

ABSTRACT
Radiation-induced lung injury (RILI) occurs most often in radiotherapy of lung cancer, esophageal cancer, and other thoracic cancers. The occurrence of RILI is a complex process that includes a variety of cellular and molecular interactions, which ultimately result in carcinogenesis. However, the underlying mechanism is unknown. Here we show that Ras-related C3 botulinum toxin substrate 2 (RAC2) and transcription factor jun-B (JUNB) were upregulated in non-small cell carcinoma (NSCLC) tissues and were associated with poor prognoses for NSCLC patients. Ionizing radiation also caused increased expression of RAC2 in quiescent stage cells, and the reentry of quiescent cells into a new cell cycle. The activity of the serum response factor (SRF) was activated by RAC2 and other Rho family genes (RhoA, ROCK, and LIM kinase). Consequently, JUNB acted as an oncogene and induced abnormal proliferation of quiescent cells. Together, the results showed that RAC2 can be used as a target gene for radiation protection. A better understanding of the RAC2 and JUNB mechanisms in the molecular etiology of lung cancer will be helpful in reducing cancer risks and side effects during treatment of this disorder. Our study therefore provides a new perspective on the involvement of RAC2 and JUNB as oncogenes in the tumorigenesis of NSCLC.
KEYWORDS: Radiation-induced lung injury, RAC2, Tumorigenesis
Introduction
The lung is one of several moderately radiosensitive organs. Radiation-induced lung injury (RILI) occurs most often in radiotherapy of lung cancer, esophageal cancer, and other thoracic cancers. The occurrence of RILI is a complex process that includes a variety of cellular and molecular interactions, which ultimately lead to fibroblast accumulation, proliferation, and differentiation, and even resulting in carcinogenesis [1]. Radiation-induced cancer has been related to the radiation dose following exposures. The dose-response relationship suggests that each ionization increases the risk of cancer risk [2]. The role of cell and molecular changes in the development of cancer, including mutations and chromosome aberrations, have been thought to be a critical step in the pathway from normal cells to cancer cells. High dose exposures have been shown to lead to chromosomal aberrations and increased cell killing via apoptosis that leads to increased cell turnover [3].
The risk assessment of ionizing radiation is important in the protection of normal tissues during radiotherapy. The majority of cells in the human body remain in the G0 phase (quiescent stage) of the cell cycle. They exist as quiescent or non-cycling cells, and may remain at this stage for days or even years until the resumption of cell division. For example, muscle and nerve cells are permanently arrested in the G0 phase, whereas other cell types such as liver cells resume the cell cycle and enter the G1 phase in response to exogenous stimuli such as injury [4,5]. Therefore, it is more reasonable to use quiescent cells to assess the radiation risk during X-ray treatments.
RAC2 is a GTPase of ~21 kDa containing the catalytic subunit of NADPH oxidase, which is the main component of the respiratory chain. GTPases are important regulators of diverse cell processes including cell growth, cytoskeletal reorganization, and the activation of protein kinases. Several studies have reported that RAC2 plays an essential role in the regulation of cytoskeleton remodeling [6,7], and in the gene expression of oncogenes [8]. Although recent reports also suggest the contribution of RAC2 to host defense responses in vivo [9], some laboratory reported that RAC2 is important in macrophage and endothelial cell migration on specific provisional matrix proteins. RAC2 promotes tumor growth, angiogenesis and invasion [10]. However, its role in the regulation of cellular responses to ionizing radiation is less known.
Although JUNB has historically been studied primarily in the contexts of cell cycle regulation and differentiation, several recent studies have defined it to be a oncogene [11,12]. Many works have reported the relationship between JUNB and invasion/metastasis in non-small-cell lung cancer (NSCLC) [13], prostate cancer [14], breast cancer [11], head and neck squamous cell carcinoma [15], et al. Serum response factor (SRF) activities in many cancers [16]. Genes that rely on SRF correlate with aggressive disease, and are associated with biochemical recurrence. Thus, understanding the mechanism(s) by which SRF pathways may provide novel opportunities to target clinically relevant signaling. RAC2 also mediates SRF target genes through GTPase ras homolog family member A (RhoA) [17]. Interference with expression of RhoA, activity of the RhoA effector Rho-associated coiled-coil containing protein kinase 1 (ROCK), and actin polymerization necessary for nuclear translocation of the SRF cofactor megakaryocytic acute leukemia (MAL) prevented full androgen regulation of SRF target genes [16].
In the present study, we conducted both in vitro and in vivo studies and found that RAC2 and JUNB expression was upregulated in human NSCLC tissues, which correlated with the poor prognoses of patients. Ionizing radiation also caused increased expression of RAC2 in quiescent stage cells and the reentry of quiescent cells into the cell cycle. The activity of SRF was activated by RAC2 and other Rho family genes such as RhoA, ROCK, and LIM kinase. Consequently, JUNB as an oncogene induced abnormal proliferation of quiescent cells. Overall, the results showed that RAC2 can be used as a target gene in radiation protection.
