

Abstract
Androgen receptor (AR) plays important roles in gene expression regulation, sexual phenotype maintenance and prostate cancer (PCa) development. The communications between the AR ligand-binding domain (LBD) and its coactivator are critical to the activation of AR. It is still unclear how the ligand binding would affect the AR-coactivator interactions. In this work, the effects of the ligand binding on the AR-coactivator communications were explored by molecular dynamics (MD) simulations. The results showed that the ligand binding regulates the residue interactions in the function site AF-2. The ligand-to-coactivator allosteric pathway, which involves the coactivator, helix 3 (H3), helix 4 (H4), loop between H3 and H4 (L3), helix 12 (H12) and ligands, was characterized. Besides, the interactions of residues on the function site BF-3, especially on the boundary of AF-2 and BF-3, are also affected by the ligands. The MM/GBSA free energy calculations demonstrated that the binding affinity between the coactivator and apo-AR is roughly weaker than those between the coactivator and antagonistic ARs but stronger than those between the coactivator and agonistic ARs. The results indicated that the long-range electrostatic interactions and the conformational entropies are the main factors affecting the binding free energies. In addition, the F876L mutation on AR-LBD affects the ligand-to-coactivator allosteric pathway, which could be the reason of point mutation induced tolerance for the antagonistic drugs such as enzalutamide. Our study would help to develop novel drug candidates against PCa.
Table of Contents
Introduction
Androgen receptor (AR) is activated by the binding of androgenic hormones, testosterone, or dihydrotestosterone (DHT). AR takes part in the regulation of expression of many important genes, which are critical for the development and maintenance of the male sexual phenotype in the healthy people.1 However, AR would promote the proliferation of cancer cells in the prostate cancer (PCa) patients.2–4 Inhibiting the activation of AR by antagonistic drugs has been regarded as a promising way to treat PCa.5, 6
As a member of nuclear receptors (NRs) family, the transcriptional activity of AR depends on the recruitments of steroid receptor coactivators (SRCs) to its ligand binding domain (LBD).7, 8 The unique coactivators recruited by AR contain the aromatic-rich motif (FXXLF) instead of the leucine-rich motif (LXXLL) for most other NRs.9, 10 Accumulated evidences showed that the transcriptional activity of AR, which is crucially important in the development of PCa, would be inhibited by breaking the AR-coactivator interactions.11–13 Understanding the detailed molecular mechanism of AR-coactivator interactions would provide deeper pharmacological insights into the development of therapies for the treatment of PCa.
The short hydrophobic residue-rich motif of coactivators interacts with the so-called activation function (AF-2) region within the AR-LBD.14 AF-2 is a hydrophobic groove on the surface of the AR-LBD that is formed by a number of residues in helices 3, 4, and 12 (Figure 1A and 1B).15, 16 The structure of AF-2 is regulated by the binding of ligands to AR. For instance, the binding of agonistic AR ligands, such as dihydrotestosterone and methyltrienolone (R1881), would induce the formation of a suitable conformation on the AF-2 region for the recruitment of coactivators and amplifying the activity of AR.17, 18 On the other side, the binding of AR antagonistic drugs, such as flutamide, bicalutamide and the second generation antiandrogen drug enzalutamide (MDV3100), to the AR LBD on the similar binding site for agonists would induce AF-2 to form a different conformation compared with the agonists bound ARs.19–21
Figure 1.
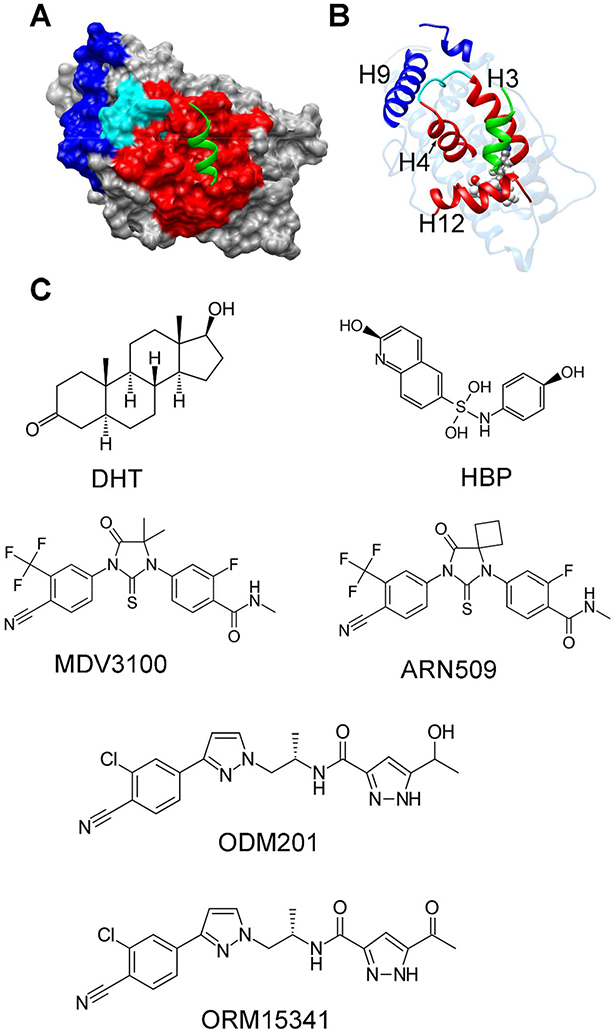
(A) The crystallographic structure of AR in complex with the coactivator peptide (PDB ID: 3V4934), AR was represented by surface and the coactivator was represented by green ribbon. The AF-2 groove on AR was colored in red, the BF-3 groove was colored in blue and the boundary between AF-2 and BF-3 was colored in cyan. (B) The same structure in (A) but was shown in ribbon. (C) The chemical structures of the ligands studied in this work.
Plenty of studies demonstrated that ligands would modulate the stability and integrity of H12.22–25 H12 takes part in the formation of the AF-2 groove after the structural rearrangement induced by the hormone binding, which provides a molecular mechanism for the regulation between ligands and AR coactivators.26 However, there still remain many unsolved problems. For instance, while the FXXFL motif of coactivators mainly interacts with H3 and H4 of AF-2 according to the X-ray crystallographic structures (Figure 1), how does the structural change of H12 induced by ligand-binding affect the interactions between coactivators and AR? In addition to the AF-2 site, the BF-3 function site was found to be important for the activity of AR.27, 28. Are there any communications between AF-2 and BF-3? Besides, some single mutations on the AR-LBD would convert antagonistic ligands into agonists and therefore confer drug resistance, e.g. the drug tolerance induced by the T877A mutation to hydroxyflutamide (HFT) and the F876L mutation to enzalutamide (MDV3100).29, 30 If the antagonist-agonist conversions are also related to the AR-coactivator interactions? Until now, computational studies have made great efforts to understand the molecular mechanisms of AR-ligand recognition and AR activity. For instance, Xu et al. proposed that the DHT binding would increase the stability of ligand binding pocket and the structure of LBD.31 Qsguthorpe and Hagler studied the antagonistic mechanism of bicalutamide (Bcu) based on molecular dynamics (MD) simulations and quantum mechanical (QM) calculations.32 By combining with MD simulations and mutation experiments, Korpal et al. found that the benzamide motif of enzalutamide extends away from H12 if F876 is mutated to a small leucine based on the computational docking models.29 Liu et al. demonstrated that the interactions between the C-ring of enzalutamide and H12 play important roles in the transcription activity of AR by simulating the wild-type (WT) AR and its mutants.33 Although progresses have been achieved, the detailed mechanism about how the agonistic/antagonistic ligands regulate the AR-coactivator communications still remains unclear.
