

ABSTRACT
Dendritic cells (DCs) play a predominant role in initiating cell immune responses. Here we generated a DC-targeting lentiviral vector (LVDC-UbHBcAg-LIGHT) and evaluated its capacity to elicit HBV-specific cytotoxic T lymphocyte (CTL) responses. DC-SIGN-mediated specific transduction using this construct was confirmed in DC-SIGN-expressing 293T cells and ex vivo-cultured bone marrow cells. LVDC-UbHBcAg-LIGHT-loaded DCs were highly effective in inducing HBV-specific CTLs. Mechanistic studies demonstrated autophagy blocking led to a significant increase in apoptosis and obvious inhibition of CD8 + T cells entry into S-phase, correspondingly attenuated LVDC-UbHBcAg-LIGHT-loaded DC-induced T cell responses. This observation was supported by accumulation of pro-apoptotic proteins and the main negative cell cycle regulator-CDKN1B that otherwise would be degraded in activated T cells where autophagy preferentially occured. Our findings revealed an important role of autophagy in the activation of T cells and suggested LVDC-UbHBcAg-LIGHT may potentially be used as a therapeutic strategy to combat persistent HBV infection with higher security.
KEYWORDS: Dendritic cell (DC)-targeting lentivector, cytotoxic T lymphocyte (CTL), autophagy
Introduction
Hepatitis B Virus (HBV) infection, responsible for approximately 250 million chronic infections globally, is one of the main causes of many dreadful consequences including cirrhosis and hepatocellular carcinoma [1–3]. Notably, viral persistence during HBV infection is mainly due to defective cytotoxic T lymphocytes (CTLs) to viral antigens and individuals who are chronically infected with HBV usually have low to undetectable CTL responses[4]. Dendritic cell (DCs) are identified as the most efficient professional antigen-presenting cells (APCs) and various efforts have been made to modify DCs to combat persistent HBV infection by genetically altering them using gene-gun, liposomes or transduction by viral vectors [5–7].
Lentivirus vectors (LVs) have been the focus of many studies because of the ability to transduce nondividing cells and permanently integrate into the genome of target cell with high efficiency [7,8]. We have previously confirmed that lentivirus-delivered and ubiquitin-fused Hepatitis B core antigen (LV-Ub-HBcAg) could promote the maturation of dendritic cells (DCs), trigger the preferential activation of HBV-specific CTL immune response and elicit therapeutic immunity in HBV transgenic mice [9–11]. Glycoprotein of vesicular stomatitis virus (VSVG) is usually used as the lentiviral vector envelope, but it has a wide spectrum of cell tropism which limits its application in vivo. An ideal lentiviral vector system for in vivo application should specifically target DCs to maximize antigen presentation, meanwhile, reduce adverse effects in the surrounding cells. To this end, Yang et al. engineered novel lentivectors with modified envelope glycoprotein of the DC-tropic Sindbis virus (SVGmu) which has lost its ability to bind to heparin sulfate structures, while retaining its specific binding to DC-specific ICAM3-grabbing Nonintegrin (DC-SIGN), a receptor that is expressed mainly on DCs[12]. The immunotherapy platform suitable for selective in vivo targeting of DCs has shown potent capacity to induce immune responses in models of many diseases [13–15].
Autophagy is a highly conserved cellular homeostasis pathway that delivers cytoplasmic material to lysosomes and is uniquely defined by the formation of autophagosomes [16,17]. In the immune system, autophagy is a mechanism to eliminate intracellular pathogens and plays an essential role in the development and function of T lymphocytes. Autophagy regulates the calcium mobilization and energy metabolism in T cells, and is critical for effector CD8 + T cell survival and memory formation [18–21]. A network of ATG genes that are essential for the formation of autophagosomes have been identified. Previous results revealed that the proliferation ability of ATG5−CD8 + T cells was significantly impaired after TCR stimulation. Furthermore, ATG5−CD8 + T cells had a decreased capacity to reach the peak effector response and were unable to maintain cell viability during the effector phase [22–25]. We have confirmed the preferential activation of HBV-specific T cells by the LVs both ex vivo and in vivo, however, whether autophagy was induced in activated T cells and the detailed mechanism by which autophagy mediated the LV-induced T cell activation still remains unknown.
LIGHT, a TNF superfamily member (TNFSF14), is expressed on activated T cells and DCs [26,27]. Experimental evidence suggested that LIGHT provided potent costimulatory activity for T cells, enhancing T cells proliferation and Th1 cytokines production independently of the B7-CD28 pathway [28,29]. Moreover, previous studies have confirmed that adenovirus vector Ad-LIGHT transduction promoted the maturation of DCs and LIGHT-modified DCs elicited more potent CTL immune responses in HBV transgenic mice[30]. Hence, in the present study we constructed a novel DC-targeting lentiviral vector encoding Ub-HBcAg and LIGHT, for the purpose of investigating whether the recombinant lentivector (LVDC-UbHBcAg-LIGHT) was able to target DCs and boost HBV-specific CTL immune responses and further assessed the molecular mechanisms underlying LVDC-UbHBcAg-LIGHT-modified DCs mediated T cell activation.
Results
Engineering and identifying the DC-targeting lentivirus encoding ubiquitin-HBcAg and LIGHT
To produce the expression plasmid H4725 (pLOV-UBC-Ub-HBcAg-EGFP-P2A-Tnfsf14-3FLAG, Tnfsf14 is another saying of LIGHT) encoding Ub-HBcAg and LIGHT (Figure 1(a)), EGFP-P2A-Tnfsf14-3FLAG gene was amplified, purified, and cloned into BamHI/XbaI sites of the expression plasmid CN1043 (pLOV-UBC-UB-HBcAg-EGFP-3FLAG) we have assembled previously [28,29]. Fusion gene UB-HBcAg-EGFP and Tnfsf14-3FLAG could be expressed independently by the self-splicing of P2A. Plasmid H4725 was confirmed by direct sequencing (data not shown) and western blot (Figure 1(c)).
Figure 1.
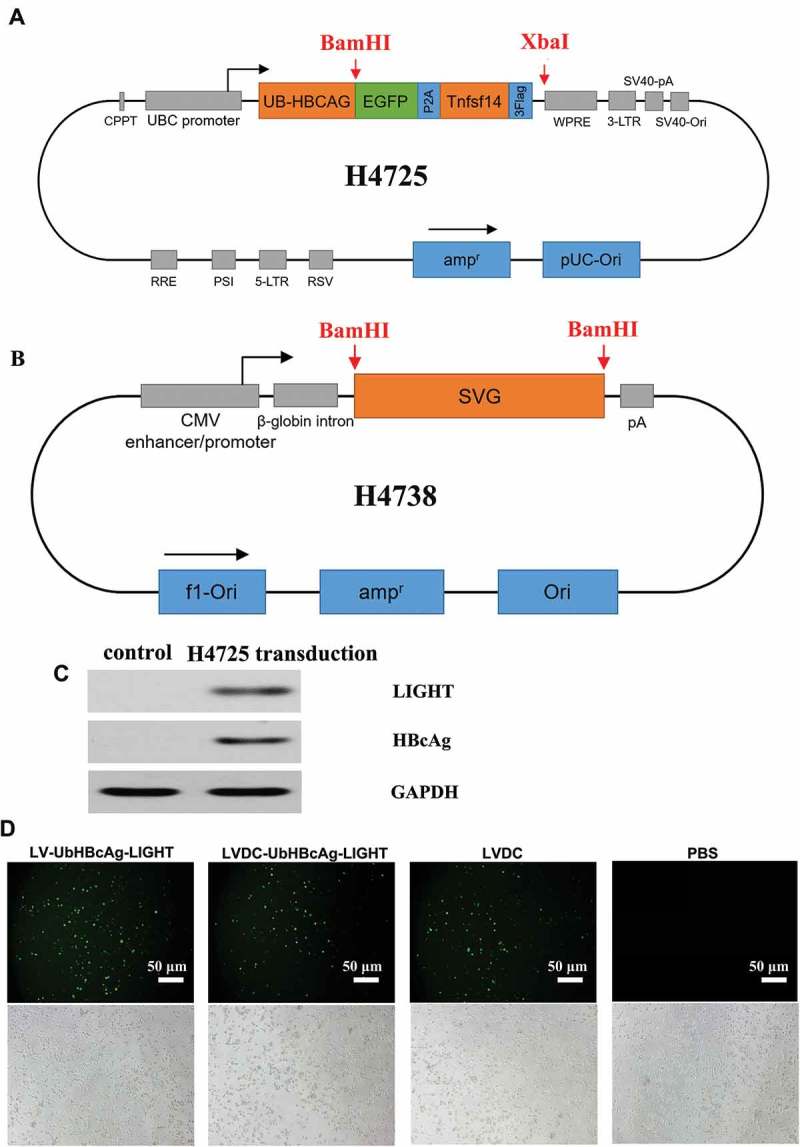
Schematic representation, HBcAg and LIGHT protein expression and GFP expression in DCs transduced with the lentivectors. (a,b) Schematic representations of H4725 and H4738 vector. (c) The 293T cells were transduced with H4725 and cultured for 48 h. The protein lysates were analyzed by western blot with anti-HBcAg and anti-LIGHT antibodies. (d) DCs were cultured at 2 × 105 cells/well in six-well plates and LVDC-UbHBcAg-LIGHT, LV-UbHBcAg-LIGHT, or LVDC was added to transduce DCs at an MOI of 20. GFP expression in DCs was observed using an inverted fluorescence microscope. (Original magnification: × 100). The scale bars: 20 μm.
