

Abstract
Humans have co-evolved alongside numerous other organisms, some having a profound effect on health and nutrition. As the earliest pharmaceutical subject, pharmacognosy has evolved into a meta-discipline devoted to natural biomedical agents and their functional properties. While the acquisition of expanding data volumes is ongoing, contextualization is lagging. Thus, we assert that the establishment of an integrated and open databases ecosystem will nurture the discipline. After proposing an epistemological framework of knowledge acquisition in pharmacognosy, this study focuses on recent computational and analytical approaches. It then elaborates on the flux of research data, where good practices could foster the implementation of more integrated systems, which will in turn help shaping the future of pharmacognosy and determine its constitutional societal relevance.
Graphical abstract
Introduction
For healthcare, humankind has always depended on natural resources. Traditional medicines continue to represent the main source of therapy for the majority of the world’s population and rely on written documents, verbal teachings, and (in)formally transmitted practices. This study considers traditional medicines as being based on the outcomes of holistic experimentation (Figure 1, Corner A). Written pharmacognosy knowledge has evolved from documents such as the Eber’s Papyrus into contemporary publication formats that describe the discovery of bioactive natural products, usually based on reductionist experimental approaches (Figure 1, Corner B). Indeed, the main paradigm of the last 200+ years of pharmacognosy and related research consisted in studying complex solvent extracts and ideally characterizing its individual chemical principle. Coupled to bioassay, this method, known as bioactivity-guided fractionation (BGF), has contributed significantly to the fact that almost half of currently marketed drugs are related to natural products (NPs) [1].
Figure 1.
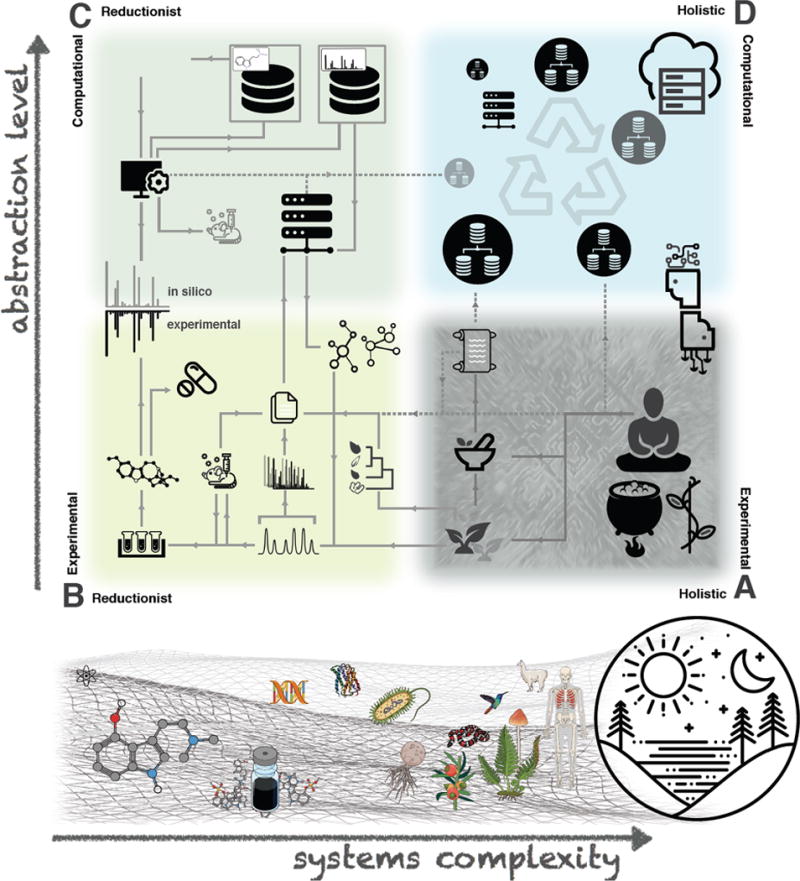
Proposed epistemological framework of knowledge acquisition methods in pharmacognosy. At the bottom are depicted the main subjects of study of the discipline in order of increasing systems complexity: single chemical entities, natural product extracts, genetic material, proteins, microorganisms, plants, animals (including humans), the whole planet ecosystem and their possible interactions. Above are schematized knowledge acquisition methods in the discipline. They were divided four ways according to their tendency to rely on: holistic and experimental approaches (Corner A, e.g. traditional medicines), reductionist and experimental approaches (Corner B, e.g. bio-guided fractionation), reductionist and computational approaches (Corner C, e.g. in silico fragmentation of a single chemical entity) and holistic and computational approaches (Corner D, a hypothetical ecosystem of open databases aggregating pharmacognostic knowledge). Grey links represent examples of information fluxes (solid lines as existing fluxes and dashed lines as potential ones). Actually, these knowledge acquisition methods are not mutually exclusive. For example: the use of an in silico annotated molecular network organizing fragmentation data acquired on plants selected trough an ethnopharmacological survey to highlight metabolites involved in the reported traditional use. This approach would feed from three knowledge acquisition methods (Corner A, B and C).
