

Abstract
In primary bovine fibroblasts with an hspa1b/luciferase transgene, we examined the intensity of heat-shock response (HSR) following four types of oxidative stress or heat stress (HS), and its putative relationship with changes to different cell parameters, including reactive oxygen species (ROS), the redox status of the key molecules glutathione (GSH), NADP(H) NAD(H), and the post-translational protein modifications carbonylation, S-glutathionylation, and ubiquitination. We determined the sub-lethal condition generating the maximal luciferase activity and inducible HSPA protein level for treatments with hydrogen peroxide (H2O2), UVA-induced oxygen photo-activation, the superoxide-generating agent menadione (MN), and diamide (DA), an electrophilic and sulfhydryl reagent. The level of HSR induced by oxidative stress was the highest after DA and MN, followed by UVA and H2O2 treatments, and was not correlated to the level of ROS production nor to the extent of protein S-glutathionylation or carbonylation observed immediately after stress. We found a correlation following oxidative treatments between HSR and the level of GSH/GSSG immediately after stress, and the increase in protein ubiquitination during the recovery period. Conversely, HS treatment, which led to the highest HSR level, did not generate ROS nor modified or depended on GSH redox state. Furthermore, the level of protein ubiquitination was maximum immediately after HS and lower than after MN and DA treatments thereafter. In these cells, heat-induced HSR was therefore clearly different from oxidative stress-induced HSR, in which conversely early redox changes of the major cellular thiol predicted the level of HSR and polyubiquinated proteins.
Electronic supplementary material
The online version of this article (10.1007/s12192-018-0909-y) contains supplementary material, which is available to authorized users.
Keywords: Heat-shock response, Oxidative stress, Redox state, Protein modification, Glutathione oxidation, Glutathionylation, Ubiquitination, Carbonylation
Introduction
Heat-shock response (HSR) is one of the pathways that are induced by oxidative stress. However, it is well known that different types of oxidative stress differ in their capacity to induce HSR (Wallen et al. 1997; Wirth et al. 2002). Moreover, a same oxidative stress may lead to differential responses, depending on the cells. For instance, peroxide has been reported to successfully induce a high HSR level in one of the human cervical cancer HeLa cell lines (Doulias et al. 2007), but not in another one (Steels et al. 1992) nor in the human malignant glioma T98G cell line (Adachi et al. 2009). These data point out that HSR may primarily depend on cell physiology and how any given cell reacts to stress. The underlying cellular processes, however, remain poorly characterized although several candidate pathways have been proposed.
HSR is well illustrated by the rapid induction of heat-shock proteins (HSP) HSPA (also known as HSP70) gene expression (Kampinga et al. 2009)—here referred to as the group of inducible HSPA (iHSPA). Several HSP genes, including HSPA, encode proteins that are major molecular chaperones of cell proteins (Kim et al. 2013), helping them to (re)fold or assemble correctly, or to be degraded. In cellular and animal models, it has been found that the increased level of iHSPA correlates with cytoprotection level upon a proteotoxic stress (Horowitz and Assadi 2010).
Heat-shock transcriptional factors like HSF1 in mammalian cells are “sensing” factors of the rate of cellular protein denaturation. An increase in the rate of denaturation in cellular proteins frees HSF1 from chaperone repression and leads to the formation of HSF1 trimers, which is competent for DNA binding activity and transcriptional activation of target genes like iHSPA (Gidalevitz et al. 2011; Vabulas et al. 2010; Westerheide et al. 2012).
Oxidative stress is also able to increase the rate of protein denaturation (Freeman et al. 1995; Gosslau et al. 2001; McDuffee et al. 1997; Senisterra et al. 1997), which leads to a transient increase in protein degradation during the recovery period (Freeman et al. 1995; Shang and Taylor 2011). It has been shown that the level of HSR and hydrophobic segments exposed after stress are correlated (Gosslau et al. 2001) and oxidative stress-induced HSR is associated with the disruption of HSPA-HSF1 interactions (Jacobs and Marnett 2007). An increase in protein denaturation following heat as well as oxidative stress generally leads to an increase in protein (poly)-ubiquitination (Figueiredo-Pereira et al. 1998; Nivon et al. 2012; Taylor et al. 2002).
Taken together, these facts fit well with the hypothesis that oxidative stress-induced HSR is due to protein denaturation triggered by direct oxidation of proteins due to diverse reactive species that can be detected by some popular fluorescent probes (Cossarizza et al. 2009). However, it has been shown that even in the case of HSR induced by iodoacetamide treatment, which leads to the formation of adducts on proteins, the key event is a reduced level of the major cellular redox and anti-oxidant regulator, glutathione, resulting in a large increase in the rate of protein disulfide bonds (Liu et al. 1996).
Changes in redox states of GSH, the most abundant redox cell modulator, have been observed in HeLa cells after both HS and different oxidizing treatments (Zou et al. 1998). These data led the authors to propose that all treatments leading to HSR are able to modify the GSH redox state, which in turn induces changes in the redox state of key protein thiols. Because in vitro HSF1 DNA binding activity was, however, insensitive to DTT (Zou et al. 1998), the authors have proposed that the redox-sensitive step is independent of HSF1 and its direct (chaperone) partners. These treatments included in particular menadione—which can both generate superoxide and form adducts with thiols when metabolized by the cell (Giulivi and Cadenas 1994), hydrogen peroxide—which is not itself a reactive oxygen species but can generate hydroxyl radicals in the presence of metal ion in the cell (Forman et al. 2010), and diamide—a thiol oxidant which has been reportedly shown to exert its action without reactive oxygen species (ROS) production (Pias and Aw 2002). In CHO cells, diamide-induced HSR shows a 3-h delayed compared to the heat-induced response. It has been suggested that this delay corresponds to the time necessary between the formation of non-native disulfide bonds and protein denaturation and its detection by HSPA proteins (Freeman et al. 1995).
