

Abstract
The NEDD8 pathway is a known activator of the ubiquitin-protease system, a complex partially responsible for the degradation of proteins involved in cell-cycle regulation and neoplastic growth. In this study, we evaluated the anti-tumor potential of MLN4924 (pevonedistat); a potent NEDD8 inhibitor. We hypothesized MLN4924 treatment induces apoptosis in human melanoma cells. A375 and Mel39 BRAF V600E mutant melanoma cell lines were treated in vitro with MLN4924 alone or in combination with interferon-alfa (IFN-α) or vemurafenib – therapeutic agents utilized on melanoma patients. Annexin/PI flow cytometry analysis showed treatment with MLN4924 for 72 hours induced apoptosis in A375 and Mel39 melanoma cells with an IC50 of 1200nM and 143nM, respectively. Combination therapy of A375 cells with 104 Units/mL IFN-α and 1200nM MLN4924 led to a significant increase in cell death (78.2±3.7%) compared to single agent treatment by IFN-α (17.5±2.5%) or MLN4924 (50.7±1.0%; p<0.005). Treatment of A375 cells with 1 μM vemurafenib had a notable effect on cell viability. However, the addition of MLN4924 to vemurafenib had an inhibitory effect on apoptosis. Results from MTS proliferation assays indicate MLN4924 has anti-proliferative effects on melanoma cells in vitro, with the addition of IFN-α further inhibiting proliferation. Pre-treatment with MLN4924 led to A375 cell sensitization to vemurafenib treatment and immunoblot analysis of MLN4924-treated cells revealed cleavage of caspases-3, -7, -9 and poly-ADP-ribose polymerase (PARP). These results demonstrate MLN4924 does have efficacy in treating melanoma in vitro alone or in combination with IFN-α, and thus it may have potential use in patients with advanced melanoma.
Keywords: MLN4924, Pevonedistat, Melanoma, NEDD8 Inhibitor, Interferon-alfa, Vemurafenib
INTRODUCTION
Melanoma is one of the deadliest forms of cancer, with an average 10–15% 10-year survival rate for stage 4 disease [1]. While melanoma composes about 1% of all skin cancer cases in the United States, it accounts for the vast majority of skin cancer related deaths. Incidence rates of melanoma have been steadily increasing for the last 30 years, and in 2017 there will be an estimated 87,000 new melanoma cases, leading to an estimated 9,730 deaths [2]. While traditional treatments include surgical removal of the tumor, radiation therapy and chemotherapy, a new form of therapy in advanced cases is the use of immune-based treatments. The mechanism by which the immune system functions involves an intricate system of effector cells along with checks and balances that prevent misguided attacks by immune cells on normal healthy cells. Unfortunately, malignant cancers (including melanoma) take advantage of these mechanisms by secreting cytokines and other factors that suppress the anti-tumor immune response, thus allowing the tumor cells to grow and proliferate [3]. Conversely, the administration of immune stimulatory cytokines such as interferon-alfa and interleukin-2 can boost the immune system’s ability to mount an anti-tumor response [4]. Many new immunotherapeutic strategies focus on increasing the ability of the host lymphocytes to attack the malignant cells. Some of these therapeutics are immune checkpoint inhibitors such as neutralizing antibodies specific for inhibitory proteins on T cells. PD-1 and CTLA-4 are examples of clinically important T cell checkpoints. While these therapeutics have shown efficacy in the treatment of metastatic melanoma, detrimental side-effects can occur and resistance to therapy sometimes develops [5]. Therefore, new treatments with the ability to boost the effectiveness of immunotherapy are still needed in metastatic melanoma [6].
Advances in our understanding of the molecular basis of melanoma growth and metastasis have also occurred in the past decade. We now know that about 50% of melanoma tumors contain a mutation in the proto-oncogene BRAF, which causes continuous activation of the mitogen activated protein kinase (MAPK) signal transduction pathway and ultimately leads to increased proliferation and resistance to cell death (apoptotic) signals [7]. The BRAF-inhibitor vemurafenib was approved in 2011 and demonstrated initial success in containing disease progression. Unfortunately, resistance to vemurafenib was observed in about 50% of cases after an average of seven months. While the mechanism is not fully understood, resistance may involve the re-activation of the MAPK pathway via alternate signaling systems [8].