However, associating the activity of a preparation to an alleged single chemical entity (SCE) overlooks that complex matrices comprised of hundreds or (tens of) thousands of metabolites often resist physical separation and attempts seeking to explain observed biological activities. To address these challenges, progress in computational and (bio-)analytical methodology are being integrated into pharmacognosy research. These advances now allow the characterization of multiple SCEs in complex organisms (metabolomics), which re-calibrates biological interpretation towards a more holistic experimental perspective (Figure 1, Corner A) and enables the prediction of physicochemical and spectral properties associated with real or surrogate SCEs (computational pharmacognosy), using reductionist strategies as input (Figure 1, Corner C). As reductionist experiments involving physical separation foster SCE identification, the fuzzier, yet deeper, world of metabolomics coupled with computational chemistry tools becomes the best available complement to address the complexity of structurally and biologically diverse matrices. Nevertheless, precision and accuracy for the efficient annotation of extensive metabolite pools is lagging and remains a major challenge. Re-contextualizing SCEs within their organisms, pathways, and molecular targets will strengthen the metabolite identification process and promote the recognition of new potential biological activities [2,3]. Efficient ways of standardization, dissemination, and open sharing of relevant data sets are prerequisites for advancing this contextualization. They will enable the diversification of knowledge acquisition methods in pharmacognosy (Figure 1 Corner A, B, and C), the results of which could be re-assembled into holistic computational approaches (Figure 1, Corner D) and advance pharmacognosy in(to) the digital era.
Computational advances in metabolomics
Acquiring observational data at the molecular level is central to both classical reductionist and modern metabolomics experiments. Thus, acceleration and enhancement of instrumental and computational efforts to identify and characterize SCEs need to be pursued. Indeed, one of the key challenges in these fields consists in the unambiguous(!) identification of multiple SCEs in complex matrices.
Metabolomics data annotation
Metabolite qualification
When physically isolated, SCEs are classically identified through a combination of extensive 1D and 2D NMR experiments, establishing atom connectivity, and HRMS measurement assigning the molecular formula (MF). Additionally, chiroptical and/or X-ray measurements can establish the spatial conformation and configurations, including atropisomerism [4]. NMR can be considered the most appropriate tool for overall accuracy of structural elucidation, whereas MS is most suitable for metabolite annotation within complex mixtures. Both roles are central to metabolomics [5,6]. In NP research, metabolomics mainly relies on (U)HPLC hyphenated HRMS and HRMS/MS. For complex matrices, pre-separation of the extract is generally required. While C18 phases are most commonly used in (U)HPLC, a wide variety of phase chemistry can accommodate the separation of diverse NPs [7]. Mining of LC-MS data generally involves peak-picking algorithms for the identification of so-called “features”, i.e., ions of specific m/z value at a specific retention time (m/z@RT). The high number of degenerated MS features (adducts, dimers) requires deconvolution into single features [8], which can then be linked to MFs by applying various filtering steps [9]. Feature alignment across samples leads to matrices that can be mined for differential biomarkers using multi-variate data analysis (MVDA) [10]. As fragmentation spectra are related to structure, they bring valuable complementary information to the MF. MS/MS spectra are generally interpreted by applying spectral matching against experimental or theoretical spectral databases. Access to an experimental MS/MS database acquired at a standardized ionization energy and on a specific instrument would be ideal, but is unrealistic given the number of NPs reported to date (>200,000). In fact, accessible experimental databases are limited in size (typically, ca. 25,000 compounds) and diversity [11].
Metabolite annotation can also benefit greatly from direct NMR-based fingerprinting [5]. One recent methodology deconvolutes and annotates 13C data directly within complex NPs extracts [12], and minimal NMR data input approaches permit the combination of analytically orthogonal spectroscopic data (NMR plus MS) and databases [13].
Metabolite quantification
While MS and UV detectors are intrinsically limited to the quantification of metabolites for which reference standards are available, Corona Discharge (CDD) and Evaporative Light-Scattering (ELSD) detectors can sometimes overcome this limitation. MS-based approaches are usually not quantitative, unless being calibrated for every targeted SCE. Recent progress towards establishing of quantitative structure-response relationships for defined classes of analytes by means of artificial neural networks shows high predictability of the ESI-MS responsiveness [14], and might hold a possible solution for the development of truly quantitative MS-based metabolomics studies. As NMR does not require identical standards for absolute quantification and has 100% quantitation capabilities, qNMR has become applied widely [15]. Advancement in both probe and qNMR methodology, as well as the availability of 2D qNMR methods further increase the number of metabolites that can be studied concurrently [16].
