

Abstract
Pro-inflammatory and amyloidogenic S100A9 protein is an important contributor to Alzheimer’s disease (AD) pathology. Traumatic brain injury (TBI) is viewed as a precursor state for AD. Here we have shown that S100A9-driven amyloid-neuroinflammatory cascade was initiated in TBI and may serve as a mechanistic link between TBI and AD. By analyzing the TBI and AD human brain tissues, we demonstrated that in post-TBI tissues S100A9, produced by neurons and microglia, becomes drastically abundant compared to Aβ and contributes to both precursor-plaque formation and intracellular amyloid oligomerization. Conditions implicated in TBI, such as elevated S100A9 concentration, acidification and fever, provide strong positive feedback for S100A9 nucleation-dependent amyloid formation and delay in its proteinase clearance. Consequently, both intracellular and extracellular S100A9 oligomerization correlated with TBI secondary neuronal loss. Common morphology of TBI and AD plaques indicated their similar initiation around multiple aggregation centers. Importantly, in AD and TBI we found S100A9 plaques without Aβ. S100A9 and Aβ plaque pathology was significantly advanced in AD cases with TBI history at earlier age, signifying TBI as a risk factor. These new findings highlight the detrimental consequences of prolonged post-TBI neuroinflammation, which can sustain S100A9-driven amyloid-neurodegenerative cascade as a specific mechanism leading to AD development.
Introduction
Over the past decade traumatic brain injury (TBI) has become the focus of increasing attention due to frequent incidences in modern society, including sport and military injuries. Despite extensive efforts to develop short and long-term neuroprotective strategies, these are not yet satisfactory and a better understanding of underlying pathologies is required to define the specific therapeutic targets. Following primary mechanical assault, TBI leads to delayed secondary responses at the molecular and cellular levels, which occur on a longer time scale and account for post-TBI neurological deficits1. There is growing epidemiological and clinical evidence that TBI incidences, including relatively mild injuries and even repetitive ball headings2, are strong risk factors for chronic traumatic encephalopathies and Alzheimer’s disease (AD)3–5.
Massive accumulations of amyloid-β peptide (Aβ) toxic oligomers and plaques6 are among the major AD pathological hallmarks and targets for therapeutic interventions. Inflammation also plays an important role in AD, which is supported by a sharp induction of inflammatory mediators in AD-affected brain7. Importantly, non-steroidal anti-inflammatory drugs may markedly reduce age-related prevalence of AD8,9 and slow amyloid deposition by mechanisms that still remain elusive8. Recently, we have found that pro-inflammatory mediator S100A9 can serve as a critical link between the amyloid cascade and neuroinflammatory events in AD10. Specifically, being highly amyloidogenic itself S100A9 can trigger and aggravate Aβ amyloid self-assembly and significantly contribute to amyloid cytotoxicity10,11. Both Aβ pathology12,13 and neuroinflammation14,15 are the key culprits in TBI secondary events, indicating that once these processes are initiated in TBI they can be further exacerbated in AD. Here we explore how this progression may occur with the focus on pro-inflammatory S100A9 and its role in the amyloid-neuroinflammatory cascade.
S100A9 is a multifunctional calcium-binding protein with diverse roles in the inflammatory signaling pathways. S100A9 belongs to the S100 protein family, which participates in a wide range of biological processes such as proliferation, migration and/or invasion, inflammation and differentiation16–22. S100 proteins, including S100A9, lack a signal peptide for secretion via the conventional Golgi-mediated pathway, and as whether extracellular S100 proteins are actively secreted from living cells via alternative secretion pathways or passively released is still debated17,18,21.
The increasing evidence indicates that S100A9, as well as other members of the S100 family, are pro-inflammatory molecules22–25. S100A9 was classified as damage associated molecular pattern (DAMP) molecule or alarmin broadly involved in infection, cellular stress, tissue damage and cancers26–28. Concerning intracellular functions of S100A9, there is evidence that S100A9 together with S100A8 interact in a calcium-dependent manner with cytoskeletal components29. Extracellular S100A9 is able to mediate cellular responses via receptors for advanced glycation endproducts (RAGE) or Toll-like 4 (TLR4) receptors, inducing expression of pro-inflammatory cytokines26. It has been shown that S100A9 and its fibrils regulate the NLRP3 inflammasome by acting as priming agents30,31.
A widespread expression of S100A9 was reported in many ailments associated with inflammatory processes, such as AD10,32, Parkinson’s disease33, malaria34, cerebral ischemia35, TBI36, obesity37 and cardiovascular disease38, implying that S100A9 may be a universal biomarker of inflammation. The abundance of S100A9 mRNA was also identified as a strong feature of aging in various mammalian tissues, including the central nervous system, and a novel mechanism of the age-associated inflammation sustained by S100A9 was suggested39. The distinctive feature of S100A9 compared to other pro-inflammatory mediators is its ability to self-assemble into amyloids following two-step nucleation-autocatalytic growth mechanism40, which may lead to the loss of its signaling functions and acquired amyloid cytotoxicity, exceeding that of Aβ10. Therefore, the rising S100A9 level during inflammation may lead to its amyloid formation and deposition as we have shown in AD10, aging prostate41 and also in cell model for protein amyloid aggregation42. Moreover, the CSF levels of S100A9 and Aβ match each other in AD, vascular dementia and mild cognitive impairment43, emphasizing the involvement of S100A9 together with Aβ in the amyloid-neuroinflammatory cascade in all these ailments. Interestingly, S100A9 knockdown attenuated memory impairment and reduced amyloid plaque burden in an AD mouse model44. However, intranasal administration of S100A9 oligomers and fibrils to wild-type mice induced wide-spread cellular stress responses and amyloid oligomerization in the brain tissues, as well as boosted hippocampal glutamate modifying monoaminergic neurochemistry, which all together led Alzheimer’s-like memory impairment in behavioural tests45–47. Similarly, in transgenic APP mouse model the amyloid aggregation of another protein from the S100 family – S100A8 has been shown to precede Aβ amyloid plaque formation and the positive feedback was found for both S100A8 and Aβ production48. Furthermore, S100A6 protein was also found to self-assemble into amyloid oligomers and fibrils, which can promote amyloid aggregation of superoxide dismutase-1 involved in amyotropic lateral sclerosis pathology49.
Here we have elucidated the role of S100A9 in the amyloid-neuroinflammatory cascades in TBI and compared with AD, viewing this as a common mechanism linking both ailments. We compared from this perspective the human brain tissues with TBI, AD, stable mild cognitive impairment (SMCI) and AD with TBI history. These studies were complemented by the primary neuronal cell culture experiments to demonstrate S100A9 induction at the cellular level under stress conditions. Moreover, at the molecular level the effects of acidification and elevated temperature on S100A9 amyloid formation and proteinase clearance were also examined in vitro. Consequently, those effects were linked with the resulting cytotoxicity of S100A9 amyloids. Thus, we have provided the comprehensive and systematic analysis of S100A9 role in TBI as a precursor state of AD at the molecular, cellular and tissue levels.
Results
Rapid formation of S100A9 and Aβ precursor-plaques in TBI
The TBI brain tissues of 13 patients from 1 y.o. to 65 y.o. and with survival time from 4 h to 30 days (Supplementary Table S1) were subjected to immunohistochemical analysis using Aβ and S100A9 specific antibodies to examine the involvement of these antigens in TBI pathology. The Aβ immunopositive extracellular deposits were found in low numbers in 70% patients (Fig. 1A,B). These deposits were not reactive with amyloid oligomer (A11) and fibril specific (OC) antibodies (Supplementary Fig. S1), indicating the lack of amyloid structure in them, and, therefore, they were coined as precursor-plaques. Intracellular Aβ depositions were found in some axons (Fig. 1C) and they were also not reactive with A11 and OC antibodies (Supplementary Fig. S1).
Figure 1.

S100A9 and Aβ in TBI tissues. (A) Bubble chart showing counts of the precursor-plaques composed of Aβ (red) and S100A9 (light green), respectively, in the whole human TBI hippocampi and surrounding tissues in individual patients versus increasing post-TBI time (n = 13 TBI patients). Each bubble corresponds to the total counts in each individual patient tissue; the counts of S100A9 precursor-plaques per cm2 of tissue are plotted along y-axis and the scale bubbles are presented in grey. Representative images of Aβ immunopositive (B) precursor-plaques and (C) neuronal axons. Representative images of S100A9 immunopositive (D) precursor-plaques, (E) cells in blood vessel and (F,H) zoomed individual precursor-plaques. (G,I) AFM topographic images of S100A9 precursor-plaques shown in (F,H), respectively. Red arrows in (F–I) show the same position in optical and AFM images, respectively. Scale bars are 20 µm in (B,F and H), 50 µm in (C,E), 200 µm in (D) and 10 µm in (G and I). AFM z-heights correspond to a color gradient from 0 µm (dark brown) to 1.7 µm (yellow light).
