

Abstract
Neurodegeneration, a term that refers to the progressive loss of structure and function of neurons, is a feature of many neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD), Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD). There is no cure or treatment available that can prevent or reverse neurodegenerative conditions. The causes of neurodegeneration in these diseases remain largely unknown; yet, an extremely small proportion of these devastating diseases are associated with genetic mutations in proteins involved in a wide range of cellular pathways and processes. Over the past decade, it has become increasingly clear that the most notable neurodegenerative diseases, such as ALS, FTLD, and AD, share a common prominent pathological feature known as TAR DNA-binding protein 43 (TDP-43) proteinopathy, which is usually characterized by the presence of aberrant phosphorylation, ubiquitination, cleavage and/or nuclear depletion of TDP-43 in neurons and glial cells. The role of TDP-43 as a neurotoxicity trigger has been well documented in different in vitro and in vivo experimental models. As such, the investigation of TDP-43 pathomechanisms in various major neurodegenerative diseases is on the rise. Here, after a discussion of stages of TDP-43 proteinopathy during disease progression in various major neurodegenerative diseases, we review previous and most recent studies about the potential pathomechanisms with a particular emphasis on ALS, FTLD, and AD, and discuss the possibility of targeting TDP-43 as a common therapeutic approach to treat neurodegenerative diseases.
Keywords: Alzheimer’s disease, amyotrophic lateral sclerosis, frontotemporal lobar degeneration, neurodegeneration, Neurodegenerative diseases, TDP-43
Neurodegenerative diseases encompass a large number of disorders characterized by progressive loss or dysfunction of neurons in the central nervous system (CNS) or peripheral nervous system (PNS) during aging. Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and frontotemporal lobar degeneration (FTLD) are all widely known neurodegenerative diseases. Although progressive neuronal impairment is the common hallmark of all neurodegenerative diseases, specific neurodegenerative disorders demonstrate distinct pathological and clinical features and can be distinguished by their characteristic neuronal loss occurring in specific brain regions or spinal cord. The prevalence of neurodegenerative diseases has increased rapidly in the past decades. However, effective treatments for these devastating diseases remain very limited.
TAR DNA-binding protein 43 (TDP-43, encoded by the TARDBP gene and also referred to as ALS10) was first identified as a host cell protein that binds specifically to pyrimidine-rich DNA motifs in a long terminal repeat, known as TAR of human immunodeficiency virus type 1 and represses virus type 1 gene transcription (Ou et al. 1995). However, subsequent studies have demonstrated that TDP-43 more frequently binds RNA and regulates messenger ribonucleic acid (mRNA) splicing, translation, transportation, and even degradation (Buratti and Baralle 2001; Buratti et al. 2001, 2005; Hefferon et al. 2004; Ayala et al. 2005; Mercado et al. 2005). The link between TDP-43 and neurodegenerative diseases was established by the initial identification of TDP-43 as the major component of the pathological hallmark, ubiqui-tin-positive protein inclusions, in patients with ALS and FTLD (Arai et al. 2006; Neumann et al. 2006). The discovery of genetic mutations in TDP-43 associated with both ALS and FTLD soon followed (Kabashi et al. 2008; Kwiatkowski et al. 2009; Vance et al. 2009). Since then, TDP-43 has become a novel rising star in the field of neurodegenerative diseases.
Despite different etiologies, clinical symptoms, and pathological hallmarks, various major neurodegenerative diseases demonstrate similar TDP-43 pathological manifestations in neurons and even glia including the accumulation of detergent-resistant, ubiquitinated or hyperphosphorylated TDP-43 inclusions in the cytoplasm, usually accompanied by the depletion of TDP-43 from the nucleus. These characteristic TDP-43-related pathological features are usually referred to as TDP-43 proteinopathy. Interest in understanding the path-omechanisms underlying TDP-43 proteinopathy has increased substantially in the past decade, largely because of the presence of TDP-43 proteinopathy as a common key pathological feature in a wide range of different neurodegenerative diseases including ALS (Kabashi et al. 2008; Sreedharan et al. 2008), FTLD (Kabashi et al. 2008; Sreedharan et al. 2008), AD (Amador-Ortiz et al. 2007; Josephs et al. 2014), PD (Chanson et al. 2010), and HD (Schwab et al. 2008; Davidson et al. 2009). In this review, we first describe the stages and progression of TDP-43 proteinopathy in various major neurodegenerative diseases with a particular emphasis on ALS, FTLD, and AD, extensively discuss the potential pathomechanisms of TDP-43, and finally provide insight into the possibility of targeting TDP-43 as a common therapeutic approach to treat neurodegenerative diseases.
Stages of TDP-43 proteinopathy in ALS, FTLD, and AD
Amyotrophic lateral sclerosis
ALS, also known as Lou Gehrig’s disease, is the most common motor neuron disease characterized by progressive and fatal degeneration of both upper and lower motor neurons, causing progressive muscle denervation, weakness, atrophy, spasticity, paralysis, and eventually death (Pasinelli and Brown 2006). The vast majorities of ALS cases have no family history and are usually referred to as sporadic ALS (sALS). Less than 10% of ALS cases are familial (fALS), of which approximately 40% are caused by repeat expansions of the C9ORF72 gene, 20% by mutations in the gene encoding copper–zinc super-oxide dismutase (SOD1), 4% each by mutations in TDP-43 or fused in sarcoma (FUS), and less than 1% each by mutations in many other genes encoding p62 (SQSTM1), optineurin, TANK-binding kinase 1, ubiliquin-2 (UBQLN2), vesicle-associated membrane protein-associated protein B, senataxin (SETX), angiogenin (ANG), valosin-containing protein (VCP), SIGMAR1, or dynactin (DCTN1) (Chen et al. 2013; Picher-Martel et al. 2016).
Although a very small subset of sALS and fALS patients are associated with TDP-43 mutations, TDP-43 proteinopathy can be present in up to 97% of ALS patients (Arai et al. 2006; Neumann et al. 2006; Mackenzie et al. 2007), suggesting the likely critical role of TDP-43 in the pathogenesis of this devastating disease. Based on TDP-43 proteinopathy in neurons, ALS can be effectively divided into different stages with the first stage characterized by the appearance of lesions in the motor cortex, brainstem, and spinal cord; the second stage by increased lesions in the prefrontal neocortex, brainstem, precerebellar nuclei, and the red nucleus; the third stage by the spreading of pathology to the prefrontal and postcentral neocortex and striatum, and the advanced stage by the greatest burden of lesions in anteromedial portions of the temporal lobe (Brettschneider et al. 2013, 2014), further highlighting the role of TDP-43 in disease progression. Although TDP-43 pro-teinopathy is unlikely epiphenomena in ALS, its role in the degeneration of motor neurons remains debatable. Notably, TDP-43 proteinopathy was also seen in non-neural cells such as astrocytes and microglia in patients with ALS (Brettschnei-der et al. 2012; Sloan and Barres 2013), providing the possibility that TDP-43 proteinopathy may trigger motor neuron death in both cell-autonomous and non-cell-autonomous models.
Frontotemporal lobar degeneration
FTLD, characterized by the progressive decline in behavior or language associated with degeneration of the frontal and anterior temporal lobes of the brain, is the second most frequent form of dementia in people under the age of 65 years after AD (Ratnavalli et al. 2002). The most common clinical presentations of FTLD typically include behavioral variant frontotemporal dementia, semantic variant primary progressive aphasia, and nonfluent variant primary progressive aphasia, with behavioral variant frontotemporal dementia as the most common form accounting for nearly 60% of FTLD patients (Onyike and Diehl-Schmid 2013; Pan and Chen 2013). As a group of clinically heterogeneous disorders, FTLD may also comprise progressive supranuclear palsy and corticobasal syndrome (CBS), and clinically and neuropathologically overlap with ALS and other motor neuron diseases (MND) (Tsai and Boxer 2014). Approximately 30–50% of FTLD cases are familial and associated commonly with genetic mutations in C9ORF72, progranulin (GRN), and microtubule-associated protein tau, and rarely with VCP and Chromatin-modifying protein 2B, TDP-43, FUS, p62/SQSTM1, and ubiquilin 2 (UBQLN2) (Galimberti et al. 2015). The non-coding G4C2 hexanucleotide repeat expansion in C9orf72 has been identified as the most common causal mutation in patients with concomitant FTLD and ALS (FTLD-ALS) (Mori et al. 2013). Of note, unlike C9ORF72, mutations in other ALS-associated proteins such as TDP-43 and FUS have only been linked to FTLD-ALS but not FTLD lacking MND (Mackenzie et al. 2010).