Metabolite localization and dynamics
The spatial localization of specific metabolites in tissue can provide important clues when studying interactions between organisms or when surveying the distribution of metabolites within an organism up to the scale of ecosystems. A recent review has covered the use of Mass Spectrometry Imaging from the nano to the macro range [17]. Beyond spatial dimensions, time is also critical, as certain metabolites may be produced only during specific developmental stages of the organism [18].
Metabolite properties prediction
Structural, spectral, and biological data have been used to annotate, sort, and predict the roles of metabolites, with objectives ranging from dereplication [19] to prediction of biological properties [2], and prioritization of high-added value molecules [3]. Additionally, bioactives can be inferred from traditional knowledge [20] or genomic mining aimed at predicting metabolic products [21]. Spectral prediction [22,23] of metabolites is becoming an essential dereplication tool for unknowns, or when original data are missing. One critical step when annotating metabolites via experimental or theoretical spectral matching is the establishment of a scoring system that rates the confidence of the annotation. For this, statistical methods to estimate false discovery rates of annotations have been developed [24] and applied to high-resolution imaging mass spectrometry data [25].
Metabolomics data contextualization
Analyte specificity and “individualization” for effective detection is a common denominator in modern analytical methods. Thus, contextualizing these singleton molecular data is a means of accessing the reality of the studied organism and helps with the metabolite annotation process. One game-changing tool in this field is the Global Natural Products Social molecular networking (MN) platform [26]. MN organizes untargeted MS/MS data and allows for the visualization of the analytes’ relationships in the form of clusters of structurally-related molecules. Thus, MN offers a way to propagate annotation information among generated networks, providing an efficient tool for NP dereplication [27]. The annotation potential can be extended by automatically querying constituents of the MN against vast theoretical spectral NP DBs [28]. Further contextualization by overlaying MN of large NP extract libraries with biological and taxonomical information can lead to prioritization of bioactive NPs [3]. An additional layer of biological significance can be obtained by gathering information about the metabolic reactions expressed in the studied biological system and placing the detected features into a relevant biochemical context [29]. The in silico metabolic expansion of structural DBs, followed by conversion of these expanded DBs to theoretical spectral DBs used to automatically annotated experimental data organized as MN becomes achievable. When followed by re-injection of the annotated structures at the metabolic expansion phase and feeding of structural/spectral pairs for improvement during the in silico fragmentation phase, this approach could lead to a virtuous cycle of metabolite identification [30], provided that solid annotation scoring systems can be established. Recently, such an integrated approach was devised for the annotation of unknowns in GC-MS data [19]. Regarding NMR data contextualization, an algorithm comparing 2D NMR spectra was trained on >2,000 HSQC spectra using deep convolutional neural networks [31]. This tool, SMART, assisted in NP discovery efforts and detected several new compounds of known skeletons.
Integrative solutions are essential for NP studies, and to grasp the complexity of the biochemical interactions involved in pharmacognosy. For their efficiency, the development of open and integrated DBs capable of linking structural, spectral, genetic, phylogenetic, ethnomedical, biological, clinical, and regulatory information is critically important. (Figure 1, Corner D) The creation of such a DB ecosystem depends on the efficient accessibility, diffusion, and sharing of accurately curated data.
Evolution of Pharmacognosy through enhanced data integration
Almost all pharmacognosy research endeavors start, and are guided, with collected and taxonomically identified organisms, possibly annotated by traditional use. At this initial stage of documentation and data production, existing resources are typically consulted (Figure 2). As Open Access and continuous data reuse models are being taken seriously [32], studies show the positive impact of data-sharing on scientific productivity [33] – despite newly emerging issues [34]. Additionally, access to biological resources and traditional knowledge today must follow a strict legal framework within a country [35]. Furthermore, most of the published research knowledge that has accumulated over the past 200 years is owned by third-parties, and access is tightly regulated. In comparison with other fields, current data-sharing practices are sub-standard in pharmacognosy. The behavior of researchers towards data-sharing is not only dictated by norms (however, efficient policies should be implemented [36]), but is also driven by personal attitude towards the behavior. Thus, data reuse should also be encouraged by demonstration of it’s continuing intrinsic value [37].
Figure 2.
