

Abstract
Recently we purified and identified a previously uncharacterized transcription factor from rat liver binding to the carbohydrate responsive element of the L-type pyruvate kinase (L-PK) gene. This factor was named carbohydrate responsive element binding protein (ChREBP). ChREBP, essential for L-PK gene transcription, is activated by high glucose and inhibited by cAMP. Here, we demonstrated that (i) nuclear localization signal and basic helix-loop-helix/leucine-zipper domains of ChREBP were essential for the transcription, and (ii) these domains were the targets of regulation by cAMP and glucose. Among three cAMP-dependent protein kinase phosphorylation sites, Ser196 and Thr666 were the target sites. Phosphorylation of the former resulted in inactivation of nuclear import, and that of the latter resulted in loss of the DNA-binding activity and L-PK transcription. On the other hand, glucose activated the nuclear import by dephosphorylation of Ser196 in the cytoplasm and also stimulated the DNA-binding activity by dephosphorylation of Thr666 in the nucleus. These results thus reveal mechanisms for regulation of ChREBP and the L-PK transcription by excess carbohydrate and cAMP.
The liver is the principal organ responsible for conversion of excess dietary carbohydrate to triglycerides. A high carbohydrate diet leads to activation of several regulatory enzymes of glycolysis and lipogenesis including L-type pyruvate kinase (L-PK), acetyl CoA carboxylase, and fatty acid synthase (1). Excess carbohydrate also results in post-translational activation of several key enzymes involved in carbohydrate metabolism and lipogenesis (1). Until recently it was thought that hormones such as insulin and glucagon regulate the transcription of genes. It has only been appreciated recently that nutrients themselves play an important role in the regulation.
Glucose-stimulated L-PK gene expression in liver is mediated through the glucose or carbohydrate response element (ChRE) that is located in the region −183 to −96 base pairs upstream from the cap site of the L-PK gene (2). The binding site for the ChRE contains an E-box sequence, CACGGG, separated by 5 bases that corresponds to the consensus binding site (CACGTG) for upstream stimulatory factors and their related family members. Several transcription factors binding the ChRE have been reported previously (3–5), but none of these factors vary with diet, and the mechanisms of regulation are still unclear.
We recently purified, identified, and cloned a transcription factor that binds specifically to the ChRE of the L-PK gene (6). We termed this new transcription factor ChRE-binding protein (ChREBP; ref. 6). ChREBP is expressed specifically in liver and is responsive to excess carbohydrate, i.e., ChREBP is activated by high glucose diet and inhibited by high fat diet. Overexpression of ChREBP in primary cultured hepatocytes results in increased transcription activity of the L-PK gene in response to high glucose.
ChREBP is a member of the basic helix–loop–helix/leucine zipper (bHLH/ZIP) family of transcription factors of Mr = 94,600 and contains several potentially functional domains including a nuclear localization signal (NLS), a proline-rich stretch (PRO), a bHLH/ZIP, and a ZIP-like domain (Fig. 1; ref. 6). It contains three consensus phosphorylation sites for PKA. Previously we demonstrated that a partially purified ChREBP from rat liver nuclei is phosphorylated by PKA in vitro resulting in loss of the DNA-binding activity (6), but the site of the phosphorylation and mechanisms of loss of the DNA-binding activity are not known.
Figure 1.

Schematic representation of domain structure of mouse ChREBP. The locations of ChREBP, NLS, PRO, bHLH/ZIP, and ZIP-like domains are indicated. The locations of three putative phosphorylation sites of cAMP-dependent protein kinase (PKA) are indicated as P1, P2, and P3.
The objectives of the current investigation were to characterize ChREBP further by identifying the functional domains and to determine the phosphorylation sites, regulated negatively by cAMP and PKA and positively by high glucose.
Experimental Procedures
Materials.
All reagents were from Sigma unless otherwise indicated.
Plasmids, Domain Deletion, and Mutagenesis.
The constructs were verified by nucleotide sequencing. Full-length wild-type (WT) ChREBP cDNA (GenBank accession no. AF156604) was ligated into the Invitrogen mammalian expression vector pcDNA3 (ChREBP/pcDNA3; ref. 6) or CLONTECH vector pEGFP-N3 (ChREBP/pEGFP) encoding enhanced green fluorescent protein (GFP). The promoter region between positions −206 and −7 of the L-PK gene was ligated into the luciferase expression plasmid, pGL-3 basic vector (Promega), as described previously (7). Plasmids containing the NLS, PRO, bHLH/ZIP, ZIP-like deletion mutants, or point mutants in the putative phosphorylation sites of PKA of ChREBP were constructed by using the QuickChange site-directed mutagenesis kit (Stratagene). Oligonucleotides used to introduce a new restriction site immediately upstream or downstream of each domain are listed in Table 1. Oligonucleotides used to introduce the desired mutations are listed in Table 2. The double mutant plasmids were constructed by using the same method as that used for making single mutants.