The SVG gene was engineered with the alterations as described in the Materials and methods to blind its canonical binding receptor heparin while leave its intact ability to interact with DC-SIGN. We cloned the mutant gene into BamHI sites of the traditional envelope plasmid pHCMV-VSV-G to replace the VSVG gene and achieved the novel targeting envelope plasmid H4738 (Figure 1(b)). The engineered envelope plasmid was confirmed by direct sequencing (data not shown) and incorporated onto the surface of H4725 to produce the recombinant lentiviral vector LVDC-UbHBcAg-LIGHT. Transduction efficiency of LVDC-UbHBcAg-LIGHT in DCs was assessed by detecting GFP expression using an inverted fluorescence microscope (Figure 1(d)).
Targeting of DC-SIGN-expressing 293T cell lines by the DC-targeting lentivector
To facilitate our study of targeted transduction, 293T cells were transduced with a designed LV-DCSIGN at the MOI of 1, 5 and 20, respectively, and the obtained lines (293T.DCSIGN.1, 293T.DCSIGN.5, 293T.DCSIGN.20) over-expressed murine DC-SIGN on the cell membrane. The DC-SIGN protein level in each group was analyzed by western blot (Figure 2(a)). Then the above 293T cell lines were transduced by LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT respectively. The results showed that LV-UbHBcAg-LIGHT had similar transduction efficiency (51.7–63.7%) towards the four target cell lines (293T, 293T.DCSIGN.1, 293T.DCSIGN.5, 293T.DCSIGN.20) (Figure 2(b)), In contrast, LVDC-UbHBcAg-LIGHT could specifically transduce 293T.DCSIGN.1, 293T.DCSIGN.5 and 293T.DCSIGN.20 cells, with 24.6%, 34.6% and 40.3% transduction efficiencies, respectively, but not the untreated 293T cells (Figure 2(c)). Strikingly, adding a neutralizing anti-mouse DC-SIGN antibody to the viral supernatant before its exposure to 293T.DCSIGN.20 cells could significantly reduce the LVDC-UbHBcAg-LIGHT transduction efficiency, but not the LV-UbHBcAg-LIGHT.
Figure 2.
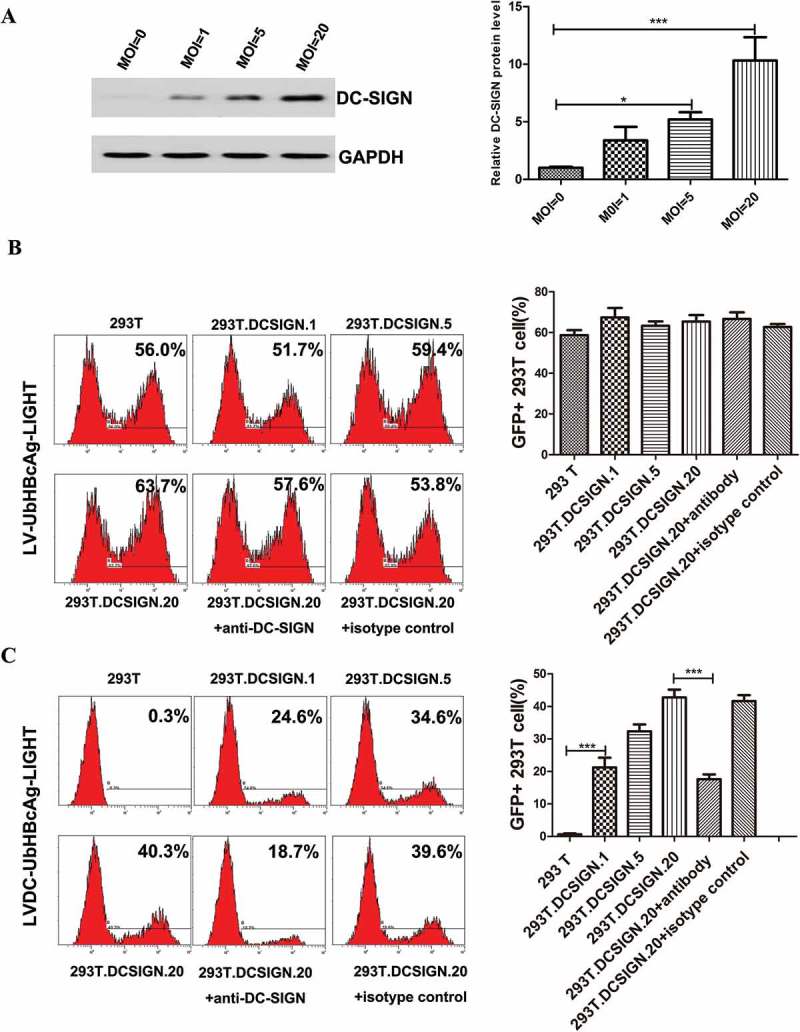
Lentivector bearing engineered SVG targeted to DC-SIGN-expressing 293T cells. (a) We transduced the 293T cells with a designed LV-DCSIGN at the MOI of 1, 5 and 20, respectively. The expression levels of DC-SIGN were analyzed by western blot. Left: representative immunoblots. Right: densitometric analysis. Bars represent the mean ± SD of three independent experiments. *p < 0.05, ***p < 0.001. (b,c) LV-Ub-HBcAg-LIGHT and LVDC-Ub-HBcAg-LIGHT were used to transduce 293T cell lines (MOI = 20) with increasing protein levels of murine DC-SIGN, respectively. An anti-murine DC-SIGN antibody was added into viral supernatant during the transduction for 8 h when necessary, isotype-matched antibody was used as a control. 72h later, the transduction efficiency was determined by flow cytometry. Left: representative flow cytometric analysis. Right: statistical analysis.***p < 0.001.
Targeting of DCs ex vivo by the DC-targeting lentivector
We next tested the capacity of the lentivectors to transduce ex vivo cultured bone marrow cells. Flow cytometry analysis showed that in a mouse bone marrow culture, approximately 11% of the cells were CD11c positive (data not shown). After LVDC-UbHBcAg-LIGHT transduction, about 10% of the cells were GFP+, within the GFP+ cells, up to 82.5% of the transduced cells were CD11c+DC-SIGN+ (Figure 3(a)), moreover, the neutralizing anti-mouse DC-SIGN antibody sharply reduced the percentage of GFP+ cells (from 36.6% to 14.4%) (Figure 3(c)). In contrast, although 53.6% of the cells were GFP+ after LV-UbHBcAg-LIGHT transduction, only 15.7% of the transduced cells were CD11c+DC-SIGN+ (Figure 3(b)). Noticeably, the blocking DC-SIGN antibody did not make any difference to the transduction efficiency of LV-UbHBcAg-LIGHT (Figure 3(d)). Additionally, the lentivectors were used to transduce primary T and B cells harvested from mouse spleen. The results showed that LV-UbHBcAg-LIGHT could transduced both T and B cells with an efficiency of about 13%, while LVDC-UbHBcAg-LIGHT had a low to undetectable transduction efficiency (Figure 3(e)).
Figure 3.