Evidence, however, now exists in favor of a direct redox regulation of HSR (Rudolph and Freeman 2009; West et al. 2012). Indeed, several cysteine residues accessible to redox regulation have been identified in the recent years to play a role in HSR induced by heat stress (Ahn and Thiele 2003) and by diverse molecules associated with oxidative stress (Mahmood et al. 2012). Depending on the treatment and/or cell type, these cysteine residues belong either to HSF1 or to one of its associated HSP chaperones (West et al. 2012). These data are therefore more in favor of a specific redox signaling pathway (Dinkova-Kostova 2012; Go et al. 2004). These latter data fit well with the idea of a redox code as proposed in the recent years (Jones and Sies 2015), in which the different redox couples present in the cell, including the thiol redox regulation of glutathione (GSH), thioredoxin (Trx) (Gorrini et al. 2013), and free cysteine and cysteine residues in proteins (Jones et al. 2004), and key metabolites like NAD+/NADH and NADP+/NADPH, are major actors of a complex network of redox circuits (Jones and Sies 2015; Ying 2008). Some of these circuits are interconnected as illustrated by GSH and NADPH redox states because NADPH is required to reduce oxidized GSH, but individual redox signaling may also be targeted in absence of global changes in the balance between anti-oxidants and oxidants in favor of the latter, generally illustrated by changes in major redox couples such as reduced (GSH) and oxidized (GSSG) GSH (Jones and Sies 2015).
In this work, we re-examined these questions in an attempt to provide key cell parameters to predict HSR in a unique, non-cancerous, non-immortalized cell system equipped with a sensitive HSR reporter locus. By submitting these cells to sub-lethal, reversible stress conditions associated with four types of oxidative stress treatments known to induce HSR in at least some cell types, we looked for cellular factors correlated to HSR level since these treatments induced very different levels of this response.
Material and methods
Chemicals and antibodies
Hydrogen peroxide (H2O2), tert-butyl hydroxide (t-BOOH), N-acetyl cysteine (NAC), 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), 2-methyl-1,4-naphthoquinone (menadione, MN), 1,4-benzoquinone (BQ), diamide (DA), thiazolyl tetrazolium blue (MTT), buthionine sulfoximine (BSO), trichloroethanol (TCE), and D-Luciferin were purchased from Sigma-Aldrich, France. Dihydroethidium, DHE (Marker Gene technologies Inc., OR, USA), was purchased from Tebu-bio (France) and prepared as described by the manufacturer. Dihydrorhodamine 123 (DHR123) and 5-(and 6-)chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) were purchased from Life Technologies SAS, France. The primary antibodies used in this study were anti-heat-inducible HSP70 (StressMarq SMC-100A/B), anti-luciferase (Sigma L 2164), anti-β-actin (Sigma AC15), anti-glutathione (Santa Cruz D8 clone) and anti-lamin A/C (Santa Cruz, #sc-376248) mouse monoclonal antibodies, and anti-HSF1 (Cell Signaling Technology, #4356,), anti-phospho-HSF1(Ser326) (Epitomics, #2092-1), and anti-ubiquitin (Cell Signaling Technology, #3933) rabbit polyclonal antibodies. All secondary anti-mouse and anti-rabbit antibodies were conjugated to horseradish peroxidase (HRP) (Jackson Immun. Res.).
Cell cultures, treatments, and cell viability assay
The origin and culture of the male transgenic bovine primary fibroblasts referred to as “OV7060” were described previously (Lelievre et al. 2010). The hspa1b/Luciferase transgene comprises 574 bp of the flanking region and 232 bp of the non-coding 5′ extremity of the mRNA region, including the initiation codon ATG (Lelièvre unpublished, and Menck et al. 1998). Consensus HS elements redefined recently (Jaeger et al. 2014) are found at two places in the promoter region and recover the sites experimentally evidenced (Christians et al. 1997). During this study, cells from passage 6 to 15 were cultured in “complete” DMEM, namely DMEM high glucose (Lonza or Gibco) supplemented with penicillin and streptomycin (100 U ml−1), 10% fetal bovine serum, and 2 mM glutamine at 37 °C. As indicated in the text, we also used the MRC5Vi cell line that are immortalized human fibroblasts and cultured as previously described (Graindorge et al., 2015). For cell viability experiments, cells were at 40–50% confluent on the day of the treatments, while for all other experiments, the treatments were performed when cells were 80–90% confluent and in serum-free, phenol red-free, and glutamine-free DMEM medium in which stock solutions of the different chemicals were diluted. UVA treatment was performed as described previously (Girard et al. 2013). Intracellular depletion of GSH was obtained by incubating the cells in the presence of 0.5 mM BSO for 16–20 h (Girard et al. 2013). Immediately following the treatments, the cells were rinsed twice with complete medium and then cultured in fresh complete medium for various times. Luciferase activity and cell viability were determined simultaneously, generally 5 h after the end of the treatments or as mentioned in the text. Cell viability was assessed by MTT assay as described elsewhere (Girard et al. 2016). Spectroscopic measurements were performed at 560 nm.
Measurement of luciferase activity
Extraction of luciferase protein was performed as described previously (Lelievre et al. 2010). In brief, the culture medium was removed, the cells were rinsed in PBS, and 0.4 ml of luciferase extraction buffer was added per 2-cm2 well (corresponding to 100 to 150 × 103 cells). The cells were scraped with the tip and the whole cell lysate was transferred to a microtube and stored at − 20 °C until measurement. Dosage of luciferase activity was performed in 96-well microplates in the TriStar LB 941 Multimode Microplate Reader (Berthold Tech GmbH, Bad Wildbad, Germany). Samples with an activity level lower than 1080 relative luminescence units (RLU) mn−1 (background level) were considered null. Luciferase activity was adjusted according to the number of viable cells.