Table 1.
Oligonucleotides used to construct plasmids containing deletions of domain
| Deletion domain | Location | Restriction site | Orientation | Sequence (position) |
|---|---|---|---|---|
| NLS | Upstream | DraI | Forward | 5′-CACATGAGCACTTTAAACCTGAGGCTGT-3′ (bp 461–488) |
| Reverse | 5′-ACAGCCTCAGGTTTAAAGTGCTCATGTG-3′ (bp 461–488) | |||
| Downstream | MscI | Forward | 5′-TGGTGATGCGCTGGCCACACGTGTGGAG-3′ (bp 527–554) | |
| Reverse | 5′-CTCCACACGTGTGGCCAGCGCATCACCA-3′ (bp 527–554) | |||
| PRO | Upstream | AflII | Forward | 5′-TTCCTGAAGACCTTAAGACCAAGATCCC-3′ (bp 1148–1175) |
| Reverse | 5′-GGGATCTTGGTCTTAAGGTCTTCAGGAA-3′ (bp 1148–1175) | |||
| Downstream | AflII | Forward | 5′-TCTCTGTAGACCTTAAGCCCCATGGGTA-3′ (bp 1418–1445) | |
| Reverse | 5′-TACCCATGGGGCTTAAGGTCTACAGAGA-3′ (bp 1418–1445) | |||
| bHLH/ZIP | Upstream | AflII | Forward | 5′-ACAACAAGATGCTTAAGCGACGTATCAC-3′ (bp 1967–1994) |
| Reverse | 5′-GTGATACGTCGCTTAAGCATCTTGTTGT-3′ (bp 1967–1994) | |||
| Downstream | AflII | Forward | 5′-TCAACTTGTGCCTTAAGCAGCTACCGGC-3′ (bp 2219–2246) | |
| Reverse | 5′-GCCGGTAGCTGCTTAAGGCACAAGTTGA-3′ (bp 2219–2246) | |||
| ZIP-like | Upstream | DraI | Forward | 5′-CCGCCAGCTTGTTTAAACTCCGCCAGAC-3′ (bp 2399–2426) |
| Reverse | 5′-GTCTGGCGGAGTTTAAACAAGCTGGCGG-3′ (bp 2399–2426) | |||
| Downstream | MscI | Forward | 5′-CCAGCCTTGTGTGGCCACAAGCCACACG-3′ (bp 2531–2558) | |
| Reverse | 5′-CGTGTGGCTTGTGGCCACACAAGGCTGG-3′ (bp 2531–2558) |
Table 2.
Oligonucleotides used to construct plasmids containing point mutations in the putative phosphorylation sites of PKA
| Mutant | Orientation | Sequence (position) |
|---|---|---|
| S196A | Forward | 5′-CTCCGTAAGTCCGCCAGGGAAGGGGAT-3′ (bp 574–600) |
| Reverse | 5′-ATCCCCTTCCCTGGCGGACTTACGGAG-3′ (bp 574–600) | |
| S196D | Forward | 5′-CTCCGTAAGTCCGACAGGGAAGGGGAT-3′ (bp 574–600) |
| Reverse | 5′-ATCCCCTTCCCTGTCGGACTTACGGAG-3′ (bp 574–600) | |
| S626A | Forward | 5′-GAGCGGCGACTAGCCGGGGATCTCAAC-3′ (bp 1864–1890) |
| Reverse | 5′-GTTGAGATCCCCGGCTAGTCGCCGCTC-3′ (bp 1864–1890) | |
| S626D | Forward | 5′-GAGCGGCGACTAGACGGGGATCTCAAC-3′ (bp 1864–1890) |
| Reverse | 5′-GTTGAGATCCCCGTCTAGTCGCCGCTC-3′ (bp 1864–1890) | |
| T666A | Forward | 5′-AACCGACGTATCGCCCACATCTCCGCG-3′ (bp 1981–2007) |
| Reverse | 5′-CGCGGAGATGTGGGCGATACGTCGGTT-3′ (bp 1981–2007) | |
| T666D | Forward | 5′-AACCGACGTATCGACCACATCTCCGCG-3′ (bp 1981–2007) |
| Reverse | 5′-CGCGGAGATGTGGTCGATACGTCGGTT-3′ (bp 1981–2007) |
The mutated nucleotides are underlined.