Results
RAC2 and JUNB expression was upregulated in human NSCLC tissues and correlated with poor prognoses
We analyzed the expression levels of RAC2 and JUNB in human NSCLC tissues using sequencing data downloaded from the Cancer Genome Atlas (TCGA). To evaluate the relationship between RAC2 and JUNB expression levels and NSCLC prognoses, we used Kaplan-Meier survival analyses and the log-rank test. Overall survival (OS) curves were plotted according to RAC2 and JUNB expression levels. Figure 1(a,b) show that the OS for patients with high RAC2 and JUNB expressions was significantly better than low RAC2 and JUNB expression patients (p = 0.0318 for the RAC2 group; p = 0.0020 for the JUNB group). There were 99 high expression RAC2 patients and 112 low expression RAC2 patients; and there were 86 high expression JUNB patients and 88 low expression JUNB patients. Figure 1(a) shows that the OS of 3 years for patients with high RAC2 expression was 48.90% (n = 88), but was 70.45% for low RAC2 expression patients (n = 88); and Figure 1(b) shows that the OS of 3 years for patients with high JUNB expression was 58.11% (n = 74), but was 78.95% for the low JUNB expression patients (n = 57). These results indicated that overexpression of RAC2 and JUNB represented a novel predicator of poor prognosis and/or a progression marker for NSCLC. Furthermore, we analyzed the expression levels of RAC2 and JUNB in human NSCLC tissues using RT-PCR assays. Figure 1(c) shows that RAC2 expression was upregulated in human NSCLC tissues (n = 23) when compared with normal lung tissues (n = 4) (p = 0.013), and Figure 1(d) shows that JUNB expression was upregulated in human NSCLC tissues (n = 23) when compared with normal lung tissues (n = 4) (p = 0.004). Together, these results were consistent with the TCGA sequencing data.
Figure 1.
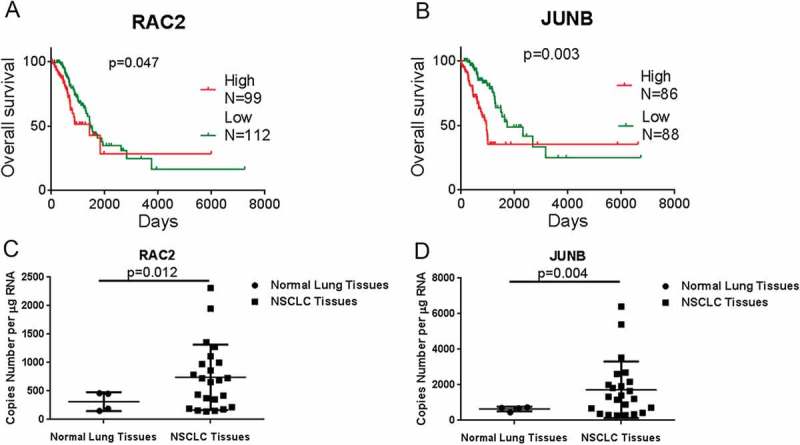
RAC2 and JUNB are upregulated in non-small cell carcinoma (NSCLC) tissues and were correlated with poor prognoses.
(a, b) The relative expressions of RAC2 and JUNB (b) in NSCLC tissues were analyzed using the Cancer Genome Atlas data set. Kaplan-Meier progression-free survival and overall survival curves were used to analyze RAC2 and JUNB expression levels. X = 1095. (c, d) The absolute expression levels of RAC2 (c) and JUNB (d) were determined in 23 clinical lung cancer tissues and four normal lung tissues using quantitative RT-PCR. The data are expressed as the mean±SEM.
Ionizing radiation caused increased expression of RAC2 in quiescent stage cells and reentry of quiescent cells into the cell cycle
To investigate the functional role of RAC2 in lung cells, we first performed western blot analyses to examine the expression of RAC2 in quiescent cells. Figures 2(a,b) show that when quiescent cells were exposed to 2 Gy X-ray irradiation, RAC2 expression levels were upregulated (p = 0.034). The expression of JUNB was also upregulated after 2 Gy of X-rays exposure (p = 0.011). In order to counteract the phenotypic changes caused by the upregulated RAC2, we used RAC2 shRNA in the following experiments. To examine whether the knockdown of RAC2 affected quiescent cell proliferation or cell cycle progression, flow cytometric analyses were performed. Figures 2(d–g) shows that after exposure to 2 Gy X-ray irradiation, the proportions of S phase cells increased from 9.46%±1.72% to 13.28%±0.60%, while in shRAC2 cells, the proportion of S phase cells was 10.63 ± 0.72%. After exposure to 2 Gy of X-ray irradiation, the proportions of G2/M phase cells increased from 4.00%±1.19% to 9.16%±1.34%, while in shRAC2 cells, the proportion of G2/M phase cells was 6.30 ± 0.91%. These results showed that X-ray exposure caused reentry of the quiescent cells into the cell cycle, but knockdown of RAC2 mitigated this process. We then performed cell proliferation experiments. Cells were incubated in 0.5% fetal bovine serum in MEM medium after treatment. The CCK-8 assay, measuring the absorbance at 490 nm, was used on the third day after irradiation. Figures 3(h,i) shows that the quiescent cells underwent abnormal proliferation. The relative cell activity increased 1.45 times compared with the control group (p = 0.009), but the relative cell activity decreased in shRAC2 cells when compared to the IR group (p = 0.049). A marker of cell proliferation, Ki67, significantly increased after X-ray exposure (p = 0.009) (Figures. 2(j, k)).