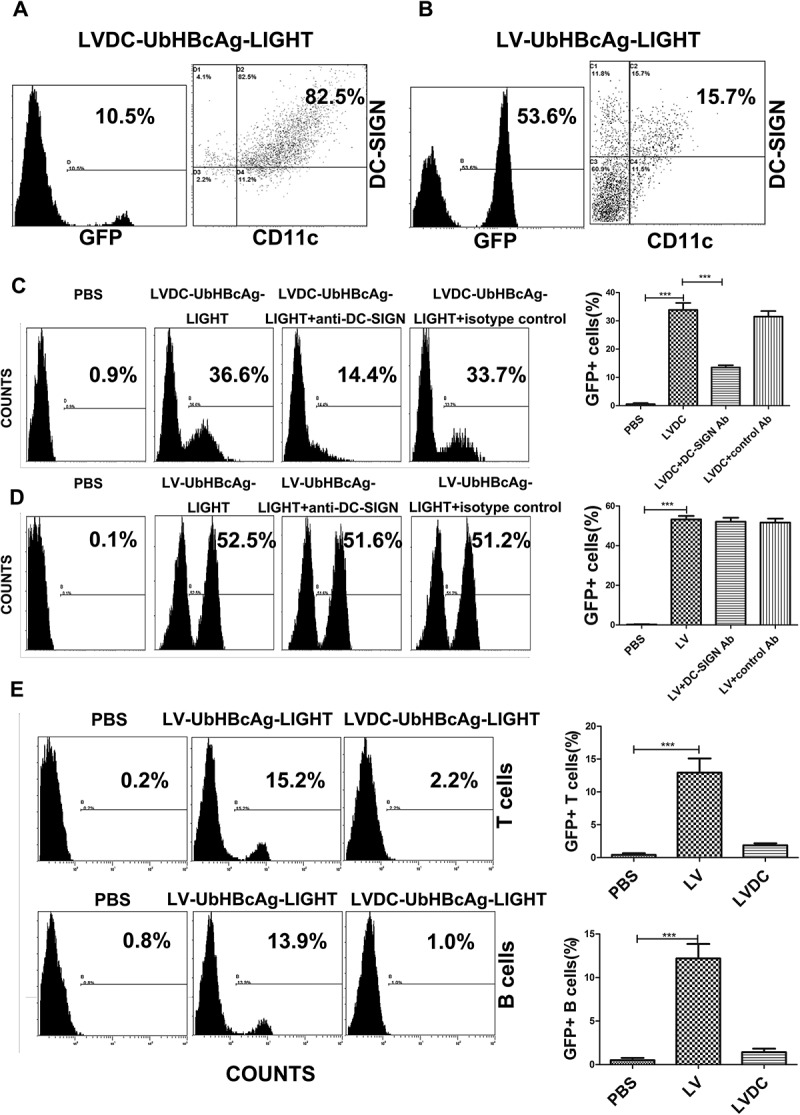
LVDC-Ub-HBcAg-LIGHT could selectively transduce DCs ex vivo. (a,b) The ex vivo cultured-bone marrow cells were exposed to LVDC-Ub-HBcAg-LIGHT and LV-Ub-HBcAg-LIGHT, respectively on day 5. Three days after transduction, the cells were collected and stained with anti-CD11c and anti-DC-SIGN antibodies. The percentages of GFP+ cells (left) and DC-SIGN+CD11c+ cells within the GFP+ gate (right) were assessed by flow cytometry. (c,d) We transduced BMDCs with LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT, respectively with or without the presence of anti-murine DC-SIGN antibody. Transduction efficiency was evaluated by flow cytometry. Left: representative flow cytometric analysis. Right: statistical analysis. LVDC is short for LVDC-UbHBcAg-LIGHT and LV is short for LV-UbHBcAg-LIGHT. ***p < 0.001. (e) Primary B cells or T cells were transduced with LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT, respectively. The percentages of GFP+ cells were detected by flow cytometry. Left: representative flow cytometric analysis. Right: statistical analysis. LVDC is short for LVDC-UbHBcAg-LIGHT and LV is short for LV-UbHBcAg-LIGHT. ***p < 0.001.
LVDC-UbHBcAg-light induced DCs maturation and increased IL-12p70 secretion
Here we explored whether the DC-targeting lentivector could activate and mature BMDCs. Flow cytometry analysis of mBMDCs 3 days after transduction showed that both LV-UbHBcAg-LIGHT and LVDC-UbHBcAg-LIGHT could significantly elevate the expression of DC activation markers (CD80, CD83, CD86 and MHC-II) when compared with the alternatives (Figure 4(a,b)). However, there was no difference between LV-UbHBcAg-LIGHT and LVDC-UbHBcAg-LIGHT. In this study, we also discovered that LV-UbHBcAg-LIGHT and LVDC-UbHBcAg-LIGHT transduction led to obvious upregulation of IL-12p protein levels in the medium compared with the alternatives (Figure 4(c)). The results confirmed that the engineered DC-targeting lentivectors could significantly mature DCs.
Figure 4.
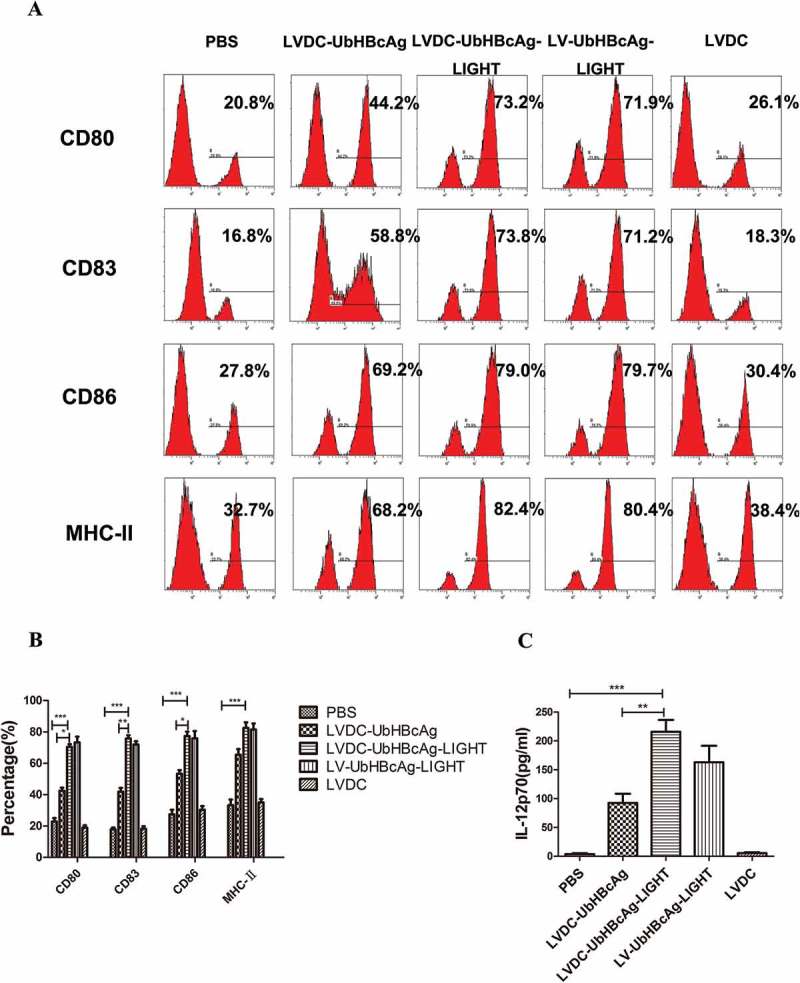
LVDC-UbHBcAg-LIGHT promoted DCs maturation and enhanced IL-12p70 secretion. (a) The expression levels of DC activation markers (CD80, CD83, CD86 and MHC-II) were determined by flow cytometry. (b) The percentage of DCs surface molecules (c) ELISA analysis of IL-12p70 secretion levels in the supernatants. **p < 0.01, ***p < 0.001.
LVDC-UbHBcAg-LIGHT induced strong cytotoxic T lymphocyte immune activities largely through upregulating autophagy in T cells
The secretion of cytokines in the supernatant (including IL-2, IL-6, TNF-α and IFN-γ) was detected in all groups after 72 h of incubation. As shown in Figure 5(c), T cells in the LVDC-UbHBcAg-LIGHT group secreted higher levels of IFN-γ than those in the LVDC-UbHBcAg group, however, there was no significant difference between LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT group. Similar results were observed for the examination of IL-2, IL-6 and TNF-α (Figure 5(c)). Furthermore, intracellular IFN-γ and TNF-α production in CD8 + T cells was measured by flow cytometry. As shown in Figure 6(a,b), the LVDC-UbHBcAg-LIGHT group had obviously increased percentages of IFN-γ+ or TNF-α+CD8 + T cells than the LVDC-UbHBcAg group suggesting the additional expression of LIGHT in the vector significantly enhanced the ability of lentiviral particles to activate T cells, however, there was no significant difference between LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT group. In addition, the HBcAg-specific cytotoxicity of T lymphocytes against P815/c cells in each group was evaluated by LDH release assays. As shown in Figure 6(d), the percentages of specific cytolysis in the LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT group were always higher than other groups at effector: target (E:T) ratios of 20:1, 10:1, 5:1. ATG5 is a known autophagy associated gene and has been verified to exert enormous function on the formation of autophagosomes. To investigate the role autophagy played in T cell activation, we transduced T cells with ATG5 siRNA for 24 h or pretreated T cells with autophagy inhibitors 3-MA (10 mM) for 1 h before the incubation with LV-modified DCs. The interference efficiency of the ATG5 siRNA was examined by PCR and western blot (Figure 5(a,b)). Our results showed that both 3-MA and ATG5 siRNA treatment significantly reduced LVDC-UbHBcAg-LIGHT or LV-UbHBcAg-LIGHT-induced secretion of cytokines, the percentages of IFN-γ+ or TNF-α+CD8 + T cells and the activities of HBV-specific cytotoxic T lymphocytes (Figures 5(c) and 6(a-d)).
Figure 5.
