

Abstract
Honeybee propolis and its bioactive component, caffeic acid phenethyl ester (CAPE), are known for a variety of therapeutic potentials. By recruiting a cell-based reporter assay for screening of hypoxia-modulating natural drugs, we identified CAPE as a pro-hypoxia factor. In silico studies were used to probe the capacity of CAPE to interact with potential hypoxia-responsive proteins. CAPE could not dock into hypoxia inducing factor (HIF-1), the master regulator of hypoxia response pathway. On the other hand, it was predicted to bind to factor inhibiting HIF (FIH-1). The active site residue (Asp201) of FIH-1α was involved in hydrogen bond formation with CAPE and its analogue, caffeic acid methyl ester (CAME), especially in the presence of Fe and 2-oxoglutaric acid (OGA). We provide experimental evidence that the low doses of CAPE, that did not cause cytotoxicity or anti-migratory effect, activated HIF-1α and inhibited stress-induced protein aggregation, a common cause of age-related pathologies. Furthermore, by structural homology search, we explored and found candidate compounds that possess stronger FIH-1 binding capacity. These compounds could be promising candidates for modulating therapeutic potential of CAPE, and its recruitment in treatment of protein aggregation-based disorders.
Electronic supplementary material
The online version of this article (10.1007/s12192-018-0915-0) contains supplementary material, which is available to authorized users.
Keywords: Caffeic acid phenethyl ester, Hypoxia inducible factor, Factor inhibiting HIF-1α, Pro-hypoxia, Anti-stress molecules
Introduction
Hypoxia-inducible transcription factor (HIF) is the key regulator of hypoxia signaling associated deregulation of which has been associated with several pathological conditions. Activated hypoxia signaling has been linked to malignant transformation and tumor aggressiveness; its deactivation has been shown to trigger brain disorders (Choi et al. 2003, 2010; Vaupel 2004; Gordan and Simon 2007; Masoud and Li 2015; Mohme et al. 2017; Soni and Padwad 2017). HIF is a transcription factor that acts as a heterodimer (HIF-1α and HIF-1β); whereas HIF-1α is regulated by oxygen, HIF-1β is constitutively expressed (Liu and Simon 2004). A Per-ARNT-Sim (PAS) domain is present in both the subunits. It is centrally involved in oxygen homeostasis and activated in a large majority of tumors (Marin-Hernandez et al. 2009; Wang et al. 2013). Under normoxia conditions, α-subunit undergoes hydroxylation by proline-hydroxylase-2 (HPH-2). The hydroxylated HIF-1α undergoes degradation by proteasome mediated degradation pathway involving tumor suppressor VHL (von Hippel-Lindau protein) (Huang et al. 1998). Another oxygen-dependent modification (asparaginyl hydroxylation) catalyzed by dioxygenase called factor inhibiting HIF-1α (FIH-1) occurs in the C-terminal (Asn803) transactivation domain of HIF-1α (Ranasinghe et al. 2015; Soni and Padwad 2017). Under hypoxia conditions that prevent HIF-1α hydroxylation and degradation, HIF-1α accumulates, translocates to the nucleus, dimerizes with HIF-1β, and transactivates several effector proteins (Lee et al. 2004; Calzada and del Peso 2007). It is also regulated by CREB binding protein (CBP) and p300 that interact with the carboxy-terminal transactivation domain of HIF-1α and act as its transcriptional co-activators (Berlow et al. 2017). It has been shown that hydroxylation at Asn803 under normoxic conditions inhibits interactions of HIF-1α to CBP and p300 (Lando et al. 2002). Since HIF-1 is a key driver of hypoxia signaling, involved in cancer progression in one hand and several brain disorders on the other, the identification of HIF-modulating drugs/factors and molecular mechanism of their action has been warranted for disease therapeutics (Masoud and Li 2015). Cancer is generally characterized as a disease of cell proliferation and spreading of abnormally growing cells to other parts of the body. It continues to be a killer throughout the world although past decade has seen rapid developments in cancer diagnostics and treatment. The primary reason for treatment failure and high mortality has been attributed to the highly invasive nature of cancer cells resulting in rapid cancer progression and metastasis, the phenotypes that often involve hyperactive hypoxia signaling. In view of the vital role of hypoxia in several diseases, we performed a screening of natural drugs to search for hypoxia-modulating effects using cell-based HIF-1α reporter assay, and identified caffeic acid phenethyl ester (CAPE) as one of the pro-hypoxia factors.
CAPE is a bioactive compound found in many plants (Metzner et al. 1979). It is a key component of propolis obtained from honeybee hives (Borrelli et al. 2002; Markiewicz-Zukowska et al. 2013; Tolba et al. 2013, 2016). The presence of hydroxyl groups in the catechol ring of this polyphenol has been ascribed to be responsible for many of its biological activities, such as anti-cancer, anti-microbial, anti-viral, neuroprotective, and anti-inflammatory properties (Özyurt et al. 2004; Markiewicz-Zukowska et al. 2013; Tolba et al. 2013, 2016; Murtaza et al. 2014). Choi et al. (2010) reported that CAPE is a potent inhibitor of HIF prolyl hydroxylase (HPH-2) raising the question on its relevance to modulate hypoxia signaling in cancer cells. Roos et al. (2011) also reported the stabilization of HIF-1α and subsequent induction of heme oxygenase-1 (HO-1) during CAPE induced growth arrest in PDGF-activated vascular smooth muscle cells. In view of these information and identification of CAPE as a candidate pro-hypoxia factor in our screenings, we explored the docking potential of CAPE with HIF-1α and FIH-1 (inhibits HIF-1α by hydroxylation at Asn803). We found that although CAPE was incapable of interacting with HIF-1α, it docked efficiently into the active site of FIH-1. We provide experimental evidence for the pro-hypoxia activity of CAPE that may be the result of inhibition of FIH-1 regulated modulation of HIF-1α and its impact on stress-induced protein aggregation and cell-migration phenotypes. Furthermore, structural analogues of CAPE with predicted stronger activity were found and warrant further investigations.