Figure 2.
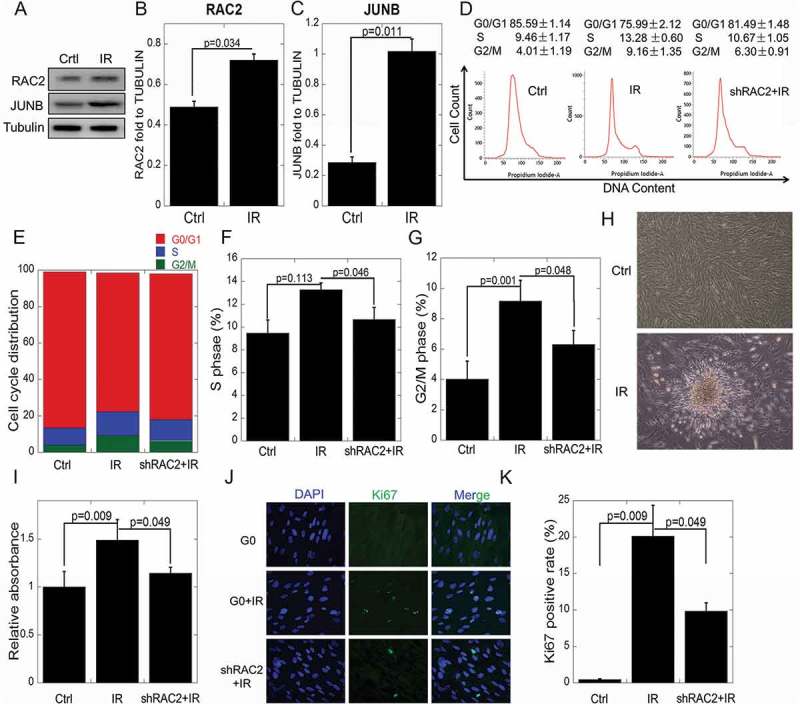
The effects of RAC2 on in vitro quiescent cell proliferation after ionizing radiation.
(a) The expression of RAC2 and JUNB were measured by western blotting. (b, c) Grayscale analyses of RAC2 and JUNB in quiescent cells and quiescent shRAC2 cells after treatment with 2 Gy of X-ray irradiation. (d–g) Flow cytometry showing significant increases in the percentages of cells in the S or G2/M phases when the cells were treated with 2 Gy X-ray irradiation. (h) The abnormal proliferation of quiescent cells after X-ray irradiation. (i) The CCK-8 assay was used to determine the viability of quiescent cells and quiescent shRAC2 cells after exposure to X-ray irradiation. (j-k) An immunofluorescent assay was used to determine the percentages of Ki67 positive quiescent cells and quiescent shRAC2 cells after exposure to X-ray irradiation. The error bars denote the mean±SE derived from three independent experiments.
Ionizing radiation increased the expression of RAC2 downstream proteins in mouse lung tissue
The proteins in the Rho GTPase family were then checked in quiescent cells. After radiation, RAC2, RhoA, ROCK, LIM kinase, phosphorylated LIM Kinase and JUNB were significantly upregulated (Figures 3(a, b)), which was decreased by downregulation of RAC2 in knockdown cells. Swinholide A, which promotes F-actin depolymerization, had the same effects with the IR group, except for JUNB. The results indicated that F-actin depolymerization blocking the signaling pathway in the up-stream of JUNB. LIM kinase, which could stabilize F-actin structure, could not promote JUNB expression in Swinhilide A treated group. So, JUNB was downstream of the Rho pathway and its expression regulated by actin morphology. To verify the expression of related proteins after lung irradiation in mice, we irradiated the right lungs of mice using computed tomography (CT) image-guided radiotherapy. The axial, sagittal, and coronal CT images are shown in Figure 3(c). The CT image was imported into the radiotherapy planning system to control the irradiation. A dose of 2 Gy was used to simulate the clinical radiotherapy of lung cancer with a single treatment. The expressions of RAC2 and JUNB were then determined in mice. Figures 3(d,e) show that RAC2 and JUNB were upregulated after 2 Gy X-ray irradiation (RAC2, p = 0.042; JUNB, p = 0.0001). After radiation, RAC2, RhoA, and LIM kinase were significantly upregulated in mice(Figures 3(f, g), n = 7). The results indicated that JUNB was downstream of the Rho pathway in mice.
Figure 3.