In this study, a serial of μs-long MD simulations were performed on the AR-LBDs in complex with a coactivator peptide, including the apo-AR-LBD without ligand, the AR-LBDs with two agonistic ligands (DHT and HBP) and the AR-LBDs with four antagonistic ligands (MDV3100, ODM201, ARN509 and ORM15341). The simulations revealed that the ligand binding would regulate the interactions between the AR AF-2 groove and the coactivator motif, i.e. the binding of agonistic ligands would facilitate the AF-2/coactivator interactions, and the antagonistic ligands would depress such interactions. Further analyses show that the allosteric regulation of ligands to the coactivator binding obeys the ligand-H12-H3/H4-coactivator pathway. The antagonist-to-agonist conversions of the ligands MDV3100 and ODM201 by single mutants were also modulated by the above allosteric pathway. Besides, the quantitative binding free energies between the coactivator and different AR-LBD systems were calculated by Molecular Mechanics/Generalized Born Surface Area (MM/GBSA). Our results revealed the detailed mechanism of how ligands regulate the coactivator recruitment of AR, and provided clues to rational pharmacological intervention to inhibit the activity of AR.
Methods
Ligands Docking and Initial Structure Models.
MD simulations were performed on a total of 11 systems, including seven WT AR-LBD systems, including the apo-AR-LBD without ligand, the AR-LBD bound with the agonistic ligand (DHT or HBP), and the AR-LBD bound with the antagonistic ligand (MDV3100, ODM201, ARN509 or ORM15341) and four AR mutants, including the F876L mutated AR-LBD bound with MDV3100, the T877A mutated AR-LBD bound with MDV3100, the F876L mutated AR-LBD bound with ODM201, and the T877A mutated AR-LBD bound with ODM201. The human WT AR-LBD in complex with a selective androgen receptor modulator PK0950 and an AR coactivator peptide (GAFQNLFQSVR) were selected as the initial template in the following molecular docking calculations (PDB entry: 3V4934). The initial structures for MD simulation were generated by the Glide module35 implemented in Schrödinger 9.0. F876L/T877A mutants of AR-LBD was established and optimized through the Build and Edit Protein module in Discovery Studio 2.5. For the protein structures, the Protein Preparation Wizard 36 in Schrödinger 9.0 was used to remove crystallographic waters, ions and DTT, add hydrogens, fix bond orders, assign partial charges with the OPLS force field, and minimize the structures until the root-mean-square deviation (RMSD) reached a maximum value of 0.3 Å. The structures of all ligands were processed by using the LigPrep module in Schrodinger 9.0. For each compound, the ionized states were generated by using Epik at pH=7.0 ±2.0. For each protein, the binding box with the size of 10 Å × 10 Å × 10 Å centered on the co-crystallized ligand was generated by using the Receptor Grid Generation component of Glide. The van der Waals radius scaling factor and the partial atomic charge cutoff were set to 1.0 and 0.25, respectively. All the prepared structures of all six ligands were docked into the WT or F876L/T877A mutated AR-LBD and scored by the extra precision (XP) scoring mode. The binding pose with the best docking score for each molecule was chosen for the following simulations. The above docking strategy was validated by re-docking PK0950 into the crystallographic structure. The re-docking RMSD of the ligand is 0.32 Å, demonstrated the high reliability of the docking scheme we used.
MD Simulation Protocols.
The AMBER99SB-ILDN force field37 and the general amber force field (gaff)38 were used for the proteins and ligands, respectively. The TIP3P water molecules were added as the solvent and the solute atoms were at least 12 Å away from the boundary of the rectangular box. The counter ions, i.e. chlorine atoms, were added to neutralize the net charge of each system. The long-range electrostatic interactions were handled by the particle mesh Ewald (PME) algorithm39, and the non-bonded cutoff for the real-space interactions was set to 10 Å. A hybrid protocol of the steepest decent method and the conjugate gradient method was employed to do the minimization. Ten thousand steps of steepest decent minimization with the restraint (the force constant of 100 kcal/mol·Å2) on the protein and ligand were firstly performed, and then the conjugate gradient method without any restraint was used until a maximum 20000 iteration steps reached or the convergence criterion (the root-mean-square of the energy gradient is less than 1.0×10−4 kcal/mol·Å) was satisfied. The systems were heated up from 0 K to 300 K linearly over the time period of 100 ps with the restraint (force constant of 10 kcal/mol·Å2) on the solute in NVT ensemble, and then the systems were equilibrated without restraint for 1 ns with Langevin thermostat40 in the NPT (P = 1 atm and T = 300 K) ensemble. Finally, the 1-μs production runs were carried out by CUDA-version Amber14 41 in the NPT (P = 1 atm and T = 300 K) ensemble. The SHAKE algorithm42 was used to restrain the covalent bonds between heavy atoms and hydrogen atoms, and the time step was set to 2 fs. The snapshots were saved every 20 ps and the conformations in the last 200 ns trajectories were used in the following analysis if no additional annotation. The RMSDs as the function of simulation time for the AR bound with coactivator, AF-2 bound with coactivator and ligand were given in Figures S1, S2 and S3, respectively. The RMSDs are well converged in the latter part of all the simulations, and the RMSD fluctuations in the last 200 ns are less than 1 Å.
Fraction of Native Contact Analysis.
The fraction of native contacts (Q) analysis43 was employed to evaluate the differences of residue interactions in the query structures to a given structure, which is usually the native structure, i.e. the X-ray or NMR experimental structure. For the residues that are more than three residues apart in sequence, their heavy atoms (each from a different residue) are considered to be in contact with each other if the distance between the atoms is less than 4.5 Å. The total number of the heavy-atom pairs that are in contact in the native state is N in Eq. 1. The fraction of the native contacts for a given snapshot s is defined as 43:
| (1) |
Where is the distance between heavy atoms i and j in the native structure and rij(S) is the distance between the same atom pair in snapshot S. The smoothing parameter β is set to 5 Å-1. and the contact fluctuation is taken into account by the parameter λ=1.8. The summation runs over the N pairs. The definition of fraction of native contacts for residue k in snapshot S is similar to Q for the whole protein,44 but only the contacts associated with residue k are calculated:
| (2) |
where Nk is the number of heavy-atom pairs in native contact between residues k and other residues (> 3 residues apart in sequence). If there is no heavy atom within 4.5 Å to the heavy atoms of residue k in the native structure, then Qk(S) = 0.
Binding Free Energy Calculation.
