

Abstract
Background
Heart failure is a growing cause of morbidity and mortality worldwide. Transforming growth factor beta (TGF-β1) promotes cardiac fibrosis, but also activates counter-regulatory pathways that serve to regulate TGF-β1 activity in heart failure. Bone morphogenetic protein 9 (BMP9) is a member of the TGFβ family of cytokines and signals via the downstream effector protein Smad1. Endoglin is a TGFβ co-receptor that promotes TGF-β1 signaling via Smad3 and binds BMP9 with high affinity. We hypothesized that BMP9 limits cardiac fibrosis by activating Smad1 and attenuating Smad3 and further that neutralizing endoglin activity promotes BMP9 activity.
Methods
We examined BMP9 expression and signaling in human cardiac fibroblasts and human subjects with heart failure. We utilized the thoracic aortic constriction (TAC) induced model of heart failure to evaluate the functional effect of BMP9 signaling on cardiac remodeling.
Results
BMP9 expression is increased in the circulation and left ventricle (LV) of human subjects with heart failure and is expressed by cardiac fibroblasts. Next, we observed that BMP9 attenuates Type I collagen synthesis in human cardiac fibroblasts using recombinant human BMP9 and an siRNA approach. In BMP9−/− mice subjected to TAC, loss of BMP9 activity promotes cardiac fibrosis, impairs LV function, and increases LV levels of phosphorylated Smad3 (pSmad3), not pSmad1. In contrast, treatment of wild-type mice subjected to TAC with recombinant BMP9 limits progression of cardiac fibrosis, improves LV function, enhances myocardial capillary density, and increases LV levels of pSmad1, not pSmad3 compared to vehicle treated controls. Since endoglin binds BMP9 with high affinity, we explored the effect of reduced endoglin activity on BMP9 activity. Neutralizing endoglin activity in human cardiac fibroblasts or in wild-type mice subjected to TAC induced heart failure limits collagen production, increases BMP9 protein levels, and increases levels of pSmad1, not pSmad3.
Conclusions
Our results identify a novel functional role for BMP9 as an endogenous inhibitor of cardiac fibrosis due to LV pressure overload and further show that treatment with either recombinant BMP9 or disruption of endoglin activity promotes BMP9 activity and limits cardiac fibrosis in heart failure, thereby providing potentially novel therapeutic approaches for patients with heart failure.
Keywords: Heart failure, Cardiac fibrosis, Transforming growth factor beta, Bone morphogenetic protein 9
Introduction
Heart failure1 is a growing public health problem expected to affect over 10 million individuals in the US by 20372, 3. Cardiac fibrosis is an independent predictor of overall mortality, sudden cardiac death, HF death, and HF hospitalization independent of left ventricular ejection fraction4–6. Cardiac fibroblasts increase extracellular matrix synthesis, which exaggerates mechanical myocardial stiffness, disorganizes contraction due to myocyte separation, disrupts electro-tonic connectivity, and worsens tissue hypoxia7–10. Cardiac fibrosis remains a significant target of therapy for patients with HF and signaling pathways that govern cardiac fibroblast activity remain a significant knowledge gap in our understanding of cardiac remodeling.
One of the most potent pro-fibrogenic cytokine systems governing cardiac fibrosis is the transforming growth factor beta (TGFβ) superfamily, which includes TGF-β1 and 22 distinct bone morphogenetic proteins (BMPs)11–14. TGF-β1 and BMPs binds directly to Type II receptors (TβRII) which dimerize with and activate Type I receptors (TβRI), also known as activin-like kinases (ALK), for signal transduction via downstream effector proteins known as Smads. One of the major challenges when targeting TGF-β1 activity is that in addition to promoting signaling down one pathway, TGF-β1 also activates counter-regulatory pathways that serve to limit it’s own activity. A classic example of TGF-β1 counter-regulation is mediated by ALK5/Smad3 and ALK1/Smad1 activity, which respectively promote and inhibit TGF-β1 activity in cardiac fibroblasts.
We recently reported that the TGF-β1 co-receptor endoglin (CD105) promotes TGF-β1 signaling by preferentially phosphorylating Smad3, stimulating collagen synthesis by cardiac fibroblasts, and increasing cardiac fibrosis in models of left or right heart pressure overload15, 16. Recent reports identified that endoglin binds BMP9, also known as growth differentiation factor 2 (GDF-2) with high affinity and is required for BMP9 signaling via Smad1 in endothelium17, 18. Specifically, BMP9 promotes activin receptor-like kinase 1 (ALK-1)-mediated phosphorylation of Smads-1/5/8, which bind to Smad4 and translocate to the nucleus to activate Smad-response elements in BMP gene targets such as inhibitors of differentiation (Id 1–3) in endothelium19–21. No studies have explored a functional role for BMP9 in cardiac fibroblasts or HF nor studied an interaction between endoglin and BMP9 in cardiac fibrosis. With this background in mind, we hypothesized that BMP9 may limit cardiac fibrosis by promoting ALK1/Smad1 activity and further that neutralizing endoglin activity may serve as a novel approach to promote BMP9 activity in heart failure.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure and may be obtained by contacting the corresponding author.
Reagents
WT mice were obtained from Jackson Laboratories. Endoglin haploinsufficient (Eng+/−) mice were generously provided by Dr. Michelle Letarte, University of Toronto and BMP9−/− mice by Dr. Se-Jin Lee, John Hopkins University. Western blot analysis was performed with antibodies directed at BMP9 (R&D systems), GAPDH (Millipore), phosphorylated Smad1 (Cell Signaling), phosphorylated Smad3 (Cell Signaling), total Smad1 (Cell Signaling) and total Smad3 (Cell Signaling). For endoglin or BMP9 silencing experiments, cells were exposed to 50 nM of human endoglin siRNA (Ambion #145527), human BMP9 siRNA (Ambion #4392420), scrambled siRNA (negative control; Ambion #4390844) or GAPDH siRNA (positive control; Ambion # 4390850). To neutralize endoglin activity in human cardiac fibroblasts, we used TRC105, a human/mouse chimeric endoglin neutralizing antibody that binds human endoglin with high affinity. To neutralize endoglin activity in murine studies, we utilized M1043, a rat anti-mouse endoglin antibody. Activity of the neutralizing endoglin antibodies (M1043 and TRC105) has been previously validated by Nolan-Steveau and colleagues who demonstrated potent anti-endoglin activity in endothelial cells where BMP9 is known to promote Smad1 phosphorylation in an endoglin-dependent manner. Additional reports have previously confirmed the anti-endoglin activity of M1043 or TRC105 in hepatic fibrosis, cancer, pulmonary hypertension, and heart failure15, 22–25.
Human LV tissue Sampling
For analysis of LV BMP9 expression in patients with HF, viable LV free wall tissue was obtained from human subjects with end-stage HF (n=8) referred for left ventricular assist device implantation. Non-failing LV tissue obtained from the National Disease Research Interchange served as controls (n=8). All tissue was rinsed with sterile saline, immediately frozen in liquid nitrogen and stored at −80°C. Tissue sample collection was performed in concordance with the National Institutes of Health, approved by the Institutional Review Board of Tufts Medical Center and the subjects gave informed consent.
Human Blood Sampling
For analysis of circulating BMP9 levels, blood was collected at the time of arterial sheath insertion prior to cardiac catheterization in 45 consecutive patients referred for cardiac catheterization to evaluate suspected LV dysfunction. Patients with non-sinus rhythm, cardiogenic shock, inotrope use, presence of a ventricular assist device, acute coronary syndrome, age <18 years old, pregnancy, active or prior history of malignancy, renal failure (estimated glomerular filtration rate <= 30 ml/min/1.73 m2), liver transaminase elevation >=2 times the upper limit of normal, or perceived interference with standard clinical care were excluded from blood collection. Patients without obstructive coronary artery disease (at least 50% coronary artery stenosis) and preserved left ventricular ejection fraction served as controls (non-HF; n=10). Circulating BMP9, procollagen type I C-peptide (PIP), carboxyterminal telopeptide of type I collagen (ICTP) levels were analyzed using a commercially available enzyme linked immunosorbent assay (ELISA) (BMP9:R&D Systems, PIP and ICTP: Orion Diagnostica, Finland). Blood sample collection was performed in concordance with the National Institutes of Health, approved by the Institutional Review Board of Tufts Medical Center and subjects gave informed consent. The specificity of BMP9 reagents used in our study was tested using the BMP9 ELISA and BMP9 antibody (R&D Systems; AF3879) and failed to identify any cross reactivity for BMP10, GDF8, GDF11 (Supplemental Figure 1A–B).
