintained in complete DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. All the cell lines utilized for the presented studies were tested negative for mycoplasma contamination using universal mycoplasma detection kit (ATCC, #30–101-2K). Inhibition of PI3K activity was achieved by treatment with LY294002 (Cell Signaling, #9901) in culture media as described in figure legends.
Microarray analysis
Gene expression profile in THP-1 control and THP-1/KO cells was analyzed in three biological replicates by Affymetrix Clariom D assay (Thermo Fisher Scientific) using 500 ng RNA per sample. The analysis was performed at the Ohio State University Genomic Core Facility. Gene expression values with fold change > 2 and p-value ≤ 1 × 10−4 were considered statistically significant. Significantly affected pathways were analyzed via Ingenuity Pathway Analysis using software from Qiagen.
Immunoblotting analysis
Post indicated treatments, cells were pelleted, washed with ice-cold PBS and were lysed using 1 × cell lysis buffer (Cell signaling, #9803). Collected protein samples were normalized via BCA assay and were analyzed via SDS-PAGE followed by immunoblotting as previously described [70]. Immunoblotting was performed using the following antibodies to human proteins: SAMHD1 (1:1000, ProSci, #1224), phospho-Akt (Ser473) (1:1000, Cell signaling #4060S), Akt (1:1000, Cell signaling #9272S), PTEN (1:1000, Cell Signaling #9188T), PI3K p110α (1:1000, Cell Signaling, #4249S), phospho-p27/Kip1 (Thr157) (1:500, R&D Systems, #AF1555), p27/Kip1 (1:500, Cell Signaling #3686S), Lamin A/C (1:1000, Cell Signaling, #4777S), TNF-α (1:1000, Abcam, #9635), and GAPDH (1:3000, Bio-Rad #AHP1628).
Cell proliferation assay
Control or THP-1/KO cells were plated into 96-well plates at a density of 2.5 × 104 cells per well in 100 µl of culture media containing DMSO or 5 µM LY294002 (4 replicates per condition). On the indicated time points, MTT assay was performed as previously described [41].
Apoptosis assay
Control and THP-1/KO cells were plated at a density of 2 × 105 per well of a 12-well plate in 1 ml media with 25 µM LY294002 or equal volume of DMSO. Seven days post-seeding, cells were washed with ice-cold PBS and were resuspended in 1× binding buffer at a density of 1 × 106 cells/ml. Cells were then stained with annexin V-PE and 7-AAD for 15 min at room temperature in the dark. Percentage of cells undergoing early and late apoptosis was quantified by performing Guava Nexin Annexin V Assay (EMD Millipore, #4500–0450) according to the manufacturer’s instructions. Cells negative for both annexin V and 7-AAD were labeled as “live”, cells positive for annexin V only were labeled as “early apoptotic” and double positive cells for annexin V and 7-AAD were labeled as “late apoptotic” as described [41].
Caspase-3/7 activity assay
Control or THP-1/KO cells were plated in a 6-well plate at a density of 5 × 105 cells in 2 ml culture media containing 25 µM LY294002 or equal volume of DMSO. Seven days post-seeding, 1 × 104 cells were counted and seeded per well (in 4 replicates) in a white opaque 96-well plate. Then, 100 µl of Caspase-Glo 3/7 reagent (Promega, # G8090) was added to each well and incubated at room temperature for 1 h protected from light as described [41]. Luminescence readings were taken, were blank (media only) corrected and average of all replicates was calculated.
Isolation of cytoplasmic and nuclear factions
Control and THP-1/KO cells (1 × 107) were left untreated or treated with 10 µM or 25 µM LY294002 or DMSO for 18 h in a 25 cm2 culture flask. Post-treatment, cells were pelleted, washed with ice-cold PBS and fractions were isolated using NE-PER nuclear and cytoplasmic extraction kit (Thermo Fisher Scientific, #78833) according to manufacturer’s instructions. Isolated fractions were normalized using BCA assay and were analyzed via SDS-PAGE followed by immunoblotting for the indicated antibodies as described above. p27 levels in the nucleus and cytoplasm were quantified by densitometry and normalized to Lamin A/C (for the nuclear fraction) and GAPDH (for the cytosolic fraction) levels.