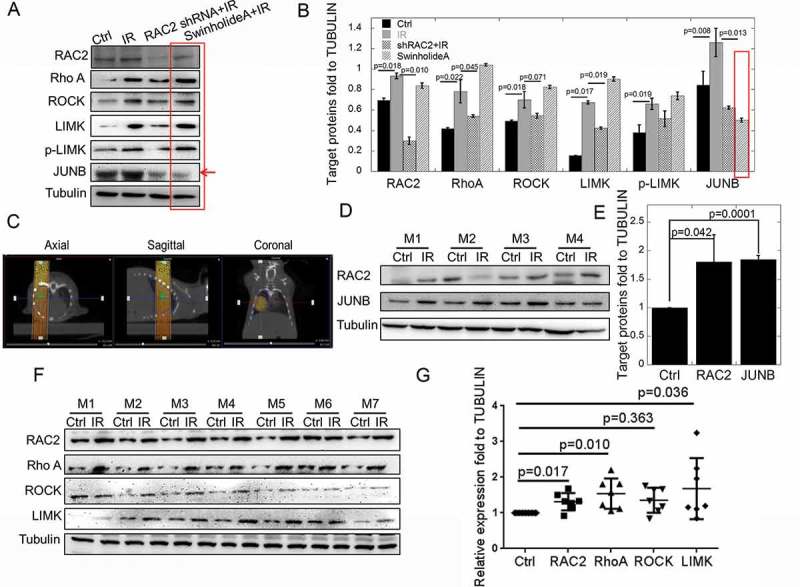
The in vivo and in vitro expressions of RAC2 and Rho GTPase pathway proteins after exposure to X-ray irradiation.
(a) The expression of RAC2, RhoA, ROCK, LIM kinase, and JUNB were measured by western blotting. (b) The grayscale analyses of RAC2, RhoA, ROCK, LIM kinase, and JUNB. (c) The target area of X-ray exposure using computed tomography as guidance. (d) The expression of RAC2 and JUNB were measured by western blotting in mice (n = 4). (e) The grayscale analyses of RAC2 and JUNB in mice (n = 4). (f) The expressions of RAC2, RhoA, ROCK, LIM kinase, and JUNB were measured by western blotting in mice (n = 7). (g) The grayscale analyses of RAC2, RhoA, ROCK, LIM kinase, and JUNB in mice (n = 7). The error bars denote the mean±SE from three independent experiments.
Ionizing radiation increased the activity of SRF by promoting the nuclear accumulation of megakaryocytic acute leukemia (MAL) protein
The Rho family of GTAases mediates extracellular signals that direct the assembly and stabilization of the actin cytoskeleton. The coordinated action of these Rho targets (RhoA, ROCK and LIM kinase) is required to regulate SRF activity by affecting actin dynamics. MAL is known as a co-activator of the SRF, and the MAL responds to G-actin. Because G-actin polymerization is regulated by LIM kinase, we determined SRF activity and the subcellular localization of MAL and SRF. Graphic representation of plasmids used to test SRF activity is shown in Figures 4(a). The CCATATTAGG sequence was constructed into the plasmid as the SRF protein binding site. Figure 4(b) shows that in the control group, MAL was evenly distributed in both the cytoplasm and nucleus. Radiation induced nuclear accumulation of endogenous MAL, whereas RAC2 shRNA efficiently inhibited the nuclear accumulation of MAL. The effects of the irradiated knockdown of JUNB cells were the same as with the IR group (Figure 4(c)). The luciferase assay showed that irradiation enhanced the activity of SRF (p = 0.046) whereas irradiated RAC2 shRNA cells reduced the activity of SRF when compared with the IR group (Figure 4(d)). Taken together, these results suggested that translocation of MAL into the nucleus and the activation of the SRF were regulated after irradiation via the RAC2 protein.
Figure 5.
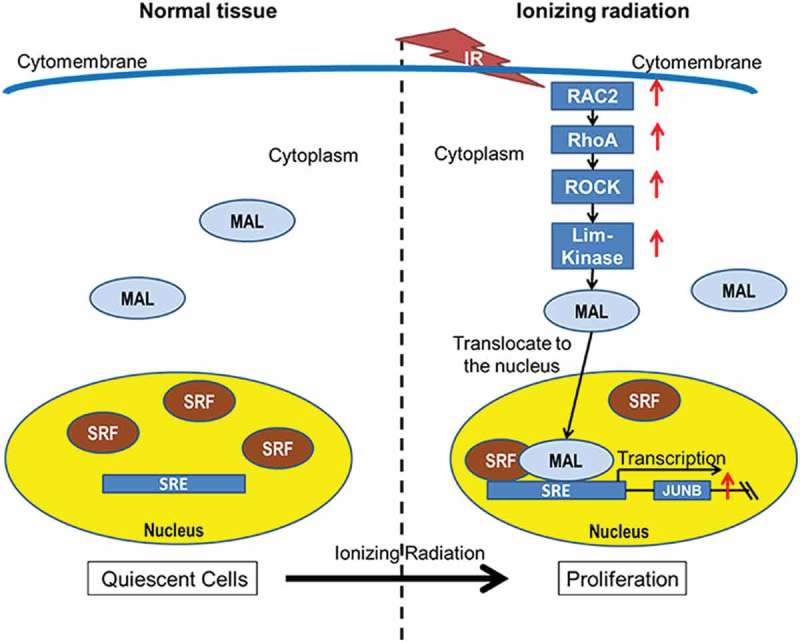
Graphical representation of the roles of the RAC2 and Rho GTPase pathways in quiescent cells involved in recycling and abnormal proliferation.
Figure 4.

Ionizing radiation increased the activity of the serum response factor (SRF) by promoting the nuclear accumulation of megakaryocytic acute leukemia (MAL) factor.
