

Abstract
Inflammatory mediators affect the brain during development. Neurodevelopmental disorders such as autism spectrum disorders, cognitive impairment, cerebral palsy, epilepsy, and schizophrenia have been linked to early life inflammation. Recent advances have shown the effects of systemic inflammation on children’s neurodevelopment. We discuss the potential mechanisms by which inflammatory molecules can exert their effects on the developing brain and consider the roles of MHC class I molecules, the HPA axis, glial cells, and monoamine metabolism. Methods to prevent the effects of cytokine imbalance may lead to the development of new therapeutics for neuropsychiatric disorders. Future research should focus on identifying at-risk individuals and early effective interventions to prevent long-term neurodevelopmental disabilities.
Cytokines and neurodevelopment
In the past several decades, inflammation has been increasingly recognized as an important contributor to central nervous system (CNS) injury in both the developing and adult brain [1,2] (Table 1). The brain is particularly vulnerable in utero as well as during infancy and early childhood, and insults that occur during these critical periods have the potential to cause long-term damage. Several neurodevelopmental disorders have been linked to early life immune activation and inflammation, including autism spectrum disorders (ASD), schizophrenia, cerebral palsy, epilepsy, cognitive impairment, and depression [3–5].
Table 1.
Key studies of inflammation and neurodevelopment
| Reference | Type of study | Key findings |
|---|---|---|
| Maternal Immune Activation (MIA) | ||
| 2 | Human | Maternal third trimester IL-6 and CRP concentrations are associated with neonatal functional connectivity and with both fetal and toddler behavior. |
| 15 | In vivo; mice | MIA induces changes in the concentrations of many cytokines in the brains and sera of offspring in a region- and age-specific manner |
| 16 | In vivo; mice | Retinoic acid receptor–related orphan nuclear receptor γt (RORγt)–dependent effector T lymphocytes [e.g., T helper 17 (TH17) cells] and the effector cytokine interleukin-17a (IL-17a) are required in mothers for MIA-induced behavioral abnormalities in offspring. |
| 22 | In vivo; mice | Pregnant mice that had been colonized with the mouse commensal segmented filamentous bacteria (SFB) or human commensal bacteria that induce intestinal Th17 cells were more likely to produce offspring with MIA-associated abnormalities. |
| 28 | Human | For the highest quintile of maternal CRP concentration, compared with the lowest quintile, there is a significant, 43% elevated risk in autism in the offspring. |
| Perinatal | ||
| 7 | Human | In children born extremely prematurely, recurrent, or persistent elevations of inflammation-related proteins in blood during in the first two postnatal weeks are associated with an attention problem at age 2 years. |
| 37 | Human | Early elevations of C-reactive protein, tumor necrosis factor-α, interleukin (IL)-8, intercellular adhesion molecule (ICAM)-1, and erythropoietin were associated with IQ values >2 SD below the expected mean at 10 years of age. |
| 38 | Human | Extremely preterm newborns with elevated concentrations of inflammation-related proteins (CRP, IL-1b, IL-6, IL-8) during the third and fourth postnatal weeks were at increased risk of ventriculomegaly during the months after birth, and of microcephaly, and low neurodevelopmental scores at 2 years of age. |
| Postnatal | ||
| 8 | Human | Elevated concentrations of the pro-inflammatory cytokines IL-1β and IL-6 were significantly associated with a decrease in motor score. Conversely, an elevated concentration of the Th-2 cytokine IL-4 was associated with increased cognitive scores. |
| 49 | Human | Inflammation-related proteins including ORM1, a-1-antichymotrypsin, reticulocalbin 1, and 3 components of the complement cascade are significantly and inversely related to cognitive scores in Nepalese children. |
| 86 | Human | Elevated concentrations of the inflammation-related proteins CRP, sCD14, IL-1β, and IL-6 in the first year of life were associated with lower neurodevelopmental outcomes at 2 years of age. |
Cytokines coordinate the host immune response and also mediate normal signaling between cells of immune and nonimmune origin, including in the CNS [1]. Cells within the developing CNS use cytokines for autocrine and paracrine signaling, and since many of these same cytokines also serve as immune modulators, normal cytokine-mediated developmental processes are susceptible to disruption by cytokine imbalances. Pro-inflammatory cytokine induction in response to maternal or early life infection has been shown to adversely affect neurodevelopment [6–8]. Conversely, anti-inflammatory or regulatory cytokines have been shown in multiple settings to diminish these adverse effects [8]. While certain cytokines are traditionally thought to have primarily pro- or anti-inflammatory properties, it should be noted that certain cytokines can exhibit both properties in different contexts [9,10].
This Review focuses on recent developments regarding perturbations in cytokine-mediated processes in early life starting in utero with maternal immune activation (MIA) and its effects on fetal brain development. We will then discuss inflammation-related perinatal insults and then the myriad of inflammatory risk factors children may be exposed to in infancy and the first few years of life (Figure 1). The emerging role of cytokines in the pathogenesis of neurodevelopmental disorders is considered, with particular attention to the careful balancing act of pro- and anti-inflammatory states required for normal healthy neurodevelopment. The dynamic roles of microglia mediated effects on neurodevelopment is considered. Finally, we discuss these cytokine-brain interactions in the context of developing new therapeutics that target the immune system to prevent or treat these neuropsychopathologies.
Figure 1.