Recombinant Protein Production.
Recombinant protein of mouse ChREBP (accession no. NP_067430) was expressed in Escherichia coli by inserting the C terminus of the ChREBP construct, encoding the amino acids 651–864 of ChREBP + His6 tag, into the BamHI and HindIII sites of the T7-based expression vector pMal-c2 (New England Biolabs) to produce an N-terminal fusion with maltose-binding protein. Expressed protein was purified according to manufacturer instructions.
Treatment of Rats with Dibutyryl (db)-cAMP and Nuclear Extract Preparation.
Male Sprague–Dawley rats weighting 250 g were used in all experiments. After 48 h of starvation, the rats were fed a high carbohydrate diet, and db-cAMP (1 mmol/kg) was injected intraperitoneally. The rats were killed 10 and 30 min after injection, and nuclear extracts were prepared immediately from rat liver as described previously (6).
Primary Hepatocyte Culture, Transfection, Luciferase Activity Assay, and DNA Binding Assay.
The primary hepatocyte culture, transfection, luciferase activity assay, and DNA binding assay were performed as described previously (6). Each GFP-ChREBP mutant expressed equally (transfected hepatocytes = ≈10% of 1 × 106 hepatocytes).
Protein Kinase and Protein Phosphatase Inhibitors.
Before exchange to 5.5 or 27.5 mM glucose medium, transfected primary culture hepatocytes were treated for 1 h with 10 μM H-89, 500 nM cantharidic acid (Calbiochem), 5 μM KN-62 (Calbiochem), 30 μM PD98059 (Calbiochem), 50 μM Genistein, or dimethyl sulfoxide vehicle.
Determination of Subcellular Localization of ChREBP.
Subcellular localization of GFP-fused WT ChREBP or its mutants was determined by using a confocal laser scanning microscope (Bio-Rad). To quantitate nuclear localization of ChREBP for each condition, 100 transfected hepatocytes from five independent experiments were scored in a blinded fashion as to whether the GFP-fused ChREBP was predominantly nuclear or cytoplasmic. The identity of the nucleus was verified by comparative phase-contrast microscopy.
Phosphorylation and Dephosphorylation of ChREBP.
The reaction mixture contained, in a final volume of 0.2 ml, 50 mM Tris⋅Cl, pH 7.5, 0.2 μM [γ-32P]ATP [3,000 Ci/mmol (1 Ci = 37 GBq)], 5 mM MgCl2, 0.2 mM EDTA, 2 mM DTT, and 5 mM potassium phosphate. The reaction was initiated with the addition of catalytic subunit of PKA (200 units) or protein phosphatase 2A (PP2A, 200 units), and the reaction mixture was incubated at 30°C. At given time intervals, aliquots were removed and DNA-binding activity was determined by a gel shift assay (6). [32P]Phosphate-bound ChREBP was also determined as described previously (8).
Statistical Analysis.
All data were expressed as mean ± SEM. The differences between two groups were analyzed by using the Mann–Whitney u test. A P value less than 0.05 was considered statistically significant.
Results
Effects of Domain Deletion of ChREBP on Transcriptional Activity of the L-PK Gene.
To determine the function of each domain of ChREBP (Fig. 1), various domain deletion mutants of ChREBP were prepared. The effects of these mutant ChREBPs on L-PK transcription activity were determined with a dual luciferase reporter system. Primary cultured hepatocytes were transfected with the WT and mutant ChREBPs, and the cells were maintained in low (5.5 mM) and high (27.5 mM) glucose. As shown in Fig. 2, the transcriptional activities in the cells transfected with empty vector were caused by the endogenous activity of ChREBP in the primary hepatocytes. The WT ChREBP showed at least 2-fold activation of the activity in high glucose compared with that in the empty vector. To confirm that this increase was caused by glucose metabolism and not osmotic stress, primary hepatocytes were incubated with 500 mM NaCl instead of 27.5 mM glucose. The increase in transcriptional activity was not seen in NaCl, and the transcriptional activation required high glucose.
Figure 2.

Effects of domain deletion of ChREBP on transcription activity of the L-PK gene. Rat primary cultured hepatocytes were transfected with the WT ChREBP or a series of domain deletion mutants. A pGL3 basic plasmid (simian virus 40 promoter driving firefly luciferase gene), carrying the promoter region between positions −206 and −7 of the L-PK gene, and pRL-TK (thymidine kinase promoter driving the Renilla luciferase gene) were also transfected into each cell as a reporter gene and an internal control, respectively. After transfection, cells were incubated under 5.5 mM (□) or 27.5 mM (■) glucose for 12 h. Relative luciferase activity was calculated as described in Experimental Procedures and are expressed as mean ± SEM (n = 5). *, P < 0.05 compared with that of WT ChREBP.