BMP9 Expression and Activity in a Mouse Model of Pressure Overload Induced Heart Failure
Adult, male, 12–14-week-old mice were used for all studies. Transverse aortic constriction (TAC) using a 27G needle generated LV pressure overload induced HF as previously described16. C57 Bl/6 wild-type (WT) mice were subjected to TAC for 2 or 8 weeks to initially quantify cardiac BMP9 expression. For BMP9 loss of function studies, BMP9−/− mice were generously provided by Se Jin Lee (Johns Hopkins Medical Center, Baltimore, MD) and subjected to TAC for 2 weeks. To further test whether recombinant murine BMP9 (rmBMP9) rescues cardiac fibrosis, WT mice were subjected to Sham operation or TAC for 4 weeks, then received daily intraperitoneal injections of vehicle or rBMP9 for 4 additional weeks with ongoing TAC induced LV pressure overload for a total of 8 weeks of TAC. At the conclusion of each experiment, terminal hemodynamics were recorded using a left ventricular conductance catheter as described26 and LV tissue was obtained for biochemical and histological analysis.
Loss of Endoglin Expression and Activity Increases BMP9 Expression in Pressure Overload Induced Heart Failure
To determine whether loss of endoglin activity rescues cardiac fibrosis and promotes BMP9 activity, WT mice were subjected to Sham operation or TAC for 4 weeks, then received twice weekly intraperitoneal injections of IgG isotype control antibody or a neutralizing endoglin antibody (Eng NAb at 15mg/kg; M1043, Amgen Inc, San Francisco, CA) for 4 additional weeks with ongoing TAC induced LV pressure overload (total of 8 weeks of TAC). At the conclusion of each experiment, terminal hemodynamics were recorded using left ventricular conductance catheter as described26 and LV tissue obtained for biochemical and histological analysis.
Histologic Evaluation of Cardiac Hypertrophy, Fibrosis and Myocardial Capillary Density
LV collagen abundance was quantified by picrosirius red staining. Cardiomyocyte cross-sectional area and capillary density were quantified as previously described16.
Real time Quantitative Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from the LV with Trizol (Life Technologies) and converted to cDNA with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR was performed in triplicate using 40 cycles at 94°C for 15 seconds, 60°C for 30 seconds and 72°C for 30 seconds with an ABI Prism 7900 Sequence Detection System. Primers for 18S ribosomal RNA, Endoglin, TGF-β1, beta-myosin heavy chain (β-MHC), sarcoplasmic endoplasmic reticulum ATPase (SERCA), Type I collagen, and calcineurin were described previously16, 26. For primer sequences see Supplemental Table 1.
Immunoblotting
Total protein was extracted and quantified from tissue homogenates or cultured cells and immunoblot analysis was performed as previously described27.
Functional Studies in Human Cardiac Fibroblasts
Human cardiac fibroblasts (hCF) were isolated from LV tissue harvested during cardiac surgery at Tufts Medical Center. For rhBMP9 studies, hCF were stimulated with TGF-β1 at 10 ng/ml or rhBMP9 at 1–10 ng/ml13. For neutralizing antibody studies, hCF were pretreated with 0.5 ng/ml of endoglin neutralizing antibody (Eng NAb; TRC105, Amgen Inc, San Francisco, CA)or control IgG isotype for 24 hours in fibroblast basal medium without supplementation prior to stimulation with TGF-β1 at 10 ng/ml. For endoglin or BMP9 silencing experiments, 50 nM siRNA stock was dilated in Optimem (Invitrogen) and combined with Lipofectamine prior to exposure to cells. After 24–48 hours, cells were harvested for RT-PCR and Western blot analysis.
Statistics
All statistical analyses were performed using Graph Pad Prism v6 (Graph Pad Software, Inc.). Comparison between two experimental groups was performed with the unpaired student’s T test and for three groups or more with a one-way ANOVA. p values less than 0.05 were accepted as statistically significant.
Study Approval
Animals were treated in compliance with the Guide for the Care and Use of Laboratory Animals (National Academy of Science). Animal protocols were approved by the Tufts Medical Center Institutional Animal Care and Use Committee. The Tufts Medical Center Institutional Review Board approved collection of human tissue for cell culture.
Results
Increased BMP9 Expression in Human Heart Failure
To explore a role for BMP9 in HF, we first determined levels of BMP9 in LV samples from patients with advanced HF referred for implantation of a left ventricular assist device. Control LV samples were obtained from postmortem subjects without HF (n=8/group). Compared to controls, LV BMP9 expression was increased in patients with advanced HF at the time of left ventricular assist device implantation (Figure 1A–B). In a separate cohort of HF patients referred for cardiac catheterization (n=45), plasma BMP9 levels were ~3 fold higher compared to patients without HF (n=10) (Figure 1C). In our human HF cohort, we found that circulating BMP9 correlated with LV ejection fraction (Pearson r = −0.45, p=0.001; Supplemental Figure 2) and not PIP levels (r = −0.12, p=0.49), ICTP levels (r = 0.26, p=0.13) or the ratio of PIP/ICTP (r = −0.24, p=0.16).
Figure 1. Bone Morphogenetic Protein 9 Expression is Increased in Heart Failure.
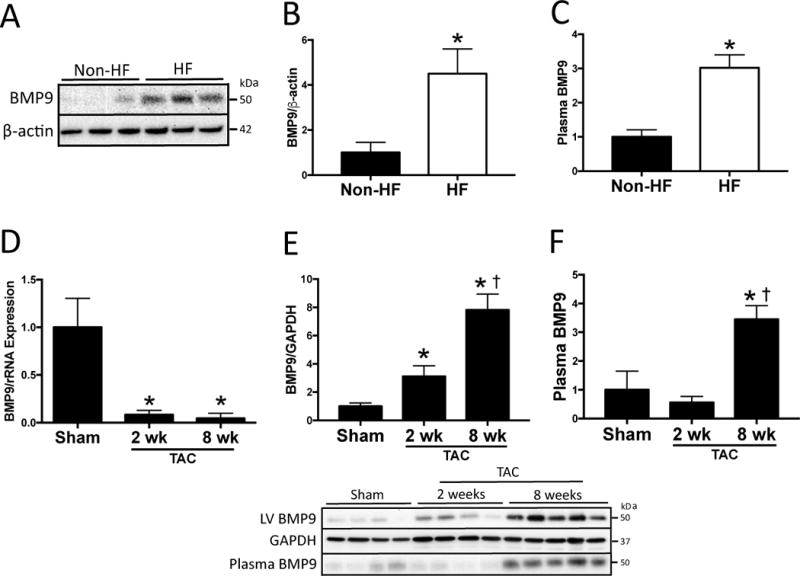
A) Representative immunoblots and B) quantitation of bone morphogenetic protein 9 (BMP9) and β-actin LV protein expression in patients with advanced heart failure referred for left ventricular assist device implantation (HF; n=8) compared to non-heart failure controls (non-HF; n=8). C) Plasma BMP9 levels measured by ELISA in patients with heart failure (HF; n=45) compared to non-heart failure controls (non-HF; n=10). D) LV BMP9 mRNA expression after two and eight weeks of transverse aortic constriction (TAC). n=3 independent experiments. E–F) Representative immunoblots and quantitation of LV BMP9, LV GAPDH and plasma BMP9 protein after two (n=4) and eight weeks (n=5) of TAC compared to Sham operated controls (n=4). Data are expressed as fold change vs. control. p<0.05: *, vs. Sham; †, vs. 2 wk TAC.
Increased BMP9 Expression in Pressure Overload Induced Heart Failure
To further explore whether BMP9 levels are increased in response to HF, WT mice were subjected to 2 and 8 weeks of pressure-overload-induced HF by TAC. After 8 weeks of TAC, WT mice exhibited increased LV filling pressures and reduced contractile function (Table 1). Compared to Sham controls, mRNA expression of BMP9 decreased while LV protein and circulating levels of BMP9 progressively increased after 2 and 8 weeks of TAC (Figure 1D–F). No change in LV BMP9 protein levels was observed in the aorta distal to the site of TAC ligature (Supplemental Figure 3A). LV end diastolic pressure correlated with LV BMP9 expression (Supplemental Figure 3B), suggesting a direct effect of pressure overload on BMP9 expression. These findings identify an association between progressive HF and increased LV and circulating BMP9 protein levels and further implicate a direct effect of LV pressure overload on BMP9 expression.