By contrast, the immunostaining with S100A9 specific antibodies revealed that S100A9 precursor-plaques were highly abundant in all early TBI cases, including even the individuals who had no Aβ deposits at all, and their amounts were also by ca. 100 times higher than those of Aβ (Fig. 1A,D). The numbers of S100A9 plaques decreased to the level of Aβ deposits with increasing post-TBI time to ca. 4 days (Fig. 1A) and, likewise Aβ deposits, they were not reactive with A11 and OC antibodies (Supplementary Fig. S1). The S100A9 accumulation in macrophages and neutrophils17,21,22 was also found by S100A9 immunostaining in numerous blood vessels (Fig. 1E), which again did not show reactivity with A11 and OC antibodies (Supplementary Fig. S1). Generally, S100A9 precursor-plaques were not recognized by Aβ antibodies and vice versa as shown by the sequential immunohistochemistry experiments with a reverse order of S100A9 and Aβ antibodies, respectively (Fig. 1B, Supplementary Fig. S1).
In order to gain a higher resolution insight into the structure of S100A9 precursor-plaques, atomic force microscopy (AFM) topography imaging was conducted, matching the corresponding immunohistochemical images (Fig. 1F–I). Based on AFM topography we have distinguished two types of S100A9 precursor-plaques. The first type was characterized by a cavernous center (Fig. 1F,G), suggesting that S100A9 can be dispersed around damaged blood vessels. The second type showed an elevated AFM topographical profile in the center (Fig. 1H,I), indicating that precursor-plaque can possess a condensed core and does not originate from a blood vessel, but, for example, from damaged cells.
Intracellular S100A9 and Aβ aggregation in TBI
For the first time we have found that S100A9 was massively present and even formed amyloid oligomers in neurons already after 4 h following TBI: the same neurons were sequentially immunostained with both S100A9 and A11 antibodies (Fig. 2A,B,D). S100A9 was not detected in microglial cells at this post-TBI time, as evident from the sequential immunohistochemistry staining, where the S100A9 and CD68 immunostaining patterns did not overlap (Fig. 2A,C,D). Activated microglial cells expressed S100A9 only after 4 to 30 days of post-TBI, but no amyloid oligomers were formed within them, by contrast to neuronal cells, as demonstrated by the superposition of the S100A9 and CD68 immunostaining and the absence of A11 immunopositive pattern (Fig. 2E–H).
Figure 2.

Analysis of intracellular S100A9/Aβ aggregation and apoptosis in TBI tissues. Representative images of sequential immunohistochemistry with (A) S100A9, (B) A11 and (C) CD68 antibodies (4 h post-TBI). (D) Superposition of the corresponding images in pseudo-colors, i.e. S100A9 staining is shown in red; CD68 – in blue and A11 – in yellow. Representative images of sequential immunohistochemistry of microglial cells with (E) S100A9, (F) A11 and (G) CD68 antibodies (5th post-TBI day). (H) Superposition of the corresponding images in pseudo-colors, i.e. S100A9 staining is shown in red and CD68 – in blue. (I,J) Bubble charts showing the counts of immunopositive cells in the whole TBI hippocampi and surrounding tissues of individual patients upon increasing post-TBI time, n = 13 TBI patients. Each bubble corresponds to the total counts in individual patient tissue; the counts of S100A9 immunopositive neurons per mm2 are plotted along y-axis and the scale bubbles are presented in grey in (I). The counts for microglial cells reactive with S100A9 antibodies are shown in red, for neurons reactive with antibodies to S100A9 – in green, to Aβ – in blue, amyloid oligomers – in cyan, Bax – in magenta and activated caspase-3 – in yellow. Representativie imagies of immunostaining of neurons with (K) Aβ, (L) Bax and (M) activated caspase-3 antibodies. (N) Graph network, showing connections between the S100A9, Aβ, A11, Bax and activated caspase-3 positive neurons. Nodes correspond to the types of immunopositive cells and their color coding is indicated in (I,J). Node sizes are proportional to cell amounts. Nodes are connected by edges, whose widths reflect the correlation strength; thick lines show strong correlation and thin lines – moderate correlation. Values of Spearman’s rho correlations between immunopositive cell and plaque counts are presented in Supplementary Table S3. Scale bars are 50 µm in (A–D and K–M) and 20 µm in (E–H).
S100A9 and A11 immunopositive neurons were counted in the hippocampi and surrounding tissues of all TBI patients (Fig. 2I,J). With increasing post-TBI time their numbers significantly reduced and between 4 to 30 post-TBI days remained at the same level. During this post-TBI period the counts of S100A9 immunopositive microglial and neuronal cells were also similar (Fig. 2I). By contrast, Aβ immunopositive neurons were not detected in 8 out of 13 TBI cases and their numbers were by ca. 8-fold lower compared to S100A9 immunopositive neurons (Fig. 2I). Sequential immunohistochemistry revealed that some Aβ immunopositive neurons were also co-stained with S100A9 and A11 antibodies (Fig. 2K, Supplementary Fig. S2), indicating that Aβ and S100A9 can be co-localized and even co-aggregate together in neuronal cells under TBI. In the brain tissues of non-TBI controls neither S100A9 nor Aβ immunopositive staining was found (Supplementary Fig. S2).
Neuronal apoptosis in TBI
Both excessive inflammatory cytokine production and amyloid oligomer formation can induce neuronal cell damage and death50,51. Here we examined the activation of apoptotic pathways by using immunohistochemistry with antibodies towards the apoptotic cascade markers – Bax and activated caspase-352,53. Sequential immunohistochemistry revealed that some S100A9 and A11 immunopositive neurons were also stained with Bax or caspase-3 antibodies (Fig. 2L,M, Supplementary Fig. S2). Bax and activated caspase-3 immunopositive neurons were also counted in the hippocampi and surrounding tissues of all TBI patients (Fig. 2J). To identify the association between the intra-neuronal amyloid, neuroinflammatory and apoptotic events, Spearman’s rho correlations were calculated and the graph network was built up, demonstrating strong and moderate associations between them (Fig. 2N, Supplementary Table S3). The strong correlation (Spearman’s rho correlation coefficient equals 0.73) was found between intra-neuronal S100A9 presence and amyloid oligomerization, manifested in co-immunostaining with A11 antibodies. The correlation between intra-neuronal Aβ presence and amyloid oligomerization was also strong (0.62), despite the significantly lower number of Aβ immunopositive neurons. The moderate and strong associations were observed between the A11 and Bax (0.53) and between the A11 and activated caspase-3 (0.60) immunopositive neurons, respectively, demonstrating that the amyloid oligomerization may indeed prompt the activation of neuronal apoptotic pathways.
Interestingly, some S100A9 immunopositive neurons were found in the proximity to S100A9 precursor-plaques and the correlation between these events was also strong – with 0.65 Spearman’s rho correlation coefficient (Supplementary Table S3). This suggests that some of the S100A9 immunopositive cells may indeed contribute to the S100A9 precursor-plaque formation.
Importantly, the amounts of both S100A9 and Aβ precursor-plaques as well as the amyloid, neuroinflammatory and apoptotic responses in neurons and microglia, as reflected in the corresponding immunostaining patterns on a patient to patient basis, did not correlate with patient’s age (Supplementary Fig. S3), indicating that they are directly linked to TBI, rather than being age-dependent.
The fact that S100A9 can be quickly induced in neurons was confirmed also in wild-type mouse primary neuronal cell culture upon the limited exposure of neurons to extracellular stress factor, specifically to Aβ42 oligomers (Fig. 3A). If before treatment the immunofluorescence of S100A9 antibodies in neuronal cells was close to noise level (Fig. 3B), it significantly increased within 24 h following addition of Aβ42 oligomers to cell culture (Fig. 3C). The relative quantification of S100A9 level in neurons was performed by an Imaris software in confocal images and demonstrated significant raise of S100A9-specific immunofluorescence intensity in neurons from the noise level to ca. 300 fold stronger signal per cell after the cellular stress induced by Aβ42 oligomers (Fig. 3D). This is congruous with trauma-associated S100A9 neuronal induction in the post-TBI human brain tissues.