Protein extraction and western blot analysis
Total soluble proteins and chromatin-bound fractions were extracted as previously described (Girard et al. 2013). Briefly, cells in dishes were washed with cold PBS and lysed on ice for 5 min in lysis buffer [10 mM Hepes, pH 7.5; 100 mM NaCl; 300 mM sucrose; 3 mM MgCl2; 1 mM EGTA; 50 mM NaF; 20 mM glycerophosphate; 0.3% triton X-100; 0.1 mM sodium orthovanadate; and complete mini EDTA-free protease inhibitors (Roche Diagnosis)]. For detecting protein glutathionylation, lysis was performed in the presence of 10 mM N-ethylmaleimide (NEM) to block free thiol groups. At this stage, supernatants containing total soluble protein extracts were transferred into 2-ml Eppendorf tubes, the protein concentration was measured using Bradford assay, and the proteins precipitated with acetone. After removal of acetone and air drying, the pellets were dissolved in 1.5 X SDS loading buffer (150 mM Tris-HCl, pH 6.8, 3% SDS, 0.15% bromophenol blue, 150 mM β-mercaptoethanol, and 15% glycerol). The remaining biological material in the dish (chromatin-bound fraction) was recovered by adding PBS and using a scraper. This material was transferred into 1.5-ml Eppendorf tubes and centrifuged, and the pellet resuspended into 1.5 X SDS loading buffer. Protein extracts were denatured for 5 min (soluble proteins) and 20 min (chromatin-bound proteins) at 95 °C. To detect glutathionylated proteins (Prot-SSG adducts), the same protocol was used except that β-mercaptoethanol was omitted in the loading buffer. About 30–35 μg of total soluble protein extracts were loaded on SDS polyacrylamide gels. For analyzing ubiquitinated and carbonylated proteins, cells were rinsed in PBS at various time points post-treatment and directly lysed in the Petri dish with 120 μl 6% SDS, 75 mM Tris-HCl pH 6.8, and 1% b-mercaptoethanol (referred to as “6% SDS solution”). The viscous suspension—because of DNA release—was immediately transferred to a microtube, heated at 90 °C for 15 min, and centrifuged for 15 min at 13,000g. To determine the level of protein carbonylation, the “6% SDS” protein extracts (15–20 μg) were treated following the guidelines of the OxyBlot™ Protein Oxidation Detection Kit (Millipore-Merck, France) to derivatize carbonyl groups to 2,4-dinitrophenylhydrazone (DNP)-labeled proteins, then detected by western analysis. Control bovine proteins (DNP-unlabeled) yielded no signal detection by anti-DNP antibodies. Gel electrophoresis and western blot analysis were performed according to standard methods as described previously (Girard et al. 2013).
To check the amount of loaded proteins, we used two methods. “Stain-free” visualization of proteins refers to fluorescent detection of proteins in gel or on blots after in-gel cross-linking of trichloroethanol (TCE) after electrophoresis as described (Ladner et al. 2004). As shown by others (Gilda and Gomes 2013), we found that this method provided useful and complementary data to the second method based on β-actin detection. It is worth noting that TCE was omitted in cast gels for the detection of carbonylated proteins as it interferes with the primary antibody (Girard and Lelièvre, personal data). Primary antibodies were diluted in Tris buffered saline (TBS) containing 5% BSA and 0.05% Tween 20. The membranes were then probed with the appropriate peroxidase-conjugated secondary antibody diluted in TBS containing 5% non-fat dry milk and 0.05% Tween 20. The signal detection was performed using the ECL western blotting Detection Reagents (Amersham Biosciences) or the ECL Prime western blotting detection reagents (GE Healthcare Europe GmbH, France) combined with a digital gel-imaging system (ChemiDoc XRS; Life Science BioRad). The intensity of the ubiquitination and DNP (OxyBlot) signals were quantified with ImageJ (imagej.nih.gov/ij).
Immunocytolocalization of HSPA1A and HSF1
Cells were fixed overnight at 4 °C in 4% formaldehyde/PBS and permeabilized in 0.5% Triton ×100. Blocking of unspecific binding sites was performed in PBS containing 0.05% Tween 20 and 1% bovine serum albumin. Incubation with primary antibodies was performed overnight at 4 °C.
Detection of intracellular ROS by DHE, DHR123, and CM-H2DCFDA probes
CM-H2DCFDA becomes H2DCFH after entering into the cell and is oxidized into DCF by a large array of ROS. DHR123 is a cell-permeable probe and reacts with ROS such as peroxide and peroxynitrite. DHE is preferentially oxidized by superoxide anion in ethidium bromide or chloride which is fluorescent when irradiated at 355 nm or 470–490 nm. However, other ROS may oxidize this probe which is then reportedly no more excited at 355 nm (Cossarizza et al. 2009). We tested two conditions: (i) probes were added to the culture media and incubated 30 min with cells before treatments; and (ii) treatments were performed first, then after washing, cells were incubated in fresh culture medium in the presence of the different probes for 15 to 30 min. After incubation with probes, cells were recovered as much as possible in the dark by trypsin treatment, and resuspended in PBS. Analysis was performed in the next hour. Cell sorting and fluorescence measurements were performed with FACS Arial (BD) or FACScalibur (BD) using standard methods.