Interferon-alfa (IFN-α) is an immune cytokine that has been used in the treatment of melanoma, primarily as an adjuvant following surgery for advanced tumors. However, previous studies have demonstrated that IFN treatment of patients with metastatic melanoma leads to complete or partial regression of disease in about 10–15% of cases [9]. Thus, IFN-α has the potential to reduce tumor burden and the development of strategies to enhance its actions seem justified. A recent study from our group demonstrated that combination treatment of melanoma cells with IFN-alfa and the novel UPS inhibitor ixazomib (MLN2238) led to enhanced levels of apoptosis [10]. A previous study performed by our group showed that IFN-alfa can act synergistically with the proteasome inhibitor bortezomib through enhancement of apoptosis via the Fas-FADD pathway. Notably, the combination treatment of IFN-alfa and bortezomib exhibited synergistic anti-tumor activity in multiple murine models of melanoma [11].
Our group has studied the pathways in melanoma that lead to degradation of proteins that are important for melanoma survival. Inhibition of these pathways could lead to reduced tumor cell viability. The NEDD8 protein (81-amino acids in length) is the sole activator of the NEDD8 pathway; a complex mechanism that leads to the targeted degradation of multiple proteins through utilization of the ubiquitin-protease system (UPS) [12]. The NEDD8 pathway is initiated upon conjugation of NEDD8 protein to the NEDD8 activating enzymes (NAE). At this point, a cascade of trans-neddylation reactions occurs, transferring NEDD8 from the NAE-NEDD8 complex to one of multiple possible E2 complexes, and subsequently transferring it to a family of E3 ubiquitin ligases that are otherwise known as cullin-RING ligases (CRLs). A neddylated CRL can then conjugate a chain of ubiquitin molecules to a substrate, thereby targeting it for proteasomal degradation. In cancer cells, the proteasome-mediated degradation of proteins is skewed toward species that enhance cell growth, survival and proliferation [12]. Studies have shown that CRLs are responsible for ubiquinating an estimated 20% of proteins targeted for degradation through the UPS [13]. If neddylation targets a protein in a pathway that is important for survival of a malignant cell (e.g., anti-apoptotic mechanisms) then the NEDD8 pathway could be seen as playing an important role in maintaining cell viability and growth in cancer.
The novel NEDD8 activating enzyme (NAE) inhibitor MLN4924 (pevonedistat) works via inhibition of the NEDD-8 pathway. It inhibits the formation of the NAE-NEDD8 complex, thereby preventing the cascade of reactions that leads to the degradation of oncogenic proteins via the recruitment of the UPS. A study in urothelial carcinoma found that MLN4924 was able to suppress cell proliferation and migration [14]. Several ongoing studies have found MLN4924 to have efficacy as a therapeutic agent in advanced cancer, but the effects of this drug on patients with malignant melanoma remain to be seen. We hypothesize that treatment of melanoma cells with MLN4924 will lead to the degradation of anti-apoptotic proteins and the persistence of apoptotic stimuli within the cell, ultimately leading to cell death. We also hypothesize that MLN4924 will work synergistically with other melanoma therapeutics, such as IFN-alfa and vemurafenib, to further decrease cell viability.
METHODS
Materials
The A375 human melanoma cell line was purchased from the American Type Culture Collection (ATCC Manassas, Virginia). The A375 vemurafenib-resistant cell line was created in Dr. Carson’s laboratory over a period of time by exposing wild-type A375 to increasing doses of vemurafenib. The human metastatic melanoma cell line Mel39 was a gift from Soldano Ferrone (Harvard Medical School, Boston, MA) and cultured in DMEM media supplemented 10% FBS, L-glutamine, and penicillin/streptomycin. Melanoma cell line M308 was kindly provided by Dr. Antoni Ribas (University of California, Los Angeles) and was maintained in RPMI-1640 supplemented with 10% FBS, L-glutamine, and penicillin/streptomycin. MLN4924 (Velcade, PS-341) was obtained from Millennium Pharmaceuticals, Inc. (Cambridge, MA) and recombinant human IFN-alfa was obtained from Schering-Plough, Inc (Kenilworth, NJ). Vemurafenib was obtained from Selleckchem.