Among the mutant ChREBPs, deletion of NLS or the bHLH/ZIP domain resulted in complete loss of the high glucose-induced transcriptional activation. However, the deletion of PRO or the ZIP-like domain at the C terminus did not affect the transcriptional activity. These results demonstrated that the NLS and bHLH/ZIP domains were essential for its glucose response, but the PRO and the ZIP-like domains were not involved in the high glucose-induced transcriptional activation of the L-PK gene.
Effects of Various Inhibitors of Protein Kinase and Phosphatase on ChREBP-Induced Transcriptional Activation of the L-PK Gene.
Protein phosphorylation and dephosphorylation is one way to regulate the activity of a transcription factor. To investigate possible involvement of phosphorylation and dephosphorylation of ChREBP in the glucose-induced activation of L-PK gene transcription, we examined the effect of a number of different inhibitors of various protein kinases and protein phosphatases. The addition of H-89, a specific inhibitor of PKA, to the culture medium increased the transcriptional activity in the ChREBP-overexpressed hepatocytes under both low and high glucose (Fig. 3). H-89 also stimulated endogenous ChREBP, but the effect was small. On the other hand, cantharidic acid, a specific inhibitor of PP2A, inhibited transcriptional activity of the L-PK gene in both 5.5 and 27.5 mM glucose. KN-62, PD98059, and Genistein, which are specific inhibitors of calcium/calmodulin-dependent protein kinase II, mitogen-activated protein kinase, and tyrosine kinase, respectively, did not affect transcriptional activity of the L-PK gene, suggesting that these protein kinases were not involved in the regulation of ChREBP. These results suggested that ChREBP activity was inhibited by phosphorylation catalyzed by PKA, and the activity was regained by PP2A.
Figure 3.

Effects of H-89 and cantharidic acid on transcription activity of L-PK gene in ChREBP-overexpressed hepatocytes. Rat primary cultured hepatocytes were transfected with the WT ChREBP. H-89, cantharidic acid, or dimethyl sulfoxide vehicle was added, and the cells were incubated in 5.5 mM (□) or 27.5 mM (■) glucose for 12 h. Relative luciferase activity was calculated as described in Experimental Procedures and is expressed as mean ± SEM (n = 5). *, P < 0.05 compared with that of WT ChREBP.
Effects of H-89 and Cantharidic Acid on Subcellular Localization of ChREBP.
NLS domains serve as signals for import of transcription factors into nuclei. We constructed a GFP-fused WT ChREBP as well as a series of domain deletion mutants of ChREBP, transfected the constructs into primary cultured hepatocytes, and observed subcellular localization of ChREBP with confocal laser scanning microscopy. Fig. 4 shows representative images of subcellular localization of ChREBP under low or high glucose. Nontransfected hepatocytes did not show any fluorescence (Fig. 4A), and endogenous ChREBP could not be seen under these conditions. Empty vector-transfected hepatocytes showed weak diffused fluorescence throughout the cells (Fig. 4B). The ChREBP-transfected hepatocytes showed cytoplasmic (Fig. 4C) and nuclear localization (Fig. 4D) in low and high glucose, respectively. The nuclei import was complete in 8 h, whereas the L-PK promoter activity took 20 h. Thus, the rate of the translocation of ChREBP was rapid enough to activate L-PK gene expression.
Figure 4.

Representative images of subcellular localization of GFP-fused ChREBP under low or high glucose. (A) Nontransfected hepatocytes; (B) Empty vector-transfected hepatocytes; (C) GFP-fused WT ChREBP transfected hepatocyte in low (5.5 mM) glucose; (D) GFP-fused WT ChREBP transfected hepatocyte in low (27.5 mM) glucose. (Bar = 10 μm.)
The WT ChREBP showed ≈40 and 80% of nuclear staining rate in 5.5 and 27.5 mM glucose, respectively (Fig. 5A). NLS-deleted ChREBP mutant showed less than 10% of nuclear staining rate in both 5.5 and 27.5 mM glucose. These results demonstrated that high glucose stimulated the translocation of ChREBP from cytosol into nuclei. By treatment of hepatocytes with H-89, the nuclear staining rate was increased in 5.5 mM glucose. On the other hand, cantharidic acid significantly decreased the nuclear staining rate, suggesting that the dephospho form of ChREBP but not the phospho form was translocated into the nuclei (Fig. 5A). Thus, NLS domain was required for nuclear translocation, and the nuclear translocation of ChREBP was regulated negatively by PKA-catalyzed phosphorylation and positively by dephosphorylation by PP2A.