Figure 3.

S100A9 expression in neuronal cell culture. (A) Fluorescence immunocytochemistry images by confocal microscopy of wild-type (WT) mouse primary neurons treated with Aβ42 oligomers. DAPI nuclei staining shown in blue and Aβ42 specific fluorescence – in green. Neurons before (B) and after Aβ42 oligomer treatment (C), respectively, with S100A9 content manifested in red fluorescence. (D) Quantification of S100A9 specific immunofluorescence signal per cell by using an Imaris software in untreated neurons (purple bar) and Aβ42 oligomer treated neurons (dark blue bar); the procedure is shown in Supplementary Fig. S7. Bars present means ± standard deviations in arbitrary units out of n = 7 and n = 6 measurements of untreated and treated cells, respectively. ***p < 0.0001 by Student’s t test. Scale bars are 30 µm (A), 15 µm (B) and 20 µm (C).
S100A9 specific amyloid plaques in SMCI and AD
In all 6 AD cases examined here we have found that most of senile plaques contained not only commonly expected Aβ, but also S100A9 as we have shown previously10. Indeed, S100A9 and Aβ immunostaining patterns were overlapped as shown over the large brain tissue area enriched with plaques and encircled by blue line (Fig. 4A,B) and in the magnified views of particular amyloid plaques (Supplementary Fig. S4). The Aβ-S100A9 plaques were also reactive with OC antibodies, demonstrating their amyloid nature (Supplementary Fig. S4). However, we have observed that within some plaques the S100A9 and Aβ immunostaining patterns were not overlapping as shown previously10, but located adjacently to each other (Fig. 4C–F), suggesting that both polypeptides may play an autonomous role in the plaque formation.
Figure 4.

S100A9 and Aβ plaques in human AD and SMCI tissues. n = 6 AD cases and n = 1 SMCI case. (A,B) Representative immunostaining with S100A9 and Aβ antibodies, respectively, of the grey and white matter areas in the frontal lobe of AD patient. The regions with S100A9 plaques at their interface and mature Aβ-S100A9 senile plaques within the grey matter are encircled by red and blue lines, respectively. Representative sequential immunohistochemistry of conjugated Aβ-S100A9 amyloid plaque with (C) S100A9, (D) Aβ, (E) A11 and (F) OC antibodies, respectively. Representative S100A9 plaques stained with sequence of (G) S100A9, (H) Aβ, (I) A11 and (J) OC antibodies, respectively. Representative sequential immunostaining of amyloid plaques in SMCI frontal lobe with (K) S100A9 and (L) Aβ antibodies, respectively. (M) Pie chart showing the amount of S100A9 (green) and Aβ (red) immunopositive plaques in SMCI (n = 1) and AD (n = 2) patients. Each circle corresponds to individual subject; outer circle shows the distribution of plaques in SMCI case and two inner circles – in two AD cases; numbers in the red circles indicate the percentage of Aβ plaques in the corresponding case. Scale bars are 500 µm in (A,B), 25 µm in (C–J) and 50 µm in (K,L).
Moreover, in 50% AD cases we have found specific type of amyloid plaques, which were reactive only with S100A9, but not Aβ antibodies (Fig. 4G,H, Supplementary Fig. S4). The S100A9 plaques were usually stained with both A11 and OC amyloid-specific antibodies (Fig. 4G,I,J) or sometimes only with A11 antibodies (Supplementary Fig. S4), thus displaying the amyloid character with different degree of amyloid maturation from oligomers to fibrils. Generally, the S100A9 plaques were randomly distributed among the Aβ-S100A9 senile plaques, but in 1 AD patient they were located with high density in distinct area at the border of the white and grey matters (Fig. 4A,B). In addition, in 1 SMCI patient, who has not yet develop AD and who had received multiple TBIs during his Ukrainian military service, we also observed distinct S100A9 and Aβ plaques, which were not cross-reactive with both S100A9 and Aβ antibodies, as well as plaques containing both antigens (Fig. 4K,L). In this SMCI patient the S100A9 immunopositive plaques not reactive with Aβ antibodies constituted 34% of all plaques (Fig. 4M), while in AD patients these numbers were 1 and 6%, the latter corresponded to AD patient with TBI history. This indicates that with progression of neurodegeneration the involvement of Aβ in plaque formation increases, but previous TBI history can be manifested in significant depositions of S100A9 alone already at the stage of SMCI. Importantly, the SMCI (43 y.o.) and AD (57 y.o) cases, both characterized by TBI history, displayed the advanced plaque pathology at relatively early age compared to an average age for other AD patients without known TBI history (82.6 y.o) (Supplementary Table S1).
To gain insight into the structures of only S100A9 (Fig. 5A–D) and joined Aβ-S100A9 (Fig. 5E–H) plaques, the combined immunofluorescence with S100A9 and Aβ antibodies, h-FTAA fluorescence and AFM topographic imaging were performed. The fluorescence images of both types of plaques showed that they were reactive with amyloid specific h-FTAA dye54, indicating their amyloid nature, and characterized by a patchy distribution of amyloid materials as evident from the corresponding patchy staining patterns (Fig. 5A,B,E,F). Interestingly, in both types of plaques the amyloid material can be deposited either at their circumference, forming donut-like structures (Fig. 5A,C,E,G) or spread from their centers (Fig. 5B,D,F,H), as shown by both fluorescence (individual and superimposed colored patterns) and AFM techniques (deposited material is shown in yellow light color). These structural similarities suggest the common mechanisms of formation for both types of plaques composed solely of S100A9 and both S100A9-Aβ.
Figure 5.

S100A9 and Aβ-S100A9 senile plaque structure in AD. n = 3 AD patient brain tissues were analyzed. (A,B) Immuno- and h-FTAA fluorescence imaging of two representative S100A9 amyloid plaques. Single channel images are shown at the top and merged images – at the bottom. S100A9 antibody staining is shown in blue, h-FTAA – in green and Aβ – in red. (C,D) AFM topography images of two representative S100A9 amyloid plaques. (E,F) Immuno- and h-FTAA fluorescence imaging of two representative Aβ-S100A9 amyloid plaques. (G,H) AFM topography images of two representative Aβ-S100A9 amyloid plaques. Scale bars are 20 µm in (A,B,E–H) and 10 µm in (C,D). AFM z-heights correspond to a color gradient from 0 µm, shown in dark brown, to (C) 1.8 µm, (D,H) 3.3 µm and (G) 3.8 µm shown in yellow light, respectively.
Modelling S100A9 amyloid formation, seeding and disaggregation in vitro
The amyloid accumulation in the brain tissues depends on both amyloid growth and clearance, including proteolytic cleavage. Alzheimer’s amyloid plaques are very protease resistant aggregates55. In vitro studies demonstrated that the insulin amyloid protease resistance is modulated by environmental conditions and amyloid aging56. Here we have examined the effect of acidification and increasing temperature, which are both relevant to traumatic brain injury conditions57,58, on S100A9 amyloid formation and proteinase disaggregation.
The S100A9 amyloid formation and proteinase K disaggregation kinetics in vitro were monitored by h-FTAA amyloid fluorescence assay in conjunction with AFM (Fig. 6, Supplementary Fig. S5). All S100A9 amyloid kinetics at different protein concentrations (2 and 5 mg/ml), pH (7.4 and 4.5) and temperatures (37 and 42 °C) were well fitted by the nucleation-dependent polymerization model59 (Fig. 6A, Supplementary Fig. S5). Following the nucleation, the amyloid assembly resulted in massive amyloid fibril formation, which were observed at time corresponding to the kinetic plateau levels, and were observed by AFM (Fig. 6C,E). The amyloid assembly at pH 4.5 and 42 °C was characterized by the highest rate constants compared to the rate constant observed at pH 7.4 and 37 °C, as well as compared to pH 7.4, 42 °C and pH 4.5, 37 °C conditions, signifying the role of acidification and raised temperature in the S100A9 amyloid formation (Fig. 6B).
Figure 6.