Table 1.
Characterization of Heart Failure After Two and Eight Weeks of TAC
| Sham | 2 wks TAC | 8 wks TAC | |
|---|---|---|---|
| Mass | |||
| Total body weight (g) | 28.9 ± 0.7 | 25.7 ± 1* | 25.1 ± 3.1* |
| LV mass (mg) | 84 ± 7 | 149 ± 9* | 208 ± 15*† |
| Lung mass (mg) | 142 ± 7 | 324 ± 66* | 471 ± 52*† |
| Tibia length (mm) | 17.5 ± 0.1 | 17.6 ± 0.2 | 17.7 ± 0.2 |
| LV/TL (mg/mm) | 4.8 ± 0.5 | 8.5 ± 0.5* | 11.8 ± 0.9*† |
| Lung/TL (mg/mm) | 8.1 ± 0.4 | 18.4 ± 3.6* | 26.7 ± 3.1*† |
| Hemodynamic data | |||
| Peak systolic pressure (mmHg) | 97 ± 7 | 138 ± 23* | 113 ± 23 |
| End diastolic pressure (mmHg) | 4 ± 2 | 20 ± 4* | 30 ± 5*† |
| dP/dT max (mmHg/sec) | 8289 ± 681 | 6178 ± 1357* | 4272 ± 1104*† |
| dP/dT min (mmHg/sec) | −8397 ± 664 | −6050 ± 1272* | −4261 ± 1223*† |
| Ejection fraction (%) | 63 ± 6 | 40 ± 2* | 25 ± 4*† |
| Heart rate (bpm) | 523 ± 10 | 526 ± 22 | 533 ± 50 |
Means±SD. p<0.05:
vs. Sham;
vs. 2 wks TAC
BMP9 Limits TGF-β1 Induced Type I Collagen Synthesis in Human Cardiac Fibroblasts
To explore which cells in the adult heart express BMP9, we isolated adult murine cardiomyocytes, cardiac fibroblasts and endothelial cells. BMP9 was abundantly expressed by fibroblasts and endothelial cells, not cardiomyocytes (Figure 2A). Next, we tested the effect of rhBMP9 on TGF-β1 induced type I collagen synthesis in human cardiac fibroblasts and identified that compared to TGF-β1 alone, co-stimulation with TGF-β1 and rhBMP9 reduced Type I collagen mRNA and protein expression (Figure 2B–C). To explore the effect of BMP9 on collagen synthesis by fibroblasts, we employed a loss of function approach using siRNA. Silencing BMP9 expression increased Type I collagen mRNA and protein expression with and without TGF-β1 stimulation in human cardiac fibroblasts (Figure 2D–E). These data identify that BMP9 is expressed by cardiac fibroblasts and negatively regulates TGF-β1 induced Type I collagen synthesis.
Figure 2. Bone Morphogenetic Protein 9 Attenuates TGFβ1 Induced Collagen Production in Human Cardiac Fibroblasts.
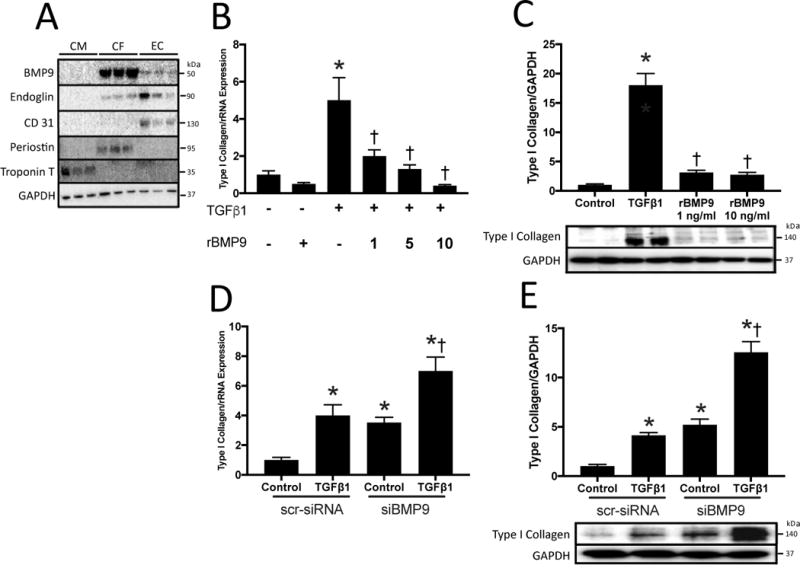
A) Representative immunoblots for bone morphogenetic protein 9 (BMP9), Endoglin, CD31, Periostin, Troponin T and GAPDH in human cardiomyocytes (CM), cardiac fibroblasts (CF), and endothelial cells (EC). B) Type I collagen mRNA expression in human cardiac fibroblasts stimulated with TGF-β1 at 10 ng/ml, recombinant human BMP9 (rhBMP9) at 1, 5, 10 ng/ml, or both C) Quantitation of type I collagen protein expression normalized to GAPDH expression with representative immunoblots. D) Type I collagen mRNA expression in human cardiac fibroblasts following knockdown of BMP9 expression with siBMP9 compared to scrambled siRNA (scr-siRNA). E) Quantitation and representative immunoblots of type I collagen protein expression normalized to GAPDH expression. Data for panels B–E represent an average of 3 independent experiments and are expressed as fold change vs. control. Panels B–C: p<0.05: *, vs. No TGF-β1; †, vs. TGF-β1. Panels D–E: p<0.05: *, vs. Control scr-siRNA; †, vs. Control siBMP9.
Loss of BMP9 Promotes Cardiac Fibrosis in TAC-Induced Heart Failure
To explore a functional role for BMP9 in HF, we employed BMP9 null mice (BMP9−/−) which have total body depletion of BMP9 and no known intrinsic cardiovascular abnormalities [31, 32]. After 2 weeks of TAC, both WT and BMP9−/− mice exhibited increased LV and lung mass, however BMP9−/− mice had higher LV filling pressures and lower LV dP/dT max (Table 2). No difference was observed between WT and BMP9−/− mice without TAC. Compared to Sham controls, LV fibrosis, type I collagen mRNA and protein levels were increased in both WT and BMP9−/− mice after TAC with a greater increase in collagen levels in BMP9−/− compared to WT mice after TAC (Figure 3A–C). Compared to WT mice, LV mRNA expression of TGF-β1 was higher after TAC (Supplemental Figure 4A). Compared to Sham controls, LV mRNA expression of fetal genes including endoglin, brain natriuretic peptide (BNP) calcineurin (CaN), and sarcoplasmic reticulum ATPase (SERCA) mRNA demonstrated similar patterns in both both WT and BMP9−/− mice after TAC (Supplemental Figure 4A–B). Compared to Sham controls, LV mRNA levels of the BMP receptors, ALK1 and ALK5, were unchanged in both WT and BMP9−/− mice after TAC (Supplemental Figure 4A). These data identify that loss of BMP9 is associated with worsening HF, increased LV collagen levels, and increased LV levels of TGF-β1 after TAC.
Table 2.
Characterization of Heart Failure in BMP9−/− Mice After Two Weeks of TAC
| WT Sham | WT TAC | BMP9−/− Sham | BMP9−/− TAC | Eng NAb BMP9−/− TAC | |
|---|---|---|---|---|---|
| Mass | |||||
| Total body weight (g) | 24.5 ± 0.6 | 22.2 ± 1.4* | 25.2 ± 1.5 | 21.5 ± 1.2† | 22.8 ± 3† |
| LV mass (mg) | 83 ± 7 | 149 ± 9* | 88 ± 8 | 143 ± 12† | 157 ± 15† |
| Lung mass (mg) | 143 ± 6 | 349 ± 111* | 137 ± 8 | 398 ± 58† | 379 ± 129† |
| Tibia length (mm) | 17.4 ± 0.1 | 17.5 ± 0.1 | 17.4 ± 0.2 | 17.5 ± 0.2 | 17.4 ± 0.2 |
| LV/TL (mg/mm) | 4.7 ± 0.4 | 8.5 ± 0.5* | 5.2 ± 0.1 | 8.2 ± 0.8† | 8.9 ± 0.8† |
| Lung/TL (mg/mm) | 8.2 ± 0.4 | 20 ± 6.4* | 7.9 ± 0.4 | 22.8 ± 3.2† | 21.7 ± 7.5† |
| Hemodynamic data | |||||
| Peak systolic pressure (mmHg) | 95 ± 8 | 125 ± 12* | 89 ± 2 | 96 ± 18‡ | 96 ± 10‡ |
| End diastolic pressure (mmHg) | 4 ± 2 | 22 ± 2* | 6 ± 3 | 27 ± 4†‡ | 28 ± 3†‡ |
| dP/dT max (mmHg/sec) | 8449 ± 370 | 5463 ± 990* | 8291 ± 645 | 2973 ± 517†‡ | 3232 ± 178†‡ |
| dP/dT min (mmHg/sec) | −8382 ± 577 | −5253 ± 347* | −7623 ± 637 | −3156 ± 1020†‡ | −3523 ± 628†‡ |
| Ejection fraction (%) | 62 ± 4 | 41 ± 5* | 58 ± 7 | 28 ± 6†‡ | 29 ± 5†‡ |
| Heart rate (bpm) | 508 ± 62 | 546 ± 57 | 522 ± 48 | 562 ± 48 | 546 ± 68 |
Means±SD. p<0.05:
vs. WT Sham;
vs. BMP9−/− Sham;
vs. WT TAC
Figure 3. BMP9 Deficiency Increases Fibrosis and Promotes Smad3 Signaling Following 2 weeks of TAC.