Materials and methods
Transfection and reagents
Human osteosarcoma (U2OS) was purchased from JCRB Japan and cultured in DMEM (Life Technologies). Transfection was performed using Lipofectamine 2000 (Invitrogen) in Opti-Mem (Gibco, Life Technologies) media. The pGL4-p53–3′ UTR were gifted from Dr. Chae-Ok Yun (Hanyang University, Seoul, South Korea). GFP-tagged mortalin MOT/GFP was expressed from pEGFP-C1/mot-2. NaAsO2 (sodium (meta) arsenite) was purchased from Sigma-Aldrich.
Generation of hypoxia-responsive cells
U2OS cells were transfected with the plasmid expressing luciferase driven by promoter containing hypoxia-responsive element (HRE) (Fig. 1a). Twenty-four hours post-transfections, the media was replaced with DMEM supplemented with hygromycin B (Roche) (700 μg/mL) for 10–15 days. The clones showing 100- to 1000-fold increase in luciferase in fixed and live cell assays were selected (Fig. 1b). The selected clones were amplified and established into cells stably expressing hypoxia-responsive luciferase reporter.
Fig. 1.

Screening for hypoxia modulating drugs. a Schematic diagram showing the screening assay protocol for hypoxia modulating drugs. b HIF-1α-driven Luciferase assay for U2OS cells stably transfected with HRE-luciferase plasmid. c HIF-reporter assay in cells treated with natural drugs (5.0 mM). Out of the 57 drugs tested, 5 compounds (shown by dark bars) showed strong pro-hypoxia activity and 4 compounds (shown by white bars) showed anti-hypoxia activity
Cell viability assay
Vital dye, MTT (Molecular Probes, Invitrogen) (0.5 mg/mL), was added to the U2OS cell culture medium for 4 h in a humidified incubator (37 °C and 5% CO2). MTT-containing medium was replaced with DMSO (100 μL) to dissolve purple formazan crystals. Absorbance of the blue chromogen was measured at 550 nm using spectrophotometer (TECAN, Switzerland). The standard deviation and statistical significance of the data were obtained from triplicates and three to four independent experiments, respectively.
Screening for hypoxia modulating natural drugs
U2OS-HRE cells were plated in 96-well plates at about 70% confluency. After the cells had attached well to the surface, they were treated with phytochemical library (for 48 h) at sub-toxic doses, determined by independent MTT-based cell viability assays as described above. Phytochemical library constituted of 57 compounds, isolated from a variety of natural products. All compounds were tested for 98% purity by HPLC. HIF-1 activity in the treated cells was determined by HRE-dependent luciferase by dual luciferase reporter assay system (Promega, Madison, WI). Luciferase activity was calculated per microgram protein following the manufacturer’s protocol, and reported as the relative activity normalized against untreated cells. Three independent experiments were performed for statistical significance.
Western blotting
Cells were cultured in DMEM in 6-well plates and lysed using 1% Nonidet P-40 buffer containing a protease inhibitor cocktail (WAKO). The protein concentrations of whole cell lysate were measured by bicinchonic acid assay (BCA) (Thermo Fisher Scientific, Rockford, IL). The cell lysates (15–20 μg) were separated in 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). Blocked membranes were probed with target protein-specific primary antibodies (1–2 μg/mL), including luciferase (luci17), HIF-1α, HSP70 (Santa Cruz Biotechnology Inc., CA, USA), or β-actin (Abcam, Cambridge, UK) used as an internal loading control, for overnight at 4 °C. The blots were incubated with the secondary antibodies (anti-mouse or anti-rabbit IgG; Santa Cruz Biotechnology Inc., CA, USA) (0.5–1 μg/mL) conjugated with horseradish peroxidase for 45 min. After extensive washings with Tris-buffered saline with 0.1% Tween 20 (TBS-T), the membranes were developed by enhanced chemiluminescence reaction (ECL) as per manufacturer’s instructions.
Immunofluorescence staining
Cells were cultured and fixed on a glass coverslip placed in a 12-well culture dish (TPP, Switzerland) and fixed with 4% paraformaldehyde in PBS. Fixed cells were permeabilized with 0.1% Triton X-100 for 10 min, blocked with 0.2% BSA/PBS for 1 h, and then incubated with antibodies (2–4 μg/mL) to proteins including vimentin, MMP2, MMP7, MMP9, and hnRNP-k (Santa Cruz Biotechnology Inc., CA, USA) for overnight at 4 °C. Anti-mortalin antibody was raised in our lab. Cells were washed with PBS containing 0.1% Triton-X and probed with fluorophore conjugated goat-anti-mouse secondary antibody (0.5–1 μg/mL) (Molecular Probes, Invitrogen). Counterstaining was performed with Hoechst 33342 (Sigma) for 10 min in dark, then coverslips visualized by Carl Zeiss microscope (Axiovert 200M, ×40 and ×60 magnification, Tokyo, Japan).
Docking of CAPE with HIF-1α and FIH-1 protein
Structure of FIH-1 in complex with HIF-1α fragment peptide (PDB ID 1H2K) was retrieved from Protein Data Bank (Elkins et al. 2003). Proteins were prepared by addition and optimization of hydrogen atoms, correction of bond orders, structure minimization, and separation of FIH-1 and HIF-1α using Protein Prep Wizard of Schrödinger software (Sastry et al. 2013). The possible ligand binding sites were identified using the structural studies published previously (Dann et al. 2002). Glide version 6.8 package of Schrödinger software was used to generate the grid at the identified ligand binding sites using default parameters (Friesner et al. 2006).
Structure of CAPE was retrieved from PubChem database (CID 5281787). The three-dimensional structure of CAPE was processed for docking using the LigPrep version 3.5 module of Schrödinger software (Schrödinger release 2017–2, LigPrep, Schrödinger, LLC, New York, NY 2017a). The major preparation steps involved desalting, generation of tautomers, and energy minimization. Various conformations of CAPE molecule were generated during the ligand preparation process to account for ligand flexibility that was later used for docking with protein. After the generation of CAPE conformations and the grid at active site of HIF-1α and FIH-1 proteins, Glide version 6.8 was used to dock CAPE at the identified binding site of proteins. Extra precision (XP) algorithm was used to dock CAPE (Friesner et al. 2006). FIH-1 requires 2-oxoglutarate (OGA) and Fe(II) for its functional activity; therefore, the effect of these co-factors on the binding potential of CAPE was also studied. Docking pose visualization and image generation was done using the Maestro version 10.3 Interface of the Schrödinger software (Schrödinger release 2017–2, Maestro, Schrödinger, LLC, New York, NY 2017b).