Immunofluorescence microscopy
THP-1 control or THP-1/KO cells were counted and were plated in a 12-well plate containing poly-L-lysine treated coverslips at a density of 1 × 106 cells in 2 ml culture media with DMSO or LY294002 (25 µM) for 18 h. Immunofluorescence to detect SAMHD1 and p27 was performed as described previously [71]. Briefly, one day post-seeding/treatment, cells were fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100 for 5 min and blocked with 1% bovine serum albumin in PBS for 1 h. The cells were then stained with SAMHD1 (1:500, Abcam) and p27 (1:800, Cell Signaling) primary antibodies. Alexa Fluor-488 (SAMHD1) and Alexa Fluor-550 (p27) secondary antibodies were used. All the antibodies were prepared in 2% FBS in PBS. Cells were finally stained with DAPI for nuclear staining and were imaged and processed using Delta Vision Elite deconvolution imaging system (GE Healthcare Life Sciences). Nuclear localization and expression analyzes were performed via SoftWorx software as per manufacturer’s instructions.
Cell migration assay
THP-1 control and SAMHD1 KO cells (5 × 105 cells) were pre-incubated in 100 μl of media containing 0.1% FBS and 10 µg/ml bicyclam JM-2987 (hydrobromide salt of AMD3100, (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. Catalog # 8128) or vehicle control (DMSO) for 90 min. Subsequently, cells were placed into the upper chamber of a 24-well transwell plate (Corning # 3421), and 500 μl media containing 100 ng/ml SDF-1 (PeproTech # 300-28A) were added to the lower chamber. The cells were allowed to migrate for 3 h, after which cell migration was determined by counting the cells in the bottom chamber with a hemocytometer.
Mouse xenograft experiments
All mouse experiments were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the Ohio State University. Female, 4–6 weeks old NSG mice were purchased from the Target Validation Shared Resources, the Ohio State University Comprehensive Cancer Center. THP-1 control or THP-1/KO cells (5 × 106 cells per mouse) resuspended in 200 µl serum-free culture media were injected subcutaneously on the flank into NSG mice (n = 7 per group) under anesthesia (isoflurane, USP). Injection with 200 µl serum-free media was performed as a mock control. Subcutaneous tumor sizes were regularly measured by calipers and tumor volume was quantified for ~ 6 weeks post-implantation. Mice were sacrificed upon reaching maximum tumor burden (volume of 1,600 mm3) and the tumors were harvested for further analyzes and collection of protein lysates. For protein isolation, tumor tissues were homogenized and lysed using 2 x cell lysis buffer. Clarified protein lysates were normalized and analyzed via immunoblotting.
Statistical analyzes
Error bars displayed on the bar graphs represent standard deviations. P values were calculated based on the non-parametric Student’s t-test. P < 0.05 is considered as statistically significant. Spearman’s non-parametric correlation analysis (Figure 1(b)) and Pearson’s correlation analysis (Figure 7(e)) were performed using Prism (GraphPad).
Funding Statement
This work was supported by National Cancer Institute under administrative supplement for non-AIDS defining cancers (Cancer Center Support Grant P30 CA016058) to LW. SB, NT and LW were partially supported by NIH grants (AI104483, AI120209, and GM128212) to LW.
Acknowledgments
We thank Dr. Baek Kim for sharing the THP-1 control and THP-1/KO cell lines, and the Wu lab members for valuable discussions. We thank Dr. Lianbo Yu from Center for Biostatistics at The Ohio State University for analyzing the microarray data. We also thank the Genomic Core and Facility Target Validation Shared Resources supported by the Ohio State University Comprehensive Cancer Center.