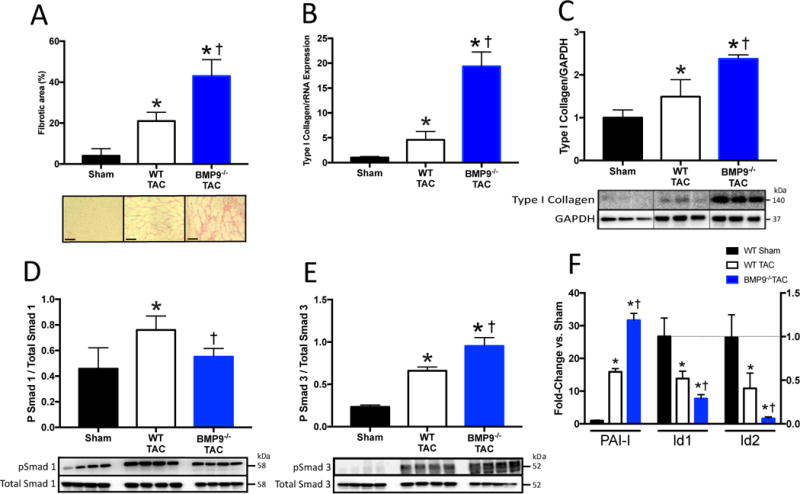
A) Representative histological staining for collagen and quantitation of LV fibrotic area in WT and BMP9−/− mice after TAC. B–C) LV mRNA and protein expression of type I collagen normalized to GAPDH expression. D–E) Representative immunoblots and quantitation of phosphorylated Smad 1 (pSmad1), phosphorylated Smad 3 (pSmad3), and GAPDH. F) LV mRNA expression of inhibitor of differentiation 1 and 2 (Id1, Id2), and plasminogen activator inhibitor-1 (PAI-I). Data are expressed as fold change vs. Sham. p<0.05: *, vs. Sham; †, vs. WT TAC.
Loss of BMP9 Promotes Smad3 and Limits Smad1 Signaling
To study the effect of BMP9 loss on downstream TGF-β1/BMP9 signaling effectors we measured LV levels of phosphorylated Smad1 (pSmad1) and pSmad3; mRNA levels of inhibitor of differentiation 1 and 2 (Id1 and Id2) as markers of Smad1 activity; and plasminogen activator inhibitor-1 (PAI-I) as a marker of Smad3 activity. Compared to Sham controls, LV levels of pSmad1 were increased in WT, but not BMP9−/− mice after TAC (Figure 3D). Compared to Sham controls, LV levels of pSmad3 were increased in both WT and BMP9−/− mice after TAC. Compared to WT mice, LV levels of pSmad3 were higher among BMP9−/− mice after TAC (Figure 3E). Consistent with the reduced pSmad1 levels, LV mRNA levels of Id1 and Id2, were reduced among WT and BMP9−/− mice after TAC with a greater loss of Id1 and Id2 among BMP9−/− mice compared to WT mice after TAC (Figure 3F). LV mRNA levels of PAI-I, a marker of Smad3 activity, was higher in BMP9−/− mice compared to WT mice (Figure 3F). These data identify that loss of BMP9 reduces Smad1 signaling and promotes Smad3 signaling in HF.
Recombinant BMP9 Prevents Progression of Cardiac Fibrosis in Heart Failure
Based on the observation that rhBMP9 attenuates Type I collagen synthesis in vitro and that loss of BMP9 activity promotes cardiac fibrosis, we next tested the hypothesis that treatment with rmBMP9 prevents progression of cardiac fibrosis in response to LV pressure overload. WT mice were subjected to four weeks of TAC and then randomized to intraperitoneal administration of rmBMP9 at 72 ng/day (rmBMP9-TAC; n=10) or vehicle control (Veh-TAC; n=10) for an additional 4 weeks of TAC exposure. After 8 weeks of TAC, 90% of rmBMP9-TAC compared to 50% of Veh-TAC survived for analysis (p=0.24 for survival comparison). Compared to Veh-TAC, rmBMP9-TAC mice demonstrated higher total body weight, lower LV and lung mass, lower LV filling pressures and higher dP/dT max and min (Table 3). Compared to Veh-TAC, treatment with rmBMP9 reduced LV fibrosis and Type I collagen mRNA and protein levels (Figure 4A–C). Compared to Veh-TAC, rmBMP9 treatment increased LV protein levels of pSmad1 and decreased levels of pSmad3 (Figure 4D–E). Compared to Sham controls, LV mRNA levels of Id1 and Id2 were decreased in Veh-TAC mice, but unchanged in rmBMP9-TAC mice, while compared to Veh-TAC, both PAI-I and Type I collagen mRNA levels were reduced in rmBMP9-TAC (Figure 4F). Compared to controls, rmBMP9 increased myocardial capillary density without affecting cardiomyocyte hypertrophy (Supplemental Figure 5A–B). LV mRNA expression of TGF-β1, ALK1 and ALK5 were similar in Veh TAC and rBMP9 TAC mice, whereas endoglin upregulation was attenuated in rBMP9 treated mice (Supplemental Figure 5C). LV mRNA levels of BNP, SERCA and CaN were similar between groups (Supplemental Figure 5D). These data support that treatment with rmBMP9 preserves LV function, reduces progression of LV fibrosis, promotes Smad1 and attenuates Smad3 signaling after development of HF.
Table 3.
Characterization of Heart Failure in Mice Treated with Recombinant Mouse BMP9
| Veh Sham | Veh TAC | rmBMP9 Sham | rmBMP9 TAC | |
|---|---|---|---|---|
| Mass | ||||
| Total body weight (g) | 31.9 ± 0.4 | 25.2 ± 3.5* | 31.9 ± 2.2 | 28.9 ± 1.4 ‡ |
| LV mass (mg) | 93 ± 2 | 221 ± 10* | 88 ± 4 | 178 ± 24 †‡ |
| Lung mass (mg) | 169 ± 19 | 432 ± 91* | 154 ± 6 | 239 ± 119 ‡ |
| Tibia length (mm) | 17.8 ± 0.2 | 17.7 ± 0.1 | 18.1 ± 0.1 | 17.9 ± 0.3 |
| LV/TL (mg/mm) | 2.9 ± 0.1 | 8.9 ± 1.1* | 2.7 ± 0.1 | 6.2 ± 0.7 †‡ |
| Lung/TL (mg/mm) | 5.2 ± 0.1 | 12.5 ± 0.5* | 4.8 ± 0.2 | 9.9 ± 1.2 †‡ |
| Hemodynamic data | ||||
| Peak systolic pressure (mmHg) | 95 ± 4 | 124 ± 24 | 97 ± 1 | 148 ± 19 ‡ |
| End diastolic pressure (mmHg) | 5 ± 3 | 28 ± 3* | 3 ± 2 | 11 ± 4 †‡ |
| dP/dT max (mmHg/sec) | 9076 ± 1481 | 5752 ± 872* | 9289 ± 755 | 7636 ± 1157 †‡ |
| dP/dT min (mmHg/sec) | −9066 ± 1140 | −5157 ± 1470* | −9541 ± 546 | −7398 ± 1318 †‡ |
| Ejection fraction (%) | 61 ± 6 | 23 ± 3* | 60 ± 9 | 44 ± 7†‡ |
| Heart rate (bpm) | 509 ± 69 | 587 ± 67 | 556 ± 39 | 572 ± 16 |
Means±SD. p<0.05:
vs. Vehicle Sham;
vs. rBMP9 Sham;
vs. Vehicle TAC
Figure 4. Recombinant BMP9 Reduces Fibrosis and Promotes Smad1 Signaling in Heart Failure.