Annexin V/propidium iodine (PI) staining
Human melanoma cell lines A375 and Mel39 were plated in a 6-well plate at a concentration of 200,000 cells/well and treated with vehicle or MLN4924 for various amounts of time. Cells were harvested and apoptosis-induced phosphatidylserine exposure and cell membrane integrity was measured in tumor cells by flow cytometric analysis using APC-Annexin V and PE-propidium iodide (PI; BD Pharmingen, San Jose, CA), as previously described [16]. Each analysis was performed using 10,000 – 20,000 events and provides the percentage of the cell population that is viable or apoptotic [16]. The externalization of phosphatidylserine (PS) residues on the cell membrane occurs first during apoptosis. Annexin V has a high affinity for PS and monitors it once PS transfers from the inner membrane to the outer membrane. Propidium iodide is not able to penetrate viable cell membranes. Once the integrity of the cell membrane begins to degrade during apoptosis, propidium iodide can then penetrate the cell. The combination of these two agents together provides a useful assay to determine whether a cell population is viable, apoptotic, or necrotic [16]. The full method is described in Appendix A.
Immunoblot analysis
Immunoblots were prepared to probe with antibodies specific for caspase-3, cleaved caspase-3, caspase-7, cleaved caspase-7, caspase-9, poly(ADP-ribose) polymerase (PARP; Cell Signaling Technology, Danvers, MA), or β-actin (Sigma, St. Louis, MO). Following incubation with the appropriate horseradish peroxidase-conjugated secondary antibody, immune complexes were detected using an enhanced chemiluminescence detection kit (Thermo Scientific, Waltham, MA).
Proliferation assays
The proliferation of melanoma cells was measured as absorbance at 490 nm using the Cell Titer 96 Aqueous One Solution Cell Proliferation MTS Assay (Promega). All assays were performed in triplicate. Cells were also stained with trypan blue (Life Technologies, Carlsbad, CA) and live/dead cell counting was used to confirm proliferation assay results.
Statistical analysis
Statistical significance of differences between groups was analyzed by ANOVA and Student’s t-test, and p ≤ 0.05 was considered to be statistically significant. P values were adjusted for multiple comparisons using the Holm-Bonferroni method.
RESULTS
Treatment of BRAF (V600E) mutant human melanoma cell lines with MLN4924 leads to apoptotic cell death
The potential of MLN4924 to induce apoptosis was explored in the BRAF V600E mutant cell lines A375 and Mel39. Cell death was measured via Annexin V/PI staining over a time period of 72 hours. Since only sparse data was available prior to the start of this project, it was necessary to perform a titration over a wide range of concentrations to determine the IC50 of MLN4924 for each cell line. A375 cells underwent titration treatment, starting with an untreated (negative) control and proceeding with increasing concentrations of MLN4924 from 25 nM to 3200 nM (Figure 1A). Following the titration, A375 cells underwent another treatment at the calculated IC50 concentration of 1200 nM, yielding 54.1% cell death compared to untreated cells exhibiting 9.58% cell death (Figure 1B). Mel39 cells also underwent a titration with MLN4924 concentrations ranging from 50 nM to 400 nM (Figure 1C). The calculated IC50 concentration of MLN4924 in Mel39 was found to be 143 nM. Treatment of Mel39 cells at near IC50 concentration of 150 nM yielded 52.2% cell death compared to 14.9% cell death of control untreated cells (Figure 1D).
Figure 1. MLN4924 induces apoptosis in the BRAF mutant A375 and Mel39 human melanoma cell lines.