Authors’ contributions
LW conceived the study and designed experiments with KMK. KMK, SB and NT performed experiments and analyzed data with LW. KMK, SB and LW wrote the manuscript. All the authors read and approved the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Juliusson G, Antunovic P, Derolf A, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood. 2009;113(18):4179–4187. PubMed PMID: 19008455. [DOI] [PubMed] [Google Scholar]
- [2].Ossenkoppele G, Löwenberg B. How I treat the older patient with acute myeloid leukemia. Blood. 2015;125(5):767–774. PubMed PMID: 25515963. [DOI] [PubMed] [Google Scholar]
- [3].Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. PubMed PMID: 19629070. [DOI] [PubMed] [Google Scholar]
- [4].Park S, Chapuis N, Tamburini J, et al. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica. 2010;95(5):819–828. PubMed PMID: 19951971. PubMed Central PMCID: PMCPMC2864389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203. PubMed PMID: 22358332. [DOI] [PubMed] [Google Scholar]
- [6].Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol Cell Ther. 2015;3:2 PubMed PMID: 26056603. PubMed Central PMCID: PMCPMC4452048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lindblad O, Cordero E, Puissant A, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene. 2016;35(39):5119–5131. PubMed PMID: 26999641. PubMed Central PMCID: PMCPMC5399143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bortul R, Tazzari PL, Billi AM, et al. Deguelin, A PI3K/AKT inhibitor, enhances chemosensitivity of leukaemia cells with an active PI3K/AKT pathway. Br J Haematol. 2005;129(5):677–686. PubMed PMID: 15916691. [DOI] [PubMed] [Google Scholar]
- [9].Martelli AM, Evangelisti C, Chiarini F, et al. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1(2):89–103. PubMed PMID: 20671809. PubMed Central PMCID: PMCPMC2911128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Polivka J, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142(2):164–175. PubMed PMID: 24333502. [DOI] [PubMed] [Google Scholar]
- [11].Sampath D, Cortes J, Estrov Z, et al. Pharmacodynamics of cytarabine alone and in combination with 7-hydroxystaurosporine (UCN-01) in AML blasts in vitro and during a clinical trial. Blood. 2006;107(6):2517–2524. doi: 10.1182/blood-2005-08-3351 PubMed PMID: 16293603. PubMed Central PMCID: PMCPMC1895741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sato S, Fujita N, Tsuruo T. Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine). Oncogene. 2002;21(11):1727–1738. PubMed PMID: 11896604. [DOI] [PubMed] [Google Scholar]
- [13].Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8(4):253–267. PubMed PMID: 18354415. [DOI] [PubMed] [Google Scholar]
- [14].Lloyd RV, Erickson LA, Jin L, et al. p27kip1: a multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am J Pathol. 1999;154(2):313–323. PubMed PMID: 10027389. PubMed Central PMCID: PMCPMC1850003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Min YH, Cheong JW, Kim JY, et al. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res. 2004;64(15):5225–5231. PubMed PMID: 15289327. [DOI] [PubMed] [Google Scholar]
- [16].Motti ML, Califano D, Troncone G, et al. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol. 2005;166(3):737–749. PubMed PMID: 15743786. PubMed Central PMCID: PMCPMC1602368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prasad SB, Yadav SS, Das M, et al. PI3K/AKT pathway-mediated regulation of p27(Kip1) is associated with cell cycle arrest and apoptosis in cervical cancer. Cell Oncol (Dordr). 2015;38(3):215–225. PubMed PMID: 25821107. [DOI] [PubMed] [Google Scholar]
- [18].Yokozawa T, Towatari M, Iida H, et al. Prognostic significance of the cell cycle inhibitor p27Kip1 in acute myeloid leukemia. Leukemia. 2000;14(1):28–33. PubMed PMID: 10637473. [DOI] [PubMed] [Google Scholar]
- [19].Goldstone DC, Ennis-Adeniran V, Hedden JJ, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480(7377):379–382. PubMed PMID: 22056990. [DOI] [PubMed] [Google Scholar]
- [20].Franzolin E, Pontarin G, Rampazzo C, et al. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc Natl Acad Sci U S A. 2013;110(35):14272–14277. PubMed PMID: 23858451. PubMed Central PMCID: PMC3761606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hrecka K, Hao C, Gierszewska M, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474(7353):658–661. PubMed PMID: 21720370. PubMed Central PMCID: PMC3179858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Laguette N, Sobhian B, Casartelli N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474(7353):654–657. PubMed PMID: 21613998. PubMed Central PMCID: PMC3595993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Powell RD, Holland PJ, Hollis T, et al. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem. 2011;286(51):43596–43600. PubMed PMID: 22069334. PubMed Central PMCID: PMC3243528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. PubMed PMID: 22810696. PubMed Central PMCID: PMC3401966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Clifford R, Louis T, Robbe P, et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. 2014;123(7):1021–1031. PubMed PMID: 24335234. PubMed Central PMCID: PMC3924925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schuh A, Becq J, Humphray S, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood. 2012;120(20):4191–4196. PubMed PMID: 22915640. [DOI] [PubMed] [Google Scholar]