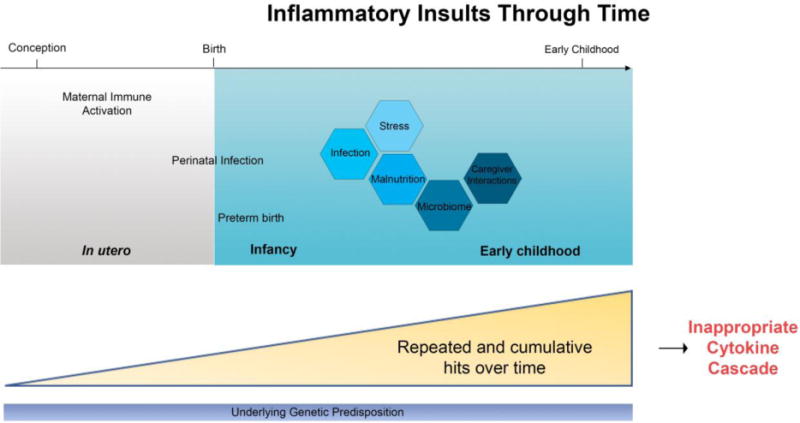
An overview of the inflammatory insults the developing brain may be exposed to from conception through early childhood. An individual’s response to inflammatory insults are a result of interactions between their underlying genetic disposition and the various factors they are exposed to. The cumulative effect of these insults through time can result in various “hits” that synergistically act on one another.
In utero: maternal immune activation (MIA)
Maternal immune activation (MIA) is the activation of the innate and adaptive immune systems due to infection, stress, autoimmunity, asthma, allergies, or inflammation. MIA has been associated with several neuropathologies in the progeny particularly with ASD and schizophrenia with emerging evidence for its role in epilepsy and cerebral palsy [4,11,12]. Most triggers of MIA in pregnant women, however, do not lead to neuropathologies in the offspring, thus it is thought that MIA acts as a disease primer or first “hit”, predisposing susceptible individuals for further exposures or “hits” later in life. The idea that MIA plays a role in fetal neurodevelopment was first proposed when epidemiologic studies linked infection during pregnancy with increased rates of ASD and schizophrenia in the offspring [4]. Animal models of MIA have supported this epidemiologic data and demonstrate that MIA alone is enough to cause neuropathology in the offspring [11]. It is thought that MIA may influence the developing fetal CNS through the increased production of inflammatory cytokines. Here, we summarize current literature suggesting a role for cytokines in mediating the fetal neurodevelopmental effects of MIA.
Animal evidence of role of cytokines in MIA
A causal relationship between MIA and ASD- and schizophrenia-related behavioral abnormalities has been demonstrated using rodent and nonhuman primate (NHP) animal models. In these MIA models, pregnant animals are exposed to mimics of viral infection [dsRNA poly(I:C)] or bacterial infection [lipopolysaccharide (LPS)] [4]. The offspring of the pregnant animals subjected to MIA develop behaviors and brain pathology similar to those seen in human ASD and schizophrenia [4,12–14]. Evidence in these models has pointed to cytokines as critical mediators of the effects of MIA on fetal brain development.
In rodent models, MIA has been shown to disrupt normal concentrations of cytokines throughout the brain, with region-specific changes seen throughout development [15]. Within a matter of hours, MIA can induce pro-inflammatory cytokines not only in protein and mRNA maternal serum, but also increases of IL-1β, IL-6, IL-10, and TNF-α expression within the fetal brain in mice as compared with pregnant dams in which MIA was not induced. Maternal intraperitoneal injection of a single inflammatory cytokine (IL-6, IL-17, or IL-2) in wild-type pregnant rodents is sufficient to induce several ASD- and schizophrenia-like behaviors in the offspring, deficits in working memory and cognitive flexibility, abnormal new motor learning, decreased sensorimotor gating, increased anxiety, and enhanced sensitivity to amphetamines [14,16,17]. MIA by administration of poly(I:C) in IL-6 knockout pregnant mice does not result in these behavioral effects in the progeny [4,16]. Furthermore, co-administration of poly(I:C) with an IL-6 or IL-17 blocking antibody partially prevents the effects of MIA in mice models. Conversely, overexpression of the anti-inflammatory cytokine IL-10 partially prevents the effects of MIA in offspring [11].
The role of IL-6 in MIA has been particularly well-studied, and IL-6 appears to be a key mediator of the effects of MIA. Maternally-derived IL-6 activates the JAK/STAT3 pathway in the spongiotrophoblast layer of the placenta resulting in downstream expression of acute phase genes [17]. Additionally, maternally-derived IL-6 appears to disrupt of the growth hormone-insulin-like growth factor (GH-IGF) axis along with alterations in other placental endocrine factors important for promoting embryonic development in mice placentas [18]. Mice with restricted deletion of the receptor for IL-6 (IL-6Rα) in placental trophoblasts did not exhibit MIA-induced inflammatory responses in the placenta and fetal brain. The behavioral abnormalities in the offspring were also not observed in these mothers that lacked IL-6 signaling in trophoblasts. It appears that IL-6 activation in placenta during MIA is required for relaying inflammatory signals to the fetal brain and impacting behaviors in the offspring [19]. Additionally, IL-6 is a key factor for (T helper 17) Th17 differentiation and the production of IL-17 [16]. Dysregulation of maternal IL-17 may serve as a causal factor for ASD. IL-17a from maternal Th17 cells in pregnant mice was found to induce behavioral and cortical abnormalities in the progeny exposed to MIA [20–22]. Genetic removal of the transcription factor RORγt to selectively inhibit Th17 differentiation in pregnant mice prevented ASD-like behaviors in the offspring [16].