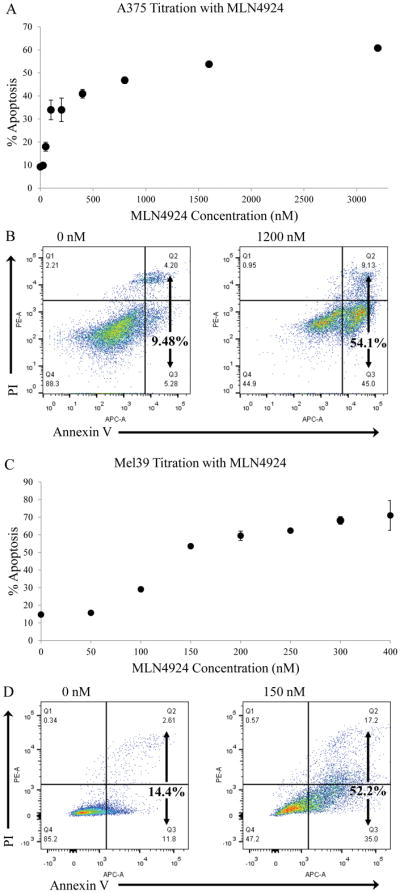
The A375 BRAF mutant (V600E) human melanoma cell line was treated for 72 hours with either vehicle or varying concentrations of MLN4924 ranging from 25 nM to 3200 nM. Following incubation, cells were harvest and Annexin V/propidium iodide flow cytometric analysis was utilized to determine the percentage of apoptotic cells (combined percentage of right quadrants). A) Graphical analysis of apoptosis (as determed by Annexin V/propridium iodide flow cytometry) of treated A375 cells. From these results, the IC50 for MLN4924 in A375 cells was determined to be 1200 nM. Data is presented as mean ± S.E.M. B) Representative flow cytometry plots of A375 cells treated with IC50 concentration of MLN4924 (1200 nM) or vehicle control (0 nM) are displayed. The same experiment for part A was repeated for the Mel39 BRAF mutant (V600E) human melanoma cell line. C) Graphical analysis of apoptosis (as determed by Annexin V/propridium iodide flow cytometry) of Mel39 cells treated for 72 hours with a titration of MLN4924 ranging from 0–400 nM. From these results, the IC50 for MLN4924 in Mel39 cells was determined to be 143 nM. D) Representative flow cytometry plots of Mel39 cells treated with IC50 concentration of MLN4924 (150 nM) or vehicle control (0 nM) are displayed.
Combination treatment of A375 BRAF (V600E) mutant human melanoma cell line with MLN4924 and IFN-alfa enhances apoptosis
To assess the capability of MLN4924 to act synergistically with other therapeutics, A375 cells were treated for 72 hours with varying concentrations of MLN4924 (0 nM, 100 nM, 1200 nM or 2000 nM), IFN-alfa (104 U/mL), vemurafenib (1 μM), or with MLN4924 in combination with IFN-alfa or the FDA-approved BRAF inhibitor vemurafenib (Figure 2). All agents were added to cultures simultaneously and apoptosis levels were measured via Annexin V/PI flow cytometric staining. No treatment and single agent treatments with IFN-alfa (104 U/mL) and vemurafenib (1 μM) induced lower levels of apoptosis than MN4924 at its IC50 concentration (1200 nM, Figure 2A). The combination of MLN4924 with IFN-alfa led to an additive level of apoptosis (78.2±3.7%) in A375 cells compared to MLN4924 treatment alone (50.7±1.0%, Figure 2B, p<0.005). However, the combination of MLN4924 and vemurafenib did not enhance apoptosis in A375 cells, as apoptosis levels were lower with the combination of vemurafenib and MLN4924 compared to MLN4924 alone treatment at higher doses (1200 nM & 2000 nM, Figure 2B).
Figure 2. The addition of IFN-alfa to MLN4924 treatment results in enhanced apoptosis of A375 BRAF(V600E) human melanoma cell line.

A) The A375 melanoma cell line was treated for 72 hours with 1200 nM MLN4924, 104 Units/mL IFN-α or 1 μM vemurafenib. Apoptosis levels were assessed by Annexin V/PI flow cytometry. B) A375 cells were treated with multiple concentrations of MLN4924 (0, 100, 1200 or 2000nM) or the combination of MLN4924 plus 104 Units/mL IFN-α or MLN4924 plus 1 μM vemurafenib and apoptosis was assessed. Data is presented as mean ± S.E.M. **p<0.005
Pre-treatment of the A375 BRAF (V600E) mutant human melanoma cell line with MLN4924 led to sensitization to vemurafenib treatment
It was previously shown that combination treatment of MLN4924 and vemurafenib in A375 cells led to lower levels of apoptosis compared to single agent MLN4924 treatment. However, a recent publication by Wand et al. has shown that significant levels of NEDD8 inhibition was present within 6–24 hours after initial treatment with MLN4924 [15]. We therefore explored the potential of MLN4924 to serve as a sensitizer of A375 BRAF (V600E) mutant melanoma cells to vemurafenib. Cells were treated with 1200 nM MLN4924 for either the first 24 hours or the full 72 hours of culture. Vemurafenib treatment (5 μM) was initiated at the 24-hour mark for a total treatment of 48 hours. For some of the treatments, the media containing MLN4924 was removed and replaced after the first 24 hours to prevent the mixing of MLN4924 with vemurafenib (as we previously observed an inhibitory effect) (R indicates the media was removed and replaced at the 24-hour mark prior to the addition of vemurafenib and NR indicates the media was not removed and replaced). Cell death levels were measured with Annexin/PI flow cytometry. Comparison of cell death percentages between all groups shows the combination ‘R’ treatment yielded the highest cell death percentage (63.4±2.3%) (Figure 3). This finding was significant (p<0.05) and indicates that pre-treatment of A375 BRAF mutant cells with MLN4924 could lead to moderate sensitization to vemurafenib treatment.