RNA extraction and RT-PCR
After treatments, about five million OV7060 transgenic cells were detached from the culture dish with trypsin and recovered in DMEM supplemented with 10% FCS. After centrifugation, cell pellets were rinsed twice in RNAse-free PBS in low-retention RNase-free microtubes and frozen before the RNA extraction. Total RNA was extracted from batches of cells using the Rneasy Mini Kit (Qiagen France Les Ulis). RNA was treated with RNAse-free DNase I (Qiagen France Les Ulis) during extraction according to the manufacturer’s instructions and eluted in elution buffer. The reverse transcription reaction was performed on 400 ng total RNA for each sample by the Superscript III enzyme and primed by random hexamers (300 ng) (Life Technologies, France) following the supplier’s protocol (25 °C for 5 min, 50 °C for 60 min, and 70 °C for 15 min). To ensure the absence of genomic DNA in each sample, minus RT control was performed under the same conditions but without reverse transcriptase. Each cDNA will be diluted to 1/100 before the qPCR. The primers used for the endogenous reference genes glyceraldehyde-3-phosphate deshydrogenase (GAPD) and succinate dehydrogenase complex flavoprotein subunit A (SDHA) have been published by Goossens et al. (2005) and by Lelievre et al. (2016) for tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ). Primers have been previously published for the genes of interest, Luciferase (Bui et al. 2009) and bovine HSPA1A (Sagirkaya et al. 2006).
| Name | Accession number | Primer sequence (final concentration nM) | Amplicon size (bp) |
|---|---|---|---|
| Luciferase (Bui et al. 2009) | M15077 | F: AGAGATACGCCCTGGTTCCT (100 nM) R: ATAAATAACGCGCCCAACAC (100 nM) |
259 |
| HSPA1A (Sagirkaya et al. 2006) | U09861 | F: GACAAGTGCCAGGAGGTGATTT (200 nM) R: CAGTCTGCTGATGATGGGGTTA (200 nM) |
117 |
| YWHAZ (Lelièvre et al. 2016) | BM446307 | F: CTGTCTTGTCACCAACCATTCCT (200 nM) R: TAGTCTGTGGGATGCAAGCAAA (200 nM) |
123 |
| GAPD (Goossens et al. 2005) | XM_618013 | F: TTCAACGGCACAGTCAAGG R: ACATACTCAGCACCAGCATCAC |
119 |
| SDHA (Goossens et al. 2005) | NM_174178 | F: GCAGAACCTGATGCTTTGTG R: CGTAGGAGAGCGTGTGCTT |
185 |
PCRs were performed on a StepOnePlus™ real-time PCR system (Applied Biosystems) in 25 μl final volume. Ten microliters of diluted cDNA (1/100) was added to the PCR mix consisting of 12.5 μl of 2× SybrGreen Mastermix (Applied Biosystems, Courtaboeuf, France) and 100 or 200 nM final of each primer. The thermal cycle profile started with 10 min at 95 °C step followed by 40 cycles consisting of a denaturation step for 15 s at 95 °C, and an annealing/extension step for 60 s at 60 °C. Dissociation curves were obtained after each PCR run to ensure that a single PCR product had been amplified. Each gene was run separately and a tenfold dilution series of bovine fibroblast cDNA was included in each run to determine the PCR efficiency and the standard curve. Assays were performed in triplicate.
The raw qRT-PCR amplification data were exported from the StepOnePlus™ software (Applied BioSystems) to the qbasePLUS software (Biogazelle, Ghent, Belgium) (Hellemans et al. 2007). All the Ct values were transformed into normalized relative quantities using qBase plus software.
Dosage of total and oxidized glutathione, NADPH, and NADH
We measured intracellular GSH and GSSH content using an acellular assay (V6611 GSH/GSSG-Glo assay, Promega). Bovine cells were plated at the same density (2 to 4 × 104 cells per well) in 24-well plates, cultured for 24 to 48 h, and then exposed to the different types of stress or mock-treated. Immediately after treatment (T0), cells were processed as recommended by the manufacturer. This yielded the calculation of total and oxidized GSH per well. The ratio of oxidized GSH vs. reduced GSSG was then calculated for each treatment. A similar protocol was followed to determine the ratio of oxidized vs. reduced NADPH and NADH using the NADP+/NADPH and NAD+/NADH Glo Assays (Promega G9081 and G9071, respectively). Finally, the normalized ratio of each experiment was obtained by dividing the ratio obtained in treated samples by the ratio obtained in mock-treated samples.
Statistical analysis
Statistical analysis was performed under SigmaPlot v11 (Systat Software, Inc., USA). Kruskal-Wallis one-way ANOVA rank test was used to compare the five or six groups of values. A statistical difference between groups was considered significant when P < 0.01. When normality (Shapiro-Wilk test) and equal variance passed, the all-pairwise multiple comparison procedures were performed according the Bonferroni t test. Otherwise, the all-pairwise multiple comparison procedures were performed according to Dunn’s method. In addition, for Figs. 7 and 8, a Friedman non-parametric data test has been applied, computed on those data using R software by considering treatments as group and time for block to determine whether a significant effect of group treatment existed. This computation has been followed by non-parametric contrast test using a Bonferroni correction. These statistics allowed us to identify the group treatment(s) responsible of the effect of group.
Fig. 7.

Comparison of the changes in protein ubiquitination profiles following the four types of oxidative stress and heat stress. Bovine cells were exposed to the different treatments as described in Fig. 3, and the total level of protein ubiquitination was detected by western blot at various time points post-treatment. a Shown is a representative image of the western blot analyses. iHSPA/HSP70 and luciferase expression were also detected by western blot. Actin was used as a loading control. b Quantification of the level of protein ubiquination after the different treatments. ImageJ was used for the quantification. The results shown are the mean ± SD of four independent experiments. Their statistical analysis showed that the weak effect of group treatment (P = 0.08) was mainly due to MN treatment (P = 0.044) compared to the control (bracket). An ANOVA for T4 values only using a non-parametric Kruskal-Wallis test indicated a significant difference between treatments (P = 0.0304), and the post hoc analysis with Bonferroni correction showed that this effect was mainly due to two significant treatments: MN and DA (P = 0.018 and 0.025, respectively) indicated by stars
Fig. 8.