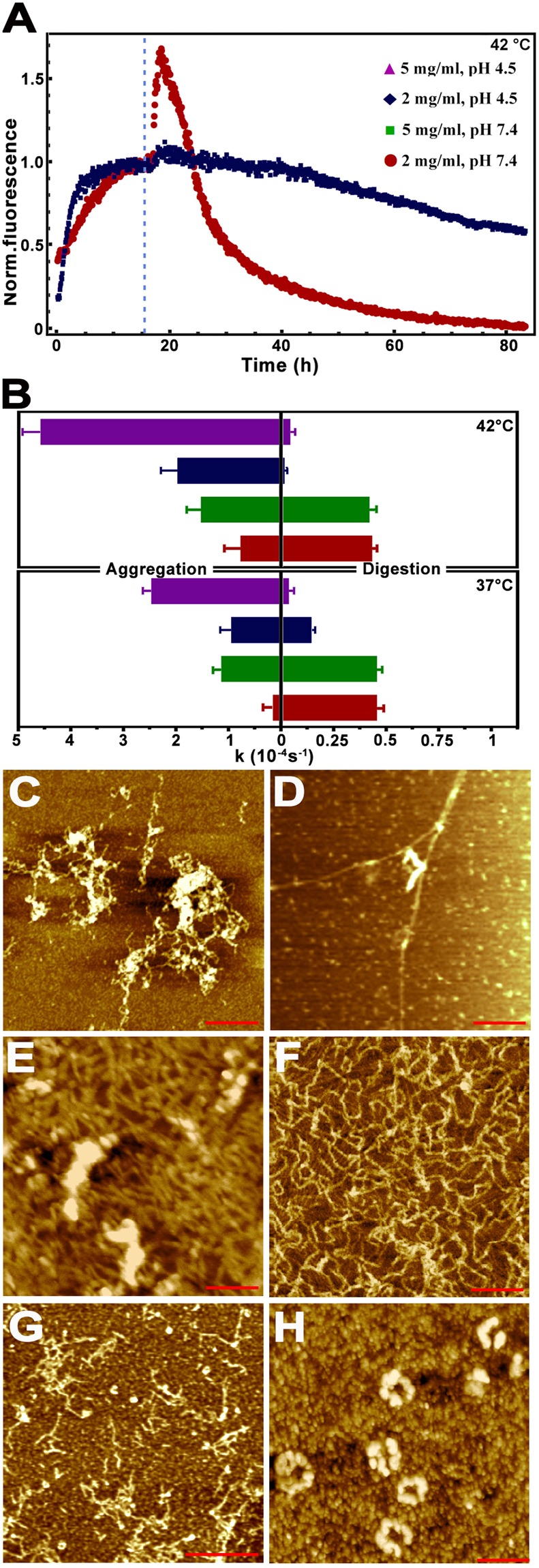
In vitro S100A9 amyloid formation and proteinase K digestion. (A) Kinetics of S100A9 amyloid formation and proteinase K digestion monitored by h-FTAA fluorescence at 42 °C. Time of protease K addition is indicated by dashed line. (B) Rate constants of S100A9 amyloid formation (left) and proteinase K digestion (right), shown by bars and corresponding to 42 °C (top) and 37 °C (bottom), represent mean ± standard deviation (n = 10 repeats for each kinetic measurement). Kinetic data observed at 5 mg/ml, pH 4.5 is shown in purple, at 2 mg/ml, pH 4.5 – in dark blue, 5 mg/ml, pH 7.4 – in green and 2 mg/ml, pH 7.4 – in red, as indicated in figure legend (A). Representative AFM height images of (C) S100A9 amyloid clumps formed in 10 mM PBS, pH 7.4, 37 °C, during 12 h under shaking with glass beads; (D) the same sample after 6 h proteinase K digestion. Representative AFM height images of (E) S100A9 amyloid clumps and fibrils formed in 20 mM sodium acetate, pH 4.5, 42 °C, during 8 h under shaking with glass beads; (F) the same sample subjected to 3 h, (G) 6 h and (H) 12 h proteinase K digestion. Scale bars are 250 nm in (C–F) and 600 nm in (G,H), respectively.
After 16 h incubation the S100A9 amyloid samples described above were subjected to proteinase K cleavage (Fig. 6A). This followed by an initial rise of h-FTAA fluorescence above the kinetic plateau level, which may be due to increased number of nuclei produced by proteinase K cleavage56. This spike was very pronounced but short lived at pH 7.4, 37 °C, when S100A9 amyloids became rapidly digested as evident both from the drop of h-FTAA fluorescence and AFM observations of their progressing disappearance after 6 and 72 h, respectively (Fig. 6D, Supplementary Fig. S5). By contrast, the proteinase K digestion of S100A9 amyloids at pH 4.5, 42 °C was much slower as evident from the lower rates of amyloid digestion kinetics (Fig. 6B), higher level of remaining h-FTAA fluorescence (Fig. 6A) and the presence of amyloids after proteinase K treatment as observed in AFM images (Fig. 6F–H, Supplementary Fig. S5). Massive S100A9 amyloid fibrils became thinner and shorter during 3 to 6 h proteinase K digestion (Fig. 6F,G), however a large quantity of ring-shaped amyloid assemblies and oligomers were formed and still present after 12 and even 72 h (Fig. 6H, Supplementary Fig. S5). This suggests that the perturbation of tissue homeostasis in the post-TBI brain manifested in its acidification and elevated temperature55,57 as well as in the significant increase of S100A9 concentration may be of a major importance in both promoting undesirable S100A9 amyloid self-assemblies, slowing their clearance and even inducing their structural rearrangement into more toxic amyloid species (Fig. 6H), which all together can be more harmful.
Since S100A9 but not Aβ was abundantly present in the precursor plaques and intracellularly in TBI tissues, the effect of S100A9 pre-formed fibrils on Aβ42 aggregation was examined in vitro (Supplementary Fig. S6). We have observed significant seeding effect on Aβ42 amyloid formation induced in a concentration-dependent manner by pre-formed amyloid fibrils of S100A9. These reflected in shortening of lag-phase of the Aβ42 amyloid kinetic curve from >2 h in the absence of S100A9 fibrils to ca. 1.4 h in the presence of 30 μM of S100A9 fibrillar sample, while the plateau was reached also faster, i.e. after 4.5 h in the absence and 2.4 h in the presence of 30 μM S100A9 fibrillar sample, respectively.
S100A9 amyloid cytotoxicity
The cytotoxic properties of S100A9 amyloid aggregates produced at pH 4.5, 42 °C were examined by adding them to SH-SY5Y neuroblastoma cells (Fig. 7). The amyloid species were collected after 0.3, 0.6, 1 and 8 h incubation periods to sample the amyloid assemblies progressing from oligomers to fibrils in accord with the kinetics of S100A9 amyloid formation presented in Fig. 6A and Supplementary S5, i.e. the oligomers were populated in the beginning of the amyloid formation process and fibrils were developed when the plateau level of the kinetic curve was reached. The morphology of S100A9 amyloid structures produced during the course of incubation at the above conditions were also examined by AFM prior subjecting them to cytotoxicity experiments and their images are presented in Fig. 7A–C. Freshly dissolved native S100A9 (zero time point) was not cytotoxic within the error of measurement (Fig. 7E). Short protofilaments and oligomers populated after 0.3 h amyloid incubation (Fig. 7A) were the most toxic, reducing the cell viability by 40%, as the amyloid oligomers are viewed to be the most cytotoxic species60. S100A9 amyloid cytotoxicity subsided, however, when filamentous structures were developed upon further incubation (Fig. 7B), and mature fibrils formed after 8 h incubation were not toxic, when added to cells.
Figure 7.