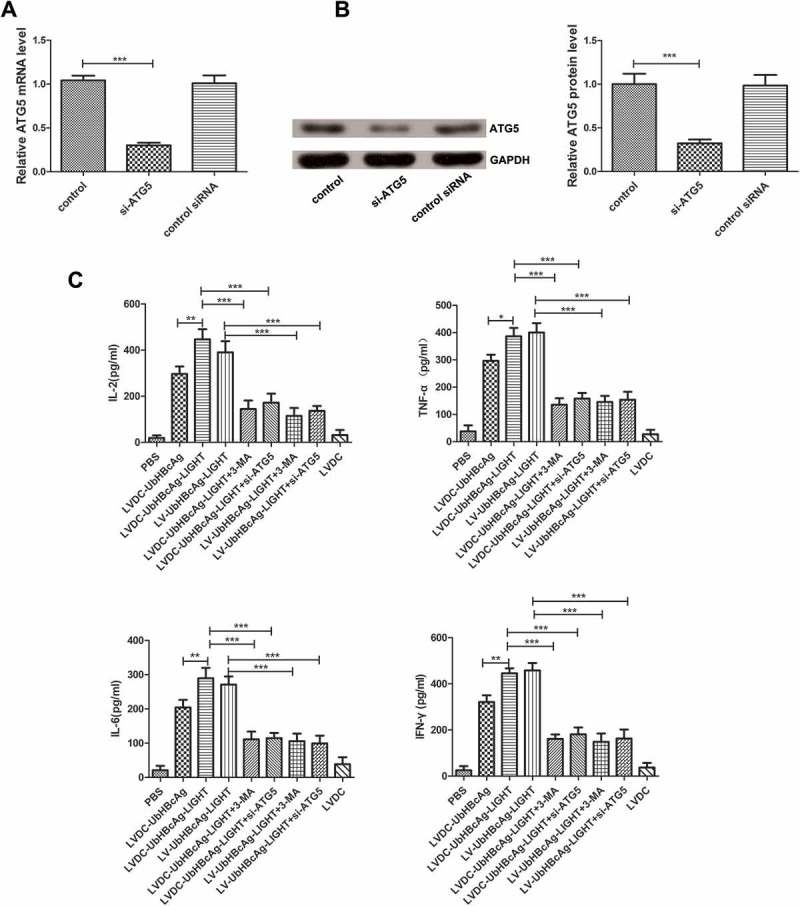
The effects of 3-MA and ATG5 siRNA on LVDC-UbHBcAg-LIGHT or LV-UbHBcAg-LIGHT-induced secretion of cytokines. (a,b) The interference efficiency of ATG5 siRNA was evaluated by PCR and western blot. (a) Total RNA was extracted from T cells and analyzed by real-time PCR for ATG5. ***p < 0.001. (b) The samples were subjected to western blot analysis for ATG5. Left: representative immunoblots. Right: densitometric analysis. ***p < 0.001. (c) The secretion of cytokines in the supernatant was detected by ELISA. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 6.
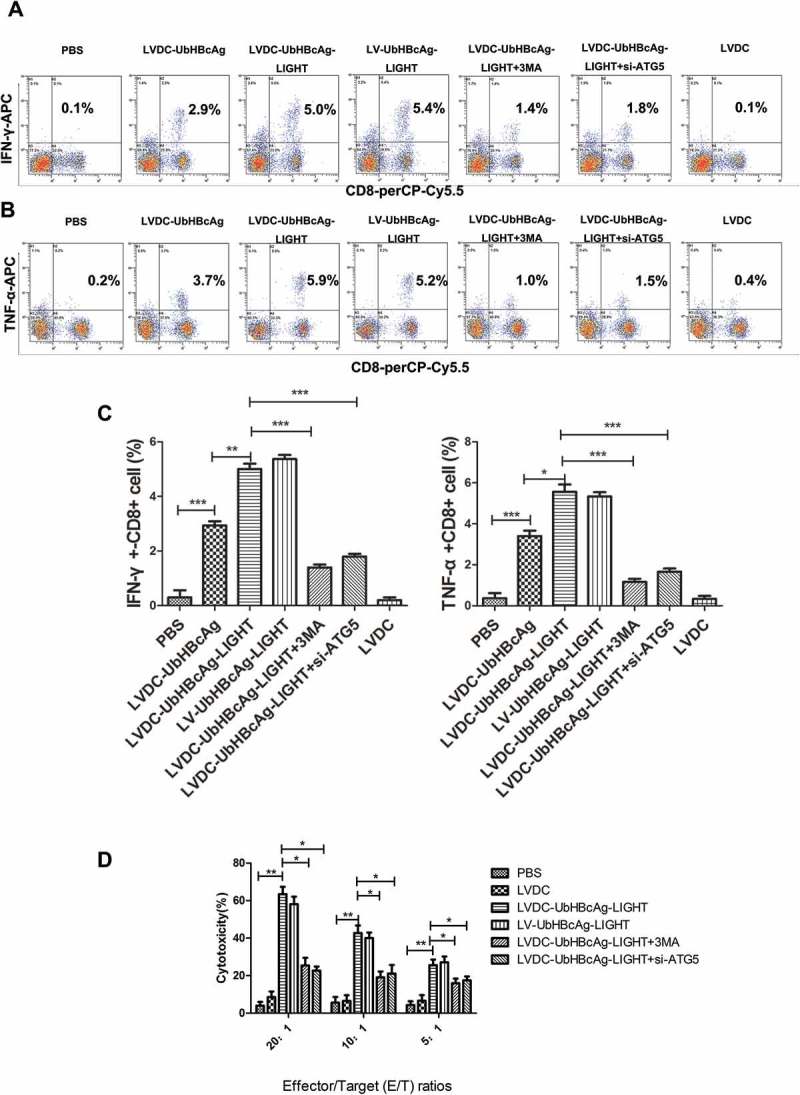
The effects of 3-MA and ATG5 siRNA on LVDC-UbHBcAg-LIGHT-induced CTL immune responses. (a,b) The whole cell population was stained with CD8-perCP-Cy5.5, IFN-γ-APC or TNF-α-APC antibodies. The percentages of IFN-γ+CD8+ and TNF-α+CD8 + T cells were examined by flow cytometry. (c) statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001. (d) The HBcAg-specific CTL activity was determined using LDH release assay. *p < 0.05, **p < 0.01.
Autophagy was notably induced in the activated T cells
To validate the induction of autophagy by LV-modified DCs in T cells, we first examined the protein levels of autophagy related membrane structures as that of LC3-II, Beclin and p62, rapamycin treatment was included as a positive control. As shown in Figure 7(a,b), western blot revealed a noticeable increase in LC3-II expression in the LVDC-UbHBcAg-LIGHT group. Meanwhile, similar results were observed for the protein expression of Beclin, however, the p62 expression varied in opposite manners. Then, the formation of autophagosomes in the T cells stably transduced with tandem mCherry-Wassabi-LC3-II was monitored by a confocal microscopy (Figure 7(d)). In these cells, early autophagosomes displayed both red (mCherry) and green (Wassabi) signal. While autophagolysosome displayed only red fluorescence because of the stability of the mCherry signal and the sensitivity of the Wassabi signal to the acidic conditions in the lysosome lumen. As shown in Figure 7(c), the total number of yellow puncta (both green and red positive, G+R+) and red puncta (G-R+) increased significantly in the LVDC-UbHBcAg-LIGHT group when compared with the control. Furthermore, more autophagic vesicles that contained engulfed organelles were observed by transmission electron microscopy in the LVDC-UbHBcAg-LIGHT group (Figure 7(e)).
Figure 7.
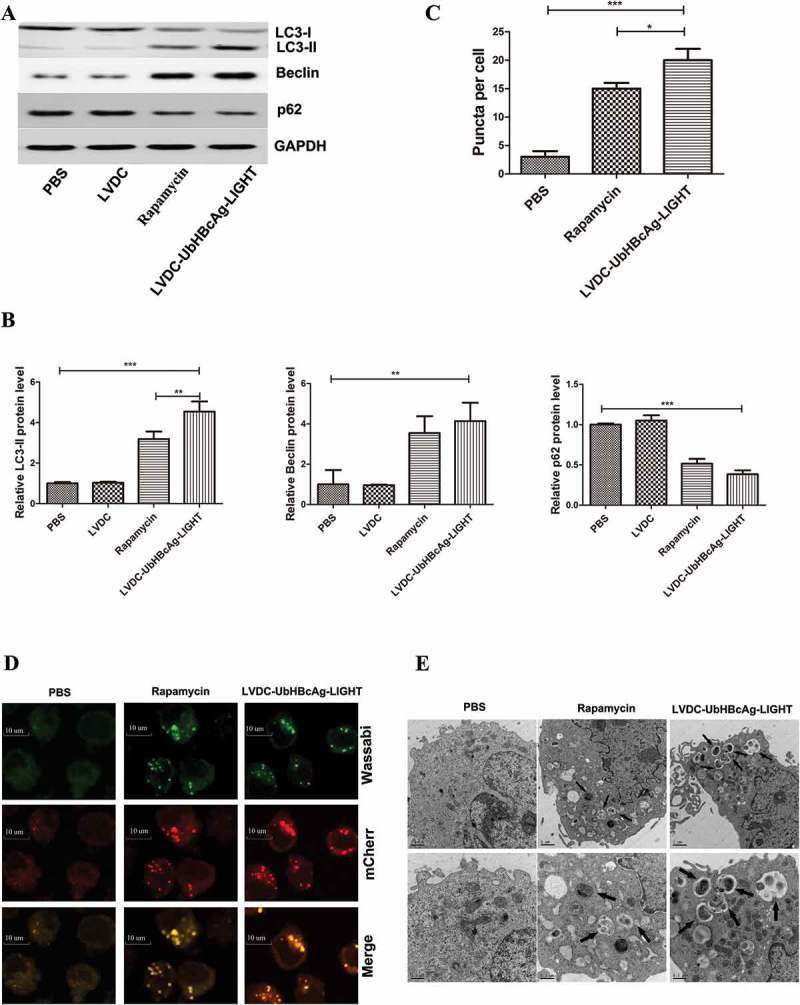
Autophagy was significantly induced in activated T cells. (a,b) T cells were co-cultured for 3 days with mBMDCs that were transduced with LVDC-UbHBcAg-LIGHT and then re-isolated by negative selection. Protein levels were detected by western blot using antibodies against LC3, Beclin and p62. The fold change of LC3 only refers to LC3-II. Rapamycin (150 nM) added for 4h was included as the positive control. (a) Representative immunoblots. (b) Densitometric analysis. **p < 0.01, ***p < 0.001. (c,d) T cells stably transduced with mCherry-Wassabi- LC3-II were incubated with DCs pulsed by LVDC-UbHBcAg-LIGHT, then subjected to confocal microscopy analysis. (Original magnification: × 1000). The scale bars: 10 μm. (c) The total number of two kinds of LC3-II puncta (yellow puncta, G + R+; red puncta, G-R+) per cell. *P < 0.05, ***p < 0.001. (e) T cells were treated as mentioned above, and then subjected to transmission electron microscopy analysis. The arrows indicate autophagosomes. The lower panels show enlarged regions of the upper panels. The arrows indicate autophagosomes. The scale bars: 1 μm (upper) and 0.5μm (lower), respectively. Original magnification: × 10,000 (upper) and × 20,000 (lower), respectively.