(a) A graphical representation of the plasmid construction to determine the SRF activity. (b) The intracellular protein hybridization of MAL and SRF. (c) The percentage of MAL in the nucleus. (d) Results of a Co-IP experiment using MAL antibody as the immunoprecipitating antibody to confirm interactions between MAL and its binding partners SRF. Normal rabbit IgG was used for Co-IP and served as a negative control, whereas normal cells lysate without Co-IP served as a positive control. The heavy chain of IgG served as a loading control. (e) The SRF activity was determined using the luciferase reporter assay. The error bars denote the mean± SE derived from three independent experiments.
A model for the mechanism of abnormal proliferation induced by ionizing radiation in quiescent stage cells
As one of the most important components of the Rho GTPase family, RAC2 and its downstream genes were significantly upregulated in our current study. MAL is predominantly cytoplasmic, where it is sequestered by actin monomers. When cells were treated with X-ray irradiation, LIM kinase upregulation caused the accumulation of F-actin and a commensurate decrease in the level of G-actin. As a consequence, MAL was no longer sequestered and was free to translocate to the nucleus, where it associated with the SRF and activated SRF-mediated gene expression. SRF bound to the promoter and induced JUNB expression. JUNB performed as an oncogenic protein, which induced reentry of quiescent stage cells into the cell cycle, resulting in abnormal proliferation.
Discussion
Carcinogenesis is a very complicated process and requires changes in many key events in the pathways leading to cancer. These changes facilitate conversion of normal cells to a cancerous phenotype. In this study, we performed both in vitro and in vivo studies and found that the Rho GTPase family genes were upregulated by treatment with ionizing radiation. RAC2-RhoA-ROCK-LIM kinase enhanced the SRF activity by regulating the actin cytoskeleton morphology. As a transcription factor, SRF promoted JUNB expression. Consequently, JUNB acted as an oncogene, and induced abnormal proliferation of quiescent cells (Figure 5).
Reactive oxygen species (ROS) are produced not only by the interaction of ionizing radiation with water and biomolecules in cells, but also by intracellular biological processes. NADPH oxidase catalyzes the production of superoxide from oxygen and NADPH [18]. RAC2 is a catalytic subunit of NADPH oxidase, which is the main component of the respiratory chain [19]. RAC2 is therefore involved in the generation of ROS [20]. ROS at physiological levels are important cell signaling molecules. However, abnormally high ROS have been associated with disease and are commonly found in cancer [21]. Excess ROS upregulate autophagy by multiple and at times counteracting mechanisms, and in turn this catabolic cellular process restores physiological ROS levels. Defective autophagy promotes the accumulation of damaged mitochondria and results in oxidative stress, which may be partly responsible for the tumor-forming abilities of autophagy-deficient cells by functioning as a genotoxic and mutagenic agent, and by triggering secondary, non-cellular autonomous effects such as inflammation [21].
The activation of SRF-mediated transcription in response to Rho GTPase-induced actin assembly involves the ability of the SRF to sense changes in the cellular level of actin monomers [16]. The Rho family of GTPases, which includes the three prototype members of Rho, Rac, and Cdc42, mediates extracellular signals that direct the assembly and stabilization of the actin cytoskeleton. The ability of Rho proteins to regulate the assembly of polymerized actin (F-actin) directly affects nuclear gene expression. The SRE is found within the promoter region of the so-called “immediate early genes,” whose expression is rapidly induced by serum, as well as several muscle specific genes. A complex of two transcription factors, SRF and ternary complex factor (TCF), binds directly to this DNA element, and substantial evidence supports a role for Rho signaling in the activation of the SRF as a transcriptional regulator at the SRE [22]. Further mechanistic studies of the Rho-SRF connection have revealed some of the downstream signaling components that link Rho to the transcriptional machinery [23]. The actin regulating functions of two of the Rho effector targets, mDia1 and ROCK, are involved. ROCK promotes F-actin stabilization by phosphorylating LIM-kinase, which in turn phosphorylates and inactivates the actin depolymerizing factor, cofilin, whereas mDia1 is a formin family protein that induces de novo F-actin assembly. The coordinated action of these Rho targets is required to regulate the SRF activity by affecting actin dynamics.
It is well-known that the epithelial-mesenchymal transition (EMT) is crucial for tumorigenesis. Some reports previously reported that SMAD3, SMAD4, and the AP1 components JUN, JUNB, FOS, and FOSL1 cooperatively regulated several established TGFβ-target genes, with a known function in the EMT and invasion, and enhanced TGFβ-induced collagen invasion [24]. ChIP-seq and RNA-seq analyses in showed that the strong and prolonged induction of JUNB by TGFβ redirected SMAD2/3 to different target sites and thereby played a major role in the activation of late TGFβ target genes as critical components of a feed-forward regulatory network[25]. TGFβ plays a dual role in tumor progression. In the late stages of tumor development, TGFβ acts as a tumor promoter by stimulating cell motility, invasion, metastasis, and tumor stem cell maintenance. This was shown by the observation that specific types of cancers are insensitive to the cytostatic effect of TGFβ because of inactivation of core components in the TGFβ pathway [25]. Knockout of JUNB in metastatic cells significantly suppressed both cell invasion and migration in vitro. In addition, the knockout of JUNB in metastatic cells significantly repressed the incidence of lung metastases and prolonged survival in vivo. These results suggested that JUNB plays an important role in promoting cell invasion, migration, and distant metastasis via pathways other than the EMT, and that the downregulation of JUNB may be an effective treatment strategy for patients [15].