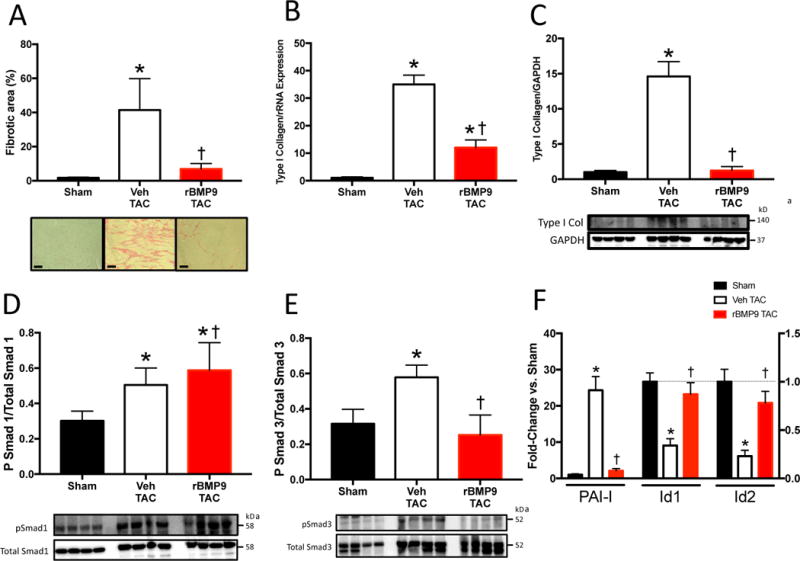
A) Representative histological staining for collagen and quantitation of LV fibrotic area in Vehicle (Veh) or recombinant mouse BMP9 treated mice (rBMP9) after TAC. B–C) LV mRNA and protein expression of type I collagen normalized to GAPDH expression in Veh and rmBMP9 treated mice after TAC. D–E) Representative immunoblots and quantiation of phosphorylated Smad 1 (pSmad1), phosphorylated Smad 3 (pSmad3), total Smad1 and total Smad3. F) LV mRNA expression of inhibitor of differentiation 1 and 2 (Id1,Id2), and plasminogen activator inhibitor-1 (PAI-I). Scale bars 20 um. Data expressed as fold change vs. Sham. p<0.05: *, vs. Sham; †, vs. Vehicle TAC.
Reduced Endoglin Activity Increases BMP9 Levels in Heart Failure
We previously reported that loss of endoglin activity prevents fibrosis and preserves LV function among Eng+/− mice after TAC. Recent reports identified endoglin as a central mediator of BMP9 activity in endothelial cells. To explore an interaction between endoglin and BMP9 in cardiac fibroblasts or HF, we first silenced endoglin expression in human cardiac fibroblasts using an siRNA approach and identified increased BMP9 protein levels from cell lysates with or without TGF-β1 stimulation (Figure 5A). Using a second loss of function approach, we blocked endoglin activity in human cardiac fibroblasts using a neutralizing anti-endoglin antibody (Eng NAb) and observed increased BMP9 protein levels with and without TGF-β1 stimulation (Figure 5B). To determine whether loss of endoglin promotes BMP9 expression in vivo, we subjected Eng+/− mice to TAC for 2 weeks. Compared to Sham controls and WT mice, LV levels of BMP9 were increased among Eng+/− mice (Figure 5C). Collectively, these data suggest that loss of endoglin activity promotes BMP9 expression in human cardiac fibroblasts and in the LV after pressure overload induced HF.
Figure 5. Endoglin Negatively Regulates BMP9 Expression in Cardiac Fibroblasts and Heart Failure.
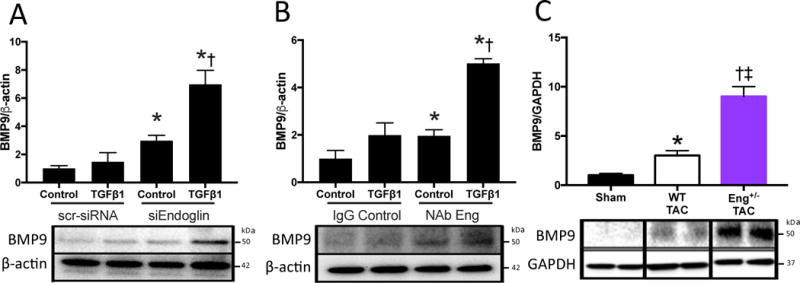
A) Representative immunoblots and quantitation of BMP9 expression normalized to β-actin expression in human cardiac fibroblasts following knockdown of Endoglin expression (siEndoglin) compared to scrambled siRNA (scr-siRNA) with and without TGF-β1 stimulation. B) Representative immunoblots and quantitation of BMP9 expression normalized to β-actin expression in human cardiac fibroblasts following treatment with a neutralizing endoglin antibody (Eng NAb) compared to isotype control (IgG) with and without TGF-β1 stimulation. C) Representative immunoblots and quantitation of LV BMP9 expression normalized to GAPDH expression following two weeks of TAC in WT and Eng+/− mice. Data expressed as fold change vs. Control or Sham. All p values<0.05. Panel A: *, vs. Control scr-siRNA; †, vs. Control siEndoglin. Panel B: p<0.05: *, vs. Control IgG; †, vs. Control NAb Eng. Panel C: *, vs. WT Sham; †, vs. Eng+/− Sham; ‡, vs. WT TAC.
Neutralizing Endoglin Activity Increases BMP9 Levels and Prevents Progression of Cardiac Fibrosis in Heart Failure
We previously reported that reduced endoglin activity prevents cardiac fibrosis due to LV pressure overload. Recent data showing that BMP9 interacts with endoglin in endothelial cells led us to hypothesize that reducing endoglin activity blocks fibrosis through a two-hit mechanism that involves attenuating TGF-β1/Smad3 signaling and promoting BMP9/Smad1 activity. To test this hypothesis, adult, male WT mice were subjected to four weeks of TAC, then randomized to treatment with a endoglin neutralizing antibody (Eng NAb; n=6) or IgG isotype control (IgG TAC; n=5) via twice weekly intraperitoneal injection. Following an additional 4 weeks of TAC, all mice survived. LV and lung mass were reduced in mice receiving Eng NAb compared to IgG controls after TAC (Table 4). Eng NAb treatment was not associated with development of mucocutaneous arteriovenous malformations on gross examination, new gastrointestinal bleeding or anemia. Eng NAb treatment reduced LV filling pressures and increased LV dp/dT max compared to IgG control after TAC. Eng NAb treatment also reduced LV fibrosis and type I collagen mRNA and protein levels (Figure 6A–C). Compared to IgG TAC, Eng NAb treatment increased LV and circulating BMP9 protein levels after TAC, which was associated with enhanced pSmad1/Id1/Id2 and reduced pSmad3/PAI-1 levels (Figure 6D–F, Supplemental Figure 6A). Compared to controls, rmBMP9 increased myocardial capillary density without affecting cardiomyocyte hypertrophy (Supplemental Figure 6B–C). LV mRNA levels of TGF-β1, ALK1 and ALK5 were similar between groups (Supplemental Figure 6D). Compared to IgG TAC, Eng Nab treatment reduced LV endoglin mRNA levels (Supplemental Figure 6D). BNP, SERCA and CaN LV mRNA levels were similar between groups (Supplemental Figure 6E). These data support that much like treatment with rmBMP9, neutralizing endoglin activity increases LV BMP9 levels and activity, preserves LV function, reduces progression of LV fibrosis, promotes Smad1 and attenuates Smad3 signaling after development of HF. To determine whether the effect of neutralizing endoglin activity is dependent on BMP9, we treated BMP9−/− mice with Eng NAb. After two weeks of TAC, BMP9−/− mice treated with Eng NAb exhibited similar LV mass, lung mass and filling pressures compared to BMP9−/− mice (Table 2). LV type I collagen mRNA and protein expression, Id1 and Id2 mRNA levels were similar in BMP9−/− mice treated with Eng NAb compared to BMP9−/− mice (Supplemental Figure 4C–E). These data indicate that the effect of Eng NAb on fibrosis and cardiac function after TAC is dependent on BMP9 expression.