Historically, FTLD patients largely show cytoplasmic aggregates of proteins, often referred to as inclusion bodies, in neurons and glial cells. From a neuropathological perspective, FTLD can be classified into different pathological FTLD subtypes such as FTLD with tau-positive inclusions (FTLD-tau), FTLD with tau and alpha-synu-clein-negative but TDP-43 and ubiquitin-positive inclusions (FTLD-TDP, previously called FTLD with ubiquitin-positive inclusions, FTLD-U), FTLD with FUS-positive inclusions (FTLD-FUS) and rare FTLD with no inclusion (FTLD-ni or previously known dementia lacking distinctive histopathology) (Arai et al. 2006; Bigio 2011; Dickson et al. 2011). Despite the very rare prevalence of TDP-43 mutations in FTLD, FTLD-TDP represents the most frequent FTLD subtype and TDP-43 proteinopathy (largely TDP-43-positive cytoplasmic inclusions and neurites and occasionally TDP-43-positive neuronal intranuclear inclusions) can be noted in up to 50% of FTLD cases (Cairns et al. 2007; Mackenzie 2007; Hasegawa et al. 2008; Weihl et al. 2008). In FTLD patients, TDP-43-positive inclusions are primarily noted in the frontotemporal cortex as well as the dentate gyrus of the hippocampus, and may also be found in the cranial nerve nuclei of the brainstem and anterior horn of the spinal cord (Davidson et al. 2007).
Alzheimer’s disease
AD is the most prevalent form of dementia in the elderly characterized by the progressive loss of neurons in brain regions critical for memory, learning, conscious thought, and language and is usually accompanied by two pathologic hallmarks, neurofibrillary tangles (NFTs) and senile plaques (SPs), and other prominent pathological changes such as neuronal loss, granulovacuolar degeneration, and dystrophic neurites (Smith 1998). NFTs are intracellular lesions composed of hyperphosphorylated tau, while SPs are made up of bundles of amyloid-β(Aβ) peptide fibrils (Smith 1998). Recently, cytoplasmic TDP-43-positive inclusions have been identified as the third predominant proteinopathy in the brains of individuals diagnosed with AD (Amador-Ortiz et al. 2007; Higashi et al. 2007; Arai et al. 2009). About 90% of AD cases are referred to as sporadic AD and are not genetically transmitted (Zhu et al. 2005). Less than 10% of AD cases are familial AD, involving a mutation in β-amyloid precursor protein (APP), presenilin 1 (PS1), or presenilin 2 (PS2), with PS1/2 associated with the majority of early-onset familial AD (Czech et al. 2000; Fraser et al. 2000; Tanahashi and Tabira 2000).
TDP-43-positive inclusions detected using a C-terminal TDP-43 antibody occur in a stereotypic manner over the latest six distinctive stages in AD (Josephs et al. 2014, 2016). The cytoplasmic accumulation of TDP-43 in neurons begins in the amygdala in stage 1 and then moves to the entorhinal cortex and subiculum in stage 2, to the dentate gyrus of the hippocampus and occipitotemporal cortex in stage 3, to the insular cortex, ventral striatum, basal forebrain, and inferior temporal cortex in stage 4, to the substantia nigra, inferior olive, and midbrain tectum in stage 5 and finally to the basal ganglia and middle frontal cortex in stage 6. It is also worth noting that the recent report that cytoplasmic TDP-43 inclusions are associated with AD-type dementia independent of pathologic hallmarks NFTs and SPs, and that patients with mixed TDP-43, Aβ, and tau proteinopathies show more severe AD-type dementia than patients with Aβand tau proteinopathies alone (James et al. 2016).
Pathomechanisms of TDP-43
RNA alternate splicing
As an ubiquitously expressed 414-amino acid (aa) protein structurally resembling the members of RNA-binding protein heterogeneous nuclear ribonucleoprotein (hnRNPs) (Buratti and Baralle 2008; Buratti and Baralle 2012; Lee et al. 2011), TDP-43 is composed of an N-terminal domain (NTD, 1-102aa), two RNA recognition motifs (RRM1, 106-177aa and RRM2, 192-259aa) and a carboxy-terminal glycine-rich domain (CTD, 274-414aa). RRM1 and RRM2 belong to a widely existing eukaryotic RNA recognition motif family (RRM is also referred to as RNA-binding domain or ribonucleoprotein domain (RNP)) (Clery et al. 2008) (Fig. 1a). Like hnRNPs, as a primarily nuclear RNA-binding protein, TDP-43 has been consistently reported to bind UG-rich domains within proximal intron regions or 3′-untranslated regions of mRNAs encoding proteins such as cystic fibrosis transmembrane conductance regulator (Buratti et al. 2001), POLDIP3 (Fiesel et al. 2012; Shiga et al. 2012; Colombrita et al. 2015), SORT1 (Prudencio et al. 2012; Mohagheghi et al. 2016), MADD/STAG2/FNIP1/BRD8 (De Conti et al. 2015), SEPT6/SULT4A1/TNIK/DICER/ELAVL3 (Colombrita et al. 2015), or even TARDBP’s own transcripts (Ayala et al. 2011) to regulate mRNA alternative splicing. Previous studies found that TDP-43 deficiency could facilitate the use of cryptic splice sites to reduce correctly spliced protein-encoding mRNAs (Mercado et al. 2005; Ayala et al. 2006). Although the global functional consequence(s) of the binding of TDP-43 to a variety of RNAs remains largely unknown, exciting recent findings of TDP-43 acting as a repressor of non-conserved cryptic exon splicing may lead to the identification of major functional RNA targets of TDP-43 (Ling et al. 2015; Tan et al. 2016; Humphrey et al. 2017).
Fig. 1.
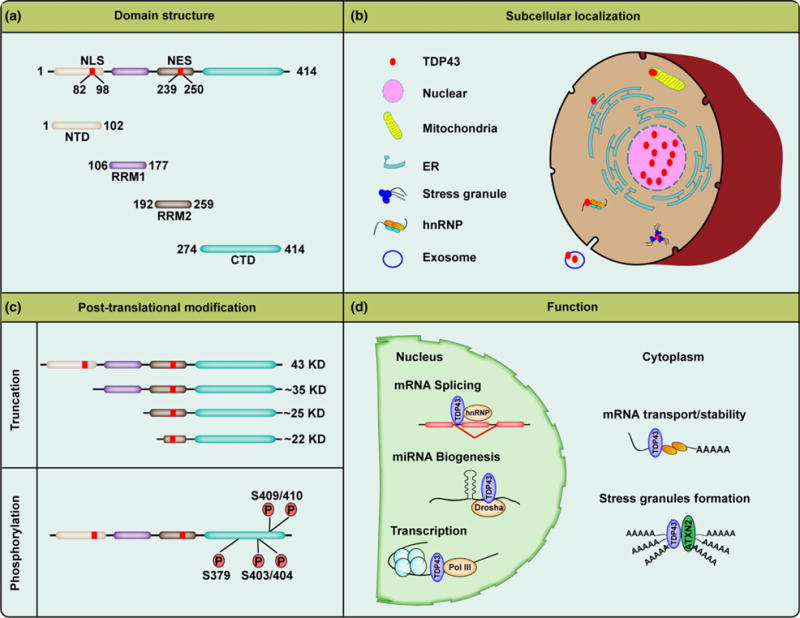
Domain structure, subcellular localization, post-translational modification, and function of TDP-43. (a) TDP-43 is a 414 amino acid protein, including an N-terminal domain (NTD, 1-102aa), two RNA recognition motifs (RRM1, 106-177aa and RRM2, 192-259aa) and a carboxy-terminal glycine-rich domain (CTD, 274-414aa). TDP-43 contains both a nuclear localization sequence (NLS, 82-98aa) and a nuclear export sequence (NES, 239-250aa). (b) The majority of TDP-43 resides in the nucleus under physiological conditions, while the remaining TDP-43 has been found to be present in other organelles, such as mitochondria, endoplasmic reticulum (ER), and exosomes. During stressful conditions, TDP-43 can be recruited to stress granules and hnRNPs. (c) The full-length 43 kDa TDP-43 is cleaved by caspases or calpain to generate ~35 kDa and 20-25 kDa fragments under disease conditions. The fragments and the full-length protein become aberrantly phosphorylated at serine residues 379, 403/404, and 409/410 in neurodegenerative disease affected brains. (d) TDP-43 exhibits multiple normal biological functions, predominantly those that regulate RNA pathways, including mRNA splicing, miRNA biogenesis, and transcription in nucleus, while it is also thought to play a role in mRNA transport, mRNA stability, and stress granule formation in the cytoplasm.