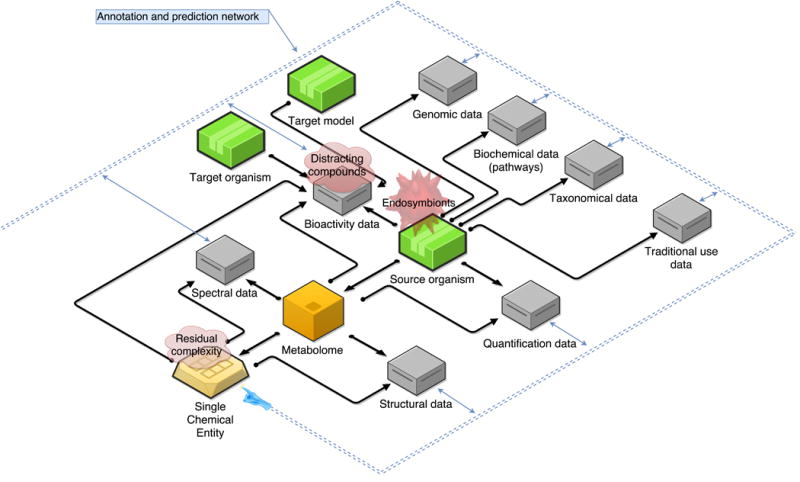
Representation of the different types of data handled in pharmacognosy (non exhaustive). The blue network that circles the graphic represents the production and consumption processes of data in order to predict and annotate existing data or entities. The transparent red items on top of Bioactivity data, Source organism and Single Chemical Entity represent some of the different issues that can complicate the interpretation or identification of these elements. See Box. 1 for details
How to manage, curate and share data
It is paramount that the data produced today will be available in the future. This requires open, properly-documented, quality-controlled formats, and, ideally, associated tools for analysis. Regrettably, contemporary analytical instruments typically produce data only in a vendor-specific, undocumented format. For MS data, functional alternatives exist, especially the mzXML format [38]. While no widely accepted format exists yet for NMR, candidates such as nmrML [39], NMReDATA (http://nmredata.org), and the Allotrope Foundation (AF) data standard (https://www.allotrope.org) have started to appear. The AF initiative consists of a network of the major instrument manufacturers, industries, and academics. AF aims at developing a common format for analytical data, including LC, UV, IR, NMR, MS, and more. The collectively built ontologies, and the use of computationally efficient, standard hierarchical data format containers (HDF5), make this a most promising project, and worthy to follow.
Best practices for data management
Data-sharing is essential to research, and gradually becomes mandatory for institutional and philanthropic funding. As stated in the FAIR principles, ideal data should be findable, accessible, inter-operable, and reusable [40]. While researchers are often tempted to establish individual repositories or indexing resources, making them sustainable and reliable requires efficient planning and business models [41]. Numerous DBs and tools address a diversity of data related to metabolites (see https://omictools.com/). Many are simply data-silos, lack standardization or links with other resources and, thereby, diffuse the efforts of both the community and the DB developers. Metabolomics [42] and plant science [43] communities are currently debating the best practices for data management, ontologies development, controlled vocabularies, and tools that make data inter-operable [44]. The Unichem project [45] demonstrated the feasibility of linked DBs. PubChem and ChEMBL allow the upload of structural and bioassay data for compounds. While these resources do not provide storage space for spectral data, they can be combined with other repositories such as Dataverse (https://www.dataverse.org), Zenodo (https://zenodo.org/), or the Open Science Framework (https://www.osf.io) to cite a few of the >650 indexed resources (http://tagteam.harvard.edu/hubs/oatp/tag/oa.repositories.data).
Data curation
The development and continuous improvement of machine learning, natural language processing, and the increase of computing power all enhance the means of extracting facts from the literature and other resources. Open initiatives, such as ContentMine (https://contentmine.org/) provide tools, support, and opportunities for extracting knowledge from publications. However, for the foreseeable future, these tools can not replace human intelligence and dispense with the need for curation. In fact, the opposite is the case, as errors produced by “artificial intelligence” tend to be multiplied across different diverging resources. Automation may accelerate this trend.
Data re-use
From a scientific perspective, restrictive licenses increase the likelihood of users favoring lower quality and less-restricted data sources. Another consequence of overly strict licensing is that researchers utilize the data without referencing the origin, adding another layer of complexity to the problem of authorship and reference tracking. In contrast, mindful data licensing also nurtures new initiatives whenever projects lose funding or discontinue due to, e.g., retirement, thereby promoting research efforts that otherwise would have been made in vain [41].
Experiment and data description and sharing
The design of scientific papers could be rethought to keep only the prosaic parts (discussions, reflections, opinions, new ideas, and new methods) in the current publication format. For reporting experimental research one alternative could be found in semantic and ontology-based publishing [46]. Both approaches can also be combined to offer the best of the two worlds. Particularly, nanopublications [47] may provide a way to reduce the time from discovery to publication and improve the quality and availability of research results, thereby fulfilling funder and societal translational expectations. Thus, the bioactivity of a previously described compound could be described as simple facts and re-usable protocols [48]. The IsaTab format (http://www.isacommons.org/), supported by several data-sharing platforms, also provides a standardized and well-supported way to describe and navigate the metadata of experimental data sets.