Comparison of the changes in protein carbonylation profiles following the four types of oxidative stress and heat stress. Bovine cells were exposed to the different treatments as described in Fig. 3, and the total level of protein carbonylation was detected by western blot at various time points post-treatment. a Shown is an example of protein carbonylation after DNPH derivatization of the same protein extracts analyzed in Fig. 7. b Quantification of the level of protein carbonylation after the different treatments. ImageJ was used for the quantification. The results are the mean ± SD of four independent experiments. Their statistical analysis showed that the significant effect of group treatment (P = 0.022) was mainly due to H2O2 and UVA treatments (P = 0.010 and P = 0.018 respectively) compared to the control
Results
Validation of the experimental system for quantifying HSR
In this study, we assessed the level of HSR in a unique tool consisting in primary bovine fibroblasts containing one locus with several copies of an hspa1b/luciferase transgene. By exposing the cells to heat shock (44–45 °C) from 5 to 30 min in a water bath (Fig. 1a) or in an incubator in an atmosphere of 95% air and 5% CO2 (data not shown), we observed a time-dependent increase of luciferase activity (Fig. 1a) that correlated with iHSPA (HSP70) and luciferase protein level (Fig. 1c). In the absence of heat stress, the induction of luciferase activity in cells left at 37 °C in the water bath or incubator was extremely limited (Fig. 1b). Luciferase activity reached a maximum in the 4–6 h following heat stress, and its level was proportional to the intensity of heat stress (Fig. 1a). An increase in iHSPA protein level paralleled the increase in luciferase activity (Fig. 1a), validating the luciferase transgene as a genuine reporter of the HSR. In these primary cells and in the conditions used, the maximum level of response (about 107 RLU min−1 10−4 cells) was reached after 20 to 30 min heat stress while cell death increased only beyond 60 min stress (Fig. 1b). For the rest of this study, we therefore used a 20- to 30-min-long heat stress at 44 °C (ΔT = 7 °C) to assess the level of HSR after heat shock.
Fig. 1.

Validation of the level of luciferase activity derived from the hspa1b/luciferase transgene as quantitative reporter of heat-induced heat-shock response. a Bovine cells were exposed to heat shock for different incubation times, and luciferase activity was recorded at various times post-treatment. b Luciferase activity and cell viability were recorded 5 h after various heat-shock treatments. The results shown are the mean ± SD of at least three independent experiments. c Bovine cells were exposed to 45 °C for the indicated periods of time and total soluble protein extracts prepared 5 h post-treatment. Induction of luciferase and HSP70/HSPA were analyzed by western blotting. Actin detection was used as loading control
Choice of the treatment conditions for the four types of oxidative stress
Proteotoxic stress is generally considered as a consequence of oxidative stress, but different kinds of oxidative stress do not lead to similar HSR levels. Moreover, HSPA proteins have also been reported as anti-apoptotic agents. We then aimed at establishing treatment conditions for four kinds of oxidative stress that we selected, differing by their redox potential and their mode of action in cells (Electronic Supplementary Material Table 1). H2O2 can lead to ROS production like hydroxyl ions and is able to change the redox cell potential and to increase the formation of disulfide bonds (Giorgio et al. 2007). UVA induces production of ROS, among them predominantly singlet oxygen, via photosensitization reactions (Graindorge et al. 2015). Diamide (DA) is a strong sulfhydryl reactant (Kosower and Kosower 1995) whose effect can be independent of ROS production. Finally, we examined the role of two naphtoquinones capable of producing superoxide anion by redox cycling (Klotz et al. 2014; Watanabe et al. 2004): DMNQ, which cannot conjugate with GSH and other cell components, and MN, which can both redox cycle and conjugate with GSH and protein thiols (arylation) (Klotz et al. 2014). In the bovine cells, we observed a very low DMNQ-induced HSR (data not shown) and a high MN-induced HSR (see below). Furthermore, BQ, which can only conjugate, induced a very low HSR production (data not shown). We therefore compared the effect of peroxide, UVA, MN, and DA with respect to heat stress (HS) on the level of HSR.
At first, bovine cells were exposed to various concentrations of H2O2, DA, and MN or doses of UVA radiations, according to previously described conditions (Girard et al. 2013; Graindorge et al. 2015). Both luciferase activity and cell viability were recorded, allowing us to define the maximal sub-lethal time × dose treatments for observing the highest level of HSR in these cells. The kinetics of luciferase activity followed a profile very similar to that observed after heat stress, leading us to measure luciferase activity and cell viability 5 h after the end of the stress treatment (Fig. 2). In contrast with what we observed after heat stress, the maximum level of induced luciferase activity after DA and MN treatment coincided with the maximum doses tolerated, i.e., between 0.2 and 0.3 mM for DA (Fig. 2a) and between 0.1 and 0.2 mM for MN (Fig. 2b), suggesting that HSR and cell mechanisms of defense were related. Conversely, after H2O2 treatment or UVA radiation, the level of activity per living cell was maintained but the percentage of cell death steadily increased (Fig. 2c, d), suggesting that after these treatments, either cell survival and HSR were unrelated or HSR signaling and luciferase expression followed antagonistic profiles. It is noteworthy that after these two treatments the luciferase activity was weaker than after the MN and DA treatments. The weak effect of H2O2 on HSR was not due to its rapid destruction by catalase since t-BOOH treatments yielded the same results (data not shown). From these data, we selected the treatment conditions for each type of stress that were used in the rest of this study: 0.25 mM DA for 30 min, 0.1 mM MN for 12 min, 160 kJ cm−2 of UVA radiation, and 0.3 mM H2O2 for 30 min.
Fig. 2.