Molecular dynamic simulations
The molecular dynamic (MD) simulations of protein-ligand complex were performed using Desmond Molecular Dynamics Simulation System (Bowers et al. 2006). All the docked complexes were solvated with SPC water model in a cubic periodic boundary box to generate required systems for MD simulations. The systems were neutralized using appropriate number of counter ions. The systems were further minimized, slowly and gradually heated up to 300-K temperature, and equilibrated until the pressure and energies of systems were stabilized. Finally, equilibrated systems were used to run 50-ns-long MD simulations. All the simulation studies were performed on Supermicro (GPU) with 8-GB DDR RAM and NVIDIA TESLA C2050 Graphics Card.
Analyses of molecular docking and molecular dynamic simulations
H-bond profiling and conformational analyses of complexes before and after the simulations were used as criterions to analyze the stability of the protein-ligand complexes. Visual Molecular Dynamics (VMD) version 1.9.2 was used to calculate root-mean-square deviations (RMSD) of the protein backbone in the simulated complex in reference to the docked complex and hydrogen bond dynamics (Humphrey et al. 1996). Residue numbering of all the discussed proteins in the present manuscript is according to PDB file.
RNA isolation and complementary DNA synthesis
Total RNA was extracted from U2OS and U2OS-HRE cell lines by using TRIzol Reagent (Life Technologies) following supplier’s protocol. Quality and quantity were determined by spectrophotometry (NanoDrop, 1000 spectrophotometer). For complementary DNA (cDNA) synthesis, total RNA (1 μg) was reverse-transcribed into cDNA using Quantitect Reverse Transcriptase Kit (QIAGEN) following the manufacturer’s protocol. cDNA was stored at − 20 °C for PCR.
Quantitative real-time PCR
Gene expression was quantified by quantitative real-time PCR using Syber Select Master mix (Applied Biosystem, Life Technologies), luciferase specific primers (F-5′-GGACTTGGACACCGGTAAGA-3′ and R-5′-CTTGTCGATGAGAGCGTTTG-3′), HIF-1α specific primers (F-5′-GTTTACTAAAGGACAAGTCACC-3′ and R-5′-TTCTGTTTGTTGAAGGGAG-3′), and EcoTM Real-Time PCR System (Illumina, San Diego, CA). Relative level of expression of target gene was normalized against the internal control 18S by ∆CT method. An amplification plot between fluorescence signals and cycle number was plotted, and the mean values in the triplicated samples of targeted genes and internal control 18S were calculated. The relative quantitative value was expressed as 2−∆CT. Statistical significance of the results was calculated from three independent experiments.
Effect of CAPE on cell migration and growth of cancer cells: wound-scratch assays
The in vitro migration capability of U2OS cells in response to CAPE treatment was observed with a wound-scratch assay. The cells were grown in a 6-well plate and wounded by uniformly scratching the cells in a line with a 200-μL pipette tip. Cells were washed several times with PBS to remove cell debris or damaged cells, and re-cultured on either the control or CAPE-supplemented medium, respectively. The time of wound scratching was designated as time 0. Movement of cells into the scratched area was recorded during next 24 h, up to 72–96 h under a phase contrast microscope with a 10× phase objective.
Effect of CAPE in stress-induced protein aggregation
Sodium arsenite-based protein aggregation model was used (Jacobson et al. 2012). U2OS cells (4 × 103/well) stably expressing mortalin-GFP (U2OS-mot-GFP) were seeded in 96-well plates for overnight, followed by treatment with sodium arsenite (20 μM) for 24 h. Cells were recovered either in normal culture media or the one supplemented with CAPE for 12–48 h. Cell viability was determined by MTT assay as described above. Statistical significance of results was determined from three to four independent experiments including triplet or quadruplet sets in each experiment. For immunofluorescence, same method was adopted in 12-well plates with coverslips.
For heat shock-induced aggregation of luciferase, cells were seeded in 6-well plate and were transfected with pGL4-p53–3′ UTR expressing luciferase from a constitutive promoter. After 24 h, cells were heat shocked at 42 °C and 5% CO2 for 2 h, followed by recovery at 37 °C either in the control or CAPE-supplemented medium for the next 48 h. Luciferase expression was determined as described above as well as by immunofluorescence using anti-luciferase antibody.
Statistical analysis
All the experiments were performed in triplicate. Data are expressed as mean ± SEM of triplicate experiments. Unpaired t test (GraphPad Prism, GraphPad Software, San Diego, CA) has been performed to determine the degree of significance between the control and experimental samples. Statistical significance was defined as p values (* and @), where *< 0.05, **< 0.01, and ***< 0.001 represent significant, very significant, and very very significant, respectively, whereas @ indicates insignificant change.
Results
We investigated hypoxia-modulating potential of 57 purified phytochemicals by HRE-driven luciferase in stably transfected cells (as described in the “Materials and methods” section and Fig. 1a, b). Cells were treated with sub-toxic doses, determined by independent experiments (MTT assay and morphological observations), of phytochemicals (data not shown). Through three rounds of screenings, we identified five pro-hypoxia and four anti-hypoxia compounds (Fig. 1c). CAPE was identified as one of the pro-hypoxia factors. We investigated dose-dependent effect of CAPE on HRE-luciferase activity in comparison to a standard pro-hypoxia drug, cobalt chloride (CoCl2). As shown in Fig. 2a, HRE promoter-driven luciferase assay showed dose-dependent increase in cells treated with sub-toxic doses of CAPE (5–10 μM) and CoCl2 (50–150 μM, an established pro-hypoxia compound). Consistent with luciferase reporter assays, qPCR for luciferase showed increase in CAPE- and CoCl2-treated cells (Fig. 2b). Detection of luciferase protein by immunofluorescence (Fig. 2c) as well as Western blotting (Fig. 2d) using anti-luciferase antibody exhibited increase in CAPE as well as CoCl2-treated cells. Consistently, expression of endogenous HIF-1α protein in control, CAPE-, and CoCl2-treated cells showed increase in a dose-dependent manner (Fig. 2e).
Fig. 2.