It remains unknown whether cytokines in maternal serum induced by MIA directly cross the placenta to act on the fetal brain or indirectly induce cytokine production elsewhere. A few studies have demonstrated permeability to IL-6 both in a healthy-term human placental perfusion model and an in vivo in a rat model during mid-gestation [23,24]. Production of cytokines from the placenta itself is another possibility to explain the presence of cytokines in amniotic fluid and fetal circulation. In vitro studies in the human first-trimester extravillous trophoblast cell line, HTR8-SVneo, have shown that human trophoblast cells respond to a viral mimetic that activates TLR-3 resulting in the secretion of pro-inflammatory factors [25].
Clinical evidence of role of cytokines in MIA
Observational and case-control studies have found associations between elevated cytokines in maternal serum in the second and third trimesters and an increased risk of neurodevelopmental disorders. Elevated expression of the cytokines IL-6, CXCL8 (IL-8), and TNF-α along with C-reactive protein (CRP) in human maternal serum is associated with an increased risk for schizophrenia in the offspring [26–29]. In addition, other known risk factors of schizophrenia including malnutrition and stress involve upregulation of inflammatory cytokines in maternal serum [1]. Similarly, birth cohort data has associated inflammatory markers in mothers with ASD. In individuals with ASD, elevated concentrations of granulocyte macrophage colony-stimulating factor, IFN-y, IL-1α, and IL-6 in maternal serum during the second trimester have been identified [12,30]. Elevated expression of C-reactive protein during pregnancy was also significantly associated with increased risk of ASD in the offspring [28].
Prenatal exposure to elevated concentrations of cytokines may contribute to structural abnormalities in the brain. In one study, adults with schizophrenia who were exposed to elevated concentrations of maternal IL-8 had volumetric decreases in entorhinal and cingulate cortices [31]. Emerging evidence also suggests that MIA is linked to functional connectivity in the brain. One recent study found that maternal third trimester IL-6 and CRP expression are negatively associated with neonatal functional connectivity and with fetal and toddler behavior [2].
Whether cytokines directly or indirectly act on the brain remains incompletely understood, and this is an area of active investigation (see cytokine targets section below). Notably, recent studies suggest that MIA disrupts inhibitory interneuron networks and suggests that the maternal gut microbiome may be a contributing factor (see gut-immune-brain axis section below) [32,33]. The underlying genetic or environmental factors that may predispose mothers or their offspring to the adverse effects of MIA have yet to be elucidated.
Perinatal: Neurodevelopment of preterm infants
Infection and inflammation are common causes of preterm birth and have been linked to brain injury in preterm infants. Ascending infection and associated inflammation are thought to initiate preterm birth as well as propagate from chorioamnioinic membranes to the amniotic fluid to reach the fetal circulation, resulting in systemic fetal inflammation. This systemic inflammation can ultimately affect several organs, including the brain. Mothers presenting with preterm labor have been found to have increased concentrations of IL-6 and IL-8 in their amniotic fluid. Epidemiologic studies have linked elevated cytokines in the umbilical cord, amniotic fluid, and fetal blood to white matter injury, cerebral palsy, and impaired development [34]. The most commonly implicated cytokines include TNF-α, IFN-gamma, IL-1, IL-6, and IL-18 [35–37].
In a multicenter study of extremely low gestational newborns (<28 weeks), it was found that elevated levels of pro-inflammatory cytokines in the first two weeks of life were associated with worse performance of developmental tests at 2 years of age. These cytokines included IL-6, TNF-α, and IL-8 [7]. In this same cohort, elevated concentrations of CRP, IL-1β, IL-6, and IL-8 in the third and fourth week of life were at increased risk of ventriculomegaly, microcephaly, and low developmental scores at 2 years of life [38]. In a follow up of this same cohort, early elevations of TNF-α, IL-8, and CRP in the first month of life were associated with IQ values >2 SD below the mean at 10 years of age [37].
Pro-inflammatory cytokines are also implicated in the pathogenesis of cerebral palsy. In a case controlled study, increased pro-inflammatory cytokines in neonatal blood (IL-1β, IL-8, IL-9, TNF-1, RANTES) during the first few days in the infant after birth had 100% sensitivity and specificity in prediction of cerebral palsy in late preterm and term infants [39]. Expression of the anti-inflammatory cytokines IL-2 and IL-3 was found to be decreased in the infants with cerebral palsy [39,40]. One study of extremely low birth weight (ELBW) infants found that significantly increased IL-8 expression in days 0-4 was highly prognostic of the development of cerebral palsy [40]. High concentrations of IL-18 in the cord blood of preterm infants has been associated with the development of periventricular leukomalacia and cerebral palsy [41]. In rodent models, IL-18 is markedly expressed in the immature rodent brain following hypoxic-ischemic brain injury as compared with controls. Interestingly, white matter injury was decreased in mice deficient in IL-18 versus wild-type mice following hypoxic-ischemic brain injury [42,43].
Despite the evidence implicating inflammation in preterm birth and its neurologic sequelae, existing tocolytic drugs do not target fetal inflammatory injury. No therapeutic molecule exists to date that has been demonstrated to prevent pathological inflammatory processes in pregnant women at risk for pre-term birth. One recent study in mice identifies rytvela (labeled 101.10), a noncompetitive inhibitor of IL-1 actions, to prevent preterm birth [44,45]. Further work in identifying possible therapeutics to ameliorate inflammation during preterm birth may serve as a promising method to prevent long-term cognitive and neurologic sequelae.