Table 4.
Characterization of Heart Failure in Mice Treated with Endoglin Neutralizing Antibody
| IgG Sham | IgG TAC | Sham Eng NAb | Eng NAb TAC | |
|---|---|---|---|---|
| Mass | ||||
| Total body weight (g) | 28.5 ± 1 | 24.6 ± 4.9 | 28.3 ± 1.4 | 27.6 ± 1.2 |
| LV mass (mg) | 83 ± 7 | 206 ± 12* | 86 ± 4 | 158 ± 12 †‡ |
| Lung mass (mg) | 143 ± 6 | 405 ± 120* | 151 ± 8 | 176 ± 22‡ |
| Tibia length (mm) | 17.3 ± 0.1 | 17.7 ± 0.2 | 17.4 ± 0.2 | 17.7 ± 0.1 |
| LV/TL (mg/mm) | 4.8 ± 0.4 | 11.6 ± 0.8* | 4.9 ± 0.2 | 8.9 ± 0.7†‡ |
| Lung/TL (mg/mm) | 8.3 ± 0.4 | 22.9 ± 6.9* | 8.7 ± 0.3 | 9.9 ± 1.3†‡ |
| Hemodynamic data | ||||
| Peak systolic pressure (mmHg) | 95 ± 8 | 119 ± 6* | 92 ± 6 | 172 ± 19† |
| End diastolic pressure (mmHg) | 3 ± 2 | 25 ± 5* | 2 ± 3 | 6 ± 4‡ |
| dP/dT max (mmHg/sec) | 8249 ± 596 | 4786 ± 985* | 9119 ± 428 | 8941 ± 1489‡ |
| dP/dT min (mmHg/sec) | 8382 ± 576 | 4432 ± 824* | 8681 ± 514 | 9250 ± 1684‡ |
| Ejection fraction (%) | 61 ± 12 | 26 ± 8* | 63 ± 7 | 49 ± 10†‡ |
| Heart rate (bpm) | 506 ± 26 | 518 ± 15 | 526 ± 23 | 490 ± 17 |
Means±SD. p<0.05:
vs. IgG Sham;
vs. Eng NAb Sham;
vs. IgG TAC
Figure 6. Neutralizing Endoglin Activity Attenuates Fibrosis and Promotes BMP9 Signaling in Heart Failure.
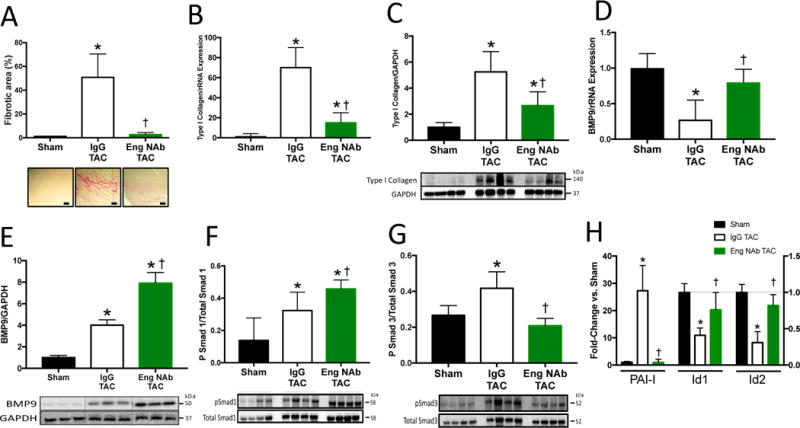
A) Representative histological staining for collagen and quantitation of LV fibrotic area in Endoglin neutralizing treated (Eng NAb) or IgG isotype control treated mice (IgG) after TAC. B–C) LV mRNA and protein expression of type I collagen after TAC. D) LV mRNA expression of BMP9. E) Representative immunoblots and quantitation of LV BMP9 expression normalized to GAPDH expression. F–G) Representative immunoblots and quantitation of phosphorylated Smad 1 (pSmad1), phosphorylated Smad 3 (pSmad3), total Smad 1, and total Smad 3. H) LV mRNA expression of inhibitor of differentiation 1 and 2 (Id1,Id2), and plasminogen activator inhibitor-1 (PAI-I). Scale bars 40 um. Data expressed as fold change vs. Sham. p<0.05: *, vs. Sham; †, vs. IgG TAC.
Discussion
Our central finding is that BMP9 expression is increased in patients with advanced HF, limits TGF-β1 induced collagen expression in human cardiac fibroblasts and that promoting BMP9 activity attenuates cardiac fibrosis in a mouse model of HF. We further introduce a new role for endoglin as a negative regulator of BMP9 expression and determine that neutralizing endoglin activity increases BMP9 levels and activity, thereby limiting TGF-β1 signaling, Type I collagen synthesis and cardiac fibrosis (Figure 7).
Figure 7. BMP9 Signaling is Attenuated by Endoglin and Limits Cardiac Fibrosis in Heart Failure.
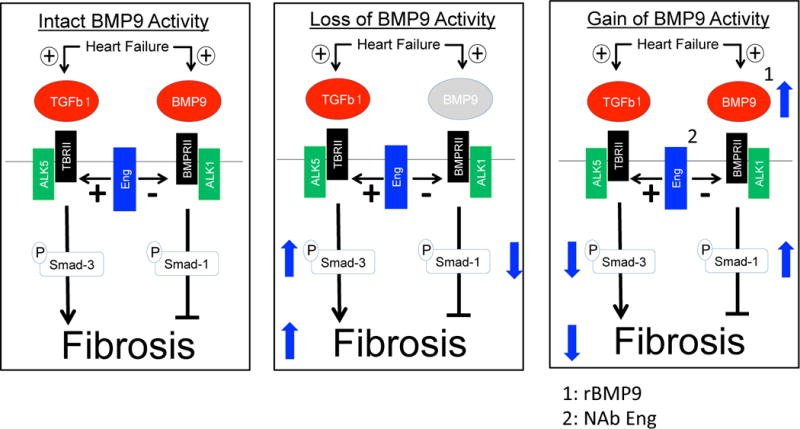
Left, Endoglin promotes cardiac fibrosis by facilitating TGFβ1 and inhibiting BMP9 signaling in heart failure
Middle, Loss of BMP9 activity promotes Smad3 dominant signaling and increases cardiac fibrosis
Right, Recombinant BMP9 or neutralizing endoglin activity increases BMP9 signaling, promotes Smad1 dominant signaling and decreases cardiac fibrosis in heart failure.
Our findings have several important clinical implications. First, most studies involving BMP9 focus on development biology and vascular remodeling with limited data in cardiac remodeling. Our findings introduce a functional role for BMP9 as an endogenous inhibitor of collagen production and cardiac fibrosis in HF. Second, in contrast to prior studies, whereby directly limiting TGF-β1 activity attenuates Smad3 phosphorylation, our findings introduce BMP9 as a master switch regulating canonical TGF-β1 signaling by promoting Smad1 and limiting Smad3 activity in HF. Third, we previously reported that limiting endoglin activity attenuates cardiac fibrosis in HF by limiting TGF-β1 activity. Since TGF-β1 signaling is an established mediator of both reparative and reactive fibrosis, we speculate that BMP9 may play a role in both processes28. We now introduce a new mechanism whereby endoglin serves as an endogenous antagonist of BMP9 expression in HF. Collectively, these findings provide new mechanistic insight into BMP signaling in HF and identify multiple novel strategies to either directly deliver BMP9 or indirectly promote BMP9 expression as potentially new therapeutic approaches to limit cardiac fibrosis in HF.
BMP signaling has been implicated in fibrosis of the kidney, lung, and liver24, 29–34. To date, the only study exploring a role for BMPs in cardiac fibrosis focused on the potential anti-fibrotic effects of BMP735. Despite initially promising data, recombinant BMP7 has not shown efficacy in treating lung, skin, or kidney fibrosis. More recently, administration of BMP9, also known as growth differentiation factor-2 (GDF2), was reported to increase Smad1 phosphorylation in vascular endothelium and to reverse pulmonary arterial hypertension in mice36. Homozygous knockout of ALK-1 or endoglin are embryonic lethal due to defects in heart and vessel development, however BMP9 knockout (BMP9−/−) mice are viable into adulthood37, 38. BMP9 expression in HF and a role for BMP9 in cardiac fibrosis remains unexplored.