Figure 3. Pre-treatment of the A375 melanoma cell line with MLN4924 sensitizes cells to vemurafenib treatment.

The A375 cell line was treated with 1200 nM MLN4924 for either an initial 24 hours or the entire 72 hours of culture. Vemurafenib treatment (5 μM) was initiated at the 24-hour mark for a total treatment of 48 hours. For some of the treatments, media containing MLN4924 was removed and replaced after the first 24 hours to prevent the mixing of MLN4924 with vemurafenib (R). NR indicates vemurafenib was added directly to MLN4924-contained culture media. Apoptosis was assessed via Annexin V/PI flow cytometry. *p<0.05
MLN4924 treatment alone and in combination with IFN-alfa or vemurafenib
Cell proliferation was assessed using the MTS assay by measuring absorbance to quantify the anti-proliferative potential of MLN4924. A cell proliferation titration curve was created by treating A375 BRAF (V600E) mutant cells with MLN4924 concentrations of 0 nM, 100 nM, 600 nM, 1200 nM and 2000 nM (Figure 4A). Results indicated a decrease in cell proliferation with increasing concentration of MLN4924 treatment with an IC50 of 408 nM. Combination treatments with 1200 nM MLN4924 and IFN-alfa (104 U/mL) or MLN4924 with 1 μM vemurafenib on A375 melanoma cells were also conducted (Figure 4B). Similar experiments were performed with the Mel39 BRAF mutant melanoma tumor cell line (Figures 4C & D). The IC50 of MLN4924 in Mel39 cells was 142 nM. Proliferation for both A375 and Mel39 cells was greatly inhibited by MLN4924 treatment, with Mel39 cells being more sensitive to the treatment compared to A375. Regarding the combination treatments, both the A375 and Mel39 cells exhibited decreased proliferation when MLN4924 was combined with IFN-alfa, yet there was no such effect when combined with vemurafenib. Overall, MLN4924 demonstrated efficacy in inhibiting melanoma tumor cell proliferation and this effect was enhanced with the addition of IFN-alfa.
Figure 4. MLN4924 in combination with IFN-alfa but not vemurafenib results in reduced cell proliferation in BRAF mutant melanoma cells as measured by an MTS assay.

A) A375 cells plated at a density of 5000 cells/well in a 96-well plate were treated with various concentrations of MLN4924 for 72 hours. Following incubation, MTS reagent was added to each well and absorbance at 490 nm was measured. The proliferation IC50 was determined to be 408 nM. B) The same procedure was repeated in A375 cells using 1200 nM MLN4924 with or without IFN-alfa (104 U/mL) or vemurafenib (1 μM). C) Proliferation was measured in Mel39 cells using same procedure as in A. The IC50 value of Mel39 for proliferation was determined to be 142 nM. D) The combination treatment was repeated in Mel39 cells using 1200 nM MLN4924 with or without IFN-alfa (104 U/mL) or vemurafenib (1 μM). Data is presented as mean ± S.E.M. *p<0.05, **p<0.005, ***p<0.0005
Treatment of A375 Vem and M308 vemurafenib-resistant human melanoma tumor cells with MLN4924 led to apoptosis
A375 vemurafenib-resistant (A375 Vem) cells were created in an in vitro setting from A375 BRAF (V600E) mutant human melanoma cells via a constant exposure to vemurafenib. M308 vemurafenib-resistant cells were isolated from a tumor of a patient who had innate resistance to vemurafenib treatment. Cells were treated with various concentrations of MLN4924 (0 nM, 100 nM, 1200 nM & 2000 nM) and incubated for 72 hours. Apoptosis levels were measured via Annexin V/PI flow cytometric staining. Following a comparison of the apoptotic cell percentages of wild-type A375, A375 Vem and M308 vemurafenib-resistant melanoma tumor cells, a significant increase in susceptibility to MLN4924 was seen in the A375 Vem cell line where vemurafenib resistance was induced in vitro. However, no such effect was observed when vemurafenib resistance was innate in vivo as in the M308 cell line (Figure 5). The mechanism for the acquisition of vemurafenib resistance is still unknown, although studies suggest it may be due to the reactivation of the MAPK or the PI3K/AKT pathways [16]. These findings ultimately do not allow us to definitively conclude whether vemurafenib resistance leads to susceptibility to MLN4924 treatment, yet it does indicate that MLN4924 may have efficacy in treating vemurafenib-resistant melanoma tumor cells.