Figure 5.

(A) Effect of NLS domain deletion, H-89, and cantharidic acid on nuclear translocation of ChREBP. Rat primary cultured hepatocytes were transfected with GFP-fused WT ChREBP or NLS domain deletion mutants. H-89, cantharidic acid, or dimethyl sulfoxide vehicle was added, and the cells were incubated under 5.5 mM (□) or 27.5 mM (■) glucose for 12 h. For each condition, 100 transfected hepatocytes from four independent experiments were scored in a blinded fashion as to whether the GFP-fused ChREBP was predominantly nuclear or cytoplasmic as described in Experimental Procedures. The values are expressed as mean ± SEM. *, P < 0.05 compared with that of WT ChREBP. (B) Putative PKA phosphorylation sites. The phosphorylation sites, Ser or Thr, are underlined. Ala or Asp introduced in the place of Ser or Thr, respectively, by site-directed mutagenesis are underlined. (C) Effects of mutation of Ser196 on subcellular localization of ChREBP. Rat primary cultured hepatocytes were transfected with GFP-fused WT ChREBP, S196A, or S196D, and the cells were incubated in 5.5 mM (□) or 27.5 mM (■) glucose for 12 h. For each condition, 100 transfected hepatocytes from four independent experiments were scored in a blinded fashion as to whether the GFP-fused ChREBP was predominantly nuclear or cytoplasmic as described in Experimental Procedures. The values are expressed as mean ± SEM. *, P < 0.05 compared with that of WT ChREBP.
Effects of Mutation of Ser196 on Subcellular Localization of ChREBP.
To determine the phosphorylation sites of ChREBP, which regulate the nuclear import, we mutated all three consensus PKA phosphorylation sites. Because a putative PKA phosphorylation site (Ser196 designated as P1 in Fig. 1) occurs near the NLS domain, we prepared Ala and Asp mutants of ChREBP (S196A and S196D; Fig. 5B) and determined the effect on nuclear localization. S196A showed ≈90% of nuclear staining rate in both 5.5 and 27.5 mM glucose (Fig. 5C), whereas the S196D mutant showed less than 10% of nuclear staining rate (Fig. 5C). To investigate the effects of phosphorylation states of the other PKA phosphorylation sites, we constructed double mutants (S196A + S626A, S196A + S626D, S196A + T666A, and S196A + T666D; Fig. 5B). All these double mutants showed ≈90% of nuclear staining rate, suggesting strongly that the nuclear translocation was regulated by phosphorylation at Ser196 regardless of the phosphorylation states of the other sites.
Effect of db-cAMP on the DNA-Binding Activity of ChREBP in Vivo.
To determine the effect of cAMP on the DNA-binding activity of ChREBP in vivo, rats were treated with i.p. injections of db-cAMP. Nuclear extracts of db-cAMP-treated rat livers were assayed for DNA-binding activity. The DNA-binding activity was inactivated readily within 10 min after the db-cAMP injection (Fig. 6). Because these rats were fed, ChREBP was localized already in nuclei, and in our preliminary experiments using GFP-ChREBP, we showed that the ChREBP was not exported from nuclei to cytoplasm within 10 min after addition of db-cAMP into 27.5 mM glucose medium (n = 3; data not shown). Moreover, it has been reported that nuclear export takes 30 min–2 h (9, 10). The loss of the DNA-binding activity may be caused by the inactivation of DNA binding, but we cannot exclude the possibility of export of ChREBP.
Figure 6.

Effect of db-cAMP on the DNA-binding activity of ChREBP in vivo. Rats were fed high carbohydrate diets after 48 h of starvation, and db-cAMP (60 mg/kg) was injected intraperitoneally. The rats were killed 10 and 30 min after the injection, and the DNA-binding activity of ChREBP was examined by using a gel shift assay. USF, upstream stimulatory factor.
Effects of PKA and PP2A on the DNA-Binding Activity of ChREBP in Vitro.