Postnatal: inflammation in children growing up in adversity
It is well-known that adverse environments (exposure to poverty, stress, war, neglect etc.) have lasting effects on child development. Tens of millions of children worldwide do not meet their full developmental potentials due to adverse environments, resulting in devastating losses in human capital [46,47]. The reasons are likely multifactorial, including exposure to infectious disease, malnutrition, psychosocial stress, and poor caregiver interactions. Cytokines may serve as a common mediator across these risk factors, as many of these factors can result in cytokine elevations.
The link between stunting, a measure of chronic malnutrition as defined as a height-for-age Z-score (HAZ) <−2, and impaired development has been consistently documented across several ages of development. Neurodevelopmental outcomes have included a decrease in exploratory behavior, impaired cognition, increased anxiety, and lower school performance. However, it is difficult to discern in these studies whether malnutrition is a causal factor or simply a proxy. Malnutrition and infection are intricately intertwined: chronic exposure to infection and inflammation can lead to malnutrition; while malnourished children are known to have chronic inflammation [48]. Thus, studying inflammatory markers may shed light on this question.
Our prospective observational study in Bangladesh found that elevated concentrations of IL-1ß and IL-6 in plasma of infants at 6 months of age were associated with worse developmental outcomes at 1 and 2 years of life, independent of linear growth and family income [8]. Similarly, inflammation was found to be negatively associated with general intelligence in a cohort of 7-9 year-old Nepalese children; [49]. Notably, elevated IL-4 concentration was found to be associated with better cognitive outcomes in our cohort. IL-4 has anti-inflammatory and regulatory properties and this finding raises the hypothesis of an imbalance of pro- and anti-inflammatory cytokines resulting in adverse outcomes. In rodent models, IL-4 has been found to be important for normal cognitive processes. In IL-4 knockout mice, cognitive impairment was exhibited; this impairment was reversed after transplantation with IL-4 competent bone marrow [50]. Furthermore, IL-4 produced by CD4+ T cells was found to be neuroprotective in two different rodent models of neuronal injury (optic nerve crush injury and spinal cord contusive injury) [51].
Psychosocial stress has been shown to activate key inflammatory pathways in peripheral blood mononuclear cells, including activation of the transcription factor nuclear factor-kB (NF-kB) and leads to marked increases in circulating concentrations of pro-inflammatory cytokines such as IL-6 [52]. Psychosocial stress has been linked to poor development in children growing up in adversity in both the developed and developing world. In response to stress, catecholamines (such as noradrenaline) released by the activated sympathetic nervous system stimulate bone marrow production and the release of myeloid cells that enter the periphery where they encounter stress-induced damage-associated molecular patterns (DAMPs) and subsequently activate inflammatory signaling pathways [53].
Much remains to be learned about the developing brain in adverse environments as most studies to date are limited to observational studies. Outstanding questions remain as to whether cytokines have a causal effect in the pathogenesis of impaired development. The development of robust animal models of adversity-induced neurocognitive deficits will be key in understanding underlying mechanisms.
Outstanding questions.
-Are there genetic factors that increase or reduce an individual’s risk of inflammation-mediated neurodevelopmental disturbances?
-What indicators (biomarkers) can we use to identify children at increased risk?
-Can we intervene early enough when a fetus or infant has been exposed to inflammation to prevent long-term sequelae?
-Are there effective therapies that combat inflammation to treat or ameliorate neurodevelopmental disorders?
-Would a vaccine prevent inflammation-mediated neurodevelopmental disorders?
How do cytokines exert their effects on the brain?
Despite the plethora of both human and animal studies implicating a role for cytokine imbalance in the pathogenesis of various neuropsychopathologies, it remains incompletely understood how these cytokines mechanistically alter long-term brain development. One question is how cytokines in the periphery affect the brain. Cytokines in the periphery are known to communicate through several ways with the CNS, including through diffusion from the circumventricular organs (outside the blood-brain barrier), via cranial nerves, through cytokine transporters, or secretion of immune-active substances (cytokines or prostaglandins) by cells of the blood-brain barrier. Importantly, cytokines do not always have to reach the brain from the periphery as most cytokines can be synthesized and released within the central nervous system by glia or neurons.
Once cytokines are in the brain, how exactly do they exert their effects? One possibility is that inappropriate activation of cytokines may lead to long-lasting changes in expression of immune molecules that regulate neural connectivity and function [11]. For example, cytokines in the CNS may act through interactions with other classes of immune molecules. One such class is the major histocompatibility complex class I (MHC I) molecules, which are also found on neurons where they negatively regulate synaptic plasticity required for activity-dependent synaptic pruning and synapse formation [11,54–56]. In MIA, there is a dramatic increase in MHC I protein expression on neurons in the brains of offspring resulting in a decreased ability to form synapses. Such alterations in synaptogenesis are thought to play a central role in the etiologies of ASD and schizophrenia [57,58]. Cytokines can also potently activate the hypothalamic-pituitary-adrenal (HPA) axis to increase adrenocorticotropic hormone (ACTH), corticotropin-releasing hormone (CRH), arginine vasopressin, and corticosterone concentrations. These hormones can stimulate the secretion of ACTH and glucocorticoids from the adrenals [59,60]. Better understanding of the mechanism by which cytokines may interact to exert its effects on the brain can give us insight into how to better intervene. Another mechanism by which cytokines can act in the CNS is through microglial activation (see below).
Microglia in neurodevelopment
Microglia are highly motile macrophage-like cells of hematopoietic origin that infiltrate and take up residence in the developing brain, where they are thought to provide a surveillance function [61]. These myeloid cells have been implicated in a wide array of CNS processes such as mediating inflammation, directly combating infection, and clearing cellular debris via phagocytosis [62]. In addition to these traditional macrophage-type roles, microglia have more recently been revealed as critical players in neurodevelopment, where they promote the survival, differentiation, and maturation of neurons during neurogenesis and strip away excess synapses in the developing brain to allow for functional neuronal circuits in a process known as pruning [63–65].