Figure 5. Treatment of the A375 vemurafenib-resistant melanoma cell line with MLN4924 leads to cell death.

Wild-type A375 cells, vemurafenib resistant A375 cells (A375 Vem, in vitro acquired resistance) or vemurafenib resistant M308 cells (innate resistance) were treated for 72 hours with increasing doses of MLN4924 (0 nM, 100 nM, 1200 nM, 2000 nM). Apoptosis levels were obtained through flow cytometric analysis. Data is presented as mean ± S.E.M. *p<0.05, ***p<0.0005
Immunoblot analysis indicates possible downstream mechanisms for MLN4924 apoptosis induction
Caspases are a family of protease enzymes that are involved in the downstream execution of apoptosis. Treatment of A375 BRAF (V600E) mutant human melanoma cells with MLN4924 for 72 hours enhanced the cleavage of caspase-3 but not caspase-7, caspase-9, or PARP (Figure 6).
Figure 6. MLN4924 treatment induces cleavage of apoptotic proteins.

A375 melanoma cells were treated for 72 hours with 0 nM, 1200 nM or 600 nM MLN4924. Cells were lysed and protein was harvested and subjected to immunoblot analysis for total or cleaved caspase-3, caspase-7, caspase-9 or PARP. β-actin was utilized as a loading control.
DISCUSSION
MLN4924 (pevonedistat) is a novel NEDD8 activating enzyme (NAE) inhibitor that ultimately leads to the inhibition of the ubiquitin protease system (UPS), which is responsible for the targeted degradation of proteins in cancer and normal cells [14]. In this study, we evaluated the efficacy of MLN4924 as a potential therapeutic agent in melanoma tumor cell lines. We hypothesized MLN4924 would stimulate apoptosis in human melanoma cells, and that it would work synergistically when utilized in combination with the immune stimulant IFN-alfa or the BRAF-inhibitor vemurafenib. Our results demonstrate that MLN4924 does induce significant levels of apoptosis and reduced proliferation in BRAF mutant A375 and Mel39 melanoma tumor cell lines. Additionally, MLN4924 does demonstrate some potential for use in the treatment of melanoma tumor cells that have acquired to resistance to vemurafenib.
Combination therapy with MLN4924 and IFN-alfa resulted in an increase in apoptosis levels for A375 BRAF mutant cell line. This effect was also observed in the ability of combination treatment to reduce tumor cell proliferation in both BRAF mutant A375 and Mel39 melanoma cell lines. On the other hand, an inhibitory effect was observed when utilizing combination therapy of vemurafenib with increasing doses of MLN4924 in A375 BRAF mutant melanoma cells for both apoptosis levels and tumor cell proliferation rates. Nevertheless, MLN4924 demonstrated an ability to act as a sensitizing agent to vemurafenib treatment in A375 cells when MLN4924 was removed from the cells prior to treatment with vemurafenib.
Our immunoblot analysis demonstrates that MLN4924-induced apoptosis was associated with the cleavage of caspase-3 but not caspase-7, caspase-9 or cleavage of PARP. These results, apart from caspase-3, contradict our original predictions, as our untreated sample should have expressed the lowest amount of cleavage. However, immunoblots from other publication show the results we expected for caspase-3, caspase-7, caspase-9 and PARP [17] [18]. The difference between our assay and these groups is that they treated tcells for 48 hours, while in the present study cells were treated for 72 hours. This difference may explain the high levels of cleavage observed in our untreated sample as at the 72-hour mark as the untreated A375 cells are close to being confluent at this time point. Nevertheless, when comparing the 600 nM and 1200 nM MLN4924 dosages, we did observe an increase in the cleavage of all the caspases and PARP with increasing MLN4924 concentration.