We first identified that circulating and myocardial protein levels of BMP9 are increased in patients with advanced HF and in a mouse model of pressure overload induced HF. Consistent with our mouse model of HF, circulating BMP9 levels correlated inversely with clinical LV ejection fraction. Little is known about cardiac expression of BMP9 in the adult heart and no studies have explored BMP9 expression in HF. Importantly, BMP9 mRNA levels were decreased after LV pressure overload in mice, suggesting post-transcriptional regulation of BMP9 protein levels in HF. Most of our understanding of BMP9 biology is derived from hepatocytes where BMP9 is produced as prepro-peptide that is converted to unprocessed pro-BMP9, which is proteolytically cleaved to form mature circulating BMP9. One potentially relevant protease known to regulate BMP9 processing is the endopeptidase furin whose levels are known to be increased in both acute and chronic HF. Future studies exploring whether furin regulates BMP9 levels in HF are required.
To explore a functional role for BMP9 in HF, we next determined that BMP9 is highly expressed by non-myocyte populations in the heart including cardiac fibroblasts and endothelial cells. Using loss of function and gain of function approaches we then identified that BMP9 attenuates TGF-β1 induced collagen expression in human cardiac fibroblasts. A role for BMP9/ALK-1 signaling remains controversial. Several studies support a pro-fibrotic role for BMP9/ALK-1 in the liver by increasing expression of hepcidin, which inhibits absorption of hepatic iron, thereby leading to iron accumulation and subsequent fibrosis. Yet studies employing whole body conditional ALK-1 knockout mice or ALK-1 heterozygous mice support that loss of ALK-1 promotes fibrosis. We recently reported that compared to wild-type controls, ALK-1 heterozygous mice exhibit impaired LV function, increased cardiac fibrosis, and reduced myocardial levels of pSmad1 after pressure overload induced HF39. Our present findings are consistent with these studies by showing that loss of BMP9 in HF replicates the pro-fibrotic phenotype associated with reduced ALK-1 activity.
There are several potential mechanisms by which a neutralizing endoglin antibody increases BMP9 levels and activity. First, prior studies have shown that neutralizing endoglin antibodies disrupt the ability of BMP9 to bind to endoglin17. As a result, endoglin blocking antibodies may increase availability of unbound BMP9. This observation is supported by our data showing that BMP9 protein levels are increased in the LV and circulation of mice treated with Eng Nab. Second, in contrast to IgG treated wild-type mice, treatment with Eng NAb increased LV mRNA and protein levels of BMP9, suggesting that Eng NAb may increase BMP9 transcription. Third, levels of BMP9 are known to be highly regulated by endopeptidases40. Neutralizing endoglin activity may decrease endoprotease levels, thereby increasing levels of intact BMP9. Future studies are required to identify mechanisms by which neutralizing endoglin activity increases BMP9 levels.
BMP targeted therapeutics for cardiac remodeling remains largely unexplored. Delivery of recombinant BMP7 limits fibrosis in models of heart or kidney injury, however in these studies, BMP7 was shown to induce Smad1 activity in endothelial cells, not cardiac fibroblasts. In contrast, we now show a direct effect of BMP9 on cardiac fibroblasts. Treatment with rBMP9 has also been previously shown to attenuate pulmonary hypertension. Given that BMP9 negatively regulates collagen expression in vitro and in vivo, we explored the therapeutic potential of delivering rBMP9 as an approach to limit progressive fibrosis by treating mice 4 weeks after induction of HF. In contrast to BMP9−/− mice, rBMP9 treatment reduced cardiac fibrosis, increased pSmad1 levels, and reduced pSmad3 levels. However, rBMP9 as a therapeutic approach may be limited by pharmacokinetics requiring frequent dosing and the risk of promoting bone formation at injection sites. Based on recent studies identifying that BMP9 binds endoglin with high affinity and our prior studies showing that reduced endoglin activity limits cardiac fibrosis, we tested the treatment effect of a potent anti-endoglin antibody (M1043; Amgen Inc). Activity of the neutralizing endoglin antibodies used in our study (M1043 and TRC105) have been previously validated by Nolan-Steveau and colleagues who demonstrated potent anti-endoglin activity in endothelial cells where BMP9 is known to promote Smad1 phosphorylation in an endoglin-dependent manner17. Similar to mice treated with rBMP9, neutralizing endoglin activity increased myocardial BMP9 levels, limited cardiac fibrosis, reduced pSmad3 levels, and increased pSmad1 levels and signaling through Id1 and Id2. While the activation of compensatory signaling pathways by cardiomyocyte dysfunction may underlie cardiac fibrosis, accumulating data suggests that excessive fibrosis driven by TGF beta mediated signaling in fibroblasts is sufficient to cause ventricular dysfunction41, 42. Our study supports that reducing pSmad 3 levels and increasing pSmad 1 levels by promoting BMP9 signaling reduces fibrosis and improves systolic function.
These findings also have potentially important clinical implications for BMP9 inhibitors under investigation as adjunctive therapy in cancer [15]. For highly vascular malignancies targeting the ALK-1 activity to limit neovascularization of tumors is an emerging new therapeutic approach [16]. An anti-human ALK-1 antibody (PF-3446962) and a ligand trap that sequesters BMP9 (ALK-1-Fc; Dalantercept) have potential to systemically disrupt BMP9 signaling and are currently under clinical investigation43, 44. Our findings now suggest that attenuating BMP9/ALK-1 signaling promotes cardiac fibrosis and may impair cardiac function. Careful monitoring for cardiovascular complications is warranted, particularly for patients at risk for developing HF.
Our study has several limitations. We used the BMP9−/− mouse model with reduced total body expression of BMP9 and cannot distinguish if our observations are, in whole or in part, due to loss of BMP9 expression in non-fibroblast cell populations. Recently, fibroblast specific loss of Smad 3 signaling has been shown to protect against cardiac fibrosis41. Future studies employing cell-specific knockouts of endoglin are required to determine the role of fibroblasts as mediators of endoglin/BMP9 interactions in cardiac fibrosis. In addition, in the rescue studies of established LV fibrosis, extra-cardiac effects of rBMP9 or Eng NAb administration may have modified the cardiac response to pressure overload induced HF. Finally studies were conducted in male mice only, thereby potentially limiting the applicability of our findings to the female sex.
HF is a significant and growing global health problem and a primary mode of death for millions of individuals, yet despite the clear importance of TGF-β1 signaling in maladaptive remodeling, no therapies specifically targeting TGF-β1 signaling exist. We have now identified an exciting new paradigm that implicates BMP9 as an endogenous inhibitor of TGF-β1 activity in HF. We further introduce that administration of rBMP9 or increasing BMP9 abundance by reducing endoglin activity enhances Smad1 signaling, rescues cardiac fibrosis, and improves cardiac function. Further investigation of strategies to promote BMP9 activity or reduce endoglin activity may lead to new approaches to selectively inhibit canonical TGF-β1 signaling and to improve the lives of millions living with HF.
Supplementary Material
Clinical Perspective.
What is new?
Cardiac fibrosis is a major component of maladaptive remodeling in the heart.
Levels of bone morphogenetic protein 9 (BMP9) are increased in patients with heart failure and we report a new role for BMP9 as an endogenous inhibitor of cardiac fibrosis.
What are the Clinical Implications?
Increasing BMP9 activity either by treatment with recombinant BMP9 or by inhibiting a high-affinity receptor for BMP9, known as endoglin, attenuates cardiac fibrosis.
These findings provide new mechanistic insight and open new therapeutic pathways for heart failure.
Acknowledgments
Sources of Funding
This work was supported by a grant from the National Institutes of Health (1R01HL133215-01) to N.K. and a grant from the National Institutes of Health (T32 HL069770) to K.M.
Footnotes
Disclosures
None.