Conclusions
As the complexity of natural medicines goes beyond their elusive bioactivities and numerous constituents, much remains to be done in order to unambiguously define the precise metabolic content of an organism under given conditions. If the developments of computational solutions for identifying metabolites should be pursued, it is also crucial that these developments go hand in hand with the contextualization of the acquired data.
Thereby, it should be necessary to consider the elaboration of an open and accessible database ecosystem. Ideally, this will allow cross-linking and foster extracting more meaning from the accumulated data, regardless of origin and acquisition method, and ultimately gaining an overall view of the studied systems.
None of the knowledge acquisition methods in the presented epistemological framework of pharmacognosy (Figure 1) should be neglected. Each method has its own advantage and disadvantage, and their combination can only be beneficial. It is timely to expand the necessary efforts within the scientific community to follow this global vision. Making research data both shared and shareable is definitely the first action item for implementation. This comprehensive view will then offer a means of preparing pharmacognosy for the digital era and, ultimately, tackling important public health issues and environmental challenges. However, more than just technical solutions and community commitment will be required to achieve this [49]. The recently emerged concept of ecopharmacognosy [50], which integrates pharmacognosy in the broader context of sustainability, offers specific thoughts on potential directions to be followed.
Highlights.
Metabolomics and Pharmacognosy are naturally connected and cross-fertilizing
Improvement in computational tools and contextualization of analytical data are needed to potentiate translational applications
Data-intensive metabolomics methods unveil the need for enhanced data practices
Establishing an ecosystem of open, interactive databases will nurture both metabolomics and pharmacognosy research
Box 1. A cautionary word regarding distracting compounds and residual complexity.
Conclusion or interpretation errors from bioassay observations are common and may be due to several neglected factors (e.g., distracting compounds, Figure 2) such as aggregation, non-specific activities and reactivity [51], fluorescence quenching or overlapping spectra, redox cycling, among others [52]. False positives can arise from misidentified and/or contaminated cell-lines [53], which are difficult to track in publications. Failure to use adequate controls and ensuring material authenticity and integrity may also result in type I and II errors and lack of reproducibility. Unsubstantiated but reported bioactivities are probably one key aspect that leads to a waste of time, effort, and money in pharmacognosy research [52]. Importantly, considering the documented impact of Residual Complexity (Figure 2), the singleton character of an SCE (purity) should be proven experimentally rather than being assumed or even taken for granted [54]. Adding another layer of complexity, insight is growing that organisms such as “a plant” represent macro- and micro-complex ecosystems rather than single biological entities [55], involving endosymbionts (Figure 2) embedded into exuberant internal and surface communities. This unveils a new universe for the metabolomics-inclined researcher, actually one that complicates standardization and understanding significantly.
Acknowledgments
PMA, AA and JLW are grateful to the Swiss National Science Foundation (SNF) for supporting their natural product metabolomics projects (grants N° 310030E-164289, 31003A_163424 and 316030_164095).
JB and GFP gratefully acknowledge support by grant U41 AT008706 from NCCIH and ODS/NIH, andby our UIC colleagues, Tina Griffin and Abigail Goben, for references and numerous helpful discussions. The members of our greater research team kindly contributed fruitful discussions and exchange over the years on various aspects covered in this work. Finally, the authors would like to thank Aaron Lav for the creative input at the initial stage of this manuscript.