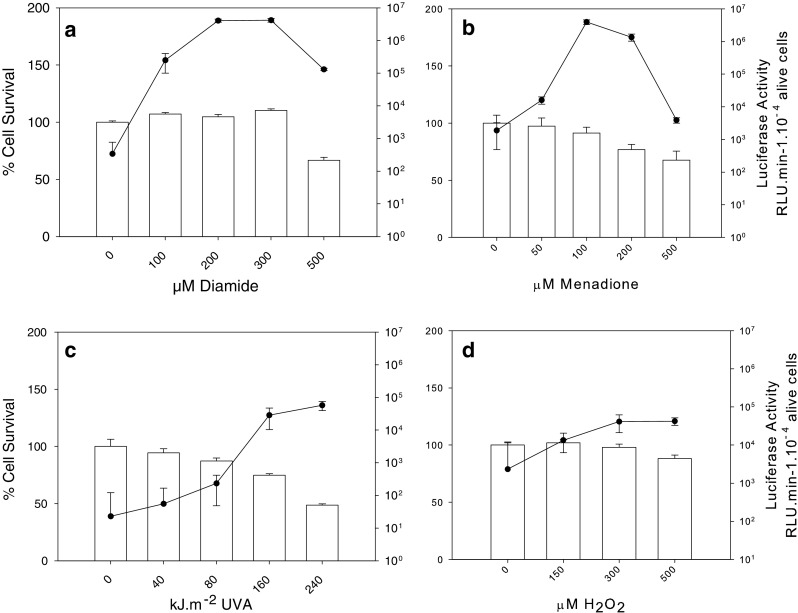
Relationship between the level of luciferase activity and cell survival following four types of oxidative stress. Each measurement was performed 5 h post-treatment. a Diamide (DA) was for 30 min, b menadione (MN) was for 12 min, c UVA irradiation (UVA) in the presence of photoactivable molecules was at various exposure times (fluency rate = 0.5 kJ m−2 s−1), d hydrogen peroxide (H2O2) was for 30 min. The results shown are the mean ± SD of at least three independent experiments. In each panel, histograms referred to percentage of survival (left y-axis) and the black dots/line to luciferase activity (right y-axis)
Induction and expression of the luciferase gene reporter parallel expression of endogenous HSP70 and HSF1 activation
Applying these different stress conditions to the primary bovine fibroblasts, we observed that DA- and MN-treated cells developed the highest level of response among the oxidative stress treatments tested in this study, while H2O2-treated cells displayed a very low level of response (Fig. 3a). The fold increase in luciferase activity induced by the four types of oxidative stress remained significantly below the level observed after heat stress (Fig. 3a). Strikingly, the difference of fold increase in luciferase activity between the treatments with low inducers (H2O2 or UVA) and each high inducer (MN or DA) was statistically significant (Fig. 3a).
Fig. 3.

Validation of the luciferase level as a quantitative reporter of oxidative stress-induced heat-shock response. Bovine cells were exposed to 0.25 mM DA for 30 min, 0.1 mM MN for 12 min, 160 kJ cm−2 of UVA radiation, 0.3 mM H2O2 for 30 min, and heat for 20 min at 44 °C. Four to five hours post-treatments, hspa1a/Luciferase activity was measured (a) and iHSPA and luciferase protein levels analyzed by western blot (b). Luciferase activity is expressed as increase fold relative to untreated samples. The blots are representative of at least five independent experiments
We checked by western blotting the change in luciferase expression in response to the different treatments and compared it with the expression of inducible HSPA/HSP70 protein. After the least efficient inducers, UVA and peroxide, an increase in iHSP70 and luciferase protein levels was barely detectable (Fig. 3b). In marked contrast, both endogenous HSP70 and inducible luciferase were detected 4 h after HS, DA, and MN treatment (Fig. 3b). The increase in luciferase after the four types of oxidative stress examined here therefore paralleled a similar increase in iHSPA protein level, at least when the fold increase was above the immunoassay detection limit.
We assessed HSF1 activation by following the stress-induced electrophoretic mobility of HSF1 since it has been shown to be one assay of HSF1 activation that is better correlated to HSR level than other assays in several cases (Park and Liu 2000; Sarge et al. 1993; Tulapurkar et al. 2009). For that, the bovine cells were exposed to HS, MN, and H2O2 as representative stresses leading to high (HS), intermediate (MN), and low (H2O2) HSR. We found a change in the electrophoretic mobility of HSF1 immediately after HS (T0), at ½ h after MN (T0.5), and almost no change after H2O2 (Electronic Supplementary Material Fig. A1a). These results therefore indicated that early changes in the electrophoretic mobility of HSF1 induced by stress were only correlated with high HSR. Alternatively, this could mean that HSF1 activation was too low to be detected after H2O2, or that H2O2-induced HSR signaling was either not or only partially dependent on HSF1 activation respectively. To discern which hypothesis was most likely, we studied HSF1 activation by monitoring its phosphorylation on Ser 326, which is a key mark of HSF1 activation (Guettouche et al. 2005). Unfortunately, HSF1 phosphorylation on Ser 326 was not detected in protein extracts from bovine cells likely due to a lack of antibody cross-reactivity with bovine specie (data not shown). Therefore, we decided to use an immortalized human fibroblast cell line, MRC5Vi. Like what was observed in bovine cells, a change in the electrophoretic mobility of HSF1 was clearly detected in soluble protein extracts after HS, to a lesser extent after MN, while it was barely detected after H2O2 (Electronic Supplementary Material Fig. A1b). HSF1 was barely detected in the chromatin-bound fraction in untreated cells (Ctr), found in it immediately after each of these treatments (T0) and its activation, as revealed by phosphorylation on Ser326, was observed as early as ½ h post-treatment (Electronic Supplementary Material Fig. A1b). These data indicated that insoluble, chromatin-associated HSF1 was activated in MRC5 cells exposed to HS and the two types of oxidative stress (Electronic Supplementary Material Fig. A1c). Together these results strongly suggest that HSF1 activation (i.e., change in electrophoretic mobility, phosphorylation on Ser346, and recruitment at the chromatin) participates in HSR signaling after these different types of stress.