In summary, our findings showed that RAC2 and JUNB were upregulated in NSCLC tissues, and that their upregulations were associated with poor prognoses for NSCLC patients. In tumor radiotherapy, RAC2 and JUNB are also highly expressed in normal lung tissues. The highly expressed RAC2 and JUNB are therefore likely to be associated with carcinogenesis. A better understanding of the RAC2 and JUNB mechanisms in the molecular etiology of lung cancer will be helpful for reducing cancer risks and the side effects associated with cancer treatments. Our study provides a new perspective on RAC2 and JUNB, which act as oncogenes in NSCLC tumorigenesis. However, other possible mechanisms by which RAC2 and JUNB participate in tumorigenesis remain to be fully understood.
Materials and methods
Cell culture and irradiation
The MRC5 human lung cell line used in this study was obtained from the American Type Culture Collection in 2010. Cells were maintained in MEM medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 1% penicillin and 100 µg/mL streptomycin, at 37°C in 5% CO2 in a humidified incubator (Thermo Scientific, NC, USA). The cell lines were used within 20 passages and freshly thawed every 2 months. These cell lines were free of mycoplasma and authenticated by quality examinations of the morphology and growth profiles. G0 cells and G1 cells were obtained as previously described [26]. In brief, 5 × 105 cells plated in T25 flasks were allowed to grow for 8 days to become quiescent (G0 phase) by contact inhibition, with one medium refreshment on day 4. The cells were then harvested, subcultured in a new flask of the same size, and incubated for 6 hours in fresh medium to allow them to reach the G1 phase. X-ray irradiation was performed using an X-ray generator (Faxitron 650; Faxitron Bioptics, Lincolnshire, IL, USA), which was operated with 100 kVp and 5 mA at room temperature.
Human NSCLC samples
Twenty-three paired human non-small lung cancer tissues used in this study were collected from the Department of Lung Cancer, Shanghai Jiao-tong University. Normal lung tissues were collected at the Judicial Expertise Center of Soochow University. All human specimens were approved by the Ethical Review Committee of the World Health Organization of the Collaborating Center for Research in Human Production authorized by the Shanghai Municipal Government, and obtained with informed consent from all patients. This study was approved by the Ethics Committee of Soochow University. All animal experiments were carried out with the approval of the Soochow University Experimental Animal Care and Use Committee.
RNA extraction and qrt-pcr analysis
Total RNA samples from the NSCLC tissue specimens and cell lines were extracted with TRIzol reagent (Invitrogen, CA) according to the manufacturer’s protocol, and quantified with Nanodrop 2000 (Thermo, Japan). First-strand cDNA was synthesized using the PrimeScript RT Reagent Kit (TaKaRa, China). The real-time polymerase chain reaction (qPCR) was performed with SYBR Green Premix Ex Taq (TaKaRa). The relative RNA expression levels were determined by qPCR and measured using an ABI Prism 7900 sequence detection system (Applied Biosystems, Foster City, CA, USA). The sequences for the gene specific primers used are: GAPDH Forward: GCACCGTCAAGGCTGAGAAC, Reverse: TGGTGAAGACGCCAGTGGA; RAC2 Forward: GGACGACAAGGACACCATCG, Reverse: CACTCCAGGTATTTCACC
GAGT; JUNB Forward: CCACCTCCCGTTTACACCAA, Reverse: GAGGTAGCTG
ATGGTGGTCG.
Immunoblotting and co- immunoprecipitating assays
The cell lysates were extracted from cultured cells with RIPA lysis buffer (or co-IP lysis buffer) containing protease inhibitors (Byotime, China), and cleared by centrifugation at 4°C for 15 minutes at 12,000 × g. The protein samples were separated by 6% and 8% SDS-PAGE and transferred to nitrocellulose filter membranes (Millipore, USA). After blocking in phosphate-buffered saline (PBS)/Tween-20 containing 5% nonfat milk, the membranes were incubated with the primary antibodies (Supplementary Table S1). Subsequent visualization used the SuperSignal West Femto Maximun Sensitivity Substrate (Thermo). In co-IP assay, 500 μg protein lysates was diluted in IP lysis buffer and incubated with 1 μg normal rabbit or mouse IgG for 2 h, to be followed by 2 h of incubation with 50 μl Protein A/G magnetic beads (Bio-tool, China) to precipitate proteins that interacted non-specifically with IgG and/or Protein A/G magnetic beads. This pre-cleared lysate was then incubated with 2 μg MAL antibody at room temperature overnight. Protein A/G magnetic beads (20 μl) was added and incubated at room temperature for 6 h, which were then washed with IP lysis buffer, and proteins in the immunocomplexes were then extracted in SDS sample buffer and used for immunoblotting to identify SRF proteins.