Microglia are both a main source of and target for cytokines in the CNS. Upon stimulation by an activating signal (e.g. cytokines, pathogen associated molecular patterns [PAMPs], DAMPs), microglia are known to retract their processes resulting in an altered morphology, and employ the molecular machinery necessary for programs such as opsonization and phagocytosis of targets, as well as the release of cytokines such as the pro-inflammatory cytokines IL-1B, IL-6, and TNF-alpha [66–68]. In addition to the production of these pro-inflammatory cytokines, microglia possess cognate receptors and are thought to respond to these signals from astrocytes, other microglia, and infiltrating immune cells upon disruption of the blood-brain barrier during cases of inflammation [68–70]. In cases of an over-exuberant microglial response, excess pro-inflammatory cytokines have been shown to induce excitotoxicity and neuronal death [70]. This phenomenon may serve as the underpinnings of developmental deficits observed when the developing brain is exposed to excessive inflammation.
The key signal during development that is necessary to launch the upregulation of complement proteins involved in synaptic pruning has been identified as the cytokine, TGF-β, produced by astrocytes [71]. The alarmin IL-33 derived from astrocytes has also recently been shown to promote the pruning process in vivo, and IL-33 knockout mice display major deficits in synaptic elimination during development [72]. While cytokines such as IFN-gamma, IL-1β, IL-6, and TNF-α can activate microglia, other anti-inflammatory cytokines have roles in restraining them. For instance, neurons produce CX3CL1, or fractalkine, for which microglia possess the fractalkine receptor, CX3CR1 [73]. Both fractalkine and CD200 signaling from neurons have been shown to keep microglia in an unactivated, non-neurotoxic state [73,74]. In mice lacking CX3CR1, microglia numbers were transiently reduced in the developing brain and synaptic pruning was delayed.
Microglia are also thought to play a role in the process of neurogenesis, or the differentiation and maturation of precursors into neurons [75]. In vitro studies suggest that secreted microglial factors are necessary for the self-renewal of neuropoietic cells [76]. During prenatal development, microglia guide neurons and their axonal elements to form functional circuits [77]. While microglial activity appears to be required for neurogenesis, some studies suggest that improper activation of microglia caused by chronic inflammation from radiation injury, or chronic expression of the pro-inflammatory cytokine IL-6 cytokine in astrocytes, have been shown to directly impede the differentiation and survival of neurons during neurogenesis [78,79].
The gut-immune-brain axis in neurodevelopmental disorders
The gut may play an important role in neurodevelopmental disorders and thus is a promising target for therapeutic interventions. During birth, the fetal gut is colonized by maternal microbiota, priming the developing immune system and directing immune homeostasis. Like individuals with ASD and other disorders, MIA offspring display altered microbiota signatures. Restoring a more typical microbiome by treating MIA offspring with Bacteroides fragilis not only restores peripheral immune homeostasis but ameliorates several aberrant behaviors [80]. As our understanding of the connection between the gut microbiome and the brain grows, so will our ability to harness such pathways to provide novel approaches to treat these disorders.
Investigators found that MIA disrupts neuronal architecture in areas of the cerebral cortex via direct intracerebral action of the pro-inflammatory cytokine IL-17a [16]. The source of IL-17 was found to be Th17 cells, a CD4+ T cell subset that plays a key role in combating bacterial and fungal infections in the maintenance of gut homeostasis. This work generated the hypothesis that maternal Th17 cells reaching fetal circulation produce IL-17a, which then affects fetal brain development. Furthermore, it was found that C57BL/6 mice from Taconic Biosciences (Tac), which contain high numbers of gut Th17 cells as they are colonized with segmented filamentous bacterium (SFB), were susceptible to MIA-induced behavioral pathology. Conversely, mice from the Jackson Laboratory (JAX), which lack gut Th17 cells and are not colonized by SFB, were not susceptible to MIA-induced behavioral pathology [22]. Such susceptibility could be conferred on JAX mice by co-housing the animals or via gastrointestinal gavage. In addition, the study demonstrated that elevated IL-17a responses during MIA were due to the activation of dendritic cells (a key antigen presenting cell type) interacting with pre-established memory Th17 cells within the SFB-colonized gut [16,22].
Another mechanism by which the gut may influence neurodevelopment is via gut inflammation and environmental enteropathy in children who grow up in adversity. Environmental enteropathy is a condition at is a result of chronic enteric infection and is characterized by gut inflammation, barrier disruption, and systemic inflammation [48,81,82]. Environmental enteropathy has been linked to poor health outcomes in children including oral vaccine failure and growth faltering [48,83]. Excessive gut inflammation can result in downstream systemic inflammation and the release of cytokines [84,85]. Systemic inflammation has been linked to poor child development in children who grow up in adversity [8]. Furthermore, markers of environmental enteropathy itself such as soluble CD14 (sCD14), a measure of gut barrier dysfunction, has also been shown to be associated with poor neurodevelopmental outcomes [86].
Potential therapeutics
The new and emerging discoveries that link immune activation and inflammation with neuropsychopathology afford opportunities for novel therapeutics with those that center around decreasing inflammation most promising. The cyclooxygenase-2 (COX-2) inhibitor celecoxib, was studied in a prospective, randomized double-blind study of acute exacerbations of schizophrenia. Patients receiving celecoxib in addition to risperidone showed a statistically significant better outcomes in terms of symptom reduction than patients receiving risperidone alone; clinical effects of COX-2 inhibition in schizophrenia were especially pronounced in cognition [87].