By using a recombinant C-terminal region of ChREBP containing the bHLH/ZIP with a single phosphorylation site (Thr666, P3 in Fig. 1), we determined the effect of PKA-dependent phosphorylation of Thr666 on the DNA-binding activity of bHLH/ZIP domain. As shown in Fig. 7, A and Inset, 32P was incorporated into the bHLH/ZIP domain; the DNA-binding activity was inactivated rapidly, and the rates of these changes were similar. The observation that both the phosphate incorporation and loss of the DNA-binding activity were similar suggested that the inactivation of DNA binding was a direct result of the phosphorylation at Thr666. Furthermore, PKA-dependent phosphorylation of the bHLH/ZIP domain was fully reversible, because the incubation of the 32P-phosphorylated bHLH/ZIP domain with catalytic subunit of PP2A resulted in complete recovering of the activity (Fig. 7, B and Inset).
Figure 7.

(A) Phosphorylation of ChREBP by PKA and effects of PKA on the DNA-binding activity of ChREBP in vitro. Recombinant C-terminal ChREBP was incubated with PKA in the presence of [γ-32P]ATP. At given time intervals, [32P]phosphate incorporation (□) and DNA-binding activity (■) were determined as described in Experimental Procedures. The values are expressed as mean ± SEM. (Insert) Representative image for effects of PKA on the DNA-binding activity of ChREBP using gel shift assay. (B) Dephosphorylation of ChREBP by PP2A and the effect of PP2A on the DNA-binding activity of ChREBP in vitro. Phosphorylated C-terminal ChREBP was incubated with PP2A. At given time intervals, remaining phosphorylated C-terminal ChREBP (□) and DNA-binding activity (■) were determined as described in Experimental Procedures. The values are expressed as mean ± SEM. (Insert) Representative image for effects of PP2A on the DNA-binding activity of ChREBP.
Effect of Mutation at Ser626 and Thr666 on Transcription Activity of L-PK Gene.
There are two PKA phosphorylation sites: Ser626, located near the bHLH domain, and Thr666, situated within the basic residues of the bHLH domain (designated as P2 and P3 in Fig. 1). To determine which phosphorylation site regulates the ChREBP DNA-binding activity, we constructed Ala and Asp mutants of these sites (S626A, S626D, T666A, and T666D). Both S626A and T666A mutants showed ≈2.5-fold activation of transcriptional activity by high glucose compared with that of WT ChREBP (Fig. 8A). On the other hand, both S626D and T666D mutants showed much lower activity under both high and low glucose (Fig. 8A). These results indicated that both Ser626 and Thr666 were the sites of phosphorylation and involved in the inhibition of the DNA-binding activity. To determine which site was more crucial for the activity, we constructed two double mutants (S196A + S626A and S196A + T666A) and examined the effects of db-cAMP on transcriptional activity. The transcriptional activity of S196A + T666A, but not S196A + S626A, was not inhibited by treatment with db-cAMP (Fig. 8B), suggesting that Thr666 was the most important regulatory site, and Ser626 may play only a supportive role in the DNA binding.
Figure 8.

(A) The effect of mutation at Ser626 and Thr666 on the transcription activity of the L-PK gene. Rat primary cultured hepatocytes were transfected with WT ChREBP, S626A, S626D, T666A, or T666D. After transfection, the cells were incubated in 5.5 mM (□) or 27.5 mM (■) glucose for 12 h. Relative luciferase activity was calculated as described in Experimental Procedures and are expressed as mean ± SEM (n = 5). *, P < 0.05 compared with that of WT ChREBP. (B) The effects of db-cAMP on L-PK gene transcription in S626A- and T666A-overexpressed hepatocytes. To exclude the effect of db-cAMP on nuclear translocation of ChREBP, double mutants (S196A + S626A and S196A + T666A) were constructed. Rat primary cultured hepatocytes were transfected with WT ChREBP, S196A + S626A, or S196A + T666A. After transfection, cells were incubated under 5.5 mM (□) or 27.5 mM (■) glucose, and cAMP was added to both low and high glucose. Relative luciferase activity was calculated as described in Experimental Procedures and are expressed as mean ± SEM (n = 5). *, P < 0.05 compared with that of WT ChREBP.
Discussion
ChREBP is a previously uncharacterized transcription factor (6) that binds to ChRE of the L-PK gene and plays an important role in the regulation of gene transcription (6). Glucagon mediation via cAMP is known to inhibit transcription of the L-PK gene, whereas high glucose or other carbohydrate activates the transcription (1). ChREBP contains several domains including NLS, PRO, bHLH/ZIP, and ZIP-like domains (Fig. 1). In the current study, we investigated the roles of each domain in L-PK transcription and studied the mechanism of regulation of ChREBP by cAMP and glucose.
Nuclear Localization via NLS Domain and Its Regulation.