References
- 1.Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B, Group E-HS Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21. [Google Scholar]
- 2.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics C and Stroke Statistics S Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG, American Heart Association Advocacy Coordinating C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention and Stroke C Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart fail. 2013;6:606–619. doi: 10.1161/HHF.0b013e318291329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, Morarji K, Brown TD, Ismail NA, Dweck MR, Di Pietro E, Roughton M, Wage R, Daryani Y, O’Hanlon R, Sheppard MN, Alpendurada F, Lyon AR, Cook SA, Cowie MR, Assomull RG, Pennell DJ, Prasad SK. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. 2013;309:896–908. doi: 10.1001/jama.2013.1363. [DOI] [PubMed] [Google Scholar]
- 5.Hsia HH, Marchlinski FE. Electrophysiology studies in patients with dilated cardiomyopathies. Card Electrophysiol Rev. 2002;6:472–481. doi: 10.1023/a:1021109130276. [DOI] [PubMed] [Google Scholar]
- 6.Iles L, Pfluger H, Lefkovits L, Butler MJ, Kistler PM, Kaye DM, Taylor AJ. Myocardial fibrosis predicts appropriate device therapy in patients with implantable cardioverter-defibrillators for primary prevention of sudden cardiac death. J Am Coll Cardiol. 2011;57:821–828. doi: 10.1016/j.jacc.2010.06.062. [DOI] [PubMed] [Google Scholar]
- 7.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J clin invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 9.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005;111:2837–2849. doi: 10.1161/CIRCULATIONAHA.104.500546. [DOI] [PubMed] [Google Scholar]
- 11.Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, Kubota T, Takeshita A. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovas res. 2004;64:526–535. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 12.Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc res. 2007;74:207–212. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 13.Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc res. 2004;63:423–432. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 14.Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, Schluter KD, Bohm M. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1) Am j physiol Heart circ physiol. 2002;283:H1253–H1262. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- 15.Kapur NK, Qiao X, Paruchuri V, Mackey EE, Daly GH, Ughreja K, Morine KJ, Levine J, Aronovitz MJ, Hill NS, Jaffe IZ, Letarte M, Karas RH. Reducing endoglin activity limits calcineurin and TRPC-6 expression and improves survival in a mouse model of right ventricular pressure overload. J Am Heart Assoc. 2014;3 doi: 10.1161/JAHA.114.000965. pii: e000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapur NK, Wilson S, Yunis AA, Qiao X, Mackey E, Paruchuri V, Baker C, Aronovitz MJ, Karumanchi SA, Letarte M, Kass DA, Mendelsohn ME, Karas RH. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation. 2012;125:2728–2738. doi: 10.1161/CIRCULATIONAHA.111.080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolan-Stevaux O, Zhong W, Culp S, Shaffer K, Hoover J, Wickramasinghe D, Ruefli-Brasse A. Endoglin requirement for BMP9 signaling in endothelial cells reveals new mechanism of action for selective anti-endoglin antibodies. PloS one. 2012;7:e50920. doi: 10.1371/journal.pone.0050920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young K, Conley B, Romero D, Tweedie E, O’Neill C, Pinz I, Brogan L, Lindner V, Liaw L, Vary CP. BMP9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood. 2012;120:4263–4273. doi: 10.1182/blood-2012-07-440784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown MA, Zhao Q, Baker KA, Naik C, Chen C, Pukac L, Singh M, Tsareva T, Parice Y, Mahoney A, Roschke V, Sanyal I, Choe S. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J biolog chem. 2005;280:25111–25118. doi: 10.1074/jbc.M503328200. [DOI] [PubMed] [Google Scholar]
- 20.David L, Feige JJ, Bailly S. Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev. 2009;20:203–212. doi: 10.1016/j.cytogfr.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Tillet E, Bailly S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Frontiers in genetics. 2014;5:456. doi: 10.3389/fgene.2014.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon MS, Robert F, Matei D, Mendelson DS, Goldman JW, Chiorean EG, Strother RM, Seon BK, Figg WD, Peer CJ, Alvarez D, Adams BJ, Theuer CP, Rosen LS. An open-label phase Ib dose-escalation study of TRC105 (anti-endoglin antibody) with bevacizumab in patients with advanced cancer. Clin cancer res. 2014;20:5918–5926. doi: 10.1158/1078-0432.CCR-14-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karzai FH, Apolo AB, Cao L, Madan RA, Adelberg DE, Parnes H, McLeod DG, Harold N, Peer C, Yu Y, Tomita Y, Lee MJ, Lee S, Trepel JB, Gulley JL, Figg WD, Dahut WL. A phase I study of TRC105 anti-endoglin (CD105) antibody in metastatic castration-resistant prostate cancer. BJU international. 2015;116:546–555. doi: 10.1111/bju.12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li P, Li Y, Zhu L, Yang Z, He J, Wang L, Shang Q, Pan H, Wang H, Ma X, Li B, Fan X, Ge S, Jia R, Zhang H. Targeting secreted cytokine BMP9 gates the attenuation of hepatic fibrosis. Biochimica et biophysica acta. 2017;1864:709–720. doi: 10.1016/j.bbadis.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Rosen LS, Hurwitz HI, Wong MK, Goldman J, Mendelson DS, Figg WD, Spencer S, Adams BJ, Alvarez D, Seon BK, Theuer CP, Leigh BR, Gordon MS. A phase I first-in-human study of TRC105 (Anti-Endoglin Antibody) in patients with advanced cancer. Clin cancer res. 2012;18:4820–4829. doi: 10.1158/1078-0432.CCR-12-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapur NK, Paruchuri V, Aronovitz MJ, Qiao X, Mackey EE, Daly GH, Ughreja K, Levine J, Blanton R, Hill NS, Karas RH. Biventricular remodeling in murine models of right ventricular pressure overload. PloS one. 2013;8:e70802. doi: 10.1371/journal.pone.0070802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapur NK, Deming CB, Kapur S, Bian C, Champion HC, Donahue JK, Kass DA, Rade JJ. Hemodynamic modulation of endocardial thromboresistance. Circulation. 2007;115:67–75. doi: 10.1161/CIRCULATIONAHA.106.640698. [DOI] [PubMed] [Google Scholar]
- 28.Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat rev Cardiol. 2013;10:15–26. doi: 10.1038/nrcardio.2012.158. [DOI] [PubMed] [Google Scholar]
- 29.De Langhe E, Cailotto F, De Vooght V, Aznar-Lopez C, Vanoirbeek JA, Luyten FP, Lories RJ. Enhanced endogenous bone morphogenetic protein signaling protects against bleomycin induced pulmonary fibrosis. Respir Res. 2015;16:38. doi: 10.1186/s12931-015-0202-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang LP, Dong JZ, Xiong LJ, Shi KQ, Zou ZL, Zhang SN, Cao ST, Lin Z, Chen YP. BMP-7 attenuates liver fibrosis via regulation of epidermal growth factor receptor. Int J Clin Exp Pathol. 2014;7:3537–3547. [PMC free article] [PubMed] [Google Scholar]
- 31.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeisberg M, Bottiglio C, Kumar N, Maeshima Y, Strutz F, Muller GA, Kalluri R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am j physiol Ren physiol. 2003;285:F1060–F1067. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- 33.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 34.Zeisberg M, Kalluri R. Reversal of experimental renal fibrosis by BMP7 provides insights into novel therapeutic strategies for chronic kidney disease. Pediat nephrol. 2008;23:1395–1398. doi: 10.1007/s00467-008-0818-x. [DOI] [PubMed] [Google Scholar]
- 35.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 36.Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat med. 2015;21:777–785. doi: 10.1038/nm.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricard N, Ciais D, Levet S, Subileau M, Mallet C, Zimmers TA, Lee SJ, Bidart M, Feige JJ, Bailly S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood. 2012;119:6162–6271. doi: 10.1182/blood-2012-01-407593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tual-Chalot S, Oh SP, Arthur HM. Mouse models of hereditary hemorrhagic telangiectasia: recent advances and future challenges. Front genet. 2015;6:25. doi: 10.3389/fgene.2015.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morine KJ, Qiao X, Paruchuri V, Aronovitz MJ, Mackey EE, Buiten L, Levine J, Ughreja K, Nepali P, Blanton RM, Oh SP, Karas RH, Kapur NK. Reduced activin receptor-like kinase 1 activity promotes cardiac fibrosis in heart failure. Cardiovasc pathol. 2017;31:26–33. doi: 10.1016/j.carpath.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bidart M, Ricard N, Levet S, Samson M, Mallet C, David L, Subileau M, Tillet E, Feige JJ, Bailly S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell molec life sci. 2012;69:313–324. doi: 10.1007/s00018-011-0751-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J clin invest. 2017;127:3770–3783. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 43.Cunha SI, Pietras K. ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood. 2011;117:6999–7006. doi: 10.1182/blood-2011-01-330142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Meeteren LA, Thorikay M, Bergqvist S, Pardali E, Stampino CG, Hu-Lowe D, Goumans MJ, ten Dijke P. Anti-human activin receptor-like kinase 1 (ALK1) antibody attenuates bone morphogenetic protein 9 (BMP9)-induced ALK1 signaling and interferes with endothelial cell sprouting. J biologic chem. 2012;287:18551–18561. doi: 10.1074/jbc.M111.338103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.