The binding free energy between the coactivator and the AR-LBD was calculated by the MM/GBSA approach45:
| (3) |
| (4) |
| (5) |
| (6) |
where ΔEint (intra-molecular interactions, including bond, angle, and dihedral energies) in Equation 5 can be completely canceled because the single trajectory strategy was used for the MM/GBSA calculations. The non-polar part of the solvation free energy (ΔGSA) was calculated by the solvent-accessible surface area (SASA) through the LCPO algorithm:46 ΔGSA=γ×SASA+β, where the surface tension constant γ and β were set to 0.0072 and 0, respectively. The polar part of the solvation energy (ΔGGB) was estimated by the Generalized Born (GB) model proposed by Onufriev et al. (GBOBC1, igb=2).47 The interior and exterior dielectric constants were set to 4 and 80, respectively. The ΔEvdw, ΔEele, ΔGGB and ΔGSA terms were computed based on the 2000 snapshots extracted from the last 200 ns MD trajectories. However, due to the expensive computational cost ofnormal mode analysis (NMA), 50 snapshots evenly extracted from the last 200 ns MD trajectories were used to calculate the conformational entropies with the MMPBSA.py 48 module in Amber14.
Results and Discussion
Ligand Binding Regulates the Configurations of AF-2 and Coactivator.
he formation of a special configuration of the functional AF-2 region is necessary for the activation of AR. It was proposed that the binding of antagonistic ligands might block the interactions between the AF-2 function site and coactivators.9 The detailed structural changes of AF-2 induced by different ligands were demonstrated by the simulations in this study. As shown in Figure 2, the representative structures of AF-2 in the different ligand bound ARs were overlapped with the X-ray crystallographic structure (PDB id: 3V49). The agonistic ligands (DHT and HBP) bound structures were given in Figure 2A, and the antagonistic ligands (MDV3100 and ODM201) bound ARs and the AR without ligand binding were given in Figure 2B. The structures of the AF-2 region of the agonistic ligand bound ARs are basically identical to that of the X-ray structure, which would facilitate the binding of coactivators (Figure 2A). On the other side, there are many differences on the AF-2 scaffold structure of the inactivate ARs compare with that of the X-ray structure, for instance, the breaking of the helix structure on the middle of H12 changes the orientation of the C-terminus of H12. Besides, the loop region between H3 and H4 shifts away from the position in the x-ray structure (Figure 2B).
Figure 2.

Comparisons of the AF-2 grooves and coactivators between the simulated structures and experimental structure. The conformations in the last 200 ns trajectories were classified into structure clusters based on the heavy-atom RMSDs. The AF-2 structures of the largest cluster center were selected as the representative structures. (A) Structure overlap of AF-2 bound with agonistic ligands (DHT: colored in cyan; HBP: colored in magenta) against the X-ray structure (colored in gray); (B) Structural overlap of AF-2 of the ODM201 bound AR (colored in cyan), MDV3100 bound AR (colored in magenta) and apo-AR (colored in green) against the X-ray structure (colored in gray). The N- and C-terminus of H12 were labeled.
Compared with the experimental structure, the overall RMSDs of the agonistic ARs are less than 3 Å, and those of the AF-2 regions in the DHT- and HBP-bound ARs are around 2 Å during the simulations (Figure 3). However, the AF-2 RMSDs of the antagonistic ARs are around 4 Å (Figure 3), which demonstrate the structural changes of AF-2 on ARs bound with antagonistic ligands during the simulations. The rearrangement of the C-terminus of H3, H4 and H12 altered the surface of the AF-2 groove, which might reduce the coactivator recruitment ability. As shown in Figure 2B, the helical structures in the C-terminus of coactivators unfolded and the coactivators preferred to depart from the ARs in the antagonistic ARs during the simulations. In fact, the modulation of the coactivator binding might be a common feature in the nuclear receptors, for instance, it was revealed that many ligands have the ability to inhibit the binding affinity of coactivator peptides to estrogen receptor (ER)49 or vitamin D receptor (VDR)50.
Figure 3.
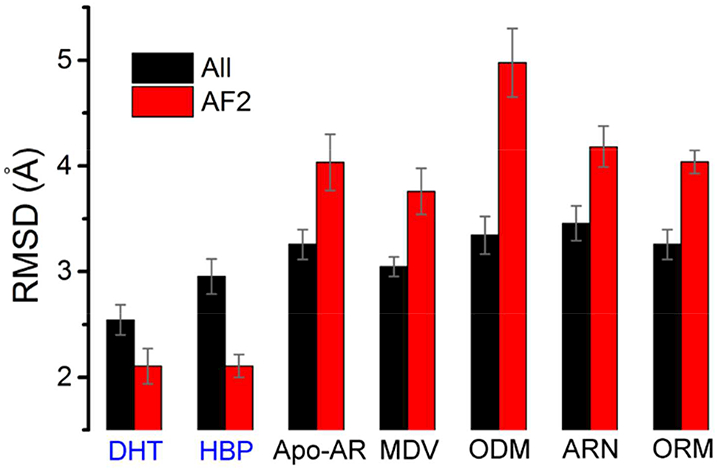
The overall RMSD and AF2 RMSD. The overall RMSD considered all the heavy atoms on AR-LBD and its coactivator except the ten N-terminal residues. The AF2 RMSD took into account the residues 709–741 (H3-H4), 892–907 (H12) and the coactivator peptide. The average RMSD values were calculated over the last 200 ns trajectories. The error bars give the errors during the last 200 ns simulations. The label of the agonistic ARs were colored in blue. The crystallographic structure (PDB ID: 3V4934) was used as the reference structure.
Ligands Regulate the Residue Interactions in AF-2.
Fraction of native contact (Q) was widely used in protein folding studies and protein structure analysis. The Q value calculates the residue contact score in the given conformation compared with a specific structure, i.e. the native structure (Eq. 1).43 And the fraction of residue native contact Qk characterizes the native-ness of residues in the dynamic conformations (Eq. 2).44 In this study, the X-ray structure of the agonistic AR (PDB id: 3V49) was used as the native structure, and the fraction of residue native contacts (Qk) was calculated to show the residue interaction properties in different ligand-bound ARs. Figure 4 gave the <Qk> on the residues of DHT-AR, apo-AR, ODM201-AR and MDV3100-AR, respectively. The angle bracket means the average value over the conformations of the simulations. The range of <Qk> is from 0 to 1. <Qk> = 1 means that the residue k in the simulated structure has the identical residue-interaction relationship with the corresponding residue in the X-ray structure. On the other side, <Qk> = 0 means that the native contacts are totally lost during the simulations for the residue k. In Figure 4, the residues were colored based on their Q values, i.e. the residues with small <Qk> were colored in red and the residues with large Q were colored in blue. For the agonistic form DHT-AR (Figure 4A), most residues have the <Qk> values close to 1, suggesting that the structure and the residue interactions for the agonistic AR during the simulations were similar to those for the crystallographic structure. However, many of the residues in the antagonistic ARs have very low <Qk> (Figure 4C and Figure 4D). The low Qk residues are mainly located on H3, L3 (the loop linking H3 and H4), H4 and H12 (Figure 4 and Figure S4). Since the AF-2 groove is consisted by H3, H4 and H12, the analysis of the Q values demonstrated that the ligand-binding greatly influences the residue interactions on the AF-2 region.
Figure 4.