HIF-1α-driven pro-hypoxia effect of CAPE on U2OS. a Effect of CAPE on HIF-1α-driven luciferase reporter assays in comparison to a standard hypoxia inducing drug, CoCl2. b–d Effect of CAPE on the luciferase transcript (b) and protein as detected by immunostaining (c) and Western blotting with anti-luciferase specific antibody (d). e Effect of CAPE on endogenous HIF-1α protein; CoCl2 was used as positive control
Being the key player of hypoxia signaling pathway, we first investigated if CAPE could dock into HIF-1α. The structure of HIF-1α (CTAD domain) was downloaded from Protein Data Bank (PDB ID 1H2L). We mainly focused on this domain because its functionality is regulated by oxygen concentration in the cells. CAPE was docked against the chain S (corresponding to HIF-1α) of the PDB structure. The binding energy obtained using Glide was − 2.904 kcal/mol. The low docking score suggested that the binding affinity of CAPE was not good enough for a stable binding at the CTAD domain of HIF-1α protein (Supplementary Fig. 1). Since HIF-1α could not be established as a candidate target for CAPE, we ensued our investigation to the next level of regulation in hypoxia signaling pathway. FIH-1 is an asparaginyl hydroxylase that inhibits the transactivation function of HIF-1α protein (Semenza et al. 1997; Kaelin Jr 2008). In normoxia condition, HIF-1α is degraded by von Hippel-Lindau tumor suppressor protein (pVHL), a part of E3 ubiquitin ligase protein complex by ubiquitination and proteasomal degradation or by FIH-1 that inhibits its interaction with co-activator p300/CBP (Kaelin Jr 2008). The catalytic function of FIH-1 is dependent on Fe(II) and OGA (Lando et al. 2002). The active site contacts of FIH-1 are precisely coordinated by three critical residues, His 199, Asp 201, and His 279. The side chains of these amino acids are also involved in co-ordination of Fe(II) (Dann et al. 2002). We next docked CAPE (Fig. 3a) against the active site of FIH-1. The docking score calculated for CAPE using Glide was − 8.744 kcal/mol, reflecting its strong binding affinity towards FIH-1 protein. Interaction analysis revealed the formation of hydrogen bond of Asp201 and OGA with the CAPE molecule. Fe(II) was also involved in co-ordinate bond formation (Fig. 3c). Binding of the ligand with Asp201, one of the critical residues of the active site of FIH-1, suggested that CAPE was occupying the active site of the protein. To further explore the role of OGA in CAPE binding to FIH-1, a similar docking study was carried out, but in the absence of OGA. Binding analysis revealed that in the absence of OGA, Asp201 was not involved in interaction with CAPE. The residues involved in hydrogen bond formation with the CAPE molecule were Ser91 and Tyr93. Fe(II) was involved in cation pi stacking with the ligand molecule (Fig. 3d). The binding energy calculated using Glide in the absence of OGA was − 6.311 kcal/mol. The binding energy was thus found to decrease by − 2.433 kcal/mol (Table 1). The data suggested that the presence of OGA favors binding of ligand at the active site of the protein. Absence of this co-factor results in a different binding orientation of CAPE molecule suggesting essential role of OGA in CAPE-FIH-1 interactions.
Fig. 3.

Chemical structures. a Caffeic acid phenethyl ester (CAPE). b Caffeic acid methyl ester (CAME). c Binding of CAPE and FIH-1. Interactions of CAPE (shown in pink) and FIH-1 in the presence of OGA (shown in green). Two hydrogen bonds are formed: one between Asp201 and CAPE molecule and the other between OGA and CAPE molecule. d Interactions of CAPE (shown in pink) and FIH-1 in the absence of OGA. CAPE is involved in hydrogen bond formation with Ser91 and Tyr93. e Binding of CAME and FIH-1. Interactions of CAME (shown in pink) and FIH-1 in the presence of OGA (shown in green). Two hydrogen bonds are formed between Asp201 and CAME molecule. f Interactions of CAME (shown in pink) and FIH-1 in the absence of OGA. Two hydrogen bonds are formed between Arg238 and CAME, and one hydrogen bond is formed between Gln239 and CAME molecule
Table 1.
Docking score of CAPE and CAME after binding with FIH-1 at its active site, in the presence and absence of its co-factor OGA
| FIH-1-CAPE complex (kcal/mol) | FIH-1-CAME complex (kcal/mol) | |
|---|---|---|
| OGA bound | − 8.744 | − 6.044 |
| OGA unbound | − 6.311 | − 4.48 |
| Difference | − 2.433 | − 1.564 |
Caffeic acid methyl ester (CAME) is a structural analogue of CAPE formed by replacing the phenethyl moiety with methyl moiety (Fig. 3a, b) (Choi et al. 2010). Docking of FIH-CAME complex was also performed in the presence/absence of OGA. Presence of OGA resulted in binding of the ligand at the active site of the protein as Asp201 was found to be involved in hydrogen bond formation with CAME and the calculated docking score was − 6.044 kcal/mol. Fe(II) was involved in coordinate bond formation with CAME only in the presence of OGA (Fig. 3e). The docking score calculated using Glide was found to decrease to − 4.48 kcal/mol in the absence of OGA. The residues involved in hydrogen bond formation in this case were Arg238 and Gln239 (Fig. 3f). These results also indicated that the OGA is required for binding of CAME at the active site of the protein as observed in case of CAPE.
MD simulations were used to study the stability of FIH-1 protein for its binding with CAPE and CAME in the presence of OGA. MD simulations were performed to check the stability of docked complexes. During the 50-ns-long MD simulations, root-mean square distance (RMSD) values of FIH-1 backbone in FIH-1-CAPE and FIH-1-CAME complex calculated in reference to the coordinates of the respective docked complexes when plotted against the simulation time revealed an almost stable trajectory. RMSD values indicate the change in the orientation of the docked complexes during the simulation run as compared to their initial orientation. The data indicated that the complexed structures were relatively stable during the complete simulation run. The protein backbone was more stable when bound to CAME as compared to CAPE (Fig. 4a). Analysis of the interacting residues showed that the hydrogen bond formed between FIH-1 and Asp201 in both the complexes was quite stable throughout the simulation run (Fig. 4b). All the results obtained were therefore in accordance with our hypothesis that CAPE and CAME could be potential inhibitors of FIH-1 functional activity (Table 2).
Fig. 4.