Upregulated autophagy contributed to decreased apoptosis and increased S phase T cells by selectively degrading proteins related to apoptosis and cell cycle
Similar co-incubation of LV-modified DCs and T cells was performed to active T cells and the samples in all groups were subjected to western blot. As shown in Figure 8(a-f), the levels of apoptotic proteins (Bim, Pro-caspase-3, Pro-caspase-8 and cleaved caspase-3, caspase-8) in the LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT group decreased considerably compared with the others, however, the inhibitory effect of the lentivectors on these proteins expression diminished upon autophagy blocking. By contrast, PCR analysis revealed no significant difference between all groups for all the targets (Pro-caspase-3, Pro-caspase-8 and Bim) (Figure 8(g)). Consistent with this idea, notably lower percentages of apoptotic CD8 + T cells were observed in the LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT group by flow cytometry analysis. Similarly, autophagy blocking significantly increased the percentages of apoptotic CD8 + T cells (Figure 9(a-c)).
Figure 8.
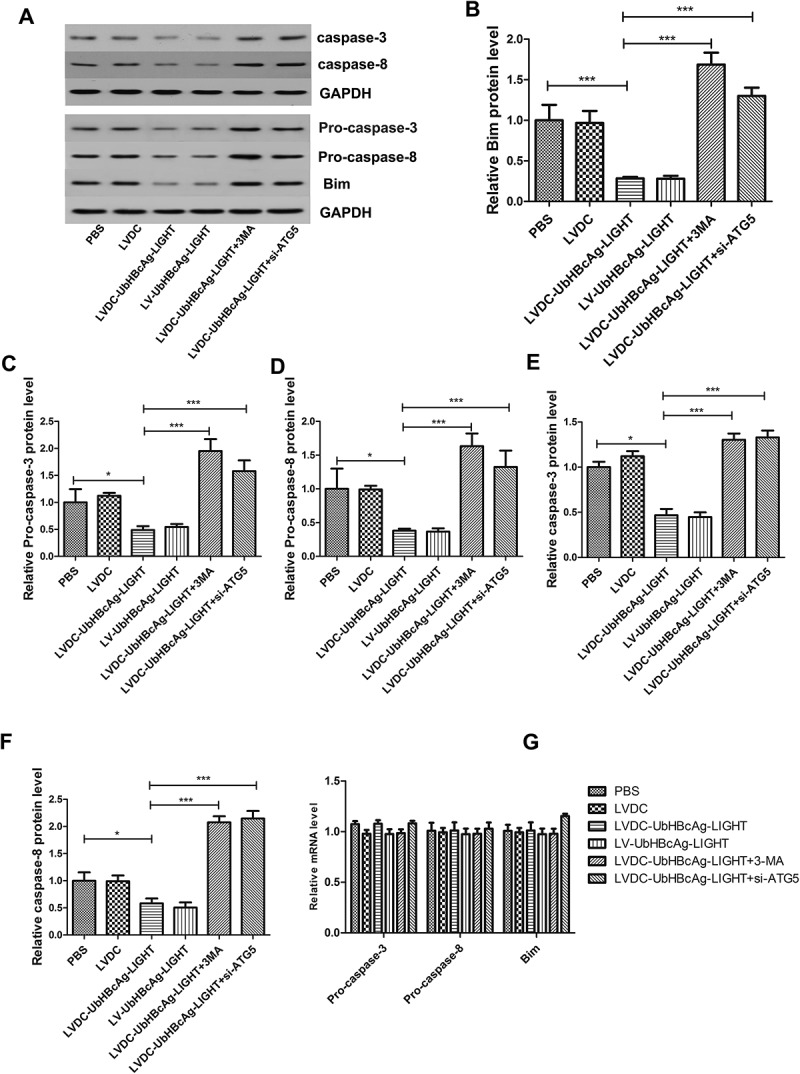
Autophagy blocking could inhibit the degradation of apoptosis-associated proteins. (a-f) T cells stimulated were re-isolated by negative selection and extract was made for western analysis of Bim, Pro-caspase-3, Pro-caspase-8 and cleaved caspase-3, caspase-8. (a) Representative immunoblots. (b-f) Densitometric analysis. *p < 0.05, ***p < 0.001. (g) Total RNA was extracted and analyzed by real-time PCR for Pro-caspase-3, Pro-caspase-8, and Bim.
Figure 9.
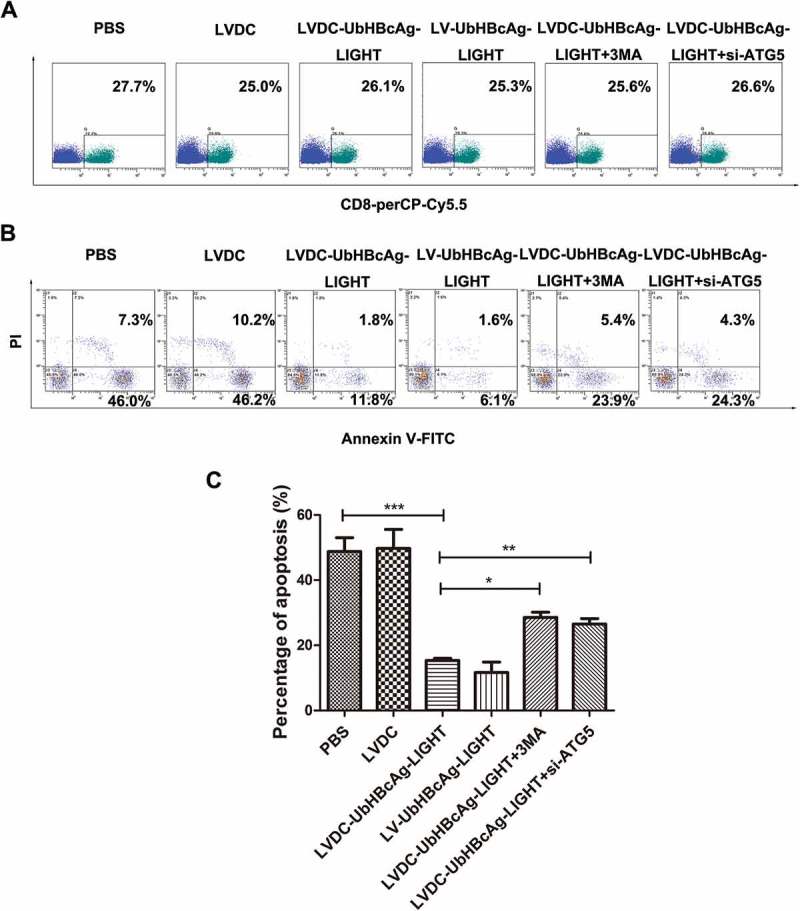
Autophagy blocking could promote the apoptosis of CD8 + T cells. (a,b) The apoptosis of CD8 + T cells was analyzed by flow cytometry. T cells were stained three times with PerCP-Cy5.5 labeled anti-CD8 antibody, Annexin V-FITC and PI. (a) The percentages of PerCP-Cy5.5+ (CD8 + T) cells in T cells. (b) Annexin V+ (apoptotic) cells within the PerCP-Cy5.5+ gate. (c) Densitometric analysis. *p < 0.05, **p < 0.01, ***p < 0.001.
T lymphocyte proliferation was also determined by CCK-8 and the data showed that the capacity of T lymphocyte proliferation in the LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT group was apparently higher than that in other groups (Figure 10(a)). Consistently, 3-MA and ATG5 siRNA treatment almost abrogated the facilitation of lentivectors on T cell proliferation. Cell cycle analysis showed that approximately 70% of CD8 + T cells progression into S-phase in the LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT group (Figure 10(b,c)). However, noticeably reduced percentages of CD8 + T cells entering into S-phase were detected after autophagy blocking by 3-MA and ATG5 siRNA. CDKN1B is the main negative cell cycle regulator and degradation of CDKN1B could facilitate the transition from G1- to S-phase in T lymphocytes. Here we found an obvious decrease in CDKN1B protein expression in the LVDC-UbHBcAg-LIGHT and LV-UbHBcAg-LIGHT group compared with the others, however, CDKN1B protein accumulated in CD8 + T cells upon autophagy blocking (Figure 10(d,e)), which coincided well with the cell-cycle and CCK-8 analysis. Again, we did not detect any difference in CDKN1B mRNA expression between all groups(Figure 10(f)).
Figure 10.