Cell cycle assay
The cells were harvested and fixed with 70% pre-chilled ethanol for > 24 hours at −20°C. Prior to analysis, fixed cells were washed twice with PBS, treated with 100 µg/mL RNase A and 50 µg/mL propidium iodide mixed buffer (BD Biosciences, San Jose, CA, USA) for 30 minutes at room temperature. The cell cycle distribution was analyzed using FACS Verse flow cytometry (Becton Dickinson). Cell cycle distribution was measured with CellQuest (Becton Dickinson). FlowJo software also used for a double check.
Immunofluorescence
Immunofluorescence assays were performed as previously described [26]. The cells were fixed in 4% paraformaldehyde for 10 minutes, permeabilized for 20 minutes in ethanol at −20ºC, washed with PBS for 30 minutes, treated with 0.5% Triton for 10 minutes, blocked for 1 hour with 5% skim milk, and stained with anti-Ki67, anti-SRF or anti-MAL antibody (1:1000) for 2 hours. The bound antibody was visualized using Alexa Fluor® 594 anti-mouse antibody (Abcam, NY, USA) and cell nuclei were counterstained with 4ʹ,6-diamidino-2-phenylindole (Invitrogen, CA, USA). The Ki67 and MAL positive rate was counted and at least 500 cells were counted under fluorescence microscope.
Gene silencing and reporter plasmids
Cells were seeded in 12-well plates (1 × 105 cells per well) with antibiotic-free MEM containing 10% serum, 48 hours before transfection. The cells were transfected with 1 × 106 RAC2 shRNA lentivirus particles (Invitrogen, Eugene, OR, USA) for 24 hours. Control shRNA and green fluorescent protein(GFP)-labelled lentivirus particles were also used according to the manufacturer’s instructions. The cells were subsequently cultured with MEM containing 1 µM puromycin (Invitrogen). Cells stably expressing RAC2, which was checked by western blotting, were used for further experiments. The SRF-lux and MLV-lacZ reporters were used to detect SRF activity using the luciferase assay, which was performed as previously described [27]. A549 cells on a 12-well plate were co-transfected with 300 ng DNA using Lipofectamine 3000. Luciferase activity was measured 48 hours later using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) with a Multiscan Spectrum (BioTek Synergy 2; USA).
Hematoxylin and eosin staining
Mouse tumor tissues were fixed with 4% formalin (v/v) and embedded with paraffin. The tissues were cut into 6 μm sections, and the sections were deparaffinized with xylene and incubated with EDTA antigenic retrieval buffer for antigenic retrieval, and then stained with hematoxylin and stain. The digital images of organs were acquired using a Nanozoomer (Hamamatsu Photonics).
Statistics
All experiments were independently repeated at least three times, and all data are expressed as the mean ± standard error. Student’s t-tests were used for statistical analyses. A value of p < 0.05 was considered to be statistically significant.
Funding Statement
This work was supported by the National Natural Science Foundations of China awarded (No. 81602794, 11405235, 81673151), China Postdoctoral Science Foundation funded project (2016M591904, 2017T100399). Natural Science Foundations of Jiangsu awarded (BK20160334). This work also supported by “A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD)”.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- [1].Huang Y, Zhang W, Yu F, et al. the cellular and molecular mechanism of radiation-induced lung injury. Medical Science Monitor: International Medical Journal of Experimental and Clinical Research. 2017. July 15;23:3446–3450. PubMed PMID: 28710886; PubMed Central PMCID: PMC5523971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tubiana M. Dose-effect relationship and estimation of the carcinogenic effects of low doses of ionizing radiation: the joint report of the academie des sciences (Paris) and of the academie nationale de medecine. Int J Radiat Oncol. 2005. October 1;63(2):317–319. PubMed PMID: WOS:000232083700001; English. [DOI] [PubMed] [Google Scholar]
- [3].Brooks AL, Hoel DG, Preston RJ. The role of dose rate in radiation cancer risk: evaluating the effect of dose rate at the molecular, cellular and tissue levels using key events in critical pathways following exposure to low LET radiation. Int J Radiat Biol. 2016. August;92(8):405–426. PubMed PMID: 27266588; PubMed Central PMCID: PMC4975094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cao W, Chen K, Bolkestein M, et al. Dynamics of proliferative and quiescent stem cells in liver homeostasis and Injury. Gastroenterology. 2017. October;153(4):1133–1147. PubMed PMID: 28716722 [DOI] [PubMed] [Google Scholar]
- [5].Menyhart J, Marcsek Z, Grof J. Proliferation related peptides in quiescent and regenerating rat liver. Acta Physiol Acad Sci Hung. 1981;58(2):119–129. PubMed PMID: 7340389 [PubMed] [Google Scholar]
- [6].Filippi MD, Harris CE, Meller J. Localization of Rac2 via the C terminus and aspartic acid 150 specifies superoxide generation, actin polarity and chemotaxis in neutrophils. Nat Immunol. 2004. July;5(7):744–751. PubMed PMID: 15170212 [DOI] [PubMed] [Google Scholar]