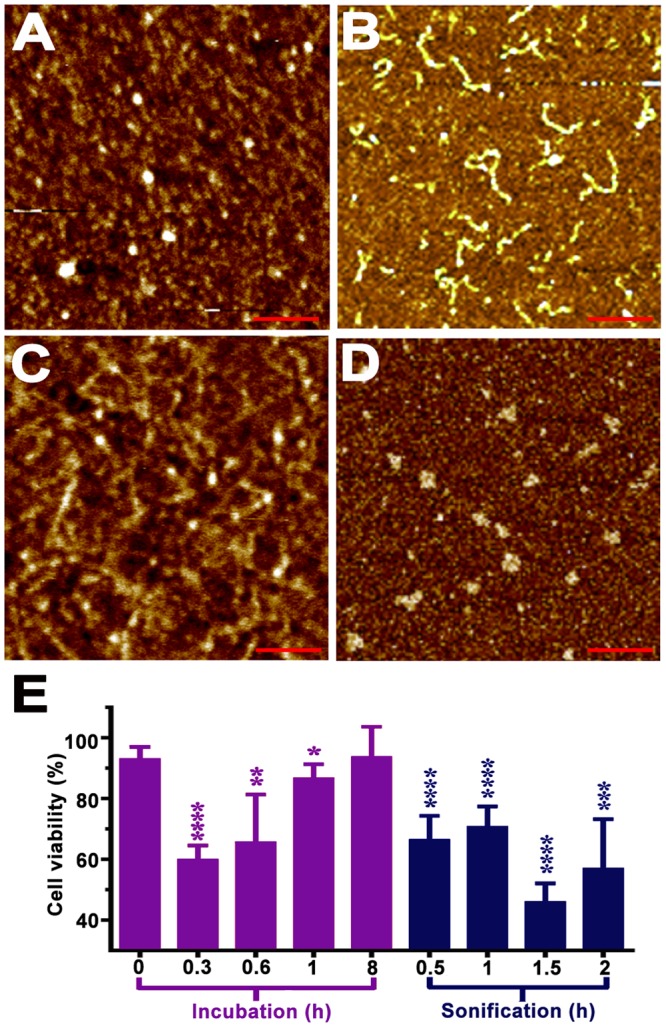
S100A9 amyloid cytotoxicity. Representative AFM height images of (A) S100A9 amyloid oligomers formed after 0.3 h and (B) protofilaments formed after 1 h incubation in 20 mM sodium acetate, pH 4.5, 42 °C under shaking with glass beads; (C) S100A9 amyloids, incubated for 8 h at the above conditions, were subjected to 0.5 h and (D) 1.5 h sonication. (E). Viability of SH-SY5Y cells measured by WST-1 assay after 24 h co-incubation with 20 μM S100A9 amyloids shown in (A–D). The cell viability depending on the added amyloid pre-incubation time is shown in magenta and the cell viability upon pre-formed amyloid sonication time – in blue. The viability of cells with added buffer is taken as 100%. Cell viability is shown by mean ± standard deviation, n = 7 repeats of each measurement. *p = 0.023, **p = 0.0023, ***p = 0.0004, ****p < 0.0001, calculated by Student’s t test.
To model the S100A9 amyloid fragmentation process occurring during proteinase K digestion, its amyloids were subjected to sonication (Fig. 7C,D). The SH-SY5Y cell viability in the presence of sonicated S100A9 protofilaments and oligomers (Fig. 7C), including ring-shaped assemblies (Fig. 7D), which are viewed to be the most toxic10, reduced by ca. 30 and 50%, respectively. This suggests that amyloid proteinase digestion indeed can potentially produce secondary highly toxic S100A9 amyloid species.
Discussion
The main goal of this research is to demonstrate that S100A9, in addition to commonly perceived Aβ peptide, may be a leading causative component of the amyloid-neuroinflammatory cascade, triggered in TBI and presenting the risk for AD and other neurodegeneration disease development. The amyloid cascade is central for understanding the molecular and cellular pathology of AD6, however there is a lack of data to demonstrate how this cascade is initiated. If to examine the advanced AD brain tissues, it is not possible to conclude beyond doubt as whether Aβ, but not some other factors, were at the origin of disease. This is why we have examined TBI, which is considered as a precursor state or risk factor for AD12,51,61. Previously TBI was linked to AD via the amyloid cascade and aggregation of Aβ peptide. However in the present research we have not found massive Aβ presence either in the precursor plaques or within the neuronal cells after TBI. Conversely, we have found an abundance of S100A9 both extracellularly in the precursor plaques and within neurons and microglial cells. Acute and chronic inflammation associated with TBI may lead to rising S100A9 level, especially as the latter can be sustained over years following TBI1,3,62. Moreover we have shown for the first time that in AD there are areas which contain only S100A9 plaques compared to those, which encompass both Aβ and S100A9. Therefore we demonstrated that S100A9 can be a common denominator in inflammation-associated conditions in TBI and AD as a driving component of the amyloid-neuroinflammatory cascade.
Previously it was suggested that the AD amyloid pathology can be initiated within hours after TBI, that manifested in appearance of Aβ plaques in ca. 30% acute cases12,61. We have demonstrated that S100A9 is dominant compared to Aβ both intra and extracellularly in post-TBI tissues, indicating that S100A9, but not Aβ, may play a leading role in amyloid aggregation. The dominance of S100A9 precursor-plaques was found in all TBI cases versus Aβ depositions in ca. 70% individuals (Fig. 1A). Moreover, on a patient to patient basis the amounts of S100A9 precursor-plaques were also overwhelmingly higher compared to those of Aβ, i.e. by 100-fold. We have not found any correlation between the accumulation of S100A9 and Aβ precursor-plaques and age, highlighting that this phenomenon is the consequence of TBI, but not ageing (Supplementary Fig. S3). In all individuals both S100A9 and Aβ precursor-plaques were relatively new lesions not reactive with amyloid-specific antibodies and dye h-FTAA, as they were developed on a time scale much shorter than required for AD senile plaque formation. This is consistent with previously reported lack of Congo red birefringence of TBI Aβ plaques, questioning their amyloid nature63, otherwise the amyloid character of Aβ deposits was not examined at all13. The staining of Aβ plaques with amyloid specific thioflavin S dye was observed only in post-TBI with long history (1 to 47 years)12. We have shown that the number of S100A9 precursor-plaques reduced to the level of Aβ deposits with increasing post-TBI time. Since pro-inflammatory S100A9 is highly amyloidogenic and its fibrils are able to seed the amyloid formation of Aβ42 as shown in vitro (Supplementary Fig. S6), if chronic inflammation would persist, sustaining its elevated level, its precursor-plaques may remain and seed further amyloid growth, representing a serious risk for AD development.
For the first time we have found specifically S100A9 amyloid plaques, which did not contain Aβ, in both SMCI with TBI history and half of AD cases, including the AD case with TBI history (Fig. 4). Remarkably, in SMCI the S100A9 plaques constituted as many as third of all deposits. Importantly, in SMCI and AD with TBI history the advanced plaque pathology was also developed at earlier age compared to an average age of the AD patients without TBI history. In the light of the amyloid hypothesis, these facts once again emphasise that high production and aggregation of S100A9 triggered by primary TBI insult, either in combination with Aβ or on its own, may provide a template for further senile plaque development leading to AD.
In AD we have shown the similarity of the plaques containing only S100A9 and mixed Aβ-S100A9 senile plaques with regards to their (a) amyloidogenic properties, such as staining with amyloid-specific h-FTAA dye, A11 and OC antibodies, and (b) morphological features shown by AFM (Fig. 5). Both types of AD plaques as well as precursor-plaques in TBI were characterized by patchy or diffused depositions of proteinaceous material as revealed by AFM, fluorescence microscopy and immunohistochemistry (Figs 1 and 5). They all displayed also similar topography, where the condensation of proteinaceous material occurred either at the circumference or spread from the center, indicating that all plaques (S100A9 and Aβ-S100A9) may originate via similar deposition mechanisms.
We have shown that the high abundance of S100A9 in TBI tissues was manifested in its intraneuronal and microglia presence (Fig. 2). The level of S100A9 immunopositive cells in TBI tissues was much higher than those of Aβ, i.e. (a) Aβ immunopositive neurons were found in only ca. 40% TBI cases and (b) their amounts were by 10-fold lower. These again highlight that the sustained high level of amyloidogenic protein in TBI is likely linked to S100A9, but not to Aβ.
By using sequential immunohistochemistry and graph analysis, we have shown that S100A9 intraneuronal oligomerization is an essential factor promoting the apoptotic pathway via Bax and caspase-3 activation (Fig. 2), which may play a major role in TBI secondary neuronal loss64. Since the number of neurons with Aβ peptide is much lower than those containing S100A9 (Fig. 2I), the potential mechanism of secondary neuronal loss in TBI is also more likely to be associated with S100A9 oligomerization and not Aβ.
The perturbations of brain tissue homeostasis manifested in acidification and fever were implicated in post-TBI, especially in the cases with unfavorable neurological outcome57,58. Here we demonstrated that S100A9 readily forms amyloids in vitro under variety of TBI relevant conditions, including increased protein concentration, acidic pH and elevated temperature, and these factors provide a strong positive feedback to amyloid self-assembly (Fig. 6). The S100A9 oligomers can be highly toxic if they are added to neuroblastoma cells (Fig. 7). Therefore S100A9 oligomerization, occurring both extra and intracellularly very rapidly after TBI, can be a target for potential therapeutic interventions aiming to prevent their harmful effects. Prospective anti-inflammatory medications can be easily implemented in post-TBI to achieve this goal9.