Evolution of the GSH, NADH, and NADPH redox states
We then wondered if the ratio between reduced (GSH) and oxidized (GSSG) glutathione was not a determinant in oxidative stress-induced HSR, given the major role of GSH in redox cell regulation. We measured the total amount of free and oxidized GSH and calculated the ratio of [GSH]/[GSSG] after the different treatments normalized by the same ratio observed in control cells. This normalized ratio was close to 1 or even higher after heat stress, indicating that HS did not give rise to a decrease in the relative amount of intracellular GSH compared to GSSG (Fig. 4a). In marked contrast, this ratio decreased after the other treatments (Fig. 4a). The lowest values for GSH redox potential were, however, observed after MN and DA treatments, the highest being after peroxide treatment (Fig. 4a). Indeed, there was a clear trend (P = 0.008, coefficient correlation = − 0.758) that the highest levels of GSHox (GSSG) be associated with the highest fold increase in luciferase activity following oxidative stress-associated treatments (Electronic Supplementary Material Fig. A2).
Fig. 4.

Evolution of the redox potential of GSH/GSSG (a), NADP+/NADPH (b), and NAD+/NADH (c). The normalized ratios were obtained by dividing the ratio of each redox couple obtained immediately after each treatment by the one obtained in the untreated condition
Next, we dosed the redox state of two key metabolites that are involved in various biological processes including cell redox state, NADPH and NADH. Regarding the NADPH/NADP+ couple (Fig. 4b), we observed that H2O2, DA, UVA, and more clearly MN treatments tended to lead to lower normalized [NADPH]/[NADP+] ratios, indicating a decrease in the relative concentration of NADPH in favor of its oxidative form. In contrast, the NADH redox state displayed profiles of very different trends following these treatments (Fig. 4c). In particular, MN treatment resulted in various levels of oxidized NADH redox states while peroxide- and UVA-treated cells displayed a redox state about five times more reductive than control cells. No changes were observed after DA treatment (Fig. 4c). Collectively, these results suggested that after oxidative stress in the bovine primary cells, there was no correlation between HSR level and the oxidative state of the redox couple NAD+/NADH, while the redox couple NADP+/NADPH followed the same trend of oxidation as the GSH/GSSG redox couple except for DA treatment.
Levels of reactive species production following oxidative stress do not correlate with HSR
To monitor the level of reactive species produced in cells after each kind of treatment, we tested two different fluorescent probes, CM-H2DCFDA and DHR123 often utilized to demonstrate the presence of oxidative stress. CM-H2DCFDA, however, mainly react with several secondary potent oxidants derived from peroxide and peroxynitrite and may participate itself in generating radical species (Kalyanaraman et al. 2012). The same is true for DHR123 (Kalyanaraman et al. 2012). To avoid direct exposure of these probes to UVA radiation and thus to allow direct comparison between the different treatments, the probes were added immediately after each treatment. Consequently, short-lived species that may be generated during each treatment should not have been detected by this approach.
Only two treatments, UVA radiation and H2O2, gave rise to a significant change in the fluorescence intensity for both CM-H2DCFDA and DHR123, with the peak intensity being more important after UVA radiation than after H2O2 (Fig. 5a). After MN and DA treatments, we observed a slight increase in the fluorescence intensity of CM-H2DCFDA but not of DHR123 (Fig. 5a). To test the role of GSH in trapping reactive species formation, we also grew cells in the presence of the well-established inhibitor of GSH biosynthesis, BSO (Griffith 1981). After 16 to 24 h of treatment, intracellular GSH content decreased by 90% or more (Electronic Supplementary Material, Fig. A3). At first, it is important to note that decreasing the endogenous level of GSH led to endogenous ROS production that can be detected by the ROS probe CM-H2DCFDA but not DHR123 (Fig. 5a). In cells expressing a low level of GSH, we observed a dramatic increase in the level of fluorescence generated by CM-H2DCFDA and DHR123 after H2O2, while the fluorescence intensities remained similar in cells with low and normal GSH content after UVA, MN, and HS (Fig. 5a). After DA treatment, we observed a slight increase in the fluorescence intensity of CM-H2DCFDA but not of DHR123 (Fig. 5a). Collectively, the results suggested a lack of correlation between ROS production and HSR in the primary bovine cells and a major role of GSH in trapping ROS production after H2O2.
Fig. 5.

Evaluation of redox changes following the four types of oxidative stress and heat shock. Bovine primary cells were grown in the presence (+) or absence (−) of 0.5 mM BSO and treated as described in Fig. 3. Immediately post-treatment, cells were either loaded with the ROS probes CM-H2DCFDA and DHR123 (N = 3 for all, except N = 2 for CM-H2DCFDA after DA) and analyzed by flow cytometry (a) or lysed in lysis buffer without β-mercaptoethanol and the profiles of protein glutathionylation (N = 3) determined by western blot analysis (b). Shown is a representative image of the western blot analyses
Another way to assess changes in oxidant production after each treatment is to look at protein S-glutathionylation, which is the reversible catalytic formation of disulfide bridges between intracellular reduced glutathione (GSH) and reactive sulfhydryl in proteins in response to oxidative stress. Indeed, it has been suggested that a connection between HSR and GSH redox status could involve transient protein glutathionylation (PSSG) (Dandrea et al. 2002). Therefore, cells were exposed to the different treatments and protein S-glutathionylation was analyzed immediately post-treatment. The results indicated that the highest level of protein S-glutathionylation was observed after UVA and MN treatment (Fig. 5b). Conversely, DA treatment and HS resulted in barely detectable increase in glutathionylation, while peroxide treatment led to slight increase in glutathionylation compared to control cells (Fig. 5b). The specificity of the signal detected was confirmed in cells with low GSH content due to the presence of the GSH biosynthesis inhibitor BSO during cell growth (Fig. 5b).
These results indicated that the level of radical species detected by current fluorescent probes and changes to protein glutathionylation induced by the four types of oxidative stress correlated poorly with the level of HSR.