- [27].Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. PubMed PMID: 21941284. PubMed Central PMCID: PMC3307543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic anal. Science. 2008;321(5897):1801–1806. PubMed PMID: 18772397. PubMed Central PMCID: PMC2848990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. PubMed PMID: 18772396. PubMed Central PMCID: PMC2820389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Parsons DW, Li M, Zhang X, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331(6016):435–439. .PubMed PMID: 21163964. PubMed Central PMCID: PMC3110744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. PubMed PMID: 16959974. [DOI] [PubMed] [Google Scholar]
- [32].Liu J, Lee W, Jiang Z, et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res. 2012;22(12):2315–2327. PubMed PMID: 23033341. PubMed Central PMCID: PMC3514662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. PubMed PMID: 23415222. PubMed Central PMCID: PMC3575604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rentoft M, Lindell K, Tran P, et al. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc Natl Acad Sci U S A. 2016;113(17):4723–4728. PubMed PMID: 27071091. PubMed Central PMCID: PMCPMC4855590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Daddacha W, Koyen AE, Bastien AJ, et al. SAMHD1 Promotes DNA End Resection to Facilitate DNA Repair by Homologous Recombination. Cell Rep. 2017;20(8):1921–1935. PubMed PMID: 28834754. PubMed Central PMCID: PMCPMC5576576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].de Silva S, Hoy H, Hake TS, et al. Promoter methylation regulates SAMHD1 gene expression in human CD4+ T cells. J Biol Chem. 2013;288(13):9284–9292. PubMed PMID: 23426363. PubMed Central PMCID: PMC3610999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kohnken R, Kodigepalli KM, Mishra A, et al. MicroRNA-181 contributes to downregulation of SAMHD1 expression in CD4+ T-cells derived from Sezary syndrome patients. Leuk Res. 2017;52:58–66. PubMed PMID: 27889686. PubMed Central PMCID: PMC5195900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].de Silva S, Wang F, Hake TS, et al. Downregulation of SAMHD1 expression correlates with promoter DNA methylation in Sezary syndrome patients. J Invest Dermatol. 2014;134(2):562–565. PubMed PMID: 23884314. PubMed Central PMCID: PMC3844041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang JL, Lu FZ, Shen XY, et al. SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem Biophys Res Commun. 2014; 455 (3–4): 229–233. PubMed PMID: 25449277. [DOI] [PubMed] [Google Scholar]
- [40].Bonifati S, Daly MB, St Gelais C, et al. SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology. 2016;495:92–100. PubMed PMID: 27183329. PubMed Central PMCID: PMCPMC4912869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kodigepalli KM, Li M, Liu SL, et al. Exogenous expression of SAMHD1 inhibits proliferation and induces apoptosis in cutaneous T-cell lymphoma-derived HuT78 cells. Cell Cycle. 2017;16(2):179–188. PubMed PMID: 27929746. PubMed Central PMCID: PMC5283819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cancer Genome Atlas Research N, TJ Ley, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England Journal of Medicine. 2013;368(22):2059–2074. PubMed PMID: 23634996. PubMed Central PMCID: PMC3767041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 2002;30(1):207–210. PubMed PMID: 11752295. PubMed Central PMCID: PMC99122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Oh C, Ryoo J, Park K, et al. A central role for PI3K-AKT signaling pathway in linking SAMHD1-deficiency to the type I interferon signature. Sci Rep. 2018;8(1):84. doi: 10.1038/s41598-017-18308-8PubMed PMID: 29311560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Semba S, Itoh N, Ito M, et al. The in vitro and in vivo effects of 2-(4-morpholinyl)-8-phenyl-chromone (LY294002), a specific inhibitor of phosphatidylinositol 3ʹ-kinase, in human colon cancer cells. Clin Cancer Res. 2002;8(6):1957–1963.PubMed PMID: 12060641. [PubMed] [Google Scholar]
- [46].Shin I, Yakes FM, Rojo F, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8(10):1145–1152. PubMed PMID: 12244301. [DOI] [PubMed] [Google Scholar]
- [47].Viglietto G, Motti ML, Bruni P, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8(10):1136–1144. PubMed PMID: 12244303. [DOI] [PubMed] [Google Scholar]
- [48].Baldauf HM, Pan X, Erikson E, et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med. 2012;18(11):1682–1687. PubMed PMID: 22972397. PubMed Central PMCID: PMC3828732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roy A, Banerjee S. p27 and leukemia: cell cycle and beyond. J Cell Physiol. 2015;230(3):504–509. PubMed PMID: 25205053. [DOI] [PubMed] [Google Scholar]