A prominent feature of TDP-43 proteinopathy, the redistribution of TDP-43 from the nucleus to the cytoplasm is generally believed to cause a considerable loss of TDP-43 nuclear function and subsequent neuronal dysfunction (Buratti and Baralle 2009). In support of the loss of function hypothesis, the significantly enhanced TDP-43-associated cryptic exon splicing was reported in ALS patients with TDP-43-positive inclusions (Ling et al. 2015). And, unlike wild-type TDP-43, exogenously expressed ALS-associated mutant TDP-43 increases exon exclusion of mRNA targets involved in neuronal transmission and function (Polymenidou et al. 2011), further indicating that mutant TDP-43 proteins may lose their mRNA splicing abilities. Along this line, TDP-43 directly binds and regulates the splicing of pre-mRNAs of FTLD-associated FUS, progranulin, or tau (Polymenidou et al. 2011; Gu et al. 2017a, b). Accordingly, FTLD-ALS patients show significantly reduced expression of FUS or increased expression of progranulin at the mRNA and protein levels (Polymenidou et al. 2011). Even though a very recent study showed that TDP-43 suppresses tau expression by binding to its 3′-UTR, and the protein level of tau is increased in ALS patients and animal models expressing ALS-associated mutant TDP-43 (Gu et al. 2017b), reduced tau protein expression has been reported to be associated with at least a subset of FTLD patients, especially FTLD patient bearing progranulin mutations (Adamec et al. 2001; Zhukareva et al. 2001; Papegaey et al. 2016). Noteworthily, despite a previous study reporting the lack of association between TDP-43 proteinopathy and tau expression and mis-splicing in AD patients (Niblock et al. 2016), two very recent studies have shown the regulation of tau mRNA stability and exon 10 alternative splicing by TDP-43 in cell and animal models (Gu et al. 2017a,b). In addition, it has been found that the cryptic exon incorporation can occur in AD cases showing TDP-43 nuclear depletion but lacking cytoplasmic TDP-43-positive inclusions (Sun et al. 2017).
Although these evidence support the possible involvement of TDP-43-related RNA splicing in neurodegeneration, the loss-of-function hypothesis is challenged by the absence of altered alternative splicing of exon 9 of cystic fibrosis transmembrane conductance regulator, one of the earliest and best-characterized targets of TDP-43 (Buratti and Baralle 2001), and other reported mRNA targets in ALS, FTLD, or AD patients or transgenic animal models. And, it still remains to be determined how the general pre-mRNA splicing activity of TDP-43 contributes to disease progression and how TDP-43 may interfere with the alternative splicing of diverse mRNAs to disrupt convergent molecular pathways.
Stress granules
TDP-43 contains both nuclear localization sequence and nuclear export sequence (NES), which are localized in the NTD and RRM2 domain, respectively (Ou et al. 1995; Lee et al. 2011) (Fig. 1a). Under physiological conditions, the majority of TDP-43 resides in the nucleus. However, likely due to the presence of NES, up to 30% of TDP-43 is also noted in the cytoplasm (Barmada et al. 2010) (Fig. 1b). Accompanying nuclear depletion, TDP-43 also shows characteristic cytoplasmic accumulation in neurons of ALS, FTLD, and AD patients. Previous studies have revealed that nuclear depletion is not required for neuronal toxicity induced by ALS-associated mutant TDP-43 (Arnold et al. 2013; Austin et al. 2014), and cytoplasmic mutant TDP-43 is sufficient to cause neurodegeneration (Barmada et al. 2010), therefore this suggests that, in addition to the loss of nuclear function, TDP-43 may also contribute to disease progression by the gain of neuronal toxicity function in the cytoplasm.
Stress granules (SGs) are cytoplasmic foci formed to suppress the translation of non-essential proteins to favor protective protein expression in response to stress. As non-membrane-bound RNA granules, SGs contain non-translating mRNA, translation initiation factors (eIF2α, eIF3 and eIF4A/B/G), and characteristic markers such as T-cell-restricted intracellular antigen-1 (TIA-1), TIA-like-1 (TIAR), Tristetraprolin, poly(A)-binding protein (PABP), and Ras-GTPase-activating protein SH3 domain-binding protein 1 (Decker and Parker 2012). In addition to SGs, mRNAs not engaged in translation can co-assemble with RNA-binding proteins to form other cytoplasmic mRNP granules such as processing bodies (P-bodies) and RNA transport granules. P-bodies consist of decapping enzymes and decapping activators to regulate RNA silencing and degradation, while RNA transport granules are responsible for the delivery of translationally repressed mRNAs (Decker and Parker 2012).
In ALS or FTLD patients, TDP-43 or phosphorylated TDP-43 has been reported to co-localize with SG makers TIA-1/PABP-1/eIF3 (Volkening et al. 2009; Liu-Yesucevitz et al. 2010; Bentmann et al. 2012; McGurk et al. 2014) (Fig. 1b), leading to one prevalent hypothesis that SG play an important role in the formation of TDP-43 inclusions. In support of this notion, TDP-43 inclusion formation can be suppressed by the inhibition of SG formation using translational inhibitors (Liu-Yesucevitz et al. 2010). Along this line, the ablation of ataxin-2, a polyglutamine protein necessary for SG assembly (Nonhoff et al. 2007; Nihei et al. 2012), reduces TDP-43 proteinopathy and neurotoxi-city (Elden et al. 2010; Becker et al. 2017). In addition, ALS-associated mutant TIA-1 has recently reported to slow down SG disassembly and enhance the accumulation of non-dynamic SGs containing TDP-43 (Mackenzie et al. 2017). Meanwhile, TDP-43 may also contribute to SG formation and maintenance in response to stress, even though it is not an essential SG component (Colombrita et al. 2009; Liu-Yesucevitz et al. 2010; McDonald et al. 2011; Aulas et al. 2012). Noteworthily, although one previous study reported enhanced SG formation by disease association mutant TDP-43 (Liu-Yesucevitz et al. 2010), other studies showed either reduced or unchanged SG formation in cells or primary human fibroblasts bearing TDP-43 mutations (McDonald et al. 2011; Bentmann et al. 2012; Orru et al. 2016). And, surprisingly, a most recent study has reported that despite the presence of widespread TDP-43 pathology in ALS/FTD with TIA1 mutations, TDP-43 inclusions do not co-localize with TIA1 (Hirsch-Reinshagen et al. 2017). While these discrepancies remain to be resolved, the functional roles of TDP-43 in translational control in stress response have been increasingly recognized. Further studies will be important to investigate the detailed mechanisms by which mislocalized cytoplasmic TDP-43 influences the dynamics of SGs and possibly other RNA granule pathways.
Axonal transport
Axonal transport is essential for neuronal function and survival (Maday et al. 2014). Abnormal vesicles or mitochondrial transport along axon has been extensively reported in ALS patients (Collard et al. 1995; Williamson and Cleveland 1999; Chevalier-Larsen and Holzbaur 2006) and AD patients (Stokin et al. 2005; Wang et al. 2009). Like another ALS causing protein FUS, ALS-associated mutations in TDP-43 suppress the microtubule-dependent axonal transport of TDP-43 granules containing mRNAs (Alami et al. 2014). In neuronal cells, TDP-43 has been reported to be enriched in dendrites to regulate localized translation (Wang et al. 2008). And, TDP-43 has also been found to form granules containing mRNAs encoding proteins such as VEGFA/GRN (Colombrita et al. 2012), survival motor neuron (SMN) protein/fragile X mental retardation protein (FMRP) (Fallini et al. 2012), neurofilament light chain (NFL) (Alami et al. 2014), actin/CAMKII (Wang et al. 2008), and MTHFSD/DDX58 (MacNair et al. 2016) to mediate mRNA degradation, transport, and translation (Fig. 1d). As the over-expression of either wild-type or mutant TDP-43 also impairs mitochondrial transport (Wang et al. 2013b; Magrane et al. 2014), another possible mechanism by which cytoplasmic TDP-43 causes neuronal dysfunction may involve the regulation of axonal transport of mRNA, mitochondria, and other cargos for localized protein translation or metabolism.
Mitochondrial dysfunction
In addition to its endoplasmic reticulum (ER) localization (Wang et al. 2017), we and at least other four groups have independently reported the localization of TDP-43 inside of mitochondria by immuno-electron microscopy and different biochemical and genetic approaches (Mori et al. 2008; Wang et al. 2016, 2017; Izumikawa et al. 2017; Ruan et al. 2017; Woo et al. 2017) (Fig. 1b). Although these data are contradictory to one study reporting the association of ALS-mutant TDP-43 with the outer mitochondrial membrane (Hibiki Kawamata et al. 2017), a number of studies have consistently indicated mitochondria as targets of either wild-type or mutant TDP-43 (Mori et al. 2008; Xu et al. 2010, 2011; Braun et al. 2011; Lu et al. 2012; Wang et al. 2013b, 2016b, 2017; Magrane et al. 2014; Stribl et al. 2014; Izumikawa et al. 2017; Ruan et al. 2017; Woo et al. 2017). It is worth noting that the study from Kawamata et al. used a high concentration of KCl at 6M for mitochondrial isolation, which has been reported to greatly disrupt mitochondrial integrity and proteome by breaching the electrostatic force between the lipids and proteins (Mishra 2015).