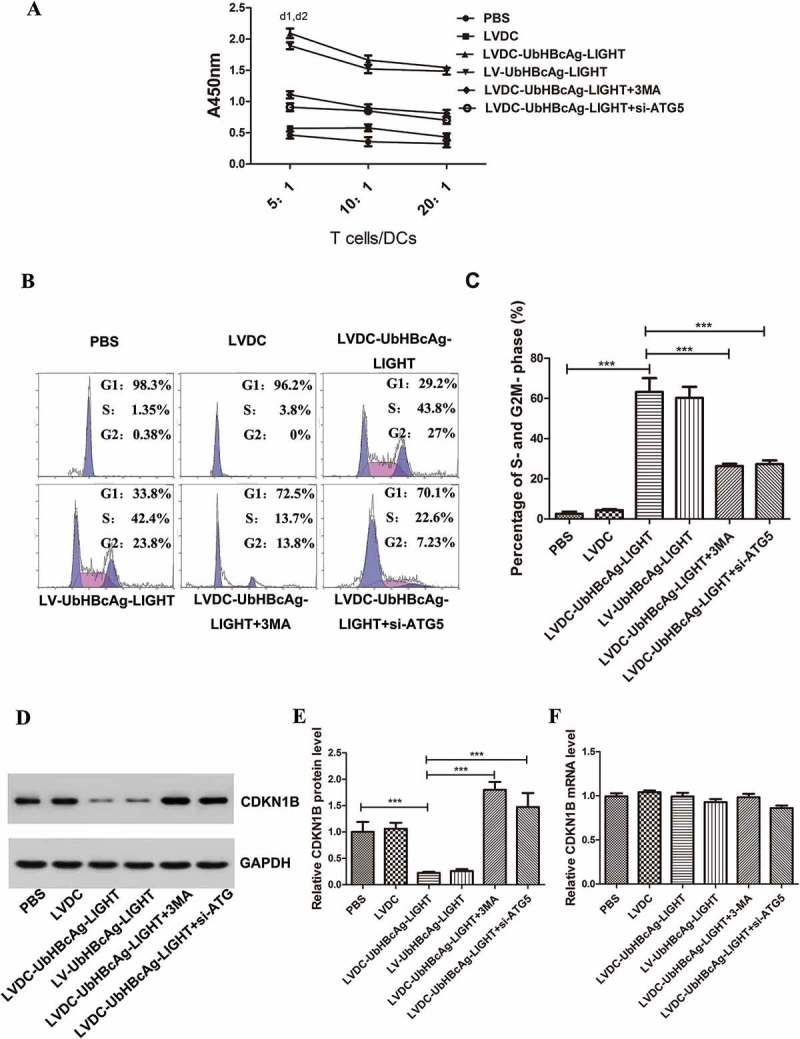
The induced autophagy promoted T cell proliferation by selectively degrading the cell-cycle associated proteins. (a) T lymphocyte proliferation activity was assessed by the CCK-8. d1: **p < 0.01 LVDC-UbHBcAg-LIGHT versus control; d2: *p < 0.05 LVDC-UbHBcAg-LIGHT versus LVDC-UbHBcAg-LIGHT/3-MA or si-ATG5. (b,c) Flow cytometry analysis was used to analyze the purified CD8 + T cells in G1, S and G2/M phase. (c) Densitometric analysis. ***p < 0.001. (d,e) The expression levels of CDKN1B in purified CD8 + T cells were determined by western blot. (e) Statistical analysis. *** p <0.001. (f) Real-time PCR analysis of CDKN1B.
Discussion
DCs are essential for the initiation of antigen-specific immune responses through antigen capture and presentation [31–33]. DC-based vaccination or immunization involving loading DCs with specific antigens have been the target of intense research [13,34–36]. Of these methods, viral vectors have been proven most effective in priming cytotoxic T lymphocytes because they can deliver antigen for MHC class I presentation to target CD8 + T cells. However, the generation of patient-specific DC vaccines through isolation and transduction of autologous DCs from patient blood is labor intensive, time consuming and costly. Therefore, the process should be optimized by directly injecting lentiviral vectors that are further standardized in the design and can target the patient’s DCs for the treatment of HBV. In the present research, we generated a new DC-targeting recombinant lentivector bearing an engineered glycoprotein derived from Sindbis virus and termed it LVDC-UbHBcAg-LIGHT.
Considering that DC-SIGN is an ideal targeting receptor expressed on DC surface and capable of rapid binding and endocytosis of materials[37], we established a cell model of DC-SIGN overexpression (293T.DCSIGN.1, 293T.DCSIGN.5, 293T.DCSIGN.20) and observed that the transduction efficiency of LVDC-UbHBcAg-LIGHT increased with the protein levels of DC-SIGN in 293T cells. In comparison, LV-UbHBcAg-LIGHT could transduce all the 293T cell lines with similar transduction efficiency suggesting that VSVG had broad tropism and DC-SIGN expression on the cell surface did not obviously alter the transduction ability. Then, we used the lentivectors to transduce ex vivo-cultured mixed bone marrow cells for the purpose of testing whether the conclusions we made from 293T cells could be of the same assessment in DCs. Interestingly, the non-targeting version transduced mixed bone marrow cells more efficiently, however, the percentage of DCs within the transduced cells was relatively low. By contrast, almost all the transduced cells were CD11c+DC-SIGN+ DCs in the LVDC-UbHBcAg-LIGHT group. Furthermore, attempts to transduce primary T and B cells harvested from mouse spleen with LVDC-UbHBcAg-LIGHT failed further confirming the specific transduction of LVDC-UbHBcAg-LIGHT.
Then we investigated the ability of the LVs to mature DCs ex vivo and explored whether LV-modified DCs could deliver antigen to T lymphocytes for stimulation of antigen-specific CD8 + T cell responses. The results indicated that both targeting (LVDC-UbHBcAg-LIGHT) and non-targeting (LV-UbHBcAg-LIGHT) constructs shared similar abilities to induce DC maturation and IL-12p production. DCs loaded with both constructs also exhibited similar capabilities to induce T cell immune responses in the form of cytokine production and cytolytic killing of HBcAg antigen-expressing target cells. This was presumably due to the wide cell tropism of LV-UbHBcAg-LIGHT and the characteristic of LVDC-UbHBcAg-LIGHT to limit transduction to DCs. Despite the relatively poor transduction capability, LVDC-UbHBcAg-LIGHT was especially advantageous for effective delivery of antigen genes into DCs, thereby inducing a more powerful antigen presentation. Notably, LIGHT expression in DCs strongly enhanced the capacity of lentiviral particles to activate T cells which was in line with previous reports [28,30].
It has been shown that autophagy played an essential role in the function of T lymphocytes and was critical for effector CD8 + T cell survival [22,37,38]. In our study, we demonstrated that autophagy blocking by ATG5 siRNA or 3-MA significantly weakened LVDC-UbHBcAg-LIGHT-induced T cell immune responses, highlighting the role of autophagy in T cell activation. Given the results observed above, we speculated that autophagy may be widely induced in the activated T cells. As expected, significantly increased numbers of characteristic double membrane autophagosomes were identified in the activated T cells populations with a transmission electron microscopy, concurring with up-regulated expression of autophagy related membrane structures such as LC3-II and Beclin as analyzed by western blot analysis. Interestingly, autophagy inhibition could significantly weaken but not completely block the LVDC-UbHBcAg-LIGHT-induced T cells responses indicating the existence of autophagy-independent pathways for the regulation of T cells activation.
The interaction between the TCR-CD3 complex and the MHC-peptide complex on APCs triggers TCR activation-induced proliferation, which expands antigen-specific T cell clones and ultimately induces the adaptive immune response. T cell expansion is regulated by the progression of the cell cycle which consists of the transition from G0 to G1 then to S-phase, and is controlled by positive regulators as well as negative regulators. Degradation of CDKN1B, the main negative cell-cycle regulator, facilitates the transition from G1- to S-phase in CD8 + T cells after TCR stimulation [39–41]. Experimental evidence suggested that autophagy promoted CD8 + T cell survival via the degradation of cell death machinery proteins. More importantly, autophagy-deficient CD8 + T cells have obvious deficiencies in survival and proliferation after TCR stimulation [23–25]. In the latter half of the study we discovered that apoptotic proteins and CDKN1B expression decreased significantly in the LVDC-UbHBcAg-LIGHT-loaded DCs activated CD8 + T cells where autophagy preferentially occured. These observations were consistent with decreased apoptosis and more CD8 + T cells entry into S-phase. As expected, autophagy blocking increased the percentages of apoptotic CD8 + T cells, decreased the percentages of CD8 + T cells progression into S phase, correspondingly resulting in remarkably compromised T cell function. These results revealed a direct role of autophagy in promoting the survival and proliferation of CD8 + T cells by selectively degrading the proteins associated with apoptosis and cell cycle after TCR stimulation.
Taken together, our study reported a DC-targeting lentivector which could stimulate robust HBV-specific CTL immune responses and provided new insights into the understanding of lentivector-mediated T cell activation. However, the precise mechanism underlying the interplay between autophagy and proteins essential for T cell activation are not yet fully understood, and more work in this direction are needed to elucidate this issue in the future. In addition, whether the targeting lentivector can specifically transduce DCs in vivo and induce therapeutic effects in HBV transgenic mice requires further investigation.