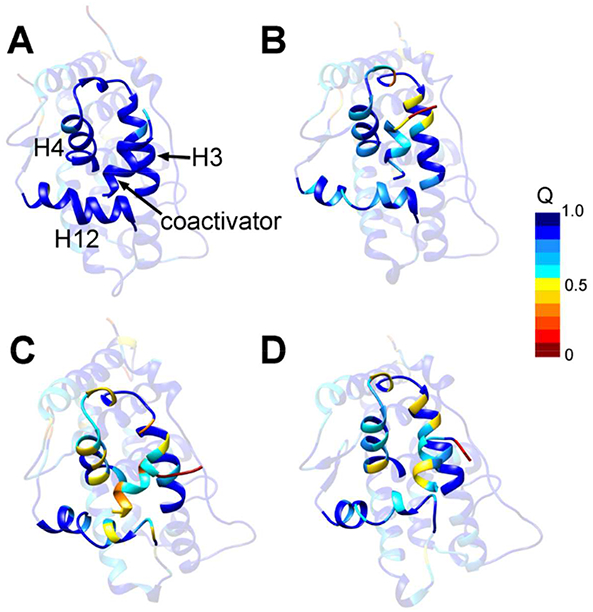
The fraction of the native contact for the residues on various ligand bound AR. (A) AR bound with DHT; (B) Apo-AR; (C) AR bound with ODM201; (D) AR bound with MDV3100. To highlight the AF-2 groove (C-terminus of H3, H4 and H12) and the coactivator, the other regions were transposed. The color scale of <Qk> values is shown in Figure.
The residues which have low <Qk> values in the antagonistic AR and high <Qk> values in the agonistic AR were listed in Figure S5, i.e. the residues 716, 717 and 720 on H3, 726–729 on L3, 731, 732, 734 and 738 on H4, 752 on H5, 823 and 826 on H9, 891, 894, 897 and 898 on H12. As an example, Figure 5 gave the interactions of the residue K720 in different ligand bound ARs. K720 was proved to be a critical residue to the FXXLF coactivator peptide binding based on different experimental evidences.51–53 The <Qk> values of K720 are around 0.5 on the non-active ARs, i.e. the apo-AR or AR bound with the antagonistic ligands (Figure 5A). In the DHT-bound agonistic AR, K720 was buried by the residues V715, F725, R726, V730, Q733 and I737 on the AR-LBD and the residues on the coactivator (Figure 5B). However, most of the contacts with K720 were lost in the ODM201-bound antagonistic AR, and K720 was exposed to the solvent (Figure 5C).
Figure 5.
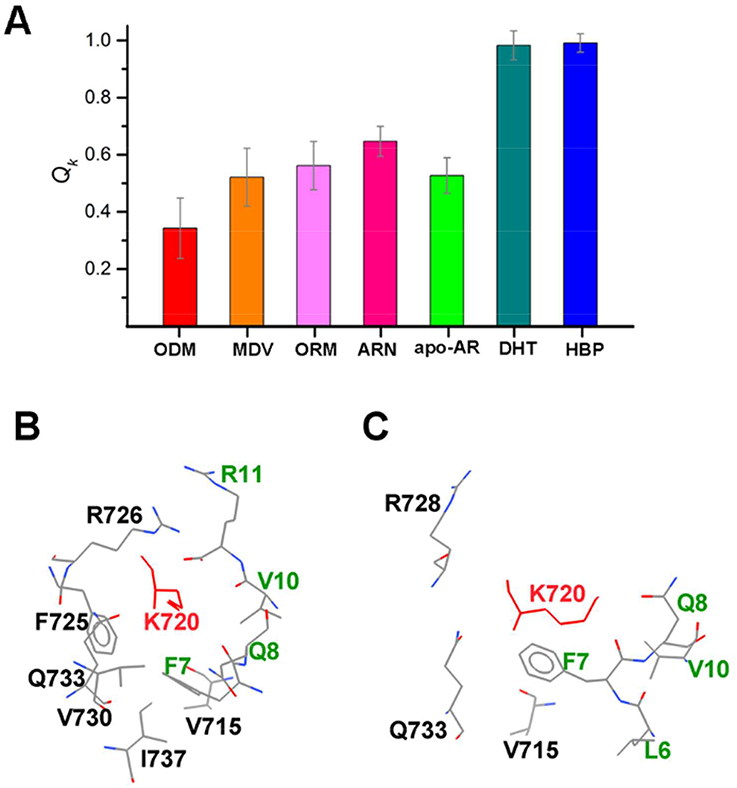
The residue interactions of the residue K720. (A) <Qk> values of K720 in different ligands-bound ARs. (B) The residue interactions K720 in the DHT-bound AR. (C) The residue interactions K720 in the ODM201-bound AR. In (B) and (C), only long-range interactions (the sequence interval with K720 larger than 3) were given. The residue names and numbers were labeled, K720 was colored in red and residues in the coactivator were colored in green.
The residues with large <Qk> difference in the agonistic/antagonistic ARs revealed by our study might be critical to the activity of AR. Actually, our observations are consistent with the in vitro experimental transcriptional assays, which demonstrated that the single mutations of the residues 726, 727 and 826 would affect the transactivation activity or N-terminus and C-terminus communications of AR.54 To further demonstrate the importance of residues with larger <Qk> differences, we analyzed the residue conservation of the nuclear receptor family. The conserved residues are usually critical in the evolution to maintain the overall structures and functions of proteins. We aligned the sequence of the AR-LBD to those of other nuclear receptors, including the estrogen receptor (ER), glucocorticoid receptor (GR), mineralocorticoid receptor (MR) and progesterone receptor (PR). The results show that most of the residues with large <Qk> differences are conserved in these proteins (Figure S6). For instance, as shown in Figure 5 the Qk difference for the residue 720 between the antagonistic ARs and agonistic ARs is larger than 0.4, meanwhile, all the residues are lysine on the positions corresponding to the residue 720 in AR for all the other four nuclear receptors. The conserved residues with large <Qk> differences show their importance to the activity of the AR functions and might be potential target sites to design new antiandrogen drugs.
BF-3 Function Site Regulated by Ligand Binding.
BF-3 is another important protein-protein interaction site on the AR-LBD. Although only some small compounds were found to bind with BF-3, researches suggested its modulating role in AR activity by interacting with other proteins.27 BF-3 adjacent to AF-2, which is consisted by helix 1, helix 9 and L3 loop. L3 loop is the boundary between BF-3 and AF-2 and links H3 and H4. Functional assays show that mutations on many residues on BF-3 would affect the AR functions. We found that the ligand-binding might not change the overall structure and backbone configuration of the BF-3 site, and the helical structures on H9 were also observed in the antagonistic ARs. Nevertheless, the binding of antagonistic ligands would significantly affect the side-chain orientations and residue interactions of many residues in this region, especially on L3 loop. As shown in Figure S5, the <Qk> differences for the residues 726 and 727 in the agonistic and antagonistic ARs are around 0.4. Besides, the <Qk> values for the residues 823 and 829 on H9 in the antagonistic ARs are slightly lower (about 0.2) than those in the agonistic ARs. The orientation changes of the charged residues and hydrophobic residues might reduce the interaction of AR to other proteins and inhibit the activity of AR.
Allosteric Regulation Pathway from Ligands to Coactivators.