Abundant evidence has revealed a prominent role for mitochondrial dysfunction in the pathogenesis of ALS (Cozzolino and Carri 2012). Multiple laboratories reported mitochondrial dysfunction as well as accumulation of SOD1 in mitochondria in various cell and animal models expressing ALS-associated mutant SOD1 (Mattiazzi et al. 2002; Liu et al. 2004; Bergemalm et al. 2006; Deng et al. 2006; Israelson et al. 2010). Mitochondrial dysfunction has also been implicated in widely studied transgenic mice expressing FTLD-associated tau (David et al. 2005). Interestingly, our recent study has revealed that TDP-43 can accumulate inside of mitochondria in neurons of ALS or FTLD patients (Wang et al. 2016). TDP-43 could be imported into mitochondria in vitro and either the loss of nuclear localization sequence signal or ALS-associated mutations in TDP-43 enhance its localization in mitochondria (Wang et al. 2013b, 2016), indicating mitochondrial localization as an intrinsic property of cytoplasmic TDP-43 (Wang et al. 2016). Noteworthily, the deletion of several TDP-43 motifs was able to suppress but not completely abolish its mitochondrial localization (Wang et al. 2016), indicating that the targeting of TDP-43 into mitochondria may need multiple sequences. Within mitochondria, TDP-43 binds a subset of mitochondria-transcribed mRNAs or transfer RNA (tRNA) to specifically impair the function of oxidative phosphorylation complex 1 (Wang et al. 2016; Izumikawa et al. 2017). The suppression of TDP-43 mitochondrial localization was sufficient to abolish TDP-43-induced mitochondrial dysfunction in TDP-43 transgenic mouse model showing ALS or FTLD-like phenotypes (Wang et al. 2016, 2017). Thus, cytoplasmic TDP-43 inclusions may target mitochondria to cause neurotoxicity in both ALS and FTLD. Nevertheless, the same discrepant study from Kawamata et al. reported unchanged mitochondrial function in mutant TDP-43 transgenic mice (Hibiki Kawamata et al. 2017), even though these mice exhibit remarkable neuronal death, mitochondrial cristae loss, and mitochondrial transport deficits (Xu et al. 2011; Wang et al. 2013b, 2017; Magrane et al. 2014). No study of TDP-43 mitochondrial localization and its relationship with mitochondrial function in AD has been reported so far. However, like ALS, mitochondrial dysfunction is a prominent and early feature of AD (Castellani et al. 2002) and complex I dysfunction has long been reported in AD (Chen and Yan 2006). Interestingly, TDP-43 can bind mitochondria-transcribed RNA and regulate the activity of oxidative phosphorylation complex 1 (Wang et al. 2016; Izumikawa et al. 2017). Mitochondria have been increasingly indicated as targets of Aβ and tau (Gao et al. 2017), therefore implicating the possible convergence of Aβ, tau, and TDP-43 pathogenic pathways at the point of impairing mitochondrial function.
Overall, while some apparent discrepancies need to be addressed, mitochondrial dysfunction has been consistently reported in cells or neurons expressing wild-type or mutant TDP-43 (Braun et al. 2011; Lu et al. 2012; Wang et al. 2013b, 2016b, 2017; Stribl et al. 2014; Izumikawa et al. 2017; Ruan et al. 2017; Woo et al. 2017), indicating that in addition to axonal transport, accumulated cytoplasmic TDP-43 may also directly interfere with mitochondrial bioenergetics to produce neuronal dysfunction and loss, and further detailed investigation of Aβ, tau, and TDP-43 functional interaction in mitochondrial bioenergetics may be warranted.
Protein quality control
Many ALS or FTLD-associated proteins such as UBQLN2, p62, VCP, and optineurin are involved in the protein quality control system (Chen et al. 2012) and the familial cases associated with genetic mutations in these proteins consistently show TDP-43-positive inclusions (Cairns et al. 2007; Mackenzie 2007; Hasegawa et al. 2008; Weihl et al. 2008). p62 has been proposed to link the ubiquitin proteasome system (UPS) and autophagy (Cohen-Kaplan et al. 2016). In addition, the disrupted physical interaction between TDP-43 and p62 has been reported in the cerebral cortex from patients with FTLD-TDP (Tanji et al. 2012). Therefore, these findings suggest that TDP-43 proteinopathy and related neurodegeneration may be consequences of systematic impaired protein quality control. To support this notion, it has indeed been shown that the suppression of either the ubiquitin proteasome system (UPS) or autophagy pathways was sufficient to cause TDP-43 cytoplasmic accumulation and neurotoxicity (Wang et al. 2010; van Eersel et al. 2011), while over-expression of p62 reduced TDP-43 cytoplasmic accumulation in both an UPS- and autophagy- dependent manner (Brady et al. 2011). In brain neurons, over-expression of mutant TDP-43 induces the unfolded protein response (response to unfolded or misfolded protein and accumulation in the lumen of the endoplasmic reticulum (ER)), ubiquitin aggregation and Golgi fragmentation preceding neuronal loss (Tong et al. 2012), further indicting the interplay between TDP-43 and the protein quality control system. However, it is worth mentioning here that, in motor neurons, only the ablation of proteasome subunit Rpt3 but not autophagy inducer Atg7 causes TDP-43 cytoplasmic accumulation and occasional nuclear inclusions in mice (Tashiro et al. 2012), suggesting that the role of both UPS and autophagy in mediating TDP-43 proteinopathy in neurons in the brain needs further detailed investigation.
Post-translational modifications
Abnormal post-translational modifications including ubiquitination, hyper-phosphorylation, and aberrant cleavage are known to be the characteristics for TDP-43 inclusions (Sreedharan et al. 2008). Although the ubiquitination sites in TDP-43 remain largely elusive, many phosphorylation sites at serine, threonine, or tyrosine residues have been identified within TDP-43, among which the phosphorylation at serine 403, 404, 409, and 410 sites have been extensively studied (Gendron et al. 2010) (Fig. 1c). Although in vitro experiments have identified casein kinase as one likely kinase responsible for TDP-43 phosphorylation at serine 379, 403/S404, and 409/S410 and suggested the potential role of TDP-43 phosphorylation in regulating its oligomerization or aggregation (Hasegawa et al. 2008), the impact of TDP-43 phosphorylation on its physiological and pathological function needs to be explored by future studies. TDP-43 N-terminal fragments (NTFs) or CTD fragments with the molecular weight of 20–25 kDa or 35 kDa have been consistently reported in either patients with ALS or FTLD (Zhang et al. 2007; Hasegawa et al. 2008; Xiao et al. 2015) as well as cell and animal models expressing wild-type or mutant TDP-43 (Rutherford et al. 2008; Dormann et al. 2009; D’Alton et al. 2014; Li et al. 2015) (Fig. 1c). Previous studies have identified several proteases such as calpain, caspase 3, and caspase 7 that mediate TDP-43 cleavage (Zhang et al. 2007, 2009; Suzuki et al. 2011; Yamashita et al. 2012). Nearly all ALS/FTLD-associated mutations are found in the low-complexity CTD of TDP-43. NTFs are required for its splicing activities (Jiang et al. 2017). The CTD region is essential for hnRNP interactions and mRNA splicing activity and is generally believed to be involved in the formation of cytoplasmic TDP-43 inclusions (Conicella et al. 2016). In support of this notion, the QN-rich region of 331-369aa in CTD has been consistently reported to form amyloid-like β-sheet structures (Igaz et al. 2009; Johnson et al. 2009; Zhang et al. 2009; Fuentealba et al. 2010). The definitive pathological role for TDP-43 aggregates in triggering neurotoxicity remains elusive, though it was suggested that pathologic TDP-43 species form insoluble aggregates to exert toxicity on neurons (Lee et al. 2011). The molecular basis of cytoplasmic TDP-43 accumulation is not completely understood either, but aberrant post-translational modifications, or the presence of prion-like or amyloid-prone domains have long been implicated to mediate the mislocalization and aggregation of TDP-43 (Zhang et al. 2009; Dewey et al. 2012; Nonaka et al. 2013; Robinson et al. 2013).
Exosomes
Insoluble TDP-43 aggregates extracted from brains or cerebrospinal fluid (CSF) of patients with ALS and FTLD have been reported to induce TDP-43 phosphorylation and ubiquitination, and nucleate the aggregation of phosphorylated and ubiquitinated TDP-43 (Nonaka et al. 2013; Ding et al. 2015). Moreover, some recent studies have demonstrated that TDP-43 could be released from the cell via secreted vesicles known as exosomes to facilitate the spreading of prion-like TDP-43 aggregates from one cell to another (Feiler et al. 2015; Iguchi et al. 2016; Zondler et al. 2017) (Fig. 1b). Along this line, inhibition of exosome secretion by inactivation of neutral sphingomyelinase 2 with GW4869 exacerbated the disease phenotypes of transgenic mice expressing human TDP-43 A315T mutant (Iguchi et al. 2016). Therefore, in future studies it will be interesting to test whether and how cytoplasmic TDP-43 inclusions may use the prion-like spreading model via exosomes to affect different brain areas during the disease progression.