At the same time, acidification and raising temperature slowed proteinase K digestion of S100A9 amyloids, resulting in the prolonged life-span of short and cytotoxic amyloid species (Fig. 6), which can again cause the secondary neuronal loss. Altogether the in vitro experiments demonstrated how the environmental factors implicated in TBI paired with high expression of S100A9 in TBI tissues can produce unfavorable outcomes such as S100A9 amyloidogenicity and neuronal cytotoxicity.
The summary of our findings is schematically outlined in Fig. 8. This highlights both intra and extracellular pathways of S100A9 and Aβ amyloid self-assembly, which are intertwined together leading to cell death and neurodegeneration. For the first time we have presented here a compelling evidence for the critical role played by pro-inflammatory S100A9 protein in the amyloid-neuroinflammatory cascade in TBI, which resembles similar processes in AD and may serve as a mechanistic link between TBI and AD. While the topic of inflammation in neurodegeneration is well discussed, here we present specific culprit – S100A9, and specific mechanism – S100A9 amyloid formation, which can drive TBI-induced inflammation to the amyloid cascade in Alzheimer’s disease. Therefore, S100A9 can be viewed as a prospective therapeutic target during various post-TBI stages and far prior to AD development to halt and reverse these damaging processes.
Figure 8.

Schematic presentation of S100A9-driven amyloid-neuroinflammatory cascade in TBI. Most of S100A9 precursor-plaques (shown schematically in green) and Aβ precursor-plaques (shown in red) formed during TBI will undergo clearance (white), leading to recovery from TBI. However, some S100A9 and Aβ precursor-plaques will develop into AD senile plaques (green-red). Intracellular S100A9 oligomers (green), if they are not cleared by proteases (magenta) and other cellular clearance mechanisms, can trigger the apoptotic cascade involving Bax and activated caspase-3, leading to neurodegeneration and AD.
Methods
Regulatory Compliance
The Medical Ethics Committees of Umeå University Hospital, Sweden; Human Brain Tissue Repository for Neurobiological Studies, National Institute of Mental Health and Neurosciences, Bangalore, India; Institute of Neurology, Medical University of Vienna, Austria and Medical Institute, Sumy State University, Ukraine approved all procedures detailed in this study, that required the use of human brain tissues prior to initiating any experimental objectives. Additionally, all methods were performed in full compliance with the Medical Ethics Committees of Umeå University Hospital, Sweden; Human Brain Tissue Repository for Neurobiological Studies, National Institute of Mental Health and Neurosciences, Bangalore, India; Institute of Neurology, Medical University of Vienna, Austria and Medical Institute, Sumy State University, Ukraine. The surrogates of all patients gave informed consent for the use of patient’s post-mortem tissue samples for research studies.
Human tissues
Brain tissues from 13 TBI, 6 AD and 1 SMCI as well as 3 non-demented controls were examined. TBI patients were from 1 to 65 y.o. and without any AD history. Older patients were excluded from this study due to age-dependent neurodegenerative changes. 1 AD and 1 SMCI patients had TBI history. All tissues were paraffin embedded and microtome-sectioned to 4 µm thick slices. The characteristics of patients, including their age, sex, diagnosis and post-TBI time, if relevant, are presented in Supplementary Table S1.
Materials
S100A9 was expressed in E coli, purified as described previously and its concentrations were determined by using ε280 = 0.53 (mg/ml)−1 cm−1 65. Aβ42 was purchased from Tocris Bioscience.
Conditions of amyloid formation and proteinase K digestion
In order to produce amyloid structures S100A9 was incubated at 2 and 5 mg/ml concentrations in either 20 mM Na acetate, at pH 4.5 or 10 mM phosphate buffered saline (PBS), containing 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4, at pH 7.4. All samples were incubated both at 37 or 42 °C. Proteinase K was added to all these amyloid samples after 16 h incubation at 1:400 molar ratio to S100A9 and this reaction was stopped after 70 h. Shaking at ca. 200 rpm with glass beads in a Tecan plate reader was used for all these samples. S100A9 amyloid samples produced at 5 mg/ml, in 20 mM Na acetate, pH 4.5, 42 °C during 12 h were also sonicated from 0.5 to 2 h in a Transsonic T310 and subjected to the cytotoxicity measurements. Aβ42 added to neuronal cell culture was dissolved in 10 mM NaOH and centrifuged at 15000 rpm for 30 min at 4 °C to remove aggregates. Supernatant was collected and 10 mM PBS, pH 7.4 was added to produce required final concentration determined by both Bradford assay and absorbance at 220 nm66. Aβ42 oligomers were produced at 100 µM concentration after 30 min incubation at room temperature.
Immunohistochemistry
Single and sequential immunohistochemistry with the same tissue sections was performed as described previously10, by using reagents and antibodies summarized in Supplementary Table S2. The corresponding immunopositive plaques and cells were manually counted in the entire hippocampus region and surrounding areas.
AFM
Brain tissues and in vitro produced amyloid samples were imaged by a Bruker Bioscope Catalyst microscope operated in the peak force mode at 1 kHz frequency with 6 N/m stiff cantilevers. TBI and AD tissue samples were deparaffinized, dehydrated and immunostained with corresponding antibodies. The tissue sections were kept on the glass slides during all above procedures and AFM scanning. In vitro produced amyloid samples were deposited on the surface of freshly cleaved mica (Ted Pella) for 15 min, washed 3 × 100 μl by deionized water and dried at room temperature.
Immuno- and h-FTAA fluorescence of human brain tissues
Brain tissue sections from the TBI and AD subjects were deparaffinized, dehydrated, immunostained with antibodies towards S100A9 and Aβ and then subjected to 30 min incubation with 0.3 µM h-FTAA fluorescent dye54, which specifically binds to amyloids. Then the tissue sections were washed with PBS and mounted on glass slides. Fluorescence images were recorded by a DM6000 B fluorescence microscope (Leica Microsystems) equipped with a SpectraCube module (Applied Spectral Imaging) and bandpass filters 436/10 (LP475) – blue, 535/50 (LP590) – green and 640/30 (BP 700/75) – red. The reagents and antibodies are outlined in Supplementary Table S2.
h-FTAA fluorescence in vitro
h-FTAA was used to monitor in vitro the kinetics of amyloid formation and its digestion by proteinase K. Fluorescence of 0.3 µM h-FTAA was recorded by a Tecan P-200 fluorescence plate reader using excitation at 485 nm and emission at 590 nm.
Kinetics data fit of S100A9 amyloid aggregation and proteinase K digestion
S100A9 amyloid kinetics were described by nucleated polymerization model with some modification, accounting for the presence of pre-formed seeded aggregates59. Each data set was repeated 10 times and averaged. The kinetics were baseline-corrected and normalized, such that the relative mass concentration of amyloid aggregates was m(0) at time 0 and 1 at completion of the aggregation. The mass concentration of amyloid polymers in solution M(t) was described by eq. (1) from which the effective rate constant λ and nuclei size nc were derived by using the best fit.
| 1 |
where mtot is total mass concentration of non-aggregated protein; m(0) – initial mass concentration of amyloid seeds; λ – effective rate constant, which was defined as , k+ – elongation rate and kn – nucleation rate; β = 2/nc, , and where P(0) – initial number of pre-aggregated seeds.
Amyloid proteinase K digestion processes were described by single exponential decays
| 2 |
where m(t) is mass concentration of digested amyloids, A – arbitrary amplitude and k – digestion rate.
Cytotoxicity assay
Viability of SH-SY5Y neuroblastoma cells was measured by WST-1 assay after 24 h co-incubation with amyloids as described previously10. SH-SY5Y cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum and antibiotics in a 5% CO2 humidified atmosphere at 37 °C. Cells were plated at a density of 104 cells/well in 96-well plates; after 24 h of incubation, the medium was changed before incubation with amyloid samples. Initially S100A9 amyloid samples were incubated at 400 μM in 20 μM sodium acetate buffer, pH 4.5, 42 °C with shaking during different time periods (0, 0.3, 0.6, 1, and 8 h) to produce oligomers and fibrils, respectively. Then S100A9 samples were diluted in the culture medium and added to SH-SY5Y cells at a final concentration of 20 μM. The cell samples were co-incubated with S100A9 specimens for 24 h prior the cell viability was assessed by WST-1 assay. 10 μl of WST-1 reagent (Roche, Germany) was added to 100 μl of cell culture and the cell samples were incubated further at 37 °C for 4 h. Absorbance was measured at 450 nm by a Tecan P-200 fluorescence plate reader. Cell viability was expressed as a percentage of the absorbance in wells containing cells treated with amyloids compared to the control untreated cells.