Caspase-3 and caspase-7 are known as ‘executioner caspases’, meaning they are directly responsible for inducing apoptosis. Caspase-9 on the other hand is known as an intrinsic activator-caspase that is responsible for the activation of executioner caspases [19]. One group has presented a model mechanism for the activation of caspase-9 that begins with the inhibition of the NEDD8 pathway due to MLN4924, leading to the inhibition of the ubiquitin protease system which prevents the degradation of pro-apoptotic p21, p27, Noxa and Bik factors [17]. Since MLN4924 treatment stimulates the activation of caspase-9, there is a possibility that MLN4924 can induce apoptosis through an intrinsic (intracellular) pathway. In regards to PARP, studies performed by other groups have verified cleavage of PARP to be a direct downstream result of the activation of caspase-3 and caspase-7 [20].
While our immunoblot results indicate an intrinsic mechanism by which apoptosis is induced, another study performed in human cervical carcinoma cells revealed apoptosis was induced via activation of caspase-8 as well [18]. This is interesting as caspase-8 is activated by an extrinsic pathway, meaning an extracellular ligand binds to a death receptor outside of the cell membrane leading to cleavage of caspase-8, which then cleaves executioner caspase-3, caspase-6 and caspase-7 thereby inducing apoptosis [19].
One study found that MLN4924 treatment in lymphoma cells led to the upregulation of p27 (cyclin-dependent kinase inhibitor) and p21 (tumor suppressor protein), both of which play roles in inhibiting cell cycle progression [17]. This may explain the reduction in proliferation observed in the A375 and Mel39 BRAF mutant melanoma cells following treatment.
MLN4924 is a novel therapeutic agent and a phase I clinical trial conducted in patients with metastatic melanoma has shown that MLN4924 led to a partial response in one patient and stable disease in 15 others (4 lasting ≥ 6.5 months). It also determined the maximum tolerated dose to be 209 mg/m2 [21]. Another study has shown that treatment of various melanoma cell lines with MLN4924 led to reduced cell viability and proliferation with an IC50 <0.3 μM [22]. While our study has found similar results in assessing the potential of MLN4924 as a cytotoxic agent, our group is the first to find that MLN4924 can work with IFN-alfa to serve as a sensitizer to vemurafenib treatment. Additionally, we are the first to observe that MLN4924 has efficacy in treating vemurafenib-resistant melanoma tumor cells.
MLN4924 has shown potential in targeting other forms of cancers as well. One group found MLN4924 to be highly effective against human lymphoma cells (in vitro) compared to the classical proteasome inhibitor MG132. Interestingly, immunoblots showed the inhibition of the anti-apoptotic Bcl-2 and Mcl-1 protein families [17]. Another study performed in human urothelial carcinoma found MLN4924 to induce apoptosis and inhibit tumor cell proliferation in vitro. Studies in xenograph mouse models found dose-dependent cytotoxic effects, along with inhibition of proliferation, migration and invasion effects when mice were treated with MLN4924 [23].
MLN4924 appears to have efficacy in treating various forms of cancers both in vitro and in vivo. MLN4924 has been shown to be well-tolerated toxicity in mouse models several clinical trials are ongoing [23]. Our group will continue to assess the potential of MLN4924 as a melanoma therapeutic.
Acknowledgments
Grant Support: This work was supported by NIH Grants: P01CA095426 (WEC), P30CA016058 (WEC), T32GM068412 (MCD). One of the authors (GOS) was supported by an undergraduate Pelotonia Fellowship. This paper does not reflect the views of the Pelotonia fellowship program.