Because microglia serve as a major source of pro-inflammatory activity in the CNS, experimentally manipulating microglia may serve as a potentially powerful mechanism for addressing inflammation-mediated neurocognitive deficits. While necessary for maintaining CNS homeostasis, microglia can be targeted and depleted using multiple approaches [88].
Concluding remarks
Cytokine imbalances during the early periods of neurodevelopment may have profound long-term impacts on a variety of diseases, including ASD, schizophrenia, cerebral palsy, and cognitive impairment. These insults can occur as early as in utero via MIA up through early childhood and likely even later in life. Mechanisms by which cytokines exert their effects may include modulation of other immune mechanisms such as upregulation of MHC class I on neurons, via the HPA axis, via glial cells which then affect neurogenesis and synaptogenesis, or through monoamine metabolism. Strategies to inhibit the effects of cytokine imbalance may lead to novel therapeutics and have profound effects on prevention or treatment of neurologic disease. Recent attention has been focused on the role of microglia in neuroinflammation and neurodevelopment along with an increased interest and focus on the role of the gut microbiome in altering the immune response. Despite advances in the field in recent years, many questions still remain to be answered including the role of genetics along with how best to develop therapeutics (Outstanding Questions). Clinicians should be aware of the role that excessive inflammation in at-risk individuals may play in neurodevelopment and of potential novel therapeutics to reduce this inflammatory load (Clinician’s corner, Box 1). Research efforts should focus on identifying at-risk individuals and early effective interventions to prevent long-term neurodevelopmental disabilities.
Clinician’s Corner (Box 1).
-Neurodevelopmental disorders are a major health burden on society and have increased in prevalence. Recent advances have expanded our understanding of the impact of inflammation on the developing brain.
-Children are at increased risk of brain development disorders if their mothers had systemic inflammation during pregnancy.
-In general, elevated concentrations of pro-inflammatory cytokines are associated with poor neurodevelopmental outcomes.
-Disruption of the microbiome and intestinal barrier have been linked to inflammation and neurodevelopmental disorders.
-Research efforts should focus on identifying at-risk individuals and early effective interventions to prevent long-term neurodevelopmental disabilities.
Highlights.
-
Maternal immune activation (MIA) refers to the activation of a pregnant mother’s immune system in response to an insult such as an infection. While MIA is generally protective, MIA may also be a risk factor for neurological conditions in the child later on.
- Elevated concentrations of inflammation-related cytokines in maternal serum in utero as well as in children early in life such as IL-6 are associated with worse neurodevelopmental outcomes, while IL-4 appeared protective.
- Dysbiotic microbiota and environmental enteropathy have been connected to inflammation in the child during the first two years of life and poor neurodevelopmental outcomes.
- Microglia, macrophage-like cells in the developing brain, are a potential therapeutic target given that they are a major hub of pro-inflammatory activity.
Glossary
- Ascending infection
infection during pregnancy in which pathogens invade the uterus and fetal membranes
- Damage-associated molecular patterns (DAMPs)
host molecules that are released by stress cells that act as endogenous signals to promote the inflammatory response
- Hypothalamic-pituitary-adrenal (HPA) axis
set of interactions between the hypothalamus, pitiuitary gland, and adrenal glands that is responsible for reactions to stress as well as the regulation of body processes including the immune system
- Opsonization
The mechanism by which pathogens are marked by opsonins (e.g. antibodies) for ingestion and elimination by phagocytes
- Pathogen-associated molecular patterns (PAMPs)
molecules from pathogens that are recognized by the innate immune system, triggering the host immune response
- Tocolytic drugs
medications used to suppress premature labor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Nona M. Jiang, University of Virginia Department of Medicine.
Maureen Cowan, University of Virginia Department of Medicine
Shannon N. Moonah, University of Virginia Department of Medicine.
William A. Petri, Jr., University of Virginia Department of Medicine.