The bipartite NLS of ChREBP, located toward the N terminus (amino acids 155–174), consists of 19 aa (HRKPEAVVLEGNYWKRRIE) containing several basic residues that are separated by hydrophobic amino acids as well as acidic and hydrophobic amino acids flanking the motif. We demonstrated here that the NLS domain was essential in translocating ChREBP into nuclei (Fig. 4) and for activation of L-PK transcription (Fig. 2). Deletion of the NLS domain resulted in a complete loss of glucose response. This requirement of the NLS for ChREBP (Mr = 94,600) localization is not surprising, because it is well known that proteins larger than 45 kDa require NLS to target them to nuclei (11). We also demonstrated that the translocation of ChREBP into nuclei was stimulated by high glucose and inhibited by cAMP. The consensus phosphorylation site (193RRSS196; designated as P1 in Fig. 1) located near the NLS domain is likely to regulate translocation of ChREBP for the following reasons: (i) H-89, a specific PKA inhibitor, stimulated translocation of ChREBP into nuclei, whereas cantharidic acid, a specific PP2A inhibitor, prevented the translocation, (ii) mutation of Ser196 to Asp resulted in complete loss of localization to nuclei even in the presence of high glucose, and (iii) the S196A mutant was localized in the nuclei even in the presence of low glucose.
DNA Binding via bHLH/ZIP Domain and Its Regulation.
ChREBP belongs to the bHLH/ZIP family of transcription factors (6), and its basic domain binds an E-box sequence of CACGGG and GTGCCC separated by 5 bases. The members of this family such as MyoD bind consensus 5′-CANNTG-3′. The three-dimensional structure of this domain of MyoD complexed with an oligonucleotide containing 5′-CACGTG-3′ has been determined (12). Comparison of the amino acid sequence of the DNA binding domain of ChREBP with that of MyoD shows the following alignment:
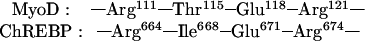 |
Arg664, Asn668, and Glu671 of ChREBP could bind one of the half sites, 5′-CAC-3′/3′-GTG-5′, of the DNA as in MyoD. The Thr666 (P3 in Fig. 1), which is located within the DNA binding site, is probably the target of the phosphorylation by PKA. Introduction of the highly negative charges of the phosphate residue on Thr666 probably breaks a salt bridge between Arg664 and the active site, Glu671, and forms a new salt bridge with Arg664. Obviously, this is the most direct and efficient way to inhibit the DNA binding. Thus, the mechanism of cAMP-mediated inhibition of L-PK transcription and the DNA binding of ChREBP is likely because of the phosphorylation of Thr666 by PKA. This conclusion is supported by the phosphorylation of the truncated ChREBP, which contained only Thr666 as a phosphorylation site, in which loss of the DNA-binding activity was correlated directly with the phosphate incorporation catalyzed by PKA (Fig. 7A).
The Ser626 (P2 in Fig. 1) also appeared to have a regulatory function, because Ala and Asp mutants of this site showed a similar response to that of Thr666. However, it may play a passive role in overall transcriptional activity. The reason for this possibility was that only the T666A, but not S626A, showed a lack of inhibition by cAMP (Fig. 8B).
Glucose Activation and Signaling.
ChREBP contains two major phosphorylation sites, P1 and P3, which are regulated by the phosphorylation/dephosphorylation mechanism mediated by cAMP and glucose. Glucose signaling activates ChREBP and the L-PK transcription at least two steps of the overall process, namely the transport of ChREBP to the nucleus from the cytoplasm and the activation of DNA binding. Both processes seem to be activated by dephosphorylation of inactive ChREBP phosphorylated at P1 and P3. The results suggest that these dephosphorylation reactions are probably catalyzed by PP2A, and that the glucose signaling involves activation of the PP2A by a glucose metabolite(s).
In summary, we propose the following scheme (Fig. 9) for the mechanism of regulation of ChREBP by cAMP and glucose. ChREBP may exist in the cytosol in an inactive form that is phosphorylated at the P3 site (P3-ChREBP), because we have not found any DNA-binding activity in the cytosol of liver (6). The first step in glucose activation is to activate by dephosphorylation of the P1 site, because only the dephospho form is imported into the nucleus. The P3 site could be either form for translocation. This first activation reaction is catalyzed by a metabolite (X)-activated PP2A to dephosphorylate the P1 site of the transport inactive P1, P3-ChREBP to P3-ChREBP. Once the ChREBP is imported into the nucleus, glucose activates the inactive form with respect to the DNA-binding activity, P3-ChREBP, by dephosphorylation of the P3 site catalyzed by a PP2A, which is activated by a metabolite (X). The metabolite (X) could be xylulose-5-P (13). For the export from the nucleus to cytosol, we suggest that ChREBP is phosphorylated probably at the P3 site in the nucleus as a first step, and the inactive P3-ChREBP is exported to the cytoplasm. The import and export of ChREBP might be far more complex, and the elucidation of these mechanisms awaits future investigation.