Decreasing GSH cell content yielded increased cell death and HSR level after oxidative stress except MN treatment
We thus assessed the role of GSH cell responses to the different stresses (Fig. 6). In GSHlow cells, (i.e., BSO-treated cells), we observed a rapid increase in cell death after H2O2, UVA, and DA (Fig. 6a). In contrast, the impact of low GSH cell content was much more limited after MN and was negligible after HS treatments (Fig. 6a). In the absence of stress or after heat stress, the level of luciferase remained identical in GSHlow and GSHnormal cells (Fig. 6b). Surprisingly, the level of luciferase activity dropped after MN treatment in GSHlow cells. This, however, was not confirmed by measuring the level of inducible HSP70, which was similarly induced by MN in GSHlow and GSHnormal cells (Electronic Supplementary Material, Fig. A4). On the other hand, we observed in GSHlow cells an increase in the level of luciferase activity after peroxide, UVA, and DA treatments. These data were confirmed by dosing the level of mRNA in GSHlow and GSHnormal cells (Electronic Supplementary Material, Fig. A5), indicating that the mRNA level for HSPA1, one of the stress-inducible HSPA genes, and for Luciferase increased several fold after peroxide and UVA treatments. Conversely, the mRNA level after MN treatment remained constant or decreased for HSPA1 and Luciferase respectively (Electronic Supplementary Material, Fig. A5). No amplification was observed without prior reverse transcription (not shown).
Fig. 6.

Combined effect of BSO treatment, an inhibitor of GSH biosynthesis, and oxidative stress or heat shock (conditions described in Fig. 3) on cell viability (a) and induction of luciferase (b). In a, the star indicates a significant difference for each treatment between cells cultured in the presence (black box) or absence (white boxes) of BSO. Numbers in parentheses indicate the number of values for each treatment. In b, the ratio (stress-induced luciferase activity in BSO-treated versus BSO-untreated cells) indicated whether HSR was stimulated or reduced
The level of protein polyubiquitination correlated with the level of oxidative stress-induced HSR but not the level of protein carbonylation
To determine the relationship between HSR and the rate of damaged proteins, we compared the level and profiles of polyubiquitination of total cellular proteins, which is generally considered as a good indicator of a proteostatic stress (Nivon et al. 2012) and that of carbonylated proteins since it is one of the irreversible protein damages observed following oxidative stress (Dalle-Donne et al. 2006; Nystrom 2005). On the average, the level of protein ubiquitination was higher than in control cells for all types of oxidative stress after 2 and 4 h of recovery (Fig. 7a), although these differences were significant from control cells only after MN treatment (Fig. 7b). However, the profiles of polyubiquitination at T4 were significantly different after indicated MN and DA treatments (Fig. 7b). For each ubiquitination profile, the level of HSR as measured by detecting the level of endogenous iHSPA and luciferase proteins (Fig. 7a) paralleled the results shown in Fig. 3, namely iHSPA level and luciferase activity were the highest at 4 h after the end of MN and DA treatments. For HS, the level of protein ubiquitination raised slightly at the end of the treatment (T0, Fig. 7b), and then returned to the control level after 2 or 4 h of recovery. From these experiments, we concluded that the level of protein polyubiquination correlated with the level of oxidative stress-induced HSR and not at all with the heat-induced HSR level.
The level of carbonylated proteins was evaluated in the same extracts. Unlike what was described for protein polyubiquitination, it turned out to be the highest (1.7 times higher than in control cells) in H2O2-treated and UVA-treated cells immediately after the end of the treatment period (T0) and up to 2 h after recovery (Fig. 8a). These results indicated that the level of carbonylated proteins did not correlate with the HSR level and that protein carbonylation and protein ubiquitination are two unrelated post-translational modifications induced by the different treatments used in this study.
Discussion
Genetic analyses have shown that inhibiting HSF1-mediated chaperone gene expression may lead to severe dysregulation of the cellular redox state in some tissues and cells during development (Bierkamp et al. 2010; Yan et al. 2002; Yan et al. 2005). This indicates that chaperones are crucial for some protein functions that play a role in the regulation of the cellular redox state. In turn, it suggests that cells have developed a redox regulation of chaperone expression more complex than the sole “sensing” of denatured proteins.
By providing a comprehensive comparison between different types of oxidative stress and heat shock, this work clearly demonstrated that in primary bovine fibroblast cells, two main HSR signaling pathways exist, although their kinetics were close (as evaluated by HSP70 and Luciferase protein and transcript levels): one induced by heat stress and another induced by oxidative stress. Nevertheless, the data we provided in this work and others suggest that the HSF1 pathway was likely activated by all the treatments tested, and its activation level related to the HSR level. Key HSF1 activation steps, like phosphorylation on Ser326, will, however, have to be measured in bovine cells to confirm these data.
First, in these cells, we failed to show any significant change in redox-associated processes and ROS production, after HS treatment conditions that were sufficient to saturate the HSR level, suggesting that HS does not trigger an oxidative-like stress response in primary bovine cells. Accordingly, a short exposure to the anti-oxidant NAC immediately before or during heat stress modified only marginally the level of heat-induced HSR (Girard and Lelièvre, unpublished). The level of HSR and of ROS production was not modified in cells with reduced GSH content (this work). Similar results have been found in several but not all cell types as mentioned in the “Introduction” section. Indeed, in HS-treated HeLa cells, an increase in GSH oxidation much higher than in H2O2-treated cells has been observed (Zou et al. 1998). The essential role of redox regulation in HS-induced HSR (Manalo and Liu 2001; Rokutan et al. 1996) and cell survival (Steels et al. 1992) has been shown in some cases. These observations also fit well with what has been shown for HSF1 activation in one HeLa cell line, namely that Cys residues in HSF1 proteins are key residues for the formation of HSF1 trimers transcriptionally active after HS (Ahn and Thiele 2003). It remains to be determined if the formation of intermolecular disulfide bonds between these Cys residues plays a similar role in all cells, as suggested by other published works (Lu et al. 2008; Manalo et al. 2002; Manalo and Liu 2001). Conversely, our work and those by others suggested that the well-demonstrated heat-