TDP-43 in other neurodegenerative diseases
TDP-43 proteinopathy has also been reported as a prominent pathological feature in many other neurodegenerative diseases including but not limited to PD and HD. PD is a neurodegenerative disorder that affects predominately dopaminergic neurons in the substantia nigra. TDP-43-positive cytoplasmic inclusions were found in the spinal cord and bulbar nuclei but not in the dentate gyrus and neocortex of PD patients with MND, accompanied by SPs and topographically limited NFTs (Nakashima-Yasuda et al. 2007). TDP-43-positive inclusions have also been reported to be associated with neuronal loss in the CA1 of familial PD patients bearing a Parkin mutant (Markopoulou et al. 2008), suggesting that there is likely a link between Parkin and TDP-43. Parkin over-expression has been reported to alleviate TDP-43-induced cell death in cell (Hebron et al. 2013, 2014) and animal models (Wenqiang et al. 2014). It is noteworthy that the co-expression of TDP-43 exacerbates dopaminergic neuron loss in transgenic mice expressing mutant α-synuclein, further suggesting the possible synergistic role of TDP-43 in PD (Tian et al. 2011).
TDP-43-positive inclusions have also been reported to coexist with huntingtin (Htt)-positive inclusions in many CNS regions of patients with HD (Schwab et al. 2008; Doi et al. 2010; DeJesus-Hernandez et al. 2011), a hereditary polyglutamine disorder caused by extension of the triplet repeat region of the Htt gene. Although it remains controversial whether TDP-43 indeed co-localizes with Htt with inclusions (Tada et al. 2012), in a transgenic worm model, the ablation of nematode orthologue of TDP-43 has been shown to reduce mutant Htt-induced neurodegeneration and behavioral defects (Tauffenberger et al. 2013), thus suggesting a possible function for TDP-43 in promoting mutant Htt toxicity that is worth further investigation.
TDP-43 as a common therapeutic target for multiple neurodegenerative diseases
The presence of TDP-43 proteinopathy in a wide variety of neurodegenerative diseases suggest that targeting the TDP-43 pathomechanism may provide a common therapeutic approach to treat these devastating diseases. Stress granule formation has been implicated as the early stage of protein aggregation (Wolozin 2012). It has been reported that ataxin-2 is important for the assembly of stress granules through the recruiting of TDP-43 (Nonhoff et al. 2007; Hart and Gitler 2012). Genetic mutations in TDP-43 only represent a very small portion of all ALS/FTLD patients. Unlike antisense oligonucleotide (ASO)-based therapy targeting mutant SOD1 (Miller et al. 2013), ASO targeting mutant TDP-43 may not have broad translational significance. However, ASO-based therapy targeting ataxin-2 has recently been reported to block the TDP-43 and ataxin-2 interaction, alleviate TDP-43-induced neurotoxicity, improve motor function, and extend survival in transgenic animal models expressing ALS-associated mutant TDP-43 (Elden et al. 2010; Becker et al. 2017), suggesting the prevention of TDP-43 inclusion formation may be a promising common therapeutic approach for TDP-43-related neurodegenerative diseases.
Our recent studies have consistently shown that the suppression of TDP-43 mitochondrial localization by a TDP-43-derived inhibitory peptide (PM1) is sufficient to abolish motor and cortical neuron loss, and prevent and even reverse behavior deficits in two mutant TDP-43 transgenic mouse models demonstrating ALS and FTLD-like phenotypes (Wang et al. 2016, 2017), further supporting the targeting of TDP-43 mitochondrial localization as another possible promising therapeutic approach. Moreover, autophagy activators such as rapamycin, fluphenazine dihydrochloride (FPZ), methotrimeprazine (MTM) and 10-(4′-(N-diethylamino)butyl)-2-chlorophenoxazine (NCP), have been consistently reported to protect neurons and improve cognitive function in TDP-43 experimental models (Caccamo et al. 2009; Wang et al. 2012, 2013; Barmada et al. 2014). Similarly, the inhibition of eIF2a signaling transduction pathway by GSK2606414, a small molecule inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) and eIF2α phosphorylation, has been reported to alleviate TDP-43 induced climbing dysfunction in flies (Kim et al. 2014). As we discussed above, TDP-43 has been linked to the protein quality control system. Therefore, TDP-43-associated neurodegeneration may also be alleviated by therapeutic approaches targeting either enhanced protein clearance or reduced protein translation.
Conclusions and perspectives
TDP-43 proteinopathy has been reported as a prominent pathological feature in a wide range of neurodegenerative diseases including ALS, FTLD, AD, PD, HD, hippocampal sclerosis (Amador-Ortiz et al. 2007; Nelson et al. 2011; Pao et al. 2011), spinocerebellar ataxia type 2 (Toyoshima et al. 2011), and Alexander disease (Walker et al. 2014). An increasing body of pathological and mechanistic evidence indicates that TDP-43 proteinopathy is likely a convergent pathological mechanism underlying neuronal loss among various neurodegenerative diseases. While it still remains controversial whether TDP-43 proteinopathy behaves through ‘gain of toxicity’ or ‘loss of function’, the pathomechanisms of TDP-43 or mutant TDP-43 in diseases seem to be more complex than originally proposed. TDP-43 nuclear depletion and cytoplasmic accumulation are two apparently unrelated conditions that appear to be associated with distinct pathomechanisms involving RNA biogenesis, protein aggregation, axonal transport, UPS, autophagy, and even mitochondrial bioenergetics. Future studies will be important to evaluate and compare these TDP-43-regulated biological and pathological processes in various major neurodegenerative diseases. Notably, it is still possible that TDP-43 proteinopathy alone is not sufficient to induce neurodegeneration. Considering the coexistence of TDP-43 proteinopathy with many other pathological hallmarks, TDP-43 likely cooperates with other factors to trigger neurodegeneration. In this respect, the investigation of TDP-43 pathomechanisms in the context of other pathological conditions may be of particular importance. Along this line, it is also important to consider if any reported or newly discovered pathomechanisms for Aβ, tau, and many other neurodegeneration-related factors also apply to TDP-43.
In the past decade, our increasing understanding of TDP-43 pathomechanisms in neurodegeneration highlights the urgent need to advance our TDP-43 translational research efforts. The consistently reported TDP-43-mediated RNA splicing may be coherent enough for the use of specific RNA targets of TDP-43 as prognostic biomarkers. Encouraging TDP-43 translational studies targeting stress granules, mitochondria, autophagy, and protein synthesis warrant further investigation of the feasibility of moving these ASO, peptides, and small molecules into phase I clinical trials. TDP-43 proteinopathy is found in an increasing number of various neurodegenerative diseases, suggesting that the cause of TDP-43 proteinopathy could be multifactorial. However, emerging studies suggest that the targeting of TDP-43 may be a common therapeutic approach to prevent neurodegeneration. Further strong efforts are needed to translate any discovered knowledge about TDP-43 pathomechanisms into clinical practice.
Acknowledgments
Conflict of interest disclosure
This work was supported by grants from the US National Institutes of Health (1R01NS089604 to X.W.) and the US Alzheimer’s Association (AARG-17-499682 to X.W.). X.W. is the inventor on patents related to PM inhibitory peptides of mitochondrial TDP-43.