We acknowledge Christoper, creative outlet, Edward Boatman, Maxim Kulikov, Deepak M, Tatina Vazest, Lloyd Humphreys, Delwar Hossain, Asimbla, Assaf Katz, n.o.o.m., Hopkins, Rockicon, Aybige, Emily van den Heever, Oksana Latysheva, Guilhem, Three Six Five, Hea Poh Lin, Eucalyp, KΛPKLΛM, Aleks, Rajive, Andrejs Kirma from the Noun Project (https://thenounproject.com/) and Mind the Graph (https://mindthegraph.com/) for icons used in the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ORCID Numbers:
Pierre-Marie Allard 0000-0003-3389-2191
Jonathan Bisson 0000-0003-1640-9989
Antonio Azzollini 0000-0003-2313-8830
Guido F. Pauli 0000-0003-1022-4326
Geoffrey A. Cordell
Jean-Luc Wolfender 0000-0002-0125-952X
Citations
- 1.Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 2*.Kurita KL, Glassey E, Linington RG. Integration of high-content screening and untargeted metabolomics for comprehensive functional annotation of natural product libraries. Proc Natl Acad Sci. 2015;112:11999–12004. doi: 10.1073/pnas.1507743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olivon F, Allard P-M, Koval A, Righi D, Genta-Jouve G, Neyts J, Apel C, Pannecouque C, Nothias L-F, Cachet X, et al. Bioactive natural products prioritization using massive multi-informational molecular networks. ACS Chem Biol. 2017;12:2644–2651. doi: 10.1021/acschembio.7b00413. [DOI] [PubMed] [Google Scholar]
- 4.Smyth JE, Butler NM, Keller PA. A twist of nature – the significance of atropisomers in biological systems. Nat Prod Rep. 2015;32:1562–1583. doi: 10.1039/c4np00121d. [DOI] [PubMed] [Google Scholar]
- 5.Markley JL, Brüschweiler R, Edison AS, Eghbalnia HR, Powers R, Raftery D, Wishart DS. The future of NMR-based metabolomics. Curr Opin Biotechnol. 2017;43:34–40. doi: 10.1016/j.copbio.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kind T, Tsugawa H, Cajka T, Ma Y, Lai Z, Mehta SS, Wohlgemuth G, Barupal DK, Showalter MR, Arita M, et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom Rev. 2017:1–20. doi: 10.1002/mas.21535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Periat A, Guillarme D, Veuthey J-L, Boccard J, Moco S, Barron D, Grand-Guillaume Perrenoud A. Optimized selection of liquid chromatography conditions for wide range analysis of natural compounds. J Chromatogr A. 2017;1504:91–104. doi: 10.1016/j.chroma.2017.05.024. [DOI] [PubMed] [Google Scholar]
- 8.Mahieu NG, Patti GJ. Systems-level annotation of a metabolomics data set reduces 25 000 features to fewer than 1000 unique metabolites. Anal Chem. 2017;89:10397–10406. doi: 10.1021/acs.analchem.7b02380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meusel M, Hufsky F, Panter F, Krug D, Müller R, Böcker S. Predicting the presence of uncommon elements in unknown biomolecules from isotope patterns. Anal Chem. 2016;88:7556–7566. doi: 10.1021/acs.analchem.6b01015. [DOI] [PubMed] [Google Scholar]
- 10.Xia J, Wishart DS. Curr Protoc Bioinformatics. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2016. Using Metaboanalyst 3.0 for comprehensive metabolomics data analysis; pp. 14.10.1–14.10.91. [DOI] [PubMed] [Google Scholar]
- 11*.Johnson SR, Lange BM. Open-access metabolomics databases for natural product research: present capabilities and future potential. Front Bioeng Biotechnol. 2015;3 doi: 10.3389/fbioe.2015.00022. An overview of current open access databases (both for MS and NMR) in natural products research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakiri A, Hubert J, Reynaud R, Lanthony S, Harakat D, Renault J-H, Nuzillard J-M. Computer-aided 13C NMR chemical profiling of crude natural extracts without fractionation. J Nat Prod. 2017;80:1387–1396. doi: 10.1021/acs.jnatprod.6b01063. [DOI] [PubMed] [Google Scholar]
- 13.Bingol K, Brüschweiler R. Knowns and unknowns in metabolomics identified by multidimensional NMR and hybrid MS/NMR methods. Curr Opin Biotechnol. 2017;43:17–24. doi: 10.1016/j.copbio.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golubović J, Birkemeyer C, Protić A, Otašević B, Zečević M. Structure–response relationship in electrospray ionization-mass spectrometry of sartans by artificial neural networks. J Chromatogr A. 2016;1438:123–132. doi: 10.1016/j.chroma.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 15.Phansalkar RS, Simmler C, Bisson J, Chen S-N, Lankin DC, McAlpine JB, Niemitz M, Pauli GF. Evolution of quantitative measures in NMR: quantum mechanical qHMNR advances chemical standardization of a red clover (Trifolium pratense) extract. J Nat Prod. 2017;80:634–647. doi: 10.1021/acs.jnatprod.6b00923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marchand J, Martineau E, Guitton Y, Dervilly-Pinel G, Giraudeau P. Multidimensional NMR approaches towards highly resolved, sensitive and high-throughput quantitative metabolomics. Curr Opin Biotechnol. 2017;43:49–55. doi: 10.1016/j.copbio.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Petras D, Jarmusch AK, Dorrestein PC. From single cells to our planet—recent advances in using mass spectrometry for spatially resolved metabolomics. Curr Opin Chem Biol. 2017;36:24–31. doi: 10.1016/j.cbpa.2016.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Link H, Fuhrer T, Gerosa L, Zamboni N, Sauer U. Real-time metabolome profiling of the metabolic switch between starvation and growth. Nat Methods. 2015;12:1091. doi: 10.1038/nmeth.3584. [DOI] [PubMed] [Google Scholar]