References
- 1.Deverman BE, Patterson PH. Cytokines and CNS Development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Spann MN, et al. Maternal Immune Activation During the Third Trimester Is Associated with Neonatal Functional Connectivity of the Salience Network and Fetal to Toddler Behavior. J Neurosci. 2018;38:2877–2886. doi: 10.1523/JNEUROSCI.2272-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim S, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. 2017 doi: 10.1038/nature23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knuesel I, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Publ Gr. 2014;10 doi: 10.1038/nrneurol.2014.187. [DOI] [PubMed] [Google Scholar]
- 5.Kuban KCK, et al. The Breadth and Type of Systemic Inflammation and the Risk of Adverse Neurological Outcomes in Extremely Low Gestation Newborns. Pediatr Neurol. 2015;52:42–48. doi: 10.1016/j.pediatrneurol.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goeden N, et al. Maternal Inflammation Disrupts Fetal Neurodevelopment via Increased Placental Output of Serotonin to the Fetal Brain. J Neurosci. 2016;36:6041–6049. doi: 10.1523/JNEUROSCI.2534-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Shea TM, et al. Elevated blood levels of inflammation-related proteins are associated with an attention problem at age 24 mo in extremely preterm infants. Pediatr Res. 2014;75:781–787. doi: 10.1038/pr.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang NM, et al. Febrile illness and pro-inflammatory cytokines are associated with lower neurodevelopmental scores in Bangladeshi infants living in poverty. BMC Pediatr. 2014;14 doi: 10.1186/1471-2431-14-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheller J, et al. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta - Mol Cell Res. 2011;1813:878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 10.Xing Z, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–20. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estes ML, McAllister AK. Maternal immune activation: Implications for neuropsychiatric disorders. Science. 2016;353:772–7. doi: 10.1126/science.aag3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patterson PH. Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav Brain Res. 2009;204:313–321. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Reisinger S, et al. The Poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol Ther. 2015;149:213–226. doi: 10.1016/j.pharmthera.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Meyer U. Prenatal Poly(I:C) Exposure and Other Developmental Immune Activation Models in Rodent Systems. Biol Psychiatry. 2014;75:307–315. doi: 10.1016/j.biopsych.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Garay PA, et al. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68. doi: 10.1016/j.bbi.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi GB, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–9. doi: 10.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponzio NM, et al. Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspring. Ann N Y Acad Sci. 2007;1107:118–28. doi: 10.1196/annals.1381.013. [DOI] [PubMed] [Google Scholar]
- 18.Hsiao EY, Patterson PH. Activation of the Maternal Immune System Induces Endocrine Changes in the Placenta via IL-6. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu WL, et al. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun. 2017;62:11–23. doi: 10.1016/j.bbi.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi GB, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–9. doi: 10.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong H, Hoeffer C. Maternal IL-17A in autism. Exp Neurol. 2018;299:228–240. doi: 10.1016/j.expneurol.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim S, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549:528–532. doi: 10.1038/nature23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaretsky MV, et al. Transfer of Inflammatory Cytokines Across the Placenta. Obstet Gynecol. 2004;103:546–550. doi: 10.1097/01.AOG.0000114980.40445.83. [DOI] [PubMed] [Google Scholar]
- 24.Dahlgren J, et al. Interleukin-6 in the Maternal Circulation Reaches the Rat Fetus in Mid-gestation. Pediatr Res. 2006;60:147–151. doi: 10.1203/01.pdr.0000230026.74139.18. [DOI] [PubMed] [Google Scholar]
- 25.Abrahams VM, et al. Expression and secretion of antiviral factors by trophoblast cells following stimulation by the TLR-3 agonist, Poly(I: C) Hum Reprod. 2006;21:2432–2439. doi: 10.1093/humrep/del178. [DOI] [PubMed] [Google Scholar]
- 26.Canetta S, et al. Elevated Maternal C-Reactive Protein and Increased Risk of Schizophrenia in a National Birth Cohort. Am J Psychiatry. 2014;171:960–968. doi: 10.1176/appi.ajp.2014.13121579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buka SL, et al. Maternal Cytokine Levels during Pregnancy and Adult Psychosis. Brain Behav Immun. 2001;15:411–420. doi: 10.1006/brbi.2001.0644. [DOI] [PubMed] [Google Scholar]
- 28.Brown AS, et al. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry. 2014;19:259–264. doi: 10.1038/mp.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown AS, et al. Elevated Maternal Interleukin-8 Levels and Risk of Schizophrenia in Adult Offspring. Am J Psychiatry. 2004;161:889–895. doi: 10.1176/appi.ajp.161.5.889. [DOI] [PubMed] [Google Scholar]
- 30.Goines PE, et al. Increased midgestational IFN-γ, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol Autism. 2011;2:13. doi: 10.1186/2040-2392-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellman LM, et al. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr Res. 2010;121:46–54. doi: 10.1016/j.schres.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13:701–712. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- 33.Kim S, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549:528–532. doi: 10.1038/nature23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dammann O, O’Shea TM. Cytokines and perinatal brain damage. Clin Perinatol. 2008;35:643–63, v. doi: 10.1016/j.clp.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malaeb S, Dammann O. Fetal inflammatory response and brain injury in the preterm newborn. J Child Neurol. 2009;24:1119–1126. doi: 10.1177/0883073809338066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dammann O, O’shea M. Cytokines and Perinatal Brain Damage. doi: 10.1016/j.clp.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuban KCK, et al. Circulating Inflammatory-Associated Proteins in the First Month of Life and Cognitive Impairment at Age 10 Years in Children Born Extremely Preterm. J Pediatr. 2017;180:116–123.e1. doi: 10.1016/j.jpeds.2016.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leviton A, et al. Systemic inflammation on postnatal days 21 and 28 and indicators of brain dysfunction 2years later among children born before the 28th week of gestation. Early Hum Dev. 2016;93:25–32. doi: 10.1016/j.earlhumdev.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson KB, et al. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol. 1998;44:665–675. doi: 10.1002/ana.410440413. [DOI] [PubMed] [Google Scholar]