- [7].Lacy P, Willetts L, Kim JD, et al. Agonist activation of f-actin-mediated eosinophil shape change and mediator release is dependent on Rac2. Int Arch Allergy Immunol. 2011;156(2):137–147. PubMed PMID: 21576984; PubMed Central PMCID: PMC3104871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang FC, Kapur R, King AJ, et al. Rac2 stimulates Akt activation affecting BAD/Bcl-XL expression while mediating survival and actin function in primary mast cells. Immunity. 2000. May;12(5):557–568. PubMed PMID: 10843388 [DOI] [PubMed] [Google Scholar]
- [9].Croker BA, Handman E, Hayball JD, et al. Rac2-deficient mice display perturbed T-cell distribution and chemotaxis, but only minor abnormalities in T(H)1 responses. Immunol Cell Biol. 2002. June;80(3):231–240. PubMed PMID: 12067410. [DOI] [PubMed] [Google Scholar]
- [10].Joshi S, Singh AR, Zulcic M, et al. Rac2 controls tumor growth, metastasis and M1-M2 macrophage differentiation in vivo. PloS One. 2014;9(4):e95893 PubMed PMID: 24770346; PubMed Central PMCID: PMC4000195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sundqvist A, Morikawa M, Ren J, et al. JUNB governs a feed-forward network of TGFbeta signaling that aggravates breast cancer invasion. Nucleic Acids Res. 2018. February 16;46(3):1180–1195. PubMed PMID: 29186616; PubMed Central PMCID: PMC5814809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Van Amsterdam JR, Wang Y, Sullivan RC, et al. Elevated expression of the junB proto-oncogene is essential for v-fos induced transformation of Rat-1 cells. Oncogene. 1994. October;9(10):2969–2976. PubMed PMID: 8084600 [PubMed] [Google Scholar]
- [13].Berghoff AS, Birner P, Streubel B, et al. ALK gene aberrations and the JUN/JUNB/PDGFR axis in metastatic NSCLC. APMIS: Acta Pathologica, Microbiologica, Et Immunologica Scandinavica. 2014. September;122(9):867–872. PubMed PMID: 24750504. [DOI] [PubMed] [Google Scholar]
- [14].Konishi N, Shimada K, Nakamura M, et al. Function of JunB in transient amplifying cell senescence and progression of human prostate cancer. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research. 2008. July 15;14(14):4408–4416. PubMed PMID: 18628455. [DOI] [PubMed] [Google Scholar]
- [15].Hyakusoku H, Sano D, Takahashi H, et al. JunB promotes cell invasion, migration and distant metastasis of head and neck squamous cell carcinoma. Journal of Experimental & Clinical Cancer Research: CR. 2016. January;12(35):6 PubMed PMID: 26754630; PubMed Central PMCID: PMC4709939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Settleman J. A nuclear MAL-function links Rho to SRF. Mol Cell. 2003. May;11(5):1121–1123. PubMed PMID: 12769835 [DOI] [PubMed] [Google Scholar]
- [17].Schmidt LJ, Duncan K, Yadav N, et al. RhoA as a Mediator of Clinically Relevant Androgen Action in Prostate Cancer Cells. Mol Endocrinology. 2012. May;26(5):716–735. PubMed PMID: WOS:000303864900003; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dworakowski R, Alom-Ruiz SP, Shah AM. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacological Reports: PR. 2008. Jan-Feb;60(1):21–28. PubMed PMID: 18276982 [PubMed] [Google Scholar]
- [19].Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006. October;16(10):522–529. PubMed PMID: 16949823 [DOI] [PubMed] [Google Scholar]
- [20].Peshavariya H. NADPH Oxidase-Derived ROS Signaling and Therapeutic Opportunities. Curr Pharm Des. 2015;21(41):5931–5932. PubMed PMID: 26537745 [DOI] [PubMed] [Google Scholar]
- [21].Kongara S, Karantza V. The interplay between autophagy and ROS in tumorigenesis. Front Oncol. 2012;2:171 PubMed PMID: 23181220; PubMed Central PMCID: PMC3502876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995. June 30;81(7):1159–1170. PubMed PMID: 7600583 [DOI] [PubMed] [Google Scholar]
- [23].Geneste O, Copeland JW, Treisman R. LIM kinase and Diaphanous cooperate to regulate serum response factor and actin dynamics. J Cell Biol. 2002. May 27;157(5):831–838. PubMed PMID: 12034774; PubMed Central PMCID: PMC2173419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sundqvist A, Zieba A, Vasilaki E, et al. Specific interactions between Smad proteins and AP-1 components determine TGF beta-induced breast cancer cell invasion. Oncogene. 2013. August 1;32(31):3606–3615. PubMed PMID: WOS:000322638400005; English. [DOI] [PubMed] [Google Scholar]
- [25].Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A.2003. July 22; 100(15):8621–8623. PubMed PMID: 12861075; PubMed Central PMCID: PMC166359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pei HL, Zhang J, Nie J, et al. RAC2-P38 MAPK-dependent NADPH oxidase activity is associated with the resistance of quiescent cells to ionizing radiation. Cell Cycle. 2017;16(1):113–122. PubMed PMID: WOS:000392737300021; English [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hu W, Xu S, Yao B, et al. MiR-663 inhibits radiation-induced bystander effects by targeting TGFB1 in a feedback mode. RNA Biology. 2014;11(9):1189–1198. PubMed PMID: 25483041; PubMed Central PMCID: PMC4615905 [DOI] [PMC free article] [PubMed] [Google Scholar]