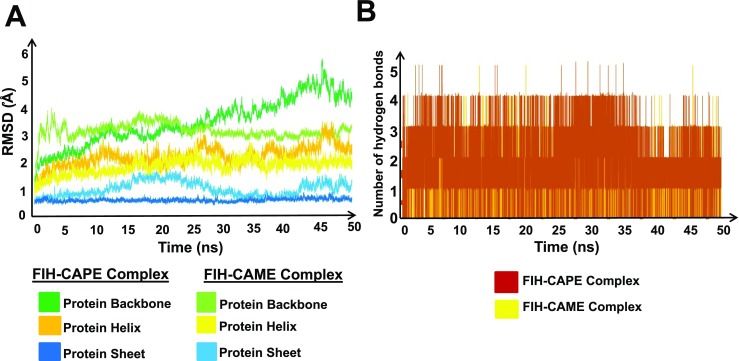
RMSD fluctuations of FIH-1 protein during 50-ns simulation. a The plot shows the variations in protein structure (backbone, helix, and sheet) after binding of CAPE and CAME with FIH-1. b Number of hydrogen bonds vs. time for FIH-1-CAPE complex (brown) and number of hydrogen bonds vs. time for FIH-CAME complex (yellow)
Table 2.
Properties of CAPE and CAME

Anti-cancer potential of CAPE has been well documented in several studies (Demestre et al. 2009; Roos et al. 2011; Murtaza et al. 2014; Paeng et al. 2015; Wadhwa et al. 2016). In view of the above findings on pro-hypoxia activity of CAPE, we next investigated the effect of low dose of CAPE on cell survival and migration. Cells treated with low doses (5–10 μM) did not show significant toxicity in 48-h treatment as detected by MTT assay (Fig. 5a). We also examined the effect of CAPE on cell migration by wound-healing assays and found that low doses of CAPE failed to affect cell migration (Fig. 5b). The observation thus made was consistent with other results where no change was observed in protein markers of cell migration studied in control and CAPE (low non-toxic dose)-treated cells (Fig. 5c).
Fig. 5.
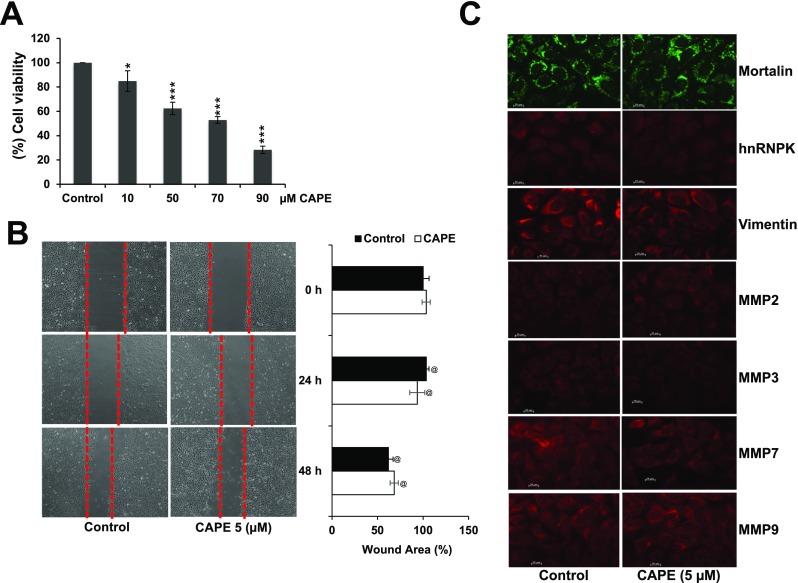
Effect of CAPE at low doses on U2OS cell viability (a), migration (b), and proteins (c) involved in cell migration
We next investigated anti-stress potential of non-toxic doses of CAPE by recruiting metal (sodium arsenite) or high-temperature-induced protein aggregation in cells exogenously transfected with either GFP or luciferase proteins, respectively. The system allowed direct observations of protein aggregation. As shown in Fig. 6a, GFP-labeled cells were treated with sodium arsenite (24 h), followed by recovery (48 h) either in the control or CAPE-supplemented medium. As expected, we found that the cells possessed cytoplasmic fluorescence of mortalin-GFP protein (Fig. 6b). Of note, whereas sodium arsenate-treated cells recovered in normal medium showed strong aggregation (Fig. 6b), protein de-aggregation was seen in the cells recovered in CAPE-supplemented medium (Fig. 6b). We next observed time-dependent de-aggregation effect of CAPE. As shown in Fig. 6c, cells treated for 24–48 h, but not 12 h, showed de-aggregation. Consistently, we found that the sodium arsenite-stressed cells treated with CAPE led to 20–30% increase in viability in 36–48 h (Fig. 6d, e). Such increase was not observed in cells treated for 12 h and was endorsed by microscopic examination of cells (Fig. 6e). We also determined the effect of CAPE on sodium arsenite-induced heat shock protein 70 (HSP70). As shown in Fig. 6f, sodium arsenite caused induction of HSP70 that was not altered in CAPE-treated cells. As expected, the latter showed induction of HIF-1α.
Fig. 6.

Effect of CAPE at low doses on sodium arsenate-induced aggregation in U2OS-mot-GFP and cell survival in U2OS cell lines. a Schematic diagram showing the protein aggregation and recovery assay. b, c Aggregation of GFP in sodium arsenate-treated and recovered (either in control or CAPE-supplemented medium). Time-dependent response of cells to CAPE treatment is shown in c. d, e Viability of cells treated with sodium arsenate followed by recovery either in control or CAPE-supplemented medium and morphological observations (e). f Western blotting of control, sodium arsenite, and CAPE-treated cells for HSP70 and HIF-1α proteins showed induction of HIF-1α, but not of HSP70, with CAPE
We, next, confirmed protein de-aggregation and anti-stress effect of CAPE by using heat shock-induced protein misfolding and aggregation of luciferase reporter (Fig. 7a) (Nguyen et al. 1989; Wang and Kurganov 2003). As shown in Fig. 7b, heat-induced aggregation of luciferase led to decrease in its activity as detected by luciferase assays. Cells recovered in normal medium for 48 h showed increase in luciferase activity that was attributed to reversal of heat-induced misfolding as also reported in another study (Wallace et al. 2015). Of note, as compared to the cells recovered in control medium, cell recovered in CAPE-supplemented medium caused 2-fold increase in luciferase activity. The results were confirmed by immunostaining of luciferase by specific antibodies (Fig. 7b). These data demonstrated that the pro-hypoxic doses of CAPE protected cells against stress-induced aggregation of proteins.