In our previous work, we demonstrated that the residues W741 and H874 play critical roles in the communication between ligands and AR-H12.25 The binding of antagonistic ligands would destabilize the structure of H12. In this study, our simulations further demonstrated the damaging of H12 integrality by the antagonistic ligands (Figure 2B). In addition, the allosteric mechanism between the ligands and the coactivator was uncovered. We found that the allosteric regulation follows the following pathway. (1) The structures of H12 were regulated by the binding of ligands. For the agonistic ligands, H12 was stabilized to be a good α-helix. For the antagonistic ligands or apo-AR, the C-terminus of H12 would move away from the ligands. (2) The orientation of the residues Q902 and K905 were regulated by the arrangement of H12. In the agonistic AR, the residues Q902 and K905 form interaction network with the polar/charged residues Q738 and W739 on the C-terminal of H5, and stabilize the structure of H5 (Figure 6A). However, in the antagonistic AR or apo-AR, the interactions between H5 and H12 were decreased due to the unstable structure of H12 (Figure 6B). (3) The increasing or decreasing of the interactions between residues on H12 and H5 would destabilize the structures of H4/H5. (4) The interactions between AF-2 and coactivator are controlled by the structures of H5 and H12. For the agonistic ARs, AF-2 contacts with the coactivator via the hydrophobic interactions between residues AR-F725, AR-I737 and F7, F3 on the coactivator motif, as well as the hydrogen bond between the K720 sidechain and K10 backbone on the coactivator motif (Figure 6A). However, the change of the AF-2 surface induced by antagonistic ligands would reduce the above hydrophobic interactions (Figure 6B), which might be the reason of the unfavorable binding between the antagonistic AR and coactivators.
Figure 6.
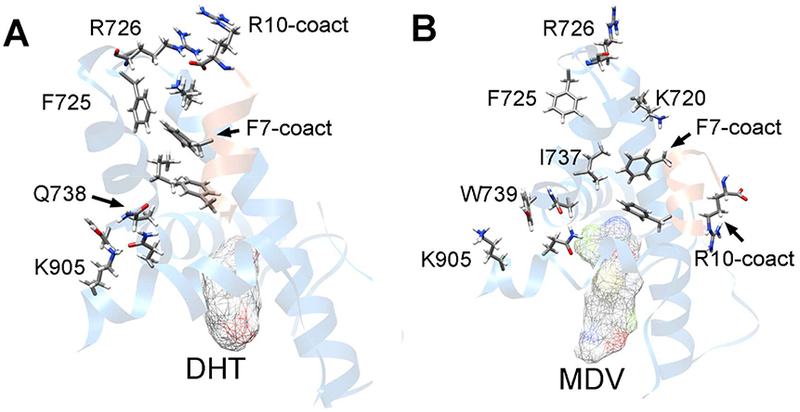
The allosteric pathway between ligand and coactivator. (A) The residue interactions in the allosteric pathway from DHT to the coactivator.; (B) The allosteric pathway between the MDV3100 and coactivator. The ligand-binding pocket and AF-2 with the coactivator were shown in ribbon. The ligands were represented by the meshed surface, ARs were colored in blue and coactivators were colored in orange. Some important residues were labeled and shown in stick. The conformations in the last 200 ns trajectories were classified into structure clusters based on the heavy-atom RMSDs. The AF-2 structures of the largest cluster center were selected as the representative structures.
Coactivator-AR Binding Energy Influenced by Ligand Binding.
The binding of antagonistic ligands decreased the interactions between residues in the AR-LBD and the C-terminus of its coactivator. Therefore, the residues in the coactivator especially its C-terminus departs from the AF-2 groove in the inactivated AR. To investigate the energetic implication between the AR and coactivator, the MM/GBSA analyses were performed on the MD trajectories. MM/GBSA was proved to be a powerful tool in the prediction of protein-ligand or protein-peptide binding free energies. Besides, the normal model analysis (NMA) technology was employed to calculate the entropy effects and characterize the contributions of the AR/coactivator fluctuation to binding free energies. The MM/GBSA and NMA calculation results are summarized in Table 1. In general, the binding free energies between the coactivator and agonistic ARs are lower than those between the coactivator and antagonistic ligands and those between the coactivator and apo-AR. The binding free energy between the coactivator and apo-AR was used as the reference, and the difference of the binding free energies (ΔΔGbinding) were also listed in Table 1. It can be seen that the ΔΔGbinding values are negative for the agonistic ARs (−1.4 kcal/mol for DHT-binding AR and −3.0 kcal/mol for HBP-binding AR), suggesting that the binding of agonistic ligands would promote the interaction between AR and its coactivator. On the other side, all the antagonistic ARs have the positive ΔΔGbinding values. Both the enthalpies and entropies contribute to the binding between the coactivator and ARs, though the entropies might have larger effects on the energy differences. Basically, the potential energy differences for the coactivator binding to the antagonistic ARs or the agonistic ARs come from the electrostatic interactions (ΔEele are around −60 kcal/mol for the binding of the coactivators to the agonistic ARs, which are much lower than those for the binding of the coactivators to the antagonistic ARs). Besides, the coactivators bind to the agonistic ARs have much higher solvation free energies (ΔGGB). The results indicate that the long-range electrostatic interactions might be critically important to the interactions and communication between the coactivators and ARs. It should be noticed that the binding free energies shown in Table 1 are only related to the interactions between the coactivator and ARs. Many other factors such as the ligand binding affinity would determine the AR activity, and therefore, ΔGbinding cannot one-to-one match with the quantitative values related to the AR activity, such as IC50 values.55
Table 1.
Binding free energies (unit in kcal/mol) between the coactivators and AR-LBDs.
| ODM_WT | MDV_WT | ORM_WT | ARN_WT | DHT_WT | HBP_WT | ODM_F876L | ODM_T877A | MDV_F876L | MDV_T877A | Apo_AR | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔEvdw | −51.5±3.8 | −50.1±4.2 | −51.9±5.6 | −52.9±3.9 | −52.8±3.6 | −52.0±3.3 | −51.2±4.7 | −49.7±5.0 | −54.9±4.8 | −51.0+5.0 | −47.2±3.9 |
| ΔEele | −55.9±4.1 | −55.4±6.3 | −50.6±4.9 | −51.7±4.8 | −57.4±4.3 | −60.6±4.0 | −56.1±5.2 | −50.6±5.1 | −59.2±5.6 | −55.9±5.7 | −52.1±5.1 |
| ΔGgb | 57.7±4.5 | 56.4±7.6 | 53.3±5.0 | 54.7±4.2 | 59.7+3.6 | 61.4±3.2 | 57.8±5.5 | 52.9±5.3 | 61.9±5.7 | 57.2±5.5 | 53.9±4.2 |
| ΔGsa | −5.8±0.3 | −5.8±0.3 | −5.9±0.3 | −5.8±0.4 | −5.9+0.3 | −5.7±0.4 | −5.6±0.3 | −5.4±0.3 | −6.1±0.4 | −5.7±0/4 | −5.8±0.3 |
| ΔEenthalpy | −55.6±4.4 | −55.1±6.8 | −55.1±5.3 | −55.7±4.8 | −56.4±4.1 | −56.9±3.5 | −55.2±4.9 | −52.8±4.9 | −58.3±5.3 | −55.4±5.6 | −53.4±4.5 |
| −TΔS | 35.7±4.1 | 36.0±3.7 | 35.4±3.5 | 35.2±3.3 | 33.1+4.1 | 32.0±4.0 | 35.6±3.9 | 33.1±4.3 | 35.7±4.1 | 34.8±5.5 | 31.5±4.6 |
| ΔGbinding | −19.8 | −19.1 | −19.7 | −20.5 | −23.3 | −24.9 | −19.6 | −19.7 | −22.6 | −20.6 | −21.9 |
| ΔΔGbindinga | 2.1 | 2.8 | 2.2 | 1.4 | −1.4 | −3.0 | 2.3 | 2.2 | −0.7 | 1.3 | 0.0 |
ΔΔGbinding(i)= ΔGbinding(i) - ΔGbinding(apo=AR)
Single Mutations Affect the Ligand-coactivator Allosteric Pathway.