Materials and methods
Reagents and antibodies
The fluorescent antibodies and their corresponding isotype controls were obtained from eBioscience (San Diego, USA), and the enzyme-linked immunosorbent assay (ELISA) kits for interleukin (IL)-12p, IL-2, IL-6, IFN-γ, TNF-α were purchased from R&D Co., Ltd (Minneapolis, USA). Antibodies against HBcAg, LIGHT, DC-SIGN, LC-3, p62, Beclin, CDKN1B, Pro-caspase-3, Pro-caspase-8, caspase-3, caspase-8 and Bim were purchased from Abcam (Cambridge, UK). The neutralizing DC-SIGN monoclonal antibody (invitrogen, Gaithersburg, USA) was used to neutralize the DC-SIGN expressed on cell surface. Phorbol 12-myristate 13-acetate (PMA), ionomycin, monensin, concanavalin A (ConA), 3-Methyladenine (3-MA) and rapamycin were purchased from Sigma-Aldrich (St Louis, USA).
Cell lines
Human Embryonic Kidney 293T cells were purchased from the American Type Culture Collection (Manassas, USA) and cultured in RPMI-1640 (Invitrogen, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA), penicillin (100 U/mL), and streptomycin (100 mg/mL) at 37°C in a humidified incubator, 5% CO2. The H-2kd mastocytoma cell line P815/c (expressing the HBcAg) was kept in our lab.
Dendritic cell generation
Mouse bone marrow-derived DCs (mBMDCs) were generated as the protocol described by Chen et al. [9]. Briefly, femurs and tibiae were removed from BALB/c mice, thereafter, the isolated bones were placed in 75% ethanol for 5 min for disinfection and washed with PBS. The ends were then cut with scissors, the bone marrow was syringed with PBS using a injector with a diameter of 0.45 mm. Clusters within the marrow suspension were removed by vigorous pipetting. The red blood cells were lysed and the bone marrow cells were seeded in complete RPMI 1640 at a density of 2 × 106 cells/mL with the addition of 10 ng/mL murine IL-4 (mIL-4; PeproTech, Rocky Hill, USA) and 20 ng/mL murine granulocyte-macrophage colony-stimulating factor (mGM-CSF, PeproTech). Non-adherent cells and half of the RPMI 1640 were removed, and the mGM-CSF, mIL-4 and fresh PRMI 1640 were supplemented on alternate days.
Small interfering RNAs
Gene-specific siRNAs and one non-targeting siRNA were purchased from Genechem Co., Ltd (Shanghai, China). ATG5 siRNA target the sequences CCUUUGGCCTAAGAAGAAA. While the sequence of the non-targeting control siRNA was 5′-UUCUCCGAACGUGUCACGUTT-3′.
Construction of the GFP labeled targeting lentiviral vectors
To achieve the targeting lentiviral particles (LVDC-UbHBcAg-LIGHT), we transduced 80% confluent 293T cells with a combination of three elements: the appropriate H4725 (pLOV-UBC-UB-HBcAg-EGFP-P2A-Tnfsf14-3FLAG) backbone plasmid (10μg), the engineered envelope plasmid (5 μg), along with 5μg of the psPAX2 packaging plasmid using Lipofectamine 2000 (Invitrogen). Plasmid H4725 was assembled by cloning the EGFP-P2A-Tnfsf14-3FLAG gene into the BamHI/XbaI sites of the CN1043 (pLOV-UBC-Ub-HBcAg-EGFP-3FLAG) which was maintained in our lab [10,11]. We generated the targeting envelope plasmid (SVGmu) by alterations including an insertion of 10-amino acid sequence (MYPYDVPDYA) between amino acids 71 and 74 of the SVG E2, mutations of 157KE158 into 157AA158 of the SVG E2. Additionally, deletion was introduced to the SVG E3 to remove amino acids 61–64 as described by Yang et al. [12]. The control lentiviral particles (LVDC) was constructed by incorporating the SVGmu onto the pLOV.UBC.EGFP.3FLAG plasmid. Lentiviral packaging was accomplished with a helper virus-free packaging system. All lentivectors in this study reached a titer of 1 × 109 transducing units/ml.
Construction of lentiviral vectors expressing the murine DC-SIGN
The primers used for murine DC-SIGN were forward: 5′-GGGTCAATATGTAATTTTCAGTG-3′ and reverse: 3′-CATAGCGTAAAAGGAGCAACA-5′. Plasmid H4723 was generated by amplifying and cloning the cDNAs into the EcoRI and NheI restriction sites of the pLenti-Ubc-EGFP-3FLAG vector (Obio Biotech Co., Ltd, Shanghai, China). The recombinant lentivirus LV-DCSIGN was generated by pseudotyping H4723 with VSVG and transducing the 293T cells similar to the methods described above.
Transduction of mixed bone marrow cells and BMDCs
Total bone marrow cells were harvested from BALB/c mice and BMDCs were generated as described above. Either bone marrow cells or immature BMDCs of day 5 were seeded in a 24-well plate (2 × 105/well) and then cultured at 37°C in the presence of the indicated lentivectors at a MOI of 20, PBS was used as a control. After 24 h, the supernatant was discarded and fresh RPMI 1640 containing 10% fetal bovine serum was added. The transduced cells were cultured for an additional 3 days and analyzed for green fluorescent protein (GFP) expression by an a Coulter Epics XL flow cytometer (Beckman, Miami, USA). A specific neutralizing monoclonal antibody (Invitrogen) was used at 10 μM to neutralize the DC-SIGN expressed on the cell surface.
Dendritic cell immunophenotyping and IL-12 production
On day 5, immature DCs were cultured for indicated time in the presence of LVDC-UbHBcAg, LVDC-UbHBcAg-LIGHT, LV-UbHBcAg-LIGHT or LVDC at a MOI of 20. On day 8, IL-12p70 secretion levels in harvested supernatants of mature DCs were assessed by a commercial mouse ELISA kit (R&D Co., Ltd, USA) according to the manufacturer’s instructions. On day 9, mature DCs were harvested and the expression levels of DC surface molecules were determined by incubation with PE-labelled anti-mouse CD80, CD83, CD86 and MHC-II antibodies (eBioscience). The stained cells were examined by flow cytometry.
Animals
H-2Kd BALB/c mice, 6–8-week-old, were purchased from Shanghai SLAC Laboratory Animal Co., Ltd of the Chinese Academy of Sciences and maintained under standard pathogen-free conditions in the Experimental Animal Center of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. Mice were treated on the basis of the criterion set up by the Shanghai Public Health Service Policy on the Humane Care and Use of Laboratory Animals.
T lymphocyte generation
T lymphocytes were generated from the above splenocytes using nylon wool columns (Wako, Tokyo, Japan) and single-cell suspensions of T lymphocytes were seeded in a 24-well plate at the density of 2 × 106 cells/well with the presence of RPMI 1640 medium containing 10% fetal bovine serum. Isolated T cells were subjected to an EasySep mouse CD8 + T cell negative-selection kit (StemCell Technologies, Canada) to harvest purified CD8 + T cells according to the manufacturer’s protocol.
Mixed leukocyte reaction
On day 9, harvested mature DCs loaded with LVDC-Ub-HBcAg, LVDC-UbHBcAg-LIGHT, LV-UbHBcAg-LIGHT, LVDC or PBS were pre-treated with 25 μg/mL mitomycin C (Sigma) for 30 min at 37 ℃. T cells collected from BALB/c mice were grown in 96 well plates and co-cultured with the mature DCs at different responder/stimulator (T cells/DCs) ratios (5:1, 10:1 or 20:1) for 72 h. Then the cells were cultured in a final volume of 200 μL complete RPMI-1640 in 96-well culture plates for 2 h and 10 μL of Cell Counting Kit-8 (CCK-8) solution (Dojindo, Kumamoto, Japan) was added to the plates for another 4h. The absorbance was recorded at 450 nm.
Ex vivo DC stimulation of T lymphocyte and functional assays
T cells were co-cultured with the different lentivectors-pulsed DCs mentioned above at a ratio of 10:1 (T cells/DCs) in 96 well plates. 3 days later, the supernatant was collected and measured for the secretion levels of different cytokines (IL-2, IL-6, TNF-α and IFN-γ) using ELISA kits.
IFN-γ and TNF-α production was also detected by intracellular staining. The above primed T cells were stimulated for 6 h in the presence of 25 μg/mL PMA, 1 μg/mL ionomycin and 1.7 μg/mL monensin. Then the stimulated cells were stained with perCP-Cy5.5 conjugated anti-CD8 McAb (eBioscience) for 30 min followed by fixation with Fix and Perm reagents A and B (eBioscience). Finally, the cells were stained with APC conjugated anti-IFN-γ or anti-TNF-α McAb (eBioscience) for 30 min and analyzed by flow cytometry.
For CTL assay, the primed lymphocytes (5 × 106 cells/ml) were used as the effector cells, P815/c cells (5 × 104 cells/well) harbouring HBcAg expression were used as target cells, and were co-cultured in 96-well plates at different effector and target (E/T) ratios (5:1, 10:1, and 20:1) for 4 h. The HBcAg-specifc CTL activity was evaluated with a CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, USA) for lactate dehydrogenase (LDH) release according to the manufacturer’s instructions.