- 19**.Lai Z, Tsugawa H, Wohlgemuth G, Mehta S, Mueller M, Zheng Y, Ogiwara A, Meissen J, Showalter M, Takeuchi K, et al. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat Methods. 2018;15:53–56. doi: 10.1038/nmeth.4512. A metabolite annotation pipeline based on the combination of state-of-the-art deconvolution and in silico fragmentation tools to a DB of hypothetical compounds and a software solution allowing to match unknowns with biological metadata: a nice example of metabolomics data contextualization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20*.Rollinger JM, Haupt S, Stuppner H, Langer T. Combining ethnopharmacology and virtual screening for lead structure discovery: COX-inhibitors as application example. J Chem Inf Comput Sci. 2004;44:480–488. doi: 10.1021/ci030031o. An interesting example of ancient pharmacognostic knowledge mining combined to advanced computational approaches. [DOI] [PubMed] [Google Scholar]
- 21.Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de los Santos ELC, Kim HU, Nave M, et al. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017;45:W36–W41. doi: 10.1093/nar/gkx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hufsky F, Böcker S. Mining molecular structure databases: identification of small molecules based on fragmentation mass spectrometry data. Mass Spectrom Rev. 2017;36:624–633. doi: 10.1002/mas.21489. [DOI] [PubMed] [Google Scholar]
- 23.Lodewyk MW, Siebert MR, Tantillo DJ. Computational prediction of 1H and 13C chemical shifts: a useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem Rev. 2012;112:1839–1862. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]
- 24*.Scheubert K, Hufsky F, Petras D, Wang M, Nothias L-F, Dührkop K, Bandeira N, Dorrestein PC, Böcker S. Significance estimation for large scale metabolomics annotations by spectral matching. Nat Commun. 2017;8:1494. doi: 10.1038/s41467-017-01318-5. Efforts towards the establishment of statistical methods for estimating the false discovery rates (FDR) of small molecule annotations in mass spectrometry libraries. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer A, Phapale P, Chernyavsky I, Lavigne R, Fay D, Tarasov A, Kovalev V, Fuchser J, Nikolenko S, Pineau C, et al. FDR-controlled metabolite annotation for high-resolution imaging mass spectrometry. Nat Methods. 2017;14:57. doi: 10.1038/nmeth.4072. [DOI] [PubMed] [Google Scholar]
- 26**.Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol. 2016;34:nbt.3597. doi: 10.1038/nbt.3597. Description of the Global Natural Products Social Molecular Networking (GNPS) platform, a precious tool for metabolomics data contextualization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang JY, Sanchez LM, Rath CM, Liu X, Boudreau PD, Bruns N, Glukhov E, Wodtke A, de Felicio R, Fenner A, et al. Molecular networking as a dereplication strategy. J Nat Prod. 2013;76:1686–1699. doi: 10.1021/np400413s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allard P-M, Péresse T, Bisson J, Gindro K, Marcourt L, Pham VC, Roussi F, Litaudon M, Wolfender J-L. Integration of molecular networking and in-silico MS/MS fragmentation for natural products dereplication. Anal Chem. 2016;88:3317–3323. doi: 10.1021/acs.analchem.5b04804. [DOI] [PubMed] [Google Scholar]
- 29.Alden N, Krishnan S, Porokhin V, Raju R, McElearney K, Gilbert A, Lee K. Biologically consistent annotation of metabolomics data. Anal Chem. 2017;89:13097–13104. doi: 10.1021/acs.analchem.7b02162. [DOI] [PubMed] [Google Scholar]
- 30*.Allard P-M, Genta-Jouve G, Wolfender J-L. Deep metabolome annotation in natural products research: towards a virtuous cycle in metabolite identification. Curr Opin Chem Biol. 2017;36:40–49. doi: 10.1016/j.cbpa.2016.12.022. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C, Idelbayev Y, Roberts N, Tao Y, Nannapaneni Y, Duggan BM, Min J, Lin EC, Gerwick EC, Cottrell GW, et al. Small Molecule Accurate Recognition Technology (SMART) to enhance natural products research. Sci Rep. 2017;7:14243. doi: 10.1038/s41598-017-13923-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Pasquetto I, Randles B, Borgman C. On the reuse of scientific data. Data Sci J. 2017;16 This essay focuses on the often-overlooked distinction between use and re-use of data inside knowledge infrastructures It is a good and short introduction of the notions of open data and data sharing. [Google Scholar]
- 33*.Piwowar HA, Day RS, Fridsma DB. Sharing detailed research data is associated with increased citation rate. PLoS ONE. 2007;2:e308. doi: 10.1371/journal.pone.0000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swauger S. Open access, power, and privilege: A response to “What I learned from predatory publishing”. Coll Res Libr News. 2017;78:603. [Google Scholar]