- 40.Carlo WA, et al. Cytokines and neurodevelopmental outcomes in extremely low birth weight infants. J Pediatr. 2011;159:919–25.e3. doi: 10.1016/j.jpeds.2011.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minagawa K, et al. POSSIBLE CORRELATION BETWEEN HIGH LEVELS OF IL-18 IN THE CORD BLOOD OF PRE-TERM INFANTS AND NEONATAL DEVELOPMENT OF PERIVENTRICULAR LEUKOMALACIA AND CEREBRAL PALSY. Cytokine. 2002;17:164–170. doi: 10.1006/cyto.2001.0988. [DOI] [PubMed] [Google Scholar]
- 42.Hedtjärn M, et al. Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci. 2002;22:5910–9. doi: 10.1523/JNEUROSCI.22-14-05910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hedtjärn M, et al. White matter injury in the immature brain: role of interleukin-18. Neurosci Lett. 2004;373:16–20. doi: 10.1016/j.neulet.2004.09.062. [DOI] [PubMed] [Google Scholar]
- 44.Nadeau-Vallée M, et al. Novel Noncompetitive IL-1 Receptor-Biased Ligand Prevents Infection- and Inflammation-Induced Preterm Birth. J Immunol. 2015;195:3402–15. doi: 10.4049/jimmunol.1500758. [DOI] [PubMed] [Google Scholar]
- 45.Girard S, et al. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J Immunol. 2010;184:3997–4005. doi: 10.4049/jimmunol.0903349. [DOI] [PubMed] [Google Scholar]
- 46.Grantham-McGregor S, et al. Developmental potential in the first 5 years for children in developing countries. Lancet. 2007;369:60–70. doi: 10.1016/S0140-6736(07)60032-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker SP, et al. Child development: risk factors for adverse outcomes in developing countries. Lancet. 2007;369:145–157. doi: 10.1016/S0140-6736(07)60076-2. [DOI] [PubMed] [Google Scholar]
- 48.Naylor C, et al. Environmental Enteropathy, Oral Vaccine Failure and Growth Faltering in Infants in Bangladesh. EBioMedicine. 2015;2:1759–66. doi: 10.1016/j.ebiom.2015.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SE, et al. General intelligence is associated with subclinical inflammation in Nepalese children: A population-based plasma proteomics study. Brain Behav Immun. 2016;56:253–263. doi: 10.1016/j.bbi.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Derecki NC, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207:1067–1080. doi: 10.1084/jem.20091419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh JT, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest. 2015;125:699–714. doi: 10.1172/JCI76210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bierhaus A, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci. 2003;100:1920–1925. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34. doi: 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Estes ML, McAllister AK. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Rev Neurosci. 2015;16:469–486. doi: 10.1038/nrn3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shatz CJ. MHC Class I: An Unexpected Role in Neuronal Plasticity. Neuron. 2009;64:40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glynn MW, et al. MHCI negatively regulates synapse density during the establishment of cortical connections. Nat Neurosci. 2011;14:442–451. doi: 10.1038/nn.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hospital TG. Original article. Methods. 2009;122:287–296. [Google Scholar]
- 59.Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci. 2000;917:608–17. doi: 10.1111/j.1749-6632.2000.tb05426.x. [DOI] [PubMed] [Google Scholar]
- 60.Turnbull AV, Rivier C. Regulation of the HPA Axis by Cytokines. Brain Behav Immun. 1995;9:253–275. doi: 10.1006/brbi.1995.1026. [DOI] [PubMed] [Google Scholar]
- 61.Sheng J, et al. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity. 2015;43:382–393. doi: 10.1016/j.immuni.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 62.Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23:1018–1027. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- 63.Paolicelli RC, et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science (80−) 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 64.Stevens B, et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 65.Schafer DP, et al. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–78. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee SC, et al. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J Immunol. 1993;150:2659–67. [PubMed] [Google Scholar]
- 68.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 69.Hickman SE, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ye L, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. 2013;125:897–908. doi: 10.1111/jnc.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16:1773–1782. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Vainchtein ID, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359:1269–1273. doi: 10.1126/science.aal3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harrison JK, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deckert M, et al. Regulation of microglial cell responses in murine Toxoplasma encephalitis by CD200/CD200 receptor interaction. Acta Neuropathol. 2006;111:548–558. doi: 10.1007/s00401-006-0062-z. [DOI] [PubMed] [Google Scholar]
- 75.Aarum J, et al. Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci U S A. 2003;100:15983–8. doi: 10.1073/pnas.2237050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walton NM, et al. Microglia instruct subventricular zone neurogenesis. Glia. 2006;54:815–825. doi: 10.1002/glia.20419. [DOI] [PubMed] [Google Scholar]
- 77.Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Monje ML, et al. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science (80−) 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 79.Vallières L, et al. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci. 2002;22:486–92. doi: 10.1523/JNEUROSCI.22-02-00486.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hsiao EY, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–63. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korpe PS, Petri WA. Environmental enteropathy: critical implications of a poorly understood condition. Trends Mol Med. 2012;18:328–336. doi: 10.1016/j.molmed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Petri WA, et al. Environmental enteropathy and malnutrition: do we know enough to intervene? BMC Med. 2014;12:187. doi: 10.1186/s12916-014-0187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Donowitz JR, et al. Small Intestine Bacterial Overgrowth and Environmental Enteropathy in Bangladeshi Children. MBio. 2016;7:e02102–15. doi: 10.1128/mBio.02102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ngobeni R, et al. Entamoeba histolytica-Encoded Homolog of Macrophage Migration Inhibitory Factor Contributes to Mucosal Inflammation during Amebic Colitis. J Infect Dis. 2017;215:1294–1302. doi: 10.1093/infdis/jix076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ngobeni R, et al. Entamoeba Species in South Africa: Correlations With the Host Microbiome, Parasite Burdens, and First Description of Entamoeba bangladeshi Outside of Asia. J Infect Dis. 2017;216:1592–1600. doi: 10.1093/infdis/jix535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jiang NM, et al. Early life inflammation and neurodevelopmental outcome in Bangladeshi infants growing up in adversity. Am J Trop Med Hyg. 2017;97 doi: 10.4269/ajtmh.17-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Müller N, Schwarz MJ. COX-2 inhibition in schizophrenia and major depression. Curr Pharm Des. 2008;14:1452–65. doi: 10.2174/138161208784480243. [DOI] [PubMed] [Google Scholar]
- 88.Kierdorf K, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]