Assessment of apoptosis
The former stimulated T cells (2 × 106 cells/mL) were washed in PBS twice and incubated with perCP-Cy5.5-labeled anti-CD8 McAb. Propidium Iodide (PI) and Annexin V–FITC staining (eBioscience) were then performed according to the manufacturer’s instructions. Thrice stained positive cells among the whole cell population were detected by flow cytometry.
Cell cycle analysis
For cell cycle analysis, the former stimulated T cells were purified with a CD8 + T cell negative-selection kit and washed with PBS twice, then a total of 5 × 105 cells were collected and fixed in 75% alcohol overnight at 4°C. Cells were incubated with 10 mg/ml RNase (Sigma) and 50 mg/ml propidium iodide (Sigma) at 37°C for 30 min in the dark. The cell cycle was measured by flow cytometry.
Real-time PCR
T cells described above were harvested for total RNA isolation by Trizol (TaKaRa Bio, Shiga, Japan); cDNA was synthesized according to the manufacturer’s instructions (TOYOBO, Japan) and quantified by PCR on an ABI 7500 FAST system (Applied Biosystems), using the SYBR Green qPCR Mix kit (TOYOBO, Japan) with appropriate primers: ATG5, 5′-GACAAAGATGT+GCTTCGAGATGTG-3′ (F) and 5ʹ-GTAGCTCAGATGCTCGCTCAG-3ʹ (R); Pro-caspase-3, 5ʹ-ATGGACAACAACGAAACCTCCGTG-3ʹ (F) and 5ʹ-CCACTCCCAGTCATTCCTTTAGTG-3ʹ (R); Pro-caspase-8, 5ʹ-GTGACAAGGGTGTCGTCTATGG-3ʹ (F) and 5ʹ-GGATGCTAAGAATGTCATCTCC-3ʹ (R); Bim, 5ʹ-TGATTACCGCGAGGCTGAA-3ʹ (F) and 5ʹ-ACCAGACGGAAGATAAGCGTAAC-3ʹ (R); CDKN1B, 5ʹ-CGCCTGGCTCGCTCCATTTGAC-3ʹ (F) and 5ʹ-GACACTCTCACGTTTGACATCTTCC-3ʹ (R). Primers were observed from Sangon Biotech (Shanghai, China). Cycling conditions were as follows: 40 cycles at 95°C for 3 min, 95°C for 15 s, and 60°C for 30 s.
Western blot
The above purified CD8 + T cells were collected and lysed with RIPA lysis buffer to obtain total protein lysates. The protein concentration of cell samples was measured using the BCA method. Equal amounts of proteins from each sample were loaded onto SDS-PAGE gels and transferred to PVDF membranes. After incubation with corresponding primary and secondary antibodies, the signals were detected with an ECL assay kit (Amersham).
Transmission electron microscopy
The stimulated T cells were treated with the indicated concentrations of triptolide for 24 h and collected by trypsinization. Cells were fixed in 2.5% phosphatebuffered glutaraldehyde at 4 °C overnight and postfixed in 1% phosphatebuffered osmium tetroxide for 1.5 h at room temperature. After being embedded and double-stained with uranyl acetate/lead citrate, the sections were determined under a H-7650 transmission electron microscope (Hitachi) at 60 kV.
Tandem mCherry-Wassabi confocal microscopy
T cells transfected with mCherry-Wassabi-LC3-II were seeded into cell culture dishes at a density of 1 × 105 cells per dish. Then the cells were washed with PBS three times and viewed under a laser-scanning confocal microscope (Nikon, Japan). Images were processed using the NIS Element Viewer software (Nikon). The average number of mCherry-Wassabi-LC3-II puncta per cell was evaluated by twenty cells randomly selected.
Statistical analysis
All values in the text and figures were obtained from three independent experiments. Results were presented as means ± SD. GraphPad Prism V5.0 (San Diego, CA, USA) was used to perform statistical analyzes. Differences between two groups was analyzed using Student’s t test, and the differences between two or more groups were using one-way analysis of variance. Differences were considered significant at P < 0.05.
Funding Statement
This study was supported by the grants from the National Natural Science Foundation of China [grants No. 81270502 and No. 81470853]; Pre-advanced research foundation of Shanghai Jiao Tong University [LY34-0003].
Acknowledgments
We thank all members of the department of Molecular Biology, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital for their advice and technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Fiel MI. Pathology of chronic hepatitis B and chronic hepatitis C. Clin Liver Dis. 2010;14:555–575. [DOI] [PubMed] [Google Scholar]
- [2].Deres K, Schroder CH, Paessens A, et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299:893–896. [DOI] [PubMed] [Google Scholar]
- [3].Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yang SH, Lee CG, Park SH, et al. Correlation of antiviral T-cell responses with suppression of viral rebound in chronic hepatitis B carriers: a proof-ofconcept study. Gene Ther. 2006;13:1110–1117. [DOI] [PubMed] [Google Scholar]
- [5].Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. [DOI] [PubMed] [Google Scholar]
- [6].Berzofsky JA, Ahlers JD, Belyakov IM. Strategies for designing and optimizing new generation vaccines. Nat Rev Immunol. 2001;1:209–219. [DOI] [PubMed] [Google Scholar]
- [7].Timmerman JM, Levy R. Dendritic cell vaccines for cancer immunotherapy. Annu Rev Med. 1999;50:507–529. [DOI] [PubMed] [Google Scholar]
- [8].Breckpot K, Escors D, Arce F, et al. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol. 2010;84:5627–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen JH, Yu YS, Chen XH, et al. Enhancement of CTLs induced by DCs loaded with ubiquitinated hepatitis B virus core antigen. World J Gastroentero. 2012;18:1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dai SL, Zhuo M, Song LL, et al. Dendritic cell-based vaccination with lentiviral vectors encoding ubiquitinated hepatitis B core antigen enhances hepatitis B virus-specific immune responses in vivo. Acta Biochim Biophys Sin. 2015;47:870–879. [DOI] [PubMed] [Google Scholar]
- [11].Dai SL, Zhuo M, Song LL, et al. Lentiviral vector encoding ubiquitinated hepatitis B core antigen induces potent cellular immune responses and therapeutic immunity in HBV transgenic mice. Immunobiology. 2016;221:813–821. [DOI] [PubMed] [Google Scholar]
- [12].Yang L, Yang H, Rideout K, et al. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol. 2008;26:326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tareen SU, Kelley-Clarke B, Nicolai CJ, et al. Design of a novel integration-deficient lentivector technology that incorporates genetic and posttranslational elements to target human dendritic cells. Mol Ther. 2014;22:575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dai BB, Yang LL, Yang HG, et al. HIV-1 Gag-specific immunity induced by a lentivector-based vaccine directed to dendritic cells. P Natl Acad Sci. 2009;106:20382–20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu Y, Tai A, Joo K-I, et al. Visualization of DC-SIGN-mediated entry Pathway of engineered lentiviral vectors in target cells. PLoS One. 2013;8:e67400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [18].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hubbard VM, Valdor R, Patel B, et al. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol. 2010;185:7349–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xu X, Araki K, Li S, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. 2014;15:1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Stephenson LM, Miller BC, Ng A, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. 2009;5:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jia W, He MX, McLeod IX, et al. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy. 2015;11:2335–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kovacs JR, Li C, Yang Q, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012;19:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schlie K, Westerback A, DeVorkin L, et al. Survival of effector CD8+ T cells during influenza infection is dependent on autophagy. J Immunol. 2015;194:4277–4286. [DOI] [PubMed] [Google Scholar]
- [26].Montgomery RI, Warner MS, Lum BJ. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. [DOI] [PubMed] [Google Scholar]
- [27].Mauri DN, Ebner R, Montgomery RI, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpes virus entry mediator. Immunity. 1998;8:21–30. [DOI] [PubMed] [Google Scholar]
- [28].Tamada K, Shimozaki K, Chapoval AI, et al. LIGHT, a TNF like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol. 2000;164:4105–4110. [DOI] [PubMed] [Google Scholar]
- [29].Wang J, Lo JC, Foster A, et al. The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J Clin Invest. 2001;108:1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jiang WZ, Chen R, Kong XB, et al. Immunization with adenovirus LIGHT-engineered dendritic cells induces potent T cell responses and therapeutic immunity in HBV transgenic mice. Vaccine. 2014;32:4565–4570. [DOI] [PubMed] [Google Scholar]
- [31].Mellman I. Dendritic cells: master regulators of the immune response. Cancer Immunol Res. 2013;1:145–149. [DOI] [PubMed] [Google Scholar]
- [32].Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hsu FJ, Benike C, Fagnoni F, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. [DOI] [PubMed] [Google Scholar]
- [34].Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. [DOI] [PubMed] [Google Scholar]
- [35].Bonifaz LC, Bonnyay DP, Charalambous A, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199:815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Geijtenbeek TB, Van Vliet SJ, Engering A, et al. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33–54. [DOI] [PubMed] [Google Scholar]
- [37].Jia W, Pua HH, Li QJ, et al. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol. 2011;186:1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol. 2011;186:5313–5322. [DOI] [PubMed] [Google Scholar]
- [39].Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. [DOI] [PubMed] [Google Scholar]
- [40].Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221–234. [DOI] [PubMed] [Google Scholar]
- [41].Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–169. [DOI] [PubMed] [Google Scholar]