- 35.Buck M, Hamilton C. The Nagoya protocol on access to genetic resources and the fair and equitable sharing of benefits arising from their utilization to the convention on biological diversity: THE NAGOYA PROTOCOL. Rev Eur Community Int Environ Law. 2011;20:47–61. [Google Scholar]
- 36.Spicer RA, Steinbeck C. A lost opportunity for science: journals promote data sharing in metabolomics but do not enforce it. Metabolomics. 2018;14:16. doi: 10.1007/s11306-017-1309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Curty RG, Crowston K, Specht A, Grant BW, Dalton ED. Attitudes and norms affecting scientists’ data reuse. PLoS ONE. 2017;12:e0189288. doi: 10.1371/journal.pone.0189288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pedrioli PGA, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 39.Schober D, Jacob D, Wilson M, Cruz JA, Marcu A, Grant JR, Moing A, Deborde C, de Figueiredo LF, Haug K, et al. nmrML: a community supported open data standard for the description, storage, and exchange of NMR data. Anal Chem. 2017;90:649–656. doi: 10.1021/acs.analchem.7b02795. [DOI] [PubMed] [Google Scholar]
- 40**.Wilkinson MD, Dumontier M, Aalbersberg IjJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, Santos LB da S, Bourne PE, et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data. 2016;3:160018. doi: 10.1038/sdata.2016.18. The FAIR principles and their rationales are part of the answer to the need for lasting and usable scholarly data They are four simple sets of rules that aim at improving data re-use and can guide data producers toward ways to make data meaningful for both humans and machines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41*.OECD. Business models for sustainable research data repositories. 2017 OECD conducted interviews of tens of repositories and studied their dependence on different categories of funding and the construction and evolution of their business models This guide is particularly important for anyone starting a new repository. [Google Scholar]
- 42.Rocca-Serra P, Salek RM, Arita M, Correa E, Dayalan S, Gonzalez-Beltran A, Ebbels T, Goodacre R, Hastings J, Haug K, et al. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics. 2016;12:1–13. doi: 10.1007/s11306-015-0879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leonelli S, Davey RP, Arnaud E, Parry G, Bastow R. Data management and best practice for plant science. Nat Plants. 2017;3:17086. doi: 10.1038/nplants.2017.86. [DOI] [PubMed] [Google Scholar]
- 44.Sansone S-A, Rocca-Serra P, Field D, Maguire E, Taylor C, Hofmann O, Fang H, Neumann S, Tong W, Amaral-Zettler L, et al. Toward interoperable bioscience data. Nat Genet. 2012;44:121–126. doi: 10.1038/ng.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chambers J, Davies M, Gaulton A, Hersey A, Velankar S, Petryszak R, Hastings J, Bellis L, McGlinchey S, Overington JP. UniChem: a unified chemical structure cross-referencing and identifier tracking system. J Cheminformatics. 2013;5:3. doi: 10.1186/1758-2946-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt N. Tackling complexity in an interdisciplinary scholarly network: Requirements for semantic publishing. First Monday. 2016;21 [Google Scholar]
- 47*.Kuhn T, Chichester C, Krauthammer M, Queralt-Rosinach N, Verborgh R, Giannakopoulos G, Ngomo A-CN, Viglianti R, Dumontier M. Decentralized provenance-aware publishing with nanopublications. PeerJ Comput Sci. 2016;2:e78. [Google Scholar]
- 48.Giraldo O, García A, López F, Corcho O. Using semantics for representing experimental protocols. J Biomed Semant. 2017;8:52. doi: 10.1186/s13326-017-0160-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49**.Cordell GA. Cognate and cognitive ecopharmacognosy — in an anthropogenic era. Phytochem Lett. 2017;20:540–549. An initiative for “Better Global Health Through Nature”, or a reflexion on the role of ecopharmacognosy as an augmented and “conscious” pharmacognosy in a context of sustainability problematics. [Google Scholar]
- 50.Cordell GA. Ecopharmacognosy and the responsibilities of natural product research to sustainability. Phytochem Lett. 2015;11:332–346. [Google Scholar]
- 51.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 52.Bisson J, McAlpine JB, Friesen JB, Chen S-N, Graham J, Pauli GF. Can invalid bioactives undermine natural product-based drug discovery? J Med Chem. 2016;59:1671–1690. doi: 10.1021/acs.jmedchem.5b01009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53*.Neimark J. Line of attack. Science. 2015;347:938–940. doi: 10.1126/science.347.6225.938. [DOI] [PubMed] [Google Scholar]
- 54.Pauli GF, Chen S-N, Friesen JB, McAlpine JB, Jaki BU. Analysis and purification of bioactive natural products: the AnaPurNa study. J Nat Prod. 2012;75:1243–1255. doi: 10.1021/np300066q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newman DJ. Predominately uncultured microbes as sources of bioactive agents. Front Microbiol. 2016;7:1–15. doi: 10.3389/fmicb.2016.01832. [DOI] [PMC free article] [PubMed] [Google Scholar]