Fig. 7.

Effect of CAPE at low doses on heat shock-induced aggregation of luciferase reporter and activity in U2OS. a Schematic diagram showing the protein aggregation and recovery assay. b Luciferase activity and expression in heat-shock treated and recovered either in control or CAPE-supplemented medium is shown
Discussion
By screening of 57 purified phytochemicals for induction of HRE-driven luciferase, we identified CAPE as one of the pro-hypoxia factors. It led to increase in HRE-driven luciferase as well as endogenous HIF-1α protein, suggesting that it may be a reliable pro-hypoxia compound. Bioinformatics and molecular docking analyses revealed that CAPE was capable of docking to FIH-1, an inhibitor of HIF-1α protein, and thereby yield activation of HIF-1α. Screening of compounds with capability to dock to FIH-1 also lead to identification of CAME, as its potential inhibitor with potential pro-hypoxia activity.
Accurate protein folding determines their subcellular niche and functional characteristics (Dobson 2003). Environmental conditions such as adverse temperature, pH, metals, and oxidative stress have been established as the prominent factors leading to protein misfolding and aggregation that are toxic for cells and have been associated with diseases including diabetes, neurodegenerative disorders, and cancer (Goldberg 2003; Tyedmers et al. 2010; Jacobson et al. 2012; Valastyan and Lindquist 2014). Endoplasmic reticulum (ER) plays an essential role in protein quality control by activating unfolded protein response (UPR) in response to altered conditions of nutrients and oxygen that trigger protein misfolding (Ron and Walter 2007). Activated UPR regulates expression of several pro-survival proteins (Tomiyama et al. 2017). Interestingly, lower doses of CAPE as well as mild hypoxia have been shown to activate UPR (Dirnagl et al. 2009; Tomiyama et al. 2017). Several studies have shown that HIF-1α stabilization provides stress protection in various neurological disease models, e.g., stroke (Siddiq et al. 2005; Nagel et al. 2011), Parkinson’s disease (Belaidi and Bush 2016), oxidative stress (Shoshani et al. 2002), Alzheimer’s disease (Crapper McLachlan et al. 1991; Ritchie et al. 2003; Belaidi and Bush 2016), and mitochondrial dysfunction (Niatsetskaya et al. 2010). The beneficial effects of HIF-1α stabilization arise mainly through the activation of HIF-1 target genes that combat oxidative stress, improve glucose metabolism, and block cell death pathways (Zhang et al. 2011). We performed sodium arsenate as well as heat-induced protein aggregation and found that CAPE caused de-aggregation effect that was correlated to induction of HIF-1α, but not HSP70.
In view of the above findings that the CAPE modulates hypoxia could be valuable for treatment of protein-aggregation-related diseases including age-related pathologies (neurodegeneration, amyloidosis, amyotrophic lateral sclerosis (ALS), and Huntington’s, Alzheimer’s, and Parkinson’s diseases), we further set out to search for structural analogues of CAPE that might enhance the therapeutic window. A virtual library of 104 compounds, similar in structure to CAPE molecule, was prepared by screening the PubChem database. Based on the docking capability as determined by docking score, number, and length of hydrogen bond with Asp201 and logP values of docked analogues, we identified three structural analogues of CAPE as potential FIH-1 inhibitors (Table 3). The rule of five was also considered while making the selection of compounds (Lipinski et al. 2001). Though the preliminary results look promising, the clinical potential of the predicted compounds for their anticipated enhanced hypoxia regulating capability can only be ensured after experimental validation that is currently underway.
Table 3.
Screening of CAPE analogues based on various parameters

We found that the pro-hypoxia function of CAPE at low doses was mediated by inhibition of FIH, resulting in attenuation of HIF-1α degradation and activation of its transcriptional activation function. Indeed, some studies have reported the prevention of hypoxia-ischemic injury (Wei et al. 2004) by CAPE and attributed it to transactivation of HIF-1 targeting genes vascular endothelial growth factors (VEGF), heme oxygenase-1 (HO-1) (Daekyu Choi et al. 2010). Our data demonstrated that CAPE caused increase in HIF-1α at protein level and was supported by computational analysis, suggesting that CAPE inhibits FIH and thereby leads to activation of HIF-1α. Choi et al. (2010) suggested that CAPE delayed the degradation of HIF-1α protein by targeting prolyl hydroxylase, the key enzyme of von Hippel-Lindau-dependent degradation. In the present study, we report that CAPE and CAME have the potential to inhibit the activity of FIH-1, and hence delay the asparaginyl hydroxylation of HIF-1α protein, required for its degradation. Consequently, an activation of HIF-1α signaling was predicted. Cell-based assays suggested that the low non-cytotoxic doses of CAPE activated pro-hypoxia signaling and protection against the protein aggregation stress reflecting on the new therapeutic aspects of CAPE and its analogues.