Graph analysis
In all TBI patient’s tissues the amounts of immunopositive plaques and cells reactive with each of studied antibodies were counted manually in the hippocampi and surrounding area. In order to reveal pair-wise relations between corresponding counts of immuno-positive cells and/or plaques, we subjected all paired data subsets to the graph theory analysis and calculations of Spearman’s rho correlations67. The nodes in graphs represent the type of immunopositive cells or plaques, which are connected by edges. The edges were drawn based on the moderate and strong Spearman’s rho correlations indicated along them (Supplementary Table S3).
Fluorescence immunocytochemistry
Wild-type mouse primary neurons non-treated and treated with 1 µM Aβ42 oligomers for 24 h were imaged by a Leica Microsystems TCS SP8 confocal microscope equipped with a HP PL APO 63×/NA1.2 objective and using Diode 405/405 nm and Argon (405, 488, 552, 638 nm) lasers. Cell nulcei were stained with 4′,6-Diamidino-2-Phenylindole (DAPI), shown by blue fluorescence; Aβ42 oligomers were detected by using Aβ specific primary antibody and DyLight 488 secondary antibodies, shown by green fluorescence, while S100A9 was recognized by S100A9 specific primary antibodies and visialised by DyLight 594 secondary antibodies (Supplementary Table S2).
S100A9 specific immunofluorescence signal per cell in the Aβ42 treated and non-treated neurons were quantified by using an Imaris (Bitplane) software. For this purpose two-dimensional images obtained by confocal microscopy were reconstructed by Imaris into three-dimensional volumetric data sets with an inclusive volume for each neuronal cell set at 16 × 16 × 3 μm3 as shown in Supplementary Fig. S7. Using the same threshold level for treated and control neurons, solid iso-surfaces were defined for red channel (S100A9) and rendered as volumes, which were used for S100A9-specific immunofluorescence quantification presented in arbitrary units per cell. Results are presented as the mean ± standard deviation.
Statistical analysis
The normality of the data sets was assessed by the Shapiro-Wilk test and the data were analyzed using Student’s t test. p value less than 0.05 was considered significant. Results are presented as the mean ± standard deviation.
Electronic supplementary material
Acknowledgements
We thank Peter Nilsson for the gift of amyloid specific h-FTAA fluorescence dye, Fredrik Clausen for experimental TBI work, Per Hammarström for helpful discussion and analysis of immunofluorescence results, Vinod K Narayana for connecting us with S.K.S. and delivering pilot samples. We thank for financial support ALF Västerbotten Läns Landsting (L.A.M.-R.), Swedish Medical Research Council (L.A.M.-R.), FP-7 Marie Curie Action “Nano-Guard” (I.A.I., R.M., L.A.M.-R.), Insamlingsstiftelsen (L.A.M.-R.), Swedish Alzheimer’s Foundation (S.N.) and Swedish Institute (R.M.).
Author Contributions
C.W., I.A.I. and L.A.M.-R. designed the project, analyzed the data and wrote the manuscript. C.W., I.A.I., J.P., S.N., O.K., G.G., J.K., I.H., R.M. and R.R. performed research and analysis ex vivo and in vitro. G.G.K. and S.K.S. collected and analyzed human brain tissues.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Chao Wang and Igor A. Iashchishyn contributed equally.
Contributor Information
Chao Wang, Email: chao.wang@wustl.edu.
Ludmilla A. Morozova-Roche, Email: Ludmilla.Morozova-Roche@umu.se
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-31141-x.
References
- 1.Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J. Neurotrauma. 2010;27:1529–1540. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- 2.Di Virgilio TG, et al. Evidence for acute electrophysiological and cognitive changes following routine soccer heading. EBioMedicine. 2016;13:66–71. doi: 10.1016/j.ebiom.2016.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breunig JJ, Guillot-Sestier MV, Town T. Brain injury, neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 2013;5:26. doi: 10.3389/fnagi.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sivanandam TM, Thakur MK. Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 2012;36:1376–1381. doi: 10.1016/j.neubiorev.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Washington PM, Villapol S, Burns MP. Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp. Neurol. 2016;275(Pt 3):381–388. doi: 10.1016/j.expneurol.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selkoe DJ. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann. N. Y. Acad. Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- 7.McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J. Alzheimers Dis. 2010;19:355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 8.Cole GM, Frautschy SA. Mechanisms of action of non-steroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets. 2010;9:140–148. doi: 10.2174/187152710791011991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.in t’ Veld BA, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014;127:507–522. doi: 10.1007/s00401-013-1208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao LN, et al. S100A9 induces aggregation-prone conformation in Abeta peptides: a combined experimental and simulation study. RSC Adv. 2013;3:24081–24089. doi: 10.1039/c3ra43665a. [DOI] [Google Scholar]
- 12.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikonomovic MD, et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76:886–899. doi: 10.1016/j.neuron.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 15.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav. Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Fritz G, Botelho HM, Morozova-Roche LA, Gomes CM. Natural and amyloid self-assembly of S100 proteins: structural basis of functional diversity. FEBS J. 2010;277:4578–4590. doi: 10.1111/j.1742-4658.2010.07887.x. [DOI] [PubMed] [Google Scholar]
- 17.Vogl T, Gharibyan AL, Morozova-Roche LA. Pro-inflammatory S100A8 and S100A9 proteins: self-assembly into multifunctional native and amyloid complexes. Int. J. Mol. Sci. 2012;13:2893–2917. doi: 10.3390/ijms13032893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat. Rev. Cancer. 2015;15:96–109. doi: 10.1038/nrc3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leclerc E, Heizmann CW. The importance of Ca2+/Zn2+ signaling S100 proteins and RAGE in translational medicine. Front. Biosci. (Schol Ed). 2011;3:1232–1262. doi: 10.2741/223. [DOI] [PubMed] [Google Scholar]
- 20.Donato, R. et al. Functions of S100 proteins. Curr. Mol. Med. 13, 24–57, CMM-EPUB-20120723-11 (2013). [PMC free article] [PubMed]
- 21.Frosch, M. et al. Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum. 43, 628–637, doi:10.1002/1529-0131(200003)43:3<628::AID-ANR20>3.0.CO;2-X (2000). [DOI] [PubMed]
- 22.Hessian PA, Edgeworth J, Hogg N. MRP-8 and MRP-14, two abundant Ca(2+)-binding proteins of neutrophils and monocytes. J. Leukoc. Biol. 1993;53:197–204. doi: 10.1002/jlb.53.2.197. [DOI] [PubMed] [Google Scholar]
- 23.Mork G, Schjerven H, Mangschau L, Soyland E, Brandtzaeg P. Proinflammatory cytokines upregulate expression of calprotectin (L1 protein, MRP-8/MRP-14) in cultured human keratinocytes. Br. J. Dermatol. 2003;149:484–491. doi: 10.1046/j.1365-2133.2003.05536.x. [DOI] [PubMed] [Google Scholar]
- 24.Nacken W, Roth J, Sorg C, Kerkhoff C. S100A9/S100A8: Myeloid representatives of the S100 protein family as prominent players in innate immunity. Microsc. Res. Tech. 2003;60:569–580. doi: 10.1002/jemt.10299. [DOI] [PubMed] [Google Scholar]
- 25.Ryckman C, et al. Monosodium urate monohydrate crystals induce the release of the proinflammatory protein S100A8/A9 from neutrophils. J. Leukoc. Biol. 2004;76:433–440. doi: 10.1189/jlb.0603294. [DOI] [PubMed] [Google Scholar]
- 26.Chen B, et al. S100A9 induced inflammatory responses are mediated by distinct damage associated molecular patterns (DAMP) receptors in vitro and in vivo. PLoS One. 2015;10:e0115828. doi: 10.1371/journal.pone.0115828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van den Bosch MH, et al. Alarmin S100A9 induces proinflammatory and catabolic effects predominantly in the M1 macrophages of human osteoarthritic synovium. J. Rheumatol. 2016;43:1874–1884. doi: 10.3899/jrheum.160270. [DOI] [PubMed] [Google Scholar]