Many mutations on AR-LBD induce the resistance to the antiandrogen therapy of PCa. For instance, the antagonistic ligand hydroxyflutamide (HFT) was found to be an agonist to the T877A AR. resistance to MDV3100 both in vitro and in vivo.20, 29 On the other side, as a AR inhibitor developed recently, ODM201 has shown its antagonistic effectivity on the T877A AR and F876L AR.55
In order to understand why a single mutation converts a AR ligand from antagonist to agonist, the effects of the T877A/F876L mutations on the structures of the AR bound with MDV3100 and ODM201 were studied. The results show that the binding of ODM201 would influence the AF-2 structures for both the F876L and T877A mutants of AR. The AF-2 RMSD of the ODM201 bound F876L and T877A mutants are 3.8 Å and 4.0 Å, respectively. The AF-2 RMSD of the MDV3100 bound T877A mutant is 3.5 Å, which is comparable with that of the MDV3100 bound WT AR (3.7 Å). However, the AF-2 RMSD for the MDV3100 bound F876L mutant is 2.3 Å, suggesting there is no large structural changes on the AF-2 groove for the T877A mutant compared with the agonistic X-ray structure. Figure 7 gives the overlapped AF-2 structures of the mutants to the agonistic experimental structure. It can be seen that the structure of the MDV3100 bound F876L mutant (colored in cyan in Figure 7A) is similar to the experimental structure, but the other mutants are distinct to the reference structure. Besides, the binding free energies of the coactivators in different AR mutants are consistent with their bioactivities, and the agonistic mutation F876L on the MDV3100 bound AR would reduce the binding free energy (Table 1).
Figure 7.

The structures of AF-2 bound with coactivator in the AR mutants. The conformations in the last 200 ns trajectories were classified into structure clusters based on the heavy-atom RMSDs. The AF-2 structures of the largest cluster center were selected as the representative structures. (A) Structure overlap of the MDV3100 bound T877A mutant (colored in magenta), MDV3100 bound F876L mutant (colored in cyan) and the X-ray structure (colored in gray); (B) Structure overlap of the ODM201 bound T877A mutant (colored in magenta), ODM201 bound F876L mutant (colored in cyan) and X-ray structure (colored in gray).
Similar to the WT AR, the communications between the ligands and the coactivator in the mutated ARs are mediated by the H12-H4/H3-coactivator allosteric pathway. The mutations such as the F876L on the MDV3100 bound AR might change the interactions in the allosteric pathway and therefore convert the protein from the antagonistic state to agonistic state. In the ligand binding pocket of the WT AR, MDV3100 was pushed toward to H12 by phenylalanine (F876) since the large volume of phenylalanine, and in consequence, the methylene group in the middle of MDV3100 has the chance to contact with the hydrophobic sidechain of the residue I899 on H12 (Figure 8A), which would break the helical structure of H12 and induce the following allosteric interaction in the H12-H4/H3-coactivator pathway. Nevertheless, the tail of MDV3100 has larger space to move away from H12 if F876 is mutated to smaller residues such as leucine (Figure 8B), and the ligand would not damage the helical structure of H12 as well as the agonistic configuration of the AF-2 interface. For the ligand ODM201, the long tail guarantees the ligand always contact with H12, and the mutations on H10 (such as T877A and F876L) would not influence the contacts in the allosteric pathway. Based on the above observations, we conclude that large ligands with long tails have higher probability to avoid the drug-tolerance induced by residue mutations on AR.
Figure 8.

The regulation of the F876L mutation to the ligand-H12 interactions. (A) The WT AR bound with MDV3100. (B) The F876L AR mutant bound with MDV3100. (C) The WT AR bound with ODM201. (D) The F876L AR mutant bound with ODM201. The hydrophobic residues on H12 were shown in stick and labeled. The sidechain of the residue 876 was shown in sphere, and the ligands were represented in ball and stick. The shortest distances between the heavy atoms on MDV3100 and I899 were given.
Conclusions
The communications between the AR-LBD and AR coactivator peptide are critical to the activation of AR. It is still unclear how the ligand binding would affect the AR-coactivator interactions. In this work, the MD simulations and MM/GBSA calculations were used to study the structural basis of the ligand binding and the mutations to the AR-coactivator communications.
The structure differences between agonistic ARs (bound with DHT or HBP), antagonistic ARs (bound with MDV3100, ODM201, ARN509 or ORM15341), as well as apo-AR were studied. The simulation results showed that the ligand binding mainly affects the structure of the function site AF-2, which is consisted by H3, H4 and H12. The ligands have some nontrivial effects on the overall scaffold of AF-2, and more importantly, the residue interactions in AF-2 are significantly regulated by the ligands. Besides, the interactions of several residues in the BF-3 interface, especially on the boundary of AF-2 and BF-3 are affected by the binding of ligands. The coactivator and AF-2 interactions follow a ligand-H12-H4/H3-coactivator allosteric pathway. The antagonistic ligands enhanced the binding free energy of coactivator with AR-LBD, while the agonistic ligands weakened the binding free energy of AR-coactivator with AR-LBD.
The structural basis of the drug-tolerance induced by point mutations on AR was investigated. We found that the activity-related mutations would affect the AR-coactivator allosteric pathway. The F876L mutation converted the MDV3100 bound AR from antagonist to agonist by changing the orientation of the MDV3100 tail and the interactions between ligands and H12, and therefore, AF-2 in the mutant would convert to the agonistic configuration. The mechanism of how ligand-binding affects the AR-coactivator communications and the structural basis of drug-tolerance provided in this study would help to develop novel PCa drug candidates.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by National Major Basic Research Program of China (2016YFA0501701, 2016YFB0201700), National Science Foundation of China (21773298, 21403291, 21575128, 81603031), and National Institutes of Health of USA (R01-GM079383, R21-GM097617).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publication website. Figure S1-Figure S3, the RMSD analysis of the MD trajectories. Figure S4, the fragment average fraction of native contacts; Figure S5, the fraction of native contacts of some important residues in different ligand bounded AR; Figure S6, sequence alignments of the ligand binding domain of nuclear receptors.
The authors declare no competing financial interest.