Electronic supplementary material
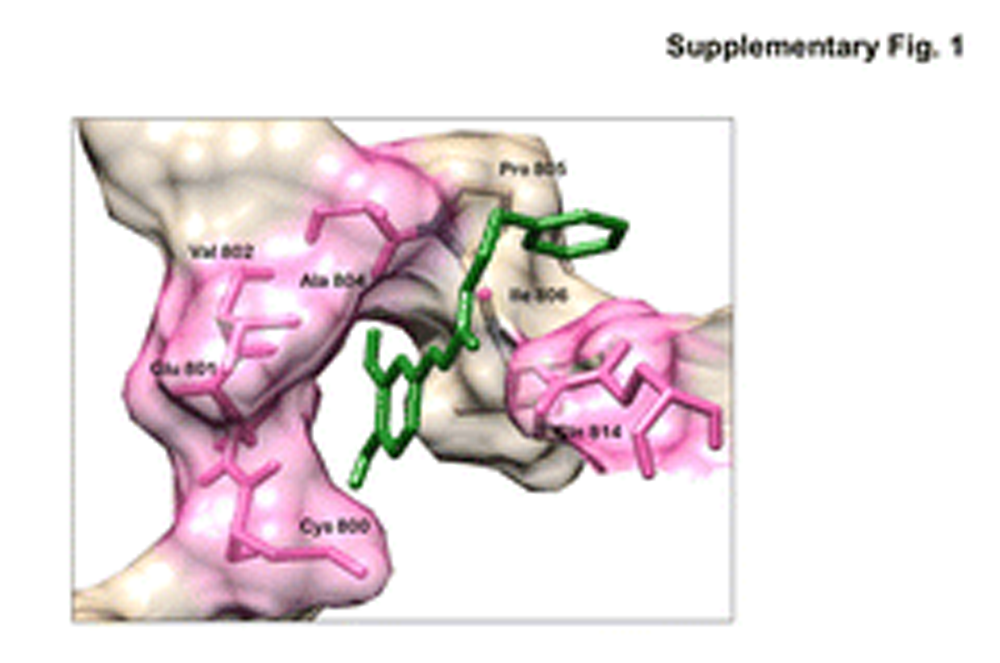{kind=link}
Molecular interaction of CAPE with C-terminal Activation Domain (CTAD) of HIF-1α: Binding of CAPE molecule (green) with CTAD of HIF-1α is shown. The amino acid residues involved in the interaction of the protein with the molecule are shown in pink. CAPE-CTAD did not show significant binding to HIF-1α. (PNG 362 kb)
Acknowledgements
Computations were performed at the Bioinformatics Centre at IIT Delhi. DAILAB is supported by grants from the Department of Biotechnology (Government of India) and AIST (Japan).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Sunil C. Kaul, Phone: 81-29-861-6713, Email: s-kaul@aist.go.jp
Durai Sundar, Phone: 91-11-26591066, Email: sundar@dbeb.iitd.ac.in.
Renu Wadhwa, Phone: 81-29-861-9464, Email: renu-wadhwa@aist.go.jp.
References
- Belaidi AA, Bush AI. Iron neurochemistry in Alzheimer's disease and Parkinson's disease: targets for therapeutics. J Neurochem. 2016;139(Suppl 1):179–197. doi: 10.1111/jnc.13425. [DOI] [PubMed] [Google Scholar]
- Berlow RB, Dyson HJ, Wright PE. Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature. 2017;543:447–451. doi: 10.1038/nature21705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli F, Maffia P, Pinto L, Ianaro A, Russo A, Capasso F, Ialenti A. Phytochemical compounds involved in the anti-inflammatory effect of propolis extract. Fitoterapia. 2002;73:S53–S63. doi: 10.1016/S0367-326X(02)00191-0. [DOI] [PubMed] [Google Scholar]
- Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA et al (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters Proceedings of the 2006 ACM/IEEE conference on Supercomputing ACM, p. 84
- Calzada MJ, del Peso L. Hypoxia-inducible factors and cancer. Clin Transl Oncol. 2007;9:278–289. doi: 10.1007/s12094-007-0055-y. [DOI] [PubMed] [Google Scholar]
- Choi KS, Bae MK, Jeong JW, Moon HE, Kim KW. Hypoxia-induced angiogenesis during carcinogenesis. J Biochem Mol Biol. 2003;36:120–127. doi: 10.5483/bmbrep.2003.36.1.120. [DOI] [PubMed] [Google Scholar]
- Choi D, Han J, Lee Y, Choi J, Han S, Hong S, Jeon H, Kim YM, Jung Y. Caffeic acid phenethyl ester is a potent inhibitor of HIF prolyl hydroxylase: structural analysis and pharmacological implication. J Nutr Biochem. 2010;21:809–817. doi: 10.1016/j.jnutbio.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, et al. Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-B. [DOI] [PubMed] [Google Scholar]
- Dann CE, Bruick RK, Deisenhofer J. Structure of factor-inhibiting hypoxia-inducible factor 1: an asparaginyl hydroxylase involved in the hypoxic response pathway. Proc Natl Acad Sci U S A. 2002;99:15351–15356. doi: 10.1073/pnas.202614999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demestre M, Messerli SM, Celli N, Shahhossini M, Kluwe L, Mautner V, Maruta H. CAPE (caffeic acid phenethyl ester)-based propolis extract (bio 30) suppresses the growth of human neurofibromatosis (NF) tumor xenografts in mice. Phytother Res. 2009;23:226–230. doi: 10.1002/ptr.2594. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, et al. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1α. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71–77. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jacobson T, Navarrete C, Sharma SK, Sideri TC, Ibstedt S, Priya S, Grant CM, Christen P, Goloubinoff P, Tamas MJ. Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J Cell Sci. 2012;125:5073–5083. doi: 10.1242/jcs.107029. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia-inducible factor (HIF-1) alpha: its protein stability and biological functions. Exp Mol Med. 2004;36:1–12. doi: 10.1038/emm.2004.1. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004;3:492–497. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- Marin-Hernandez A, Gallardo-Perez JC, Ralph SJ, Rodriguez-Enriquez S, Moreno-Sanchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini-Rev Med Chem. 2009;9:1084–1101. doi: 10.2174/138955709788922610. [DOI] [PubMed] [Google Scholar]
- Markiewicz-Zukowska R, Borawska MH, Fiedorowicz A, Naliwajko SK, Sawicka D, Car H. Propolis changes the anticancer activity of temozolomide in U87MG human glioblastoma cell line. BMC Complement Altern Med. 2013;13:50. doi: 10.1186/1472-6882-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoud GN, Li W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015;5:378–389. doi: 10.1016/j.apsb.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner J, Bekemeier H, Paintz M, Schneidewind E. On the antimicrobial activity of propolis and propolis constituents (author's transl) Die Pharmazie. 1979;34:97–102. [PubMed] [Google Scholar]
- Mohme M, Riethdorf S, Pantel K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat Rev Clin Oncol. 2017;14:155–167. doi: 10.1038/nrclinonc.2016.144. [DOI] [PubMed] [Google Scholar]
- Murtaza G, Karim S, Akram MR, Khan SA, Azhar S, Mumtaz A, et al. Caffeic acid phenethyl ester and therapeutic potentials. Biomed Res Int. 2014;2014:145342. doi: 10.1155/2014/145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel S, Papadakis M, Chen R, Hoyte LC, Brooks KJ, Gallichan D, Sibson NR, Pugh C, Buchan AM. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2011;31:132–143. doi: 10.1038/jcbfm.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Morange M, Bensaude O. Protein denaturation during heat shock and related stress. Escherichia coli beta-galactosidase and Photinus pyralis luciferase inactivation in mouse cells. J Biol Chem. 1989;264:10487–10492. [PubMed] [Google Scholar]