Abbreviations used
- AD
Alzheimer’s disease (AD)
- ALS
amyotrophic lateral sclerosis
- APP
β-amyloid precursor protein
- ASO
antisense oligonucleotide
- bvFTD
behavioral variant frontotemporal dementia
- CBS
corticobasal syndrome
- CFTR
cystic fibrosis transmembrane conductance regulator
- CHMP2B
chromatin-modifying protein 2B
- CNS
central nervous system
- CSFs
cerebrospinal fluids
- DLDH
dementia lacking distinctive histopathology
- DNs
dystrophic neurites
- ER
endoplasmic reticulum
- fALS
familial amyotrophic lateral sclerosis
- FTLD
frontotemporal lobar degeneration
- FUS
fused in sarcoma
- G3BP
Ras-GTPase-activating protein SH3 domain-binding protein 1
- GVD
granulovacuolar degeneration
- HD
Huntington’s disease
- hnRNP
heterogeneous nuclear ribonucleoprotein
- MND
motor neuron diseases
- ncRNAs
non-coding RNAs (ncRNAs)
- mRNA
messenger ribonucleic acid (mRNA)
- NES
nuclear export sequence
- NFTs
neurofibrillary tangles
- nfvPPA
nonfluent variant primary progressive aphasia
- NLS
nuclear localization sequence
- OXPHOS
oxidative phosphorylation
- PABP
poly(A)-binding protein
- P-bodies
processing bodies
- PD
Parkinson’s disease
- PNS
peripheral nervous system
- PS1
presenilin 1
- PS2
presenilin 2
- PSP
progressive supranuclear palsy
- RRM
RNA recognition motif
- sALS
sporadic amyotrophic lateral sclerosis
- SCA2
spinocerebellar ataxia type 2
- SGs
stress granules (SGs)
- SOD1
superoxide dismutase 1
- SPs
senile plaques
- svPPA
semantic variant primary progressive aphasia
- TBK1
TANK-binding kinase 1
- TDP-43
TAR DNA-binding protein 43
- TIA
T-cell-restricted intracellular antigen-1
- UPR
unfolded protein response
- UPS
ubiquitin proteasome system
- VAPB
vesicle-associated membrane protein-associated protein B (VAPB)
- VCP
valosincontaining protein
References
- Adamec E, Chang HT, Stopa EG, Hedreen JC, Vonsattel JP. Tau protein expression in frontotemporal dementias. Neurosci Lett. 2001;315:21–24. doi: 10.1016/s0304-3940(01)02314-x. [DOI] [PubMed] [Google Scholar]
- Alami NH, Smith RB, Carrasco MA, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, Iritani S, Onaya M, Akiyama H. Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:125–136. doi: 10.1007/s00401-008-0480-1. [DOI] [PubMed] [Google Scholar]
- Arnold ES, Ling SC, Huelga SC, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA. 2013;110:E736–E745. doi: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulas A, Stabile S, Velde CV. Endogenous TDP-43, but not FUS, contributes to stress granule assembly via G3BP. Molecular Neurodegeneration. 2012;7:54–68. doi: 10.1186/1750-1326-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin JA, Wright GS, Watanabe S, Grossmann JG, Antonyuk SV, Yamanaka K, Hasnain SS. Disease causing mutants of TDP-43 nucleic acid binding domains are resistant to aggregation and have increased stability and half-life. Proc Natl Acad Sci USA. 2014;111:4309–4314. doi: 10.1073/pnas.1317317111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, Pantano S, D’Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348:575–588. doi: 10.1016/j.jmb.2005.02.038. [DOI] [PubMed] [Google Scholar]
- Ayala YM, Pagani F, Baralle FE. TDP43 depletion rescues aberrant CFTR exon 9 skipping. FEBS Lett. 2006;580:1339–1344. doi: 10.1016/j.febslet.2006.01.052. [DOI] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendano-Vazquez SE, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30:277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic Mislocalization of TDP-43 Is Toxic to Neurons and Enhanced by a Mutation Associated with Familial Amyotrophic Lateral Sclerosis. J Neurosci. 2010;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677–685. doi: 10.1038/nchembio.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544:367–371. doi: 10.1038/nature22038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, Haass C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43) J Biol Chem. 2012;287:23079–23094. doi: 10.1074/jbc.M111.328757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Rehnmark A, Marklund SL. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J Neurosci. 2006;26:4147–4154. doi: 10.1523/JNEUROSCI.5461-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigio EH. TDP-43 variants of frontotemporal lobar degeneration. J Mol Neurosci. 2011;45:390–401. doi: 10.1007/s12031-011-9545-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Meng P, Zheng Y, Mao Y, Hu F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem. 2011;116:248–259. doi: 10.1111/j.1471-4159.2010.07098.x. [DOI] [PubMed] [Google Scholar]
- Braun RJ, Sommer C, Carmona-Gutierrez D, Khoury CM, Ring J, Buttner S, Madeo F. Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J Biol Chem. 2011;286:19958–19972. doi: 10.1074/jbc.M110.194852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Libon DJ, Toledo JB, et al. Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol. 2012;123:395–407. doi: 10.1007/s00401-011-0932-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Toledo JB, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Arai K, Del Tredici K, et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014;128:423–437. doi: 10.1007/s00401-014-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. The molecular links between TDP-43 dysfunction and neurodegeneration. Adv Genet. 2009;66:1–34. doi: 10.1016/S0065-2660(09)66001-6. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. TDP-43: gumming up neurons through protein-protein and protein-RNA interactions. Trends Biochem Sci. 2012;37:237–247. doi: 10.1016/j.tibs.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20:1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280:37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Deng JJ, Bai Y, Thornton FB, Oddo S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J Biol Chem. 2009;284:27416–27424. doi: 10.1074/jbc.M109.031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Neumann M, Bigio EH, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani R, Hirai K, Aliev G, et al. Role of mitochondrial dysfunction in Alzheimer’s disease. J Neurosci Res. 2002;70:357–360. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- Chanson JB, Echaniz-Laguna A, Vogel T, Mohr M, Benoilid A, Kaltenbach G, Kiesmann M. TDP43-positive intraneuronal inclusions in a patient with motor neuron disease and Parkinson’s disease. Neurodegener Dis. 2010;7:260–264. doi: 10.1159/000273591. [DOI] [PubMed] [Google Scholar]
- Chen X, Yan SD. Mitochondrial A beta: a potential cause of metabolic dysfunction in Alzheimer’s disease. IUBMB Life. 2006;58:686–694. doi: 10.1080/15216540601047767. [DOI] [PubMed] [Google Scholar]
- Chen S, Zhang X, Song L, Le W. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 2012;22:110–116. doi: 10.1111/j.1750-3639.2011.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sayana P, Zhang X, Le W. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener. 2013;8:28. doi: 10.1186/1750-1326-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier-Larsen E, Holzbaur EL. Axonal transport and neurodegenerative disease. Biochem Biophys Acta. 2006;1762:1094–1108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Clery A, Blatter M, Allain FH. RNA recognition motifs: boring? Not quite. Curr Opin Struct Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Cohen-Kaplan V, Ciechanover A, Livneh I. p62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget. 2016;7:83833–83834. doi: 10.18632/oncotarget.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collard JF, Cote F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111:1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem. 2012;287:15635–15647. doi: 10.1074/jbc.M111.333450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Onesto E, Buratti E, de la Grange P, Gumina V, Baralle FE, Silani V, Ratti A. From transcriptomic to protein level changes in TDP-43 and FUS loss-of-function cell models. Biochim Biophys Acta. 2015;1849:1398–1410. doi: 10.1016/j.bbagrm.2015.10.015. [DOI] [PubMed] [Google Scholar]
- Conicella AE, Zerze GH, Mittal J, Fawzi NL. ALS mutations disrupt phase separation mediated by alpha-helical structure in the TDP-43 low-complexity C-terminal domain. Structure. 2016;24:1537–1549. doi: 10.1016/j.str.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino M, Carri MT. Mitochondrial dysfunction in ALS. Prog Neurobiol. 2012;97:54–66. doi: 10.1016/j.pneurobio.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Czech C, Tremp G, Pradier L. Presenilins and Alzheimer’s disease: biological functions and pathogenic mechanisms. Prog Neurobiol. 2000;60:363–384. doi: 10.1016/s0301-0082(99)00033-7. [DOI] [PubMed] [Google Scholar]
- D’Alton S, Altshuler M, Cannon A, Dickson DW, Petrucelli L, Lewis J. Divergent phenotypes in mutant TDP-43 transgenic mice highlight potential confounds in TDP-43 transgenic modeling. PLoS ONE. 2014;9:e86513. doi: 10.1371/journal.pone.0086513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Hauptmann S, Scherping I, et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L Tau transgenic mice. J Biol Chem. 2005;280:23802–23814. doi: 10.1074/jbc.M500356200. [DOI] [PubMed] [Google Scholar]
- Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, Mann DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007;113:521–533. doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- Davidson Y, Amin H, Kelley T, et al. TDP-43 in ubiquitinated inclusions in the inferior olives in frontotemporal lobar degeneration and in other neurodegenerative diseases: a degenerative process distinct from normal ageing. Acta Neuropathol. 2009;118:359–369. doi: 10.1007/s00401-009-0526-z. [DOI] [PubMed] [Google Scholar]