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
References
- 1.Gao W; Bohl CE; Dalton JT, Chemistry and Structural Biology of Androgen Receptor. Chem Rev 2005, 105, 3352–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heinlein CA; Chang C, Androgen Receptor in Prostate Cancer. Endocr Rev 2004, 25, 276–308. [DOI] [PubMed] [Google Scholar]
- 3.Debes JD; Tindall DJ, Mechanisms of Androgen-Refractory Prostate Cancer. N Engl J Med 2004, 351, 1488–1490. [DOI] [PubMed] [Google Scholar]
- 4.McCarthy N, Prostate Cancer: Studying the Classics. Nat Rev Cancer 2011, 11, 386. [DOI] [PubMed] [Google Scholar]
- 5.Montgomery RB; Mostaghel EA; Vessella R; Hess DL; Kalhorn TF; Higano CS; True LD; Nelson PS, Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth. Cancer Res 2008, 68, 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong YN; Ferraldeschi R; Attard G; de Bono J, Evolution of Androgen Receptor Targeted Therapy for Advanced Prostate Cancer. Nat Rev Clin Oncol 2014, 11, 365–376. [DOI] [PubMed] [Google Scholar]
- 7.Huang P; Chandra V; Rastinejad F, Structural Overview of the Nuclear Receptor Superfamily: Insights into Physiology and Therapeutics. Annu Rev Physiol 2010, 72, 247–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helsen C; Claessens F, Looking at Nuclear Receptors from a New Angle. Mol Cell Endocrinol 2014, 382, 97–106. [DOI] [PubMed] [Google Scholar]
- 9.van de Wijngaart DJ; Dubbink HJ; van Royen ME; Trapman J; Jenster G, Androgen Receptor Coregulators: Recruitment Via the Coactivator Binding Groove. Mol Cell Endocrinol 2012, 352, 57–69. [DOI] [PubMed] [Google Scholar]
- 10.van de Wijngaart DJ; van Royen ME; Hersmus R; Pike AC; Houtsmuller AB; Jenster G; Trapman J; Dubbink HJ, Novel Fxxff and Fxxmf Motifs in Androgen Receptor Cofactors Mediate High Affinity and Specific Interactions with the Ligand-Binding Domain. J Biol Chem 2006, 281, 19407–19416. [DOI] [PubMed] [Google Scholar]
- 11.van de Wijngaart DJ; Dubbink HJ; Molier M; de Vos C; Trapman J; Jenster G, Functional Screening of Fxxlf-Like Peptide Motifs Identifies Smarcd1/Baf60a as an Androgen Receptor Cofactor That Modulates Tmprss2 Expression. Mol Endocrinol 2009, 23, 1776–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu CL; Chen YL; Ting HJ; Lin WJ; Yang Z; Zhang Y; Wang L; Wu CT; Chang HC; Yeh S; Pimplikar SW; Chang C, Androgen Receptor (Ar) Nh2- and Cooh-Terminal Interactions Result in the Differential Influences on the Ar-Mediated Transactivation and Cell Growth. Mol Endocrinol 2005, 19, 350–361. [DOI] [PubMed] [Google Scholar]
- 13.van de Wijngaart DJ; Dubbink HJ; Molier M; de Vos C; Jenster G; Trapman J, Inhibition of Androgen Receptor Functions by Gelsolin Fxxff Peptide Delivered by Transfection, Cell-Penetrating Peptides, and Lentiviral Infection. Prostate 2011, 71, 241–253. [DOI] [PubMed] [Google Scholar]
- 14.Bennett NC; Gardiner RA; Hooper JD; Johnson DW; Gobe GC, Molecular Cell Biology of Androgen Receptor Signalling. Int J Biochem Cell Biol 2010, 42, 813–827. [DOI] [PubMed] [Google Scholar]
- 15.Shang Y; Myers M; Brown M, Formation of the Androgen Receptor Transcription Complex. Mol Cell 2002, 9, 601–610. [DOI] [PubMed] [Google Scholar]
- 16.He B; Kemppainen JA; Voegel JJ; Gronemeyer H; Wilson EM, Activation Function 2 in the Human Androgen Receptor Ligand Binding Domain Mediates Interdomain Communication with the Nh(2)-Terminal Domain. J Biol Chem 1999, 274, 37219–37225. [DOI] [PubMed] [Google Scholar]
- 17.Pienta KJ; Bradley D, Mechanisms Underlying the Development of Androgen-Independent Prostate Cancer. Clin Cancer Res 2006, 12, 1665–1671. [DOI] [PubMed] [Google Scholar]
- 18.Matsumoto T; Sakari M; Okada M; Yokoyama A; Takahashi S; Kouzmenko A; Kato S, The Androgen Receptor in Health and Disease. Annu Rev Physiol 2013, 75, 201–224. [DOI] [PubMed] [Google Scholar]
- 19.Mateo J; Smith A; Ong M; de Bono JS, Novel Drugs Targeting the Androgen Receptor Pathway in Prostate Cancer. Cancer Metastasis Rev 2014, 33, 567–579. [DOI] [PubMed] [Google Scholar]
- 20.Balbas MD; Evans MJ; Hosfield DJ; Wongvipat J; Arora VK; Watson PA; Chen Y; Greene GL; Shen Y; Sawyers CL, Overcoming Mutation-Based Resistance to Antiandrogens with Rational Drug Design. Elife 2013, 2, e00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran C; Ouk S; Clegg NJ; Chen Y; Watson PA; Arora V; Wongvipat J; Smith-Jones PM; Yoo D; Kwon A; Wasielewska T; Welsbie D; Chen CD; Higano CS; Beer TM; Hung DT; Scher HI; Jung ME; Sawyers CL, Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elhaji YA; Stoica I; Dennis S; Purisima EO; Lumbroso R; Beitel LK; Trifiro MA, Impaired Helix 12 Dynamics Due to Proline 892 Substitutions in the Androgen Receptor Are Associated with Complete Androgen Insensitivity. Hum Mol Genet 2006, 15, 921–931. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J; Liu B; Geng G; Wu JH, Study of the Impact of the T877a Mutation on Ligand-Induced Helix-12 Positioning of the Androgen Receptor Resulted in Design and Synthesis of Novel Antiandrogens. Proteins 2010, 78, 623–637. [DOI] [PubMed] [Google Scholar]
- 24.Nagata N; Kawai K; Nakanishi I, Subtle Structural Changes in Tetrahydroquinolines, a New Class of Nonsteroidal Selective Androgen Receptor Modulators, Induce Different Functions. J Chem Inf Model 2012, 52, 2257–2264. [DOI] [PubMed] [Google Scholar]
- 25.Duan M; Liu N; Zhou W; Li D; Yang M; Hou T, Structural Diversity of Ligand-Binding Androgen Receptors Revealed by Microsecond Long Molecular Dynamics Simulations and Enhanced Sampling. J Chem Theory Comput 2016, 12, 4611–4619. [DOI] [PubMed] [Google Scholar]
- 26.Kattoula SR; Baker ME, Structural and Evolutionary Analysis of the Co-Activator Binding Domain in Vertebrate Progesterone Receptors. J Steroid Biochem Mol Biol 2014, 141, 7–15. [DOI] [PubMed] [Google Scholar]
- 27.Estebanez-Perpina E; Arnold LA; Nguyen P; Rodrigues ED; Mar E; Bateman R; Pallai P; Shokat KM; Baxter JD; Guy RK; Webb P; Fletterick RJ, A Surface on the Androgen Receptor That Allosterically Regulates Coactivator Binding. Proc Natl Acad Sci U S A 2007, 104, 16074–16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grosdidier S; Carbo LR; Buzon V; Brooke G; Nguyen P; Baxter JD; Bevan C; Webb P; Estebanez-Perpina E; Fernandez-Recio J, Allosteric Conversation in the Androgen Receptor Ligand-Binding Domain Surfaces. Mol Endocrinol 2012, 26, 1078–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korpal M; Korn JM; Gao X; Rakiec DP; Ruddy DA; Doshi S; Yuan J; Kovats SG; Kim S; Cooke VG; Monahan JE; Stegmeier F; Roberts TM; Sellers WR; Zhou W; Zhu P, An F876l Mutation in Androgen Receptor Confers Genetic and Phenotypic Resistance to Mdv3100 (Enzalutamide). Cancer Discov 2013, 3, 1030–1043. [DOI] [PubMed] [Google Scholar]