Footnotes
Conflict of interests: None declared
References
- 1.Survival Rates for Melanoma Skin Cancer, by Stage. American Cancer Society; 2016. May 20, Web. 21 Jan, 2017. http://www.cancer.org/cancer/melanoma-skin-cancer/detection-diagnosis-staging/survival-rates-for-melanoma-skin-cancer-by-stage.html. [Google Scholar]
- 2.Key Statistics for Melanoma Skin Cancer. American Cancer Society; 2017. Jan 6, Web. 21 Jan, 2017. http://www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html. [Google Scholar]
- 3.Immunotherapy: using the Immune System to treat Cancer. National Cancer Institute; 2015. Sep 14, Web. 21 Jan. 2017. https://www.cancer.gov/research/areas/treatment/immunotherapy-using-immune-system.html. [Google Scholar]
- 4.Cancer Immunotherapy. American Cancer Society; 2016. Aug 8, Web. 21 Jan. 2017. http://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/immunotherapy.html. [Google Scholar]
- 5.Escandon J, Peacokc S, Trabolsi A, Thomas DB, Layka A, Lutzky J. Interstitial nephritis in melanoma patients secondary to PD-1 checkpoint inhibitor. Int J Immunother Cancer Res. 2017;5:3. doi: 10.1186/s40425-016-0205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seetharamu N, Ott PA, Pavlick AC. Novel Therapeutics for Melanoma. Expert Rev Anticancer Ther. 2009;9(6):839–849. doi: 10.1586/era.09.40. [DOI] [PubMed] [Google Scholar]
- 7.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 8.Chapman PB, Hauschild A, Robert C, Larkin JMG, Haanen JBAG, Ribas A, et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAFV600Emutated melanoma. J Clin Oncol. 2012;(suppl) abstr 8502ˆ. [Google Scholar]
- 9.Lens MB, Dawes M. Interferon alfa therapy for malignant melanoma: a systematic review of randomized controlled trials. J Clin Oncol. 2002;20:1818–25. doi: 10.1200/JCO.2002.07.070. [DOI] [PubMed] [Google Scholar]
- 10.Suarez-Kelly LP, Kemper GM, Duggan MC, Stiff A, Noel TC, Markowitz J, et al. The combination of MLN2238 (Ixazomib) with interferon-alpha results in enhanced cell death in melanoma. Oncotarget. 2016;7:81172–81186. doi: 10.18632/oncotarget.12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lesinski GB, Raig ET, Guenterberg K, Brown L, Go MR, Shah NN, et al. IFN-alpha and bortezomib overcome Bcl-2 and Mcl-1 overexpression in melanoma cells by stimulating the extrinsic pathway of apoptosis. Cancer Res. 2008;68(20):8351–8360. doi: 10.1158/0008-5472.CAN-08-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soucy TA, Drick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer. 2010;1(7):10. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soucy TA, Smith PG, Milhollen MA. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 14.Kau KL, Ho IL, Shi CS, Wu JT, Lin WC, Tsai YC, et al. MNL4924, a novel protein neddylation inhibitor, suppresses proliferation and migration of human urothelial carcinoma: In vitro and in vivo studies. Cancer Lett. 2015;363(2):127–136. doi: 10.1016/j.canlet.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Zhang W, Yan Z, Liang Y, Li L, Yu X, et al. Radiosensitization by the investigational NEDD8-activating enzyme inhibitor MLN4924 (pevonedistat) in hormone-resistant prostate cancer cells. Oncotarget. 2016;7(25):38380–38391. doi: 10.18632/oncotarget.9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–11. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Luo Z, Pan Y, Wang W, Zhou X, Jeong LS, et al. Targeting protein neddylation with an NEDD8-activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biol Ther. 2015;16(3):420–429. doi: 10.1080/15384047.2014.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin WC, Kuo KL, Shi CS, Wu JT, Hsieh JT, Chang HC, et al. MLN4924, a Novel NEDD8-activating enzyme inhibitor, exhibits antitymor activity and enhances cisp;aton-induced cytotoxicity in human cervical carcinoma: in vitro and in vivo study. Am J Cancer Res. 2015;5(11):3350–3362. [PMC free article] [PubMed] [Google Scholar]
- 19.Mcllwain DR, Berger T, Mak TW. Caspase Functions in Cell Death and Disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaitanya GV, Alexander JS, Babu PP. PARP-1 cleavage fragment; signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31. doi: 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhatia S, Pavlick AC, Boasberg P, Thompson JA, Pickard MD, Faessel H, et al. A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Invest New Drugs. 2016;34:439–449. doi: 10.1007/s10637-016-0348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong KM, Micel LN, Selby HM, Tan AC, Pitts TM, Bagby SM, et al. Targeting the protein ubiquitination machinery in melanoma by the NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) Invest New Drugs. 2017;35(1):11–25. doi: 10.1007/s10637-016-0398-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo KL, Ho IL, Shi CS, Wu JT, Tsai YC, Chang HC, et al. MLN4924, a novel protein neddylation inhibitor, supresses proliferation and migration of human urothelial carcinoma: In vitro and in vivo studies. Cancer Letters. 2015;363(2):127–136. doi: 10.1016/j.canlet.2015.01.015. [DOI] [PubMed] [Google Scholar]