- [50].Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8(10):1153–1160. PubMed PMID: 12244302. [DOI] [PubMed] [Google Scholar]
- [51].Denicourt C, Saenz CC, Datnow B, et al. Relocalized p27Kip1 tumor suppressor functions as a cytoplasmic metastatic oncogene in melanoma. Cancer Res. 2007;67(19):9238–9243. PubMed PMID: 17909030. [DOI] [PubMed] [Google Scholar]
- [52].Chen G, Cheng Y, Zhang Z, et al. Prognostic significance of cytoplasmic p27 expression in human melanoma. Cancer Epidemiol Biomarkers Prev. 2011;20(10):2212–2221. .EPI-11-0472. PubMed PMID: 21828232. [DOI] [PubMed] [Google Scholar]
- [53].Zhao D, Besser AH, Wander SA, et al. Cytoplasmic p27 promotes epithelial-mesenchymal transition and tumor metastasis via STAT3-mediated Twist1 upregulation. Oncogene. 2015;34(43):5447–5459. PubMed PMID: 25684140. PubMed Central PMCID: PMC4537852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tavor S, Petit I, Porozov S, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64(8):2817–2824. .can-03-3693. PubMed PMID: 15087398. [DOI] [PubMed] [Google Scholar]
- [55].Ma M, Ye JY, Deng R, et al. Mesenchymal stromal cells may enhance metastasis of neuroblastoma via SDF-1/CXCR4 and SDF-1/CXCR7 signaling. Cancer Lett. 2011;312(1):1–10. PubMed PMID: 21906874. [DOI] [PubMed] [Google Scholar]
- [56].Li X, Ma Q, Xu Q, et al. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012;322(2):169–176. PubMed PMID: 22450749. PubMed Central PMCID: PMC3408048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mezzanotte L, Fazzina R, Michelini E, et al. In vivo bioluminescence imaging of murine xenograft cancer models with a red-shifted thermostable luciferase. Mol Imaging Biol. 2010;12(4):406–414. PubMed PMID: 19937390. [DOI] [PubMed] [Google Scholar]
- [58].Tian T, Wang M, Ma D. TNF-alpha, a good or bad factor in hematological diseases? Stem Cell Investig. 2014;1:12 PubMed PMID: 27358858. PubMed Central PMCID: PMC4923506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127(1):53–61. PubMed PMID: 26660429. PubMed Central PMCID: PMC4705610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kohnken R, Kodigepalli KM, Wu L. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer. 2015;14:176. doi: 10.1186/s12943-015-0446-6 PubMed PMID: 26416562. PubMed Central PMCID: PMC4587406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Schneider C, Oellerich T, Baldauf HM, et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat Med. 2017;23(2):250–255. PubMed PMID: 27991919. [DOI] [PubMed] [Google Scholar]
- [62].Herold N, Rudd SG, Ljungblad L, et al. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nat Med. 2017;23(2):256–263. PubMed PMID: 28067901. [DOI] [PubMed] [Google Scholar]
- [63].Herold N, Rudd SG, Sanjiv K, et al. SAMHD1 protects cancer cells from various nucleoside-based antimetabolites. Cell Cycle. 2017;16(11):1029–1038. PubMed PMID: 28436707. PubMed Central PMCID: PMCPMC5499833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Herold N, Rudd SG, Sanjiv K, et al. With me or against me: tumor suppressor and drug resistance activities of SAMHD1. Exp Hematol. 2017;52:32–39. PubMed PMID: 28502830. [DOI] [PubMed] [Google Scholar]
- [65].Bencivenga D, Caldarelli I, Stampone E, et al. p27(Kip1) and human cancers: A reappraisal of a still enigmatic protein. Cancer Lett. 2017;403:354–365. PubMed PMID: 28687353. [DOI] [PubMed] [Google Scholar]
- [66].Pagano M. Control of DNA synthesis and mitosis by the Skp2-p27-Cdk1/2 axis. Mol Cell. 2004;14(4):414–416. PubMed PMID: 15149588. [DOI] [PubMed] [Google Scholar]
- [67].Sharma SS, Pledger WJ. The non-canonical functions of p27(Kip1) in normal and tumor biology. Cell Cycle. 2016;15(9):1189–1201. PubMed PMID: 27082696. PubMed Central PMCID: PMC4889241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wu FY, Wang SE, Sanders ME, et al. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 2006;66(4):2162–2172. .CAN-05-3304.PubMed PMID: 16489017. [DOI] [PubMed] [Google Scholar]
- [69].Chen S, Bonifati S, Qin Z, et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-kappaB and interferon pathways. In Proceedings of the National Academy of Sciences of the United States of America 2018. doi: 10.1073/pnas.1801213115 PubMed PMID: 29610295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang F, St Gelais C, De Silva S, et al. Phosphorylation of mouse SAMHD1 regulates its restriction of human immunodeficiency virus type 1 infection, but not murine leukemia virus infection. Virology. 2016;487:273–284. PubMed PMID: 26580513. PubMed Central PMCID: PMCPMC4679491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].St Gelais C, Kim SH, Ding L, et al. A putative cyclin-binding motif in human SAMHD1 contributes to protein phosphorylation, localization, and stability. J Biol Chem. 2016;291(51):26332–26342. PubMed PMID: 27815502. PubMed Central PMCID: PMCPMC5159495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.