- De Conti L, Akinyi MV, Mendoza-Maldonado R, Romano M, Baralle M, Buratti E. TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic Acids Res. 2015;43:8990–9005. doi: 10.1093/nar/gkv814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Parker R. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol. 2012;4:a012286. doi: 10.1101/cshperspect.a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res. 2012;1462:16–25. doi: 10.1016/j.brainres.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau) J Mol Neurosci. 2011;45:384–389. doi: 10.1007/s12031-011-9589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Ma M, Teng J, et al. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget. 2015;6:24178–24191. doi: 10.18632/oncotarget.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi H, Koyano S, Suzuki Y, Nukina N, Kuroiwa Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci Res. 2010;66:131–133. doi: 10.1016/j.neures.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Dormann D, Capell A, Carlson AM, et al. Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J Neurochem. 2009;110:1082–1094. doi: 10.1111/j.1471-4159.2009.06211.x. [DOI] [PubMed] [Google Scholar]
- van Eersel J, Ke YD, Gladbach A, Bi M, Gotz J, Kril JJ, Ittner LM. Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS ONE. 2011;6:e22850. doi: 10.1371/journal.pone.0022850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet. 2012;21:3703–3718. doi: 10.1093/hmg/dds205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiler MS, Strobel B, Freischmidt A, et al. TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol. 2015;211:897–911. doi: 10.1083/jcb.201504057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, Weber SS, Supper J, Zell A, Kahle PJ. TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res. 2012;40:2668–2682. doi: 10.1093/nar/gkr1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser PE, Yang DS, Yu G, Levesque L, Nishimura M, Arawaka S, Serpell LC, Rogaeva E, St George-Hyslop P. Presenilin structure, function and role in Alzheimer disease. Biochem Biophys Acta. 2000;1502:1–15. doi: 10.1016/s0925-4439(00)00028-4. [DOI] [PubMed] [Google Scholar]
- Fuentealba RA, Udan M, Bell S, Wegorzewska I, Shao J, Diamond MI, Weihl CC, Baloh RH. Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J Biol Chem. 2010;285:26304–26314. doi: 10.1074/jbc.M110.125039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti D, Dell’Osso B, Altamura AC, Scarpini E. Psychiatric symptoms in frontotemporal dementia: epidemiology, phenotypes, and differential diagnosis. Biol Psychiatry. 2015;78:684–692. doi: 10.1016/j.biopsych.2015.03.028. [DOI] [PubMed] [Google Scholar]
- Gao J, Wang L, Liu J, Xie F, Su B, Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants (Basel) 2017;6:25–43. doi: 10.3390/antiox6020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Josephs KA, Petrucelli L. Review: transactive response DNA-binding protein 43 (TDP-43): mechanisms of neurodegeneration. Neuropathol Appl Neurobiol. 2010;36:97–112. doi: 10.1111/j.1365-2990.2010.01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Chen F, Iqbal K, Gong CX, Wang X, Liu F. Transactive response DNA-binding protein 43 (TDP-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. J Biol Chem. 2017a;292:10600–10612. doi: 10.1074/jbc.M117.783498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wu F, Xu W, et al. TDP-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 2017b;45:6177–6193. doi: 10.1093/nar/gkx175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MP, Gitler AD. ALS-associated ataxin 2 polyQ expansions enhance stress-induced caspase 3 activation and increase TDP-43 pathological modifications. J Neurosci. 2012;32:9133–9142. doi: 10.1523/JNEUROSCI.0996-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebron ML, Lonskaya I, Sharpe K, Weerasinghe PP, Algarzae NK, Shekoyan AR, Moussa CE. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6) J Biol Chem. 2013;288:4103–4115. doi: 10.1074/jbc.M112.419945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebron M, Chen W, Miessau MJ, Lonskaya I, Moussa CE. Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J Neurochem. 2014;129:350–361. doi: 10.1111/jnc.12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefferon TW, Groman JD, Yurk CE, Cutting GR. A variable dinucleotide repeat in the CFTR gene contributes to phenotype diversity by forming RNA secondary structures that alter splicing. Proc Natl Acad Sci USA. 2004;101:3504–3509. doi: 10.1073/pnas.0400182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibiki Kawamata PP, Konrad C, Palomo G, Bredvik K, Gerges M, Valsecchi F, Petrucelli L, Ravits JM, Starkov A, Manfredi G. Mutant TDP-43 does not impair mitochondrial bioenergetics in vitro and in vivo. Mol Neurodegener. 2017;12:37. doi: 10.1186/s13024-017-0180-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi S, Iseki E, Yamamoto R, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–294. doi: 10.1016/j.brainres.2007.09.048. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Pottier C, Nicholson AM, et al. Clinical and neuropathological features of ALS/FTD with TIA1 mutations. Acta Neuropathol Commun. 2017;5:96. doi: 10.1186/s40478-017-0493-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey J, Emmett W, Fratta P, Isaacs AM, Plagnol V. Quantitative analysis of cryptic splicing associated with TDP-43 depletion. Bmc Med Genomics. 2017;10:38–54. doi: 10.1186/s12920-017-0274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, Neumann M, Trojanowski JQ, Lee VM. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem. 2009;284:8516–8524. doi: 10.1074/jbc.M809462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi Y, Eid L, Parent M, et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain. 2016;139:3187–3201. doi: 10.1093/brain/aww237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumikawa K, Nobe Y, Yoshikawa H, et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci Rep. 2017;7:7709. doi: 10.1038/s41598-017-06953-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain. 2016;139:2983–2993. doi: 10.1093/brain/aww224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LL, Xue W, Hong JY, Zhang JT, Li MJ, Yu SN, He JH, Hu HY. The N-terminal dimerization is required for TDP-43 splicing activity. Scientific reports. 2017;7:6196–5207. doi: 10.1038/s41598-017-06263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, Petersen RC, Dickson DW. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014;127:441–450. doi: 10.1007/s00401-013-1211-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Whitwell JL, et al. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016;131:571–585. doi: 10.1007/s00401-016-1537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Raphael AR, LaDow ES, et al. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2011;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Yokoshi M, Okada H, Kawahara Y. The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat Commun. 2015;6:6183. doi: 10.1038/ncomms7183. [DOI] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349:650–655. doi: 10.1126/science.aab0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE. 2010;5:e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Duan W, Guo Y, Jiang H, Li Z, Huang J, Hong K, Li C. Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res Bull. 2012;89:185–190. doi: 10.1016/j.brainresbull.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114:49–54. doi: 10.1007/s00401-007-0223-8. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Nicholson AM, Sarkar M, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron. 2017;95(808–816):e809. doi: 10.1016/j.neuron.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNair L, Xiao S, Miletic D, Ghani M, Julien JP, Keith J, Zinman L, Rogaeva E, Robertson J. MTHFSD and DDX58 are novel RNA-binding proteins abnormally regulated in amyotrophic lateral sclerosis. Brain. 2016;139:86–100. doi: 10.1093/brain/awv308. [DOI] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292–309. doi: 10.1016/j.neuron.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23:1413–1424. doi: 10.1093/hmg/ddt528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markopoulou K, Dickson DW, McComb RD, Wszolek ZK, Katechalidou L, Avery L, Stansbury MS, Chase BA. Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson’s disease. Variability in familial Parkinson’s disease. Acta Neuropathol. 2008;116:25–35. doi: 10.1007/s00401-008-0372-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet. 2011;20:1400–1410. doi: 10.1093/hmg/ddr021. [DOI] [PubMed] [Google Scholar]
- McGurk L, Lee VM, Trojanowksi JQ, Van Deerlin VM, Lee EB, Bonini NM. Poly-A binding protein-1 localization to a subset of TDP-43 inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J Neuropathol Exp Neurol. 2014;73:837–845. doi: 10.1097/NEN.0000000000000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005;33:6000–6010. doi: 10.1093/nar/gki897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12:435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SMAR. Molecular integrity of mitochondria alters by potassium chloride. Inter J Proteomics. 2015;2015:647408–647419. doi: 10.1155/2015/647408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohagheghi F, Prudencio M, Stuani C, Cook C, Jansen-West K, Dickson DW, Petrucelli L, Buratti E. TDP-43 functions within a network of hnRNP proteins to inhibit the production of a truncated human SORT1 receptor. Hum Mol Genet. 2016;25:534–545. doi: 10.1093/hmg/ddv491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori F, Tanji K, Zhang HX, Nishihira Y, Tan CF, Takahashi H, Wakabayashi K. Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008;116:193–203. doi: 10.1007/s00401-008-0396-9. [DOI] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- Nakashima-Yasuda H, Uryu K, Robinson J, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114:221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134:1506–1518. doi: 10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Niblock M, Hortobagyi T, Troakes C, Al-Sarraj S, Spickett C, Jones R, Shaw CE, Gallo JM. Lack of association between TDP-43 pathology and tau mis-splicing in Alzheimer’s disease. Neurobiol Aging. 2016;37:45–46. doi: 10.1016/j.neurobiolaging.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihei Y, Ito D, Suzuki N. Roles of ataxin-2 in pathological cascades mediated by TAR DNA-binding protein 43 (TDP-43) and Fused in Sarcoma (FUS) J Biol Chem. 2012;287:41310–41323. doi: 10.1074/jbc.M112.398099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T, Masuda-Suzukake M, Arai T, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4:124–134. doi: 10.1016/j.celrep.