- Niatsetskaya Z, Basso M, Speer RE, McConoughey SJ, Coppola G, Ma TC, et al. HIF prolyl hydroxylase inhibitors prevent neuronal death induced by mitochondrial toxins: therapeutic implications for Huntington's disease and Alzheimer's disease. Antioxid Redox Signal. 2010;12:435–443. doi: 10.1089/ars.2009.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özyurt H, Söğüt S, Yıldırım Z, Kart L, Iraz M, Armutçu F, Temel İ, Özen S, Uzun A, Akyol Ö. Inhibitory effect of caffeic acid phenethyl ester on bleomycine-induced lung fibrosis in rats. Clin Chim Acta. 2004;339:65–75. doi: 10.1016/j.cccn.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Paeng SH, Jung WK, Park WS, Lee DS, Kim GY, Choi YH, et al. Caffeic acid phenethyl ester reduces the secretion of vascular endothelial growth factor through the inhibition of the ROS, PI3K and HIF-1alpha signaling pathways in human retinal pigment epithelial cells under hypoxic conditions. Int J Mol Med. 2015;35:1419–1426. doi: 10.3892/ijmm.2015.2116. [DOI] [PubMed] [Google Scholar]
- Ranasinghe WK, Baldwin GS, Bolton D, Shulkes A, Ischia J, Patel O. HIF1alpha expression under normoxia in prostate cancer--which pathways to target? J Urol. 2015;193:763–770. doi: 10.1016/j.juro.2014.10.085. [DOI] [PubMed] [Google Scholar]
- Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Roos TU, Heiss EH, Schwaiberger AV, Schachner D, Sroka IM, Oberan T, Vollmar AM, Dirsch VM. Caffeic acid phenethyl ester inhibits PDGF-induced proliferation of vascular smooth muscle cells via activation of p38 MAPK, HIF-1alpha, and heme oxygenase-1. J Nat Prod. 2011;74:352–356. doi: 10.1021/np100724f. [DOI] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2017–2: LigPrep, Schrödinger, LLC, New York, NY, (2017a)
- Schrödinger Release 2017–2: Maestro, Schrödinger, LLC, New York, NY, (2017b)
- Semenza GL, Agani F, Booth G, Forsythe J, Iyer N, Jiang BH, Leung S, Roe R, Wiener C, Yu A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997;51:553–555. doi: 10.1038/ki.1997.77. [DOI] [PubMed] [Google Scholar]
- Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, Kalinski H, Kamer I, Rozen A, Mor O, Keshet E, Leshkowitz D, Einat P, Skaliter R, Feinstein E. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22:2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni S, Padwad YS. HIF-1 in cancer therapy: two decade long story of a transcription factor. Acta Oncol. 2017;56:503–515. doi: 10.1080/0284186X.2017.1301680. [DOI] [PubMed] [Google Scholar]
- Tolba MF, Azab SS, Khalifa AE, Abdel-Rahman SZ, Abdel-Naim AB. Caffeic acid phenethyl ester, a promising component of propolis with a plethora of biological activities: a review on its anti-inflammatory, neuroprotective, hepatoprotective, and cardioprotective effects. IUBMB Life. 2013;65:699–709. doi: 10.1002/iub.1189. [DOI] [PubMed] [Google Scholar]
- Tolba MF, Omar HA, Azab SS, Khalifa AE, Abdel-Naim AB, Abdel-Rahman SZ. Caffeic acid Phenethyl Ester: a review of its antioxidant activity, protective effects against ischemia-reperfusion injury and drug adverse reactions. Crit Rev Food Sci Nutr. 2016;56:2183–2190. doi: 10.1080/10408398.2013.821967. [DOI] [PubMed] [Google Scholar]
- Tomiyama R, Takakura K, Takatou S, Le TM, Nishiuchi T, Nakamura Y, et al. 3,4-dihydroxybenzalacetone and caffeic acid phenethyl ester induce preconditioning ER stress and autophagy in SH-SY5Y cells. J Cell Physiol. 2017;233:1671–1684. doi: 10.1002/jcp.26080. [DOI] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- Valastyan JS, Lindquist S. Mechanisms of protein-folding diseases at a glance. Dis Model Mech. 2014;7:9–14. doi: 10.1242/dmm.013474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9(Suppl 5):10–17. doi: 10.1634/theoncologist.9-90005-10. [DOI] [PubMed] [Google Scholar]
- Wadhwa R, Nigam N, Bhargava P, Dhanjal JK, Goyal S, Grover A, Sundar D, Ishida Y, Terao K, Kaul SC. Molecular characterization and enhancement of anticancer activity of caffeic acid phenethyl ester by gamma cyclodextrin. J Cancer. 2016;7:1755–1771. doi: 10.7150/jca.15170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace EW, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, et al. Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell. 2015;162:1286–1298. doi: 10.1016/j.cell.2015.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Kurganov BI. Kinetics of heat- and acidification-induced aggregation of firefly luciferase. Biophys Chem. 2003;106:97–109. doi: 10.1016/S0301-4622(03)00134-0. [DOI] [PubMed] [Google Scholar]
- Wang X, Peralta S, Moraes CT. Mitochondrial alterations during carcinogenesis: a review of metabolic transformation and targets for anticancer treatments. Adv Cancer Res. 2013;119:127–160. doi: 10.1016/B978-0-12-407190-2.00004-6. [DOI] [PubMed] [Google Scholar]
- Wei X, Zhao L, Ma Z, Holtzman DM, Yan C, Dodel RC, et al. Caffeic acid phenethyl ester prevents neonatal hypoxic-ischaemic brain injury. Brain. 2004;127:2629–2635. doi: 10.1093/brain/awh316. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yan J, Chang Y, ShiDu Yan S, Shi H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr Med Chem. 2011;18:4335–4343. doi: 10.2174/092986711797200426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Molecular interaction of CAPE with C-terminal Activation Domain (CTAD) of HIF-1α: Binding of CAPE molecule (green) with CTAD of HIF-1α is shown. The amino acid residues involved in the interaction of the protein with the molecule are shown in pink. CAPE-CTAD did not show significant binding to HIF-1α. (PNG 362 kb)