- 28.Vogl T, et al. Alarmin S100A8/S100A9 as a biomarker for molecular imaging of local inflammatory activity. Nat. Commun. 2014;5:4593. doi: 10.1038/ncomms5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogl T, et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood. 2004;104:4260–4268. doi: 10.1182/blood-2004-02-0446. [DOI] [PubMed] [Google Scholar]
- 30.Simard JC, et al. S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-dependent activation of NF-kappaB(1.) PLoS One. 2013;8:e72138. doi: 10.1371/journal.pone.0072138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldberg EL, et al. beta-Hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep. 2017;18:2077–2087. doi: 10.1016/j.celrep.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shepherd CE, et al. Inflammatory S100A9 and S100A12 proteins in Alzheimer’s disease. Neurobiol. Aging. 2006;27:1554–1563. doi: 10.1016/j.neurobiolaging.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 33.Horvath I, et al. Co-aggregation of pro-inflammatory S100A9 with alpha-synuclein in Parkinson’s disease: ex vivo and in vitro studies. J. Neuroinflammation. 2018;15:172. doi: 10.1186/s12974-018-1210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schluesener HJ, Kremsner PG, Meyermann R. Widespread expression of MRP8 and MRP14 in human cerebral malaria by microglial cells. Acta Neuropathol. 1998;96:575–580. doi: 10.1007/s004010050938. [DOI] [PubMed] [Google Scholar]
- 35.Postler E, Lehr A, Schluesener H, Meyermann R. Expression of the S-100 proteins MRP-8 and −14 in ischemic brain lesions. Glia. 1997;19:27–34. doi: 10.1002/(SICI)1098-1136(199701)19:1<27::AID-GLIA3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 36.Engel S, et al. Dynamics of microglial activation after human traumatic brain injury are revealed by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta Neuropathol. 2000;100:313–322. doi: 10.1007/s004019900172. [DOI] [PubMed] [Google Scholar]
- 37.Nagareddy PR, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma LP, et al. S100A8/A9 complex as a new biomarker in prediction of mortality in elderly patients with severe heart failure. Int. J. Cardiol. 2012;155:26–32. doi: 10.1016/j.ijcard.2011.01.082. [DOI] [PubMed] [Google Scholar]
- 39.Swindell WR, et al. Robust shifts in S100a9 expression with aging: a novel mechanism for chronic inflammation. Sci. Rep. 2013;3:1215. doi: 10.1038/srep01215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iashchishyn IA, Sulskis D, Nguyen Ngoc M, Smirnovas V, Morozova-Roche LA. Finke-Watzky two-step nucleation-autocatalysis model of S100A9 amyloid formation: protein misfolding as “nucleation” event. ACS Chem. Neurosci. 2017;8:2152–2158. doi: 10.1021/acschemneuro.7b00251. [DOI] [PubMed] [Google Scholar]
- 41.Yanamandra K, et al. Amyloid formation by the pro-inflammatory S100A8/A9 proteins in the ageing prostate. PLoS One. 2009;4:e5562. doi: 10.1371/journal.pone.0005562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eremenko E, Ben-Zvi A, Morozova-Roche LA, Raveh D. Aggregation of human S100A8 and S100A9 amyloidogenic proteins perturbs proteostasis in a yeast model. PLoS One. 2013;8:e58218. doi: 10.1371/journal.pone.0058218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horvath I, et al. Pro-inflammatory S100A9 protein as a robust biomarker differentiating early stages of cognitive impairment in Alzheimer’s disease. ACS Chem. Neurosci. 2016;7:34–39. doi: 10.1021/acschemneuro.5b00265. [DOI] [PubMed] [Google Scholar]
- 44.Ha TY, et al. S100a9 knockdown decreases the memory impairment and the neuropathology in Tg2576 mice, AD animal model. PLoS One. 2010;5:e8840. doi: 10.1371/journal.pone.0008840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iashchishyn IA, et al. Intranasally administered S100A9 amyloids induced cellular stress, amyloid seeding, and behavioral impairment in aged mice. ACS Chem. Neurosci. 2018;9:1338–1348. doi: 10.1021/acschemneuro.7b00512. [DOI] [PubMed] [Google Scholar]
- 46.Gruden MA, et al. S100A9 protein aggregates boost hippocampal glutamate modifying monoaminergic neurochemistry: a glutamate antibody sensitive outcome on Alzheimer-like memory decline. ACS Chem. Neurosci. 2018;9:568–577. doi: 10.1021/acschemneuro.7b00379. [DOI] [PubMed] [Google Scholar]
- 47.Gruden MA, et al. The misfolded pro-inflammatory protein S100A9 disrupts memory via neurochemical remodelling instigating an Alzheimer’s disease-like cognitive deficit. Behav. Brain Res. 2016;306:106–116. doi: 10.1016/j.bbr.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 48.Lodeiro M, et al. Aggregation of the inflammatory S100A8 precedes abeta plaque formation in transgenic APP mice: positive feedback for S100A8 and abeta productions. J. Gerontol. A. Biol. Sci. Med. Sci. 2017;72:319–328. doi: 10.1093/gerona/glw073. [DOI] [PubMed] [Google Scholar]
- 49.Botelho HM, et al. S100A6 amyloid fibril formation is calcium-modulated and enhances superoxide dismutase-1 (SOD1) aggregation. J. Biol. Chem. 2012;287:42233–42242. doi: 10.1074/jbc.M112.396416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clark RS, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13:813–821. doi: 10.1096/fasebj.13.8.813. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Chen Y, Jenkins LW, Kochanek PM, Clark RS. Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury. Crit. Care. 2005;9:66–75. doi: 10.1186/cc2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klingstedt T, et al. Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org. Biomol. Chem. 2011;9:8356–8370. doi: 10.1039/c1ob05637a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soderberg L, et al. Collagenous Alzheimer amyloid plaque component assembles amyloid fibrils into protease resistant aggregates. FEBS J. 2005;272:2231–2236. doi: 10.1111/j.1742-4658.2005.04647.x. [DOI] [PubMed] [Google Scholar]
- 56.Malisauskas M, Weise C, Yanamandra K, Wolf-Watz M, Morozova-Roche L. Lability landscape and protease resistance of human insulin amyloid: a new insight into its molecular properties. J. Mol. Biol. 2010;396:60–74. doi: 10.1016/j.jmb.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 57.Sacho RH, Childs C. The significance of altered temperature after traumatic brain injury: an analysis of investigations in experimental and human studies: part 2. Br. J. Neurosurg. 2008;22:497–507. doi: 10.1080/02688690802245558. [DOI] [PubMed] [Google Scholar]
- 58.Gupta AK, et al. Extracellular brain pH and outcome following severe traumatic brain injury. J. Neurotrauma. 2004;21:678–684. doi: 10.1089/0897715041269722. [DOI] [PubMed] [Google Scholar]
- 59.Oosawa F, Kasai M. A theory of linear and helical aggregations of macromolecules. J. Mol. Biol. 1962;4:10–21. doi: 10.1016/S0022-2836(62)80112-0. [DOI] [PubMed] [Google Scholar]
- 60.Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 61.Roberts GW, et al. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br. J. Pharmacol. 2016;173:681–691. doi: 10.1111/bph.13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat. Rev. Neurol. 2013;9:211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yakovlev AG, Faden AI. Caspase-dependent apoptotic pathways in CNS injury. Mol. Neurobiol. 2001;24:131–144. doi: 10.1385/MN:24:1-3:131. [DOI] [PubMed] [Google Scholar]
- 65.Vogl T, Leukert N, Barczyk K, Strupat K, Roth J. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim. Biophys. Acta. 2006;1763:1298–1306. doi: 10.1016/j.bbamcr.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 66.Wahlstrom A, Hugonin L, Peralvarez-Marin A, Jarvet J, Graslund A. Secondary structure conversions of Alzheimer’s Abeta(1-40) peptide induced by membrane-mimicking detergents. FEBS J. 2008;275:5117–5128. doi: 10.1111/j.1742-4658.2008.06643.x. [DOI] [PubMed] [Google Scholar]
- 67.Tutte, W. T. Graph Theory. (Cambridge University Press, 2001).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.