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Nonhoff U, Ralser M, Welzel F, Piccini I, Balzereit D, Yaspo ML, Lehrach H, Krobitsch S. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell. 2007;18:1385–1396. doi: 10.1091/mbc.E06-12-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyike CU, Diehl-Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry. 2013;25:130–137. doi: 10.3109/09540261.2013.776523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orru S, Coni P, Floris A, Littera R, Carcassi C, Sogos V, Brancia C. Reduced stress granule formation and cell death in fibroblasts with the A382T mutation of TARDBP gene: evidence for loss of TDP-43 nuclear function. Hum Mol Genet. 2016;25:4473–4483. doi: 10.1093/hmg/ddw276. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XD, Chen XC. Clinic, neuropathology and molecular genetics of frontotemporal dementia: a mini-review. Transl Neurodegener. 2013;2:8. doi: 10.1186/2047-9158-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao WC, Dickson DW, Crook JE, Finch NA, Rademakers R, Graff-Radford NR. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25:364–368. doi: 10.1097/WAD.0b013e31820f8f50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papegaey A, Eddarkaoui S, Deramecourt V, et al. Reduced Tau protein expression is associated with frontotemporal degeneration with progranulin mutation. Acta Neuropathol Com. 2016;4:74–87. doi: 10.1186/s40478-016-0345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Picher-Martel V, Valdmanis PN, Gould PV, Julien JP, Dupre N. From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathol Commun. 2016;4:70. doi: 10.1186/s40478-016-0340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M, Jansen-West KR, Lee WC, et al. Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci USA. 2012;109:21510–21515. doi: 10.1073/pnas.1211577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- Robinson JL, Geser F, Stieber A, Umoh M, Kwong LK, Van Deerlin VM, Lee VMY, Trojanowski JQ. TDP-43 skeins show properties of amyloid in a subset of ALS cases. Acta Neuropathol. 2013;125:121–131. doi: 10.1007/s00401-012-1055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan LH, Zhou CK, Jin EL, Kucharavy A, Zhang Y, Wen ZH, Florens L, Li R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature. 2017;543:443–446. doi: 10.1038/nature21695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang YJ, Baker M, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Arai T, Hasegawa M, Yu S, McGeer PL. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol. 2008;67:1159–1165. doi: 10.1097/NEN.0b013e31818e8951. [DOI] [PubMed] [Google Scholar]
- Shiga A, Ishihara T, Miyashita A, et al. Alteration of POLDIP3 splicing associated with loss of function of TDP-43 in tissues affected with ALS. PLoS ONE. 2012;7:e43120. doi: 10.1371/journal.pone.0043120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan SA, Barres BA. Glia as primary drivers of neuropathology in TDP-43 proteinopathies. Proc Natl Acad Sci USA. 2013;110:4439–4440. doi: 10.1073/pnas.1301608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA. Alzheimer disease. Int Rev Neurobiol. 1998;42:1–54. doi: 10.1016/s0074-7742(08)60607-8. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Stribl C, Samara A, Trumbach D, et al. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J Biol Chem. 2014;289:10769–10784. doi: 10.1074/jbc.M113.515940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MK, Bell W, LaClair KD, et al. Cryptic exon incorporation occurs in Alzheimer’s brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol. 2017;133:923–931. doi: 10.1007/s00401-017-1701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Lee K, Matsuoka M. TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J Biol Chem. 2011;286:13171–13183. doi: 10.1074/jbc.M110.197483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M, Coon EA, Osmand AP, et al. Coexistence of Huntington’s disease and amyotrophic lateral sclerosis: a clinicopathologic study. Acta Neuropathol. 2012;124:749–760. doi: 10.1007/s00401-012-1005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan QM, Yalamanchili HK, Park J, et al. Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Hum Mol Genet. 2016;25:5083–5093. doi: 10.1093/hmg/ddw337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanahashi H, Tabira T. Alzheimer’s disease-associated presenilin 2 interacts with DRAL, an LIM-domain protein. Hum Mol Genet. 2000;9:2281–2289. doi: 10.1093/oxfordjournals.hmg.a018919. [DOI] [PubMed] [Google Scholar]
- Tanji K, Zhang HX, Mori F, Kakita A, Takahashi H, Wakabayashi K. p62/sequestosome 1 binds to TDP-43 in brains with frontotemporal lobar degeneration with TDP-43 inclusions. J Neurosci Res. 2012;90:2034–2042. doi: 10.1002/jnr.23081. [DOI] [PubMed] [Google Scholar]
- Tashiro Y, Urushitani M, Inoue H, et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem. 2012;287:42984–42994. doi: 10.1074/jbc.M112.417600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauffenberger A, Chitramuthu BP, Bateman A, Bennett HP, Parker JA. Reduction of polyglutamine toxicity by TDP-43, FUS and progranulin in Huntington’s disease models. Hum Mol Genet. 2013;22:782–794. doi: 10.1093/hmg/dds485. [DOI] [PubMed] [Google Scholar]
- Tian T, Huang C, Tong J, Yang M, Zhou H, Xia XG. TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Inter J Biol Sci. 2011;7:234–243. doi: 10.7150/ijbs.7.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong JB, Huang C, Bi FF, Wu QX, Huang B, Zhou HX. XBP1 depletion precedes ubiquitin aggregation and Golgi fragmentation in TDP-43 transgenic rats. J Neurochem. 2012;123:406–416. doi: 10.1111/jnc.12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima Y, Tanaka H, Shimohata M, Kimura K, Morita T, Kakita A, Takahashi H. Spinocerebellar ataxia type 2 (SCA2) is associated with TDP-43 pathology. Acta Neuropathol. 2011;122:375–378. doi: 10.1007/s00401-011-0862-7. [DOI] [PubMed] [Google Scholar]
- Tsai RM, Boxer AL. Treatment of frontotemporal dementia. Curr Treat Options Neurol. 2014;16:319. doi: 10.1007/s11940-014-0319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS) Brain Res. 2009;1305:168–182. doi: 10.1016/j.brainres.2009.09.105. [DOI] [PubMed] [Google Scholar]
- Walker AK, Daniels CM, Goldman JE, Trojanowski JQ, Lee VM, Messing A. Astrocytic TDP-43 pathology in Alexander disease. J Neurosci. 2014;34:6448–6458. doi: 10.1523/JNEUROSCI.0248-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Fan H, Ying Z, Li B, Wang H, Wang G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 2010;469:112–116. doi: 10.1016/j.neulet.2009.11.055. [DOI] [PubMed] [Google Scholar]
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CK. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci USA. 2012;109:15024–15029. doi: 10.1073/pnas.1206362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Tsai KJ, Shen CK. Autophagy activation ameliorates neuronal pathogenesis of FTLD-U mice: a new light for treatment of TARDBP/TDP-43 proteinopathies. Autophagy. 2013;9:239–240. doi: 10.4161/auto.22526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, Wang X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet. 2013b;22:4706–4719. doi: 10.1093/hmg/ddt319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang L, Lu J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22:869–878. doi: 10.1038/nm.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Arakawa H, Wang L, et al. Motor-coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Mol Therapy. 2017;25:127–139. doi: 10.1016/j.ymthe.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, Hanson PI, Kimonis V, Pestronk A. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79:1186–1189. doi: 10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenqiang C, Lonskaya I, Hebron ML, Ibrahim Z, Olszewski RT, Neale JH, Moussa CE. Parkin-mediated reduction of nuclear and soluble TDP-43 reverses behavioral decline in symptomatic mice. Hum Mol Genet. 2014;23:4960–4969. doi: 10.1093/hmg/ddu211. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Wolozin B. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener. 2012;7:56. doi: 10.1186/1750-1326-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo JA, Liu T, Trotter C, et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat Commun. 2017;8:15558. doi: 10.1038/ncomms15558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Sanelli T, Chiang H, Sun Y, Chakrabartty A, Keith J, Rogaeva E, Zinman L, Robertson J. Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol. 2015;130:49–61. doi: 10.1007/s00401-015-1412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Gendron TF, Zhang YJ, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Zhang YJ, Lin WL, Cao X, Stetler C, Dickson DW, Lewis J, Petrucelli L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol Neurodegener. 2011;6:73. doi: 10.1186/1750-1326-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hideyama T, Hachiga K, Teramoto S, Takano J, Iwata N, Saido TC, Kwak S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat Commun. 2012;3:1307. doi: 10.1038/ncomms2303. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA. 2009;106:7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Casadesus G, Avila J, Drew K, Perry G, Smith MA. Oxidative imbalance in Alzheimer’s disease. Mol Neurobiol. 2005;31:205–217. doi: 10.1385/MN:31:1-3:205. [DOI] [PubMed] [Google Scholar]
- Zhukareva V, Vogelsberg-Ragaglia V, Van Deerlin VM, et al. Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann Neurol. 2001;49:165–175. doi: 10.1002/1531-8249(20010201)49:2<165::aid-ana36>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Zondler L, Feiler MS, Freischmidt A, Ruf WP, Ludolph AC, Danzer KM, Weishaupt JH. Impaired activation of ALS monocytes by exosomes. Immunol Cell Biol. 2017;95:207–214. doi: 10.1038/icb.2016.89. [DOI] [PubMed] [Google Scholar]