- 30.Prekovic S; van Royen ME; Voet AR; Geverts B; Houtman R; Melchers D; Zhang KY; Van den Broeck T; Smeets E; Spans L; Houtsmuller AB; Joniau S; Claessens F; Helsen C, The Effect of F877l and T878a Mutations on Androgen Receptor Response to Enzalutamide. Mol Cancer Ther 2016, 15, 1702–1712. [DOI] [PubMed] [Google Scholar]
- 31.Xu X; Yang W; Wang X; Li Y; Wang Y; Ai C, Dynamic Communication between Androgen and Coactivator: Mutually Induced Conformational Perturbations in Androgen Receptor Ligand-Binding Domain. Proteins 2011, 79, 1154–1171. [DOI] [PubMed] [Google Scholar]
- 32.Osguthorpe DJ; Hagler AT, Mechanism of Androgen Receptor Antagonism by Bicalutamide in the Treatment of Prostate Cancer. Biochemistry 2011, 50, 4105–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H; Wang L; Tian J; Li J; Liu H, Molecular Dynamics Studies on the Enzalutamide Resistance Mechanisms Induced by Androgen Receptor Mutations. J Cell Biochem 2017, 118, 2792–2801. [DOI] [PubMed] [Google Scholar]
- 34.Nique F; Hebbe S; Peixoto C; Annoot D; Lefrancois JM; Duval E; Michoux L; Triballeau N; Lemoullec JM; Mollat P; Thauvin M; Prange T; Minet D; Clement-Lacroix P; Robin-Jagerschmidt C; Fleury D; Guedin D; Deprez P, Discovery of Diarylhydantoins as New Selective Androgen Receptor Modulators. J Med Chem 2012, 55, 8225–8235. [DOI] [PubMed] [Google Scholar]
- 35.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS, Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J Med Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 36.Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W, Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J Comput Aided Mol Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 37.Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE, Improved Side-Chain Torsion Potentials for the Amber Ff99sb Protein Force Field. Proteins 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA, Development and Testing of a General Amber Force Field. J Comput Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- 39.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG, A Smooth Particle Mesh Ewald Method. J Chem Phys 1995, 103, 8577. [Google Scholar]
- 40.Loncharich RJ; Brooks BR; Pastor RW, Langevin Dynamics of Peptides: The Frictional Dependence of Isomerization Rates of N-Acetylalanyl-N’-Methylamide. Biopolymers 1992, 32, 523–535. [DOI] [PubMed] [Google Scholar]
- 41.Gotz AW; Williamson MJ; Xu D; Poole D; Le Grand S; Walker RC, Routine Microsecond Molecular Dynamics Simulations with Amber on Gpus. 1. Generalized Born. J Chem Theory Comput 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lambrakos S; Boris J; Oran E; Chandrasekhar I; Nagumo M, A Modified Shake Algorithm for Maintaining Rigid Bonds in Molecular Dynamics Simulations of Large Molecules. Journal of Computational Physics 1989, 85, 473–486. [Google Scholar]
- 43.Best RB; Hummer G; Eaton WA, Native Contacts Determine Protein Folding Mechanisms in Atomistic Simulations. Proc Natl Acad Sci U S A 2013, 110, 17874–17879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duan M; Liu H; Li M; Huo S, Network Representation of Conformational Transitions between Hidden Intermediates of Rd-Apocytochrome B562. J Chem Phys 2015, 143, 135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kollman PA; Massova I; Reyes C; Kuhn B; Huo S; Chong L; Lee M; Lee T; Duan Y; Wang W; Donini O; Cieplak P; Srinivasan J; Case DA; Cheatham TE 3rd, Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc Chem Res 2000, 33, 889–897. [DOI] [PubMed] [Google Scholar]
- 46.Weiser J; Shenkin PS; Still WC, Approximate Solvent-Accessible Surface Areas from Tetrahedrally Directed Neighbor Densities. Biopolymers 1999, 50, 373–380. [DOI] [PubMed] [Google Scholar]
- 47.Onufriev A; Bashford D; Case DA, Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins 2004, 55, 383–394. [DOI] [PubMed] [Google Scholar]
- 48.Miller BR 3rd; McGee TD Jr.; Swails JM; Homeyer N; Gohlke H; Roitberg AE, Mmpbsa.Py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput 2012, 8, 3314–3321. [DOI] [PubMed] [Google Scholar]
- 49.Gunther JR; Du Y; Rhoden E; Lewis I; Revennaugh B; Moore TW; Kim SH; Dingledine R; Fu H; Katzenellenbogen JA, A Set of Time-Resolved Fluorescence Resonance Energy Transfer Assays for the Discovery of Inhibitors of Estrogen Receptor-Coactivator Binding. J Biomol Screen 2009, 14, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nandhikonda P; Lynt WZ; McCallum MM; Ara T; Baranowski AM; Yuan NY; Pearson D; Bikle DD; Guy RK; Arnold LA, Discovery of the First Irreversible Small Molecule Inhibitors of the Interaction between the Vitamin D Receptor and Coactivators. J Med Chem 2012, 55, 4640–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He B; Gampe RT Jr.; Kole AJ; Hnat AT; Stanley TB; An G; Stewart EL; Kalman RI; Minges JT; Wilson EM, Structural Basis for Androgen Receptor Interdomain and Coactivator Interactions Suggests a Transition in Nuclear Receptor Activation Function Dominance. Mol Cell 2004, 16, 425–438. [DOI] [PubMed] [Google Scholar]
- 52.He B; Minges JT; Lee LW; Wilson EM, The Fxxlf Motif Mediates Androgen Receptor-Specific Interactions with Coregulators. J Biol Chem 2002, 277, 10226–10235. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y; Mantravadi PK; Jobbagy S; Bao W; Koh JT, Antagonizing the Androgen Receptor with a Biomimetic Acyltransferase. ACS Chem Biol 2016, 11, 2797–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buzon V; Carbo LR; Estruch SB; Fletterick RJ; Estebanez-Perpina E, A Conserved Surface on the Ligand Binding Domain of Nuclear Receptors for Allosteric Control. Mol Cell Endocrinol 2012, 348, 394–402. [DOI] [PubMed] [Google Scholar]
- 55.Moilanen AM; Riikonen R; Oksala R; Ravanti L; Aho E; Wohlfahrt G; Nykanen PS; Tormakangas OP; Palvimo JJ; Kallio PJ, Discovery of Odm-201, a New-Generation Androgen Receptor Inhibitor Targeting Resistance Mechanisms to Androgen Signaling-Directed Prostate Cancer Therapies. Sci Rep 2015, 5, 12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.