Figure 9.

Schematic representation of possible regulatory mechanisms of ChREBP by glucose and cAMP in hepatocytes. Two PKA phosphorylation sites of ChREBP, Ser196 (P1) and Thr666 (P3), play crucial roles in high glucose-induced activation of ChREBP (P1 and P3 indicate the phosphorylated form at the sites). Glucose signaling may activate PP2A via a metabolite (X). In cytoplasm, X-activated PP2A dephosphorylates P1 site of the ChREBP, which results in stimulation of import of ChREBP into the nucleus. Once ChREBP is localized in the nucleus, glucose activates the inactive form of ChREBP (P3-ChREBP) by dephosphorylation of the P3 site catalyzed by PP2A. ChREBP, which is dephosphorylated at P1 and P3 sites, binds to ChRE of the L-PK gene and consequently activates transcription of the L-PK gene. See Discussion.
Acknowledgments
We thank Dr. Michel Roth, Dr. Lance Terada, and Young Li for technical help and Dr. Sarah McIntire for critical reading.
Abbreviations
- L-PK
L-type pyruvate kinase
- ChRE
carbohydrate response element
- ChREBP
ChRE-binding protein
- bHLH
basic helix–loop–helix
- ZIP
leucine zipper
- NLS
nuclear localization signal
- PRO
proline-rich stretch
- PKA
cAMP-dependent protein kinase
- WT
wild type
- GFP
green fluorescent protein
- db
dibutyryl
- PP2A
protein phosphatase 2A
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 13476.
References
- 1.Kahn A. Biochimie. 1997;79:113–118. doi: 10.1016/s0300-9084(97)81501-5. [DOI] [PubMed] [Google Scholar]
- 2.Shih H M, Liu Z, Towle H C. J Biol Chem. 1995;270:21991–21997. doi: 10.1074/jbc.270.37.21991. [DOI] [PubMed] [Google Scholar]
- 3.Vaulont S, Puzenat N, Levrat F, Cognet M, Kahn A, Raymondjean M J. Mol Biol. 1989;209:205–219. doi: 10.1016/0022-2836(89)90273-8. [DOI] [PubMed] [Google Scholar]
- 4.Puzenat N, Vaulont S, Kahn A, Raymondjean M. Biochem Biophys Res Commun. 1992;189:1119–1128. doi: 10.1016/0006-291x(92)92320-w. [DOI] [PubMed] [Google Scholar]
- 5.Koo S H, Towle H C. J Biol Chem. 2000;275:5200–5207. doi: 10.1074/jbc.275.7.5200. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita H, Takenoshita M, Sakurai M, Richard K B, William J H, Wendy S, David A, Uyeda K. Proc Natl Acad Sci USA. 2001;98:9116–9121. doi: 10.1073/pnas.161284298. . (First Published July 24, 2001; 10.1073/pnas.161284298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasegawa J, Osatomi K, Wu R F, Uyeda K. J Biol Chem. 1999;274:1100–1107. doi: 10.1074/jbc.274.2.1100. [DOI] [PubMed] [Google Scholar]
- 8.Kitajima S, Uyeda K. J Biol Chem. 1983;258:7352–7357. [PubMed] [Google Scholar]
- 9.Efthymiadis A, Dottorini T, Jans D A. Arch Biochem Biophys. 1998;355:254–261. doi: 10.1006/abbi.1998.0719. [DOI] [PubMed] [Google Scholar]
- 10.Mao P L, Jiang Y, Wee B Y, Porter A G. J Biol Chem. 1998;273:23621–23624. doi: 10.1074/jbc.273.37.23621. [DOI] [PubMed] [Google Scholar]
- 11.Jans D A, Hubner S. Physiol Rev. 1996;76:651–685. doi: 10.1152/physrev.1996.76.3.651. [DOI] [PubMed] [Google Scholar]
- 12.Kretzner L, Blackwood E M, Eisenman R N. Curr Top Microbiol Immunol. 1992;182:435–443. doi: 10.1007/978-3-642-77633-5_55. [DOI] [PubMed] [Google Scholar]
- 13.Nishimura M, Fedorov S, Uyeda K. J Biol Chem. 1994;269:26100–26106. [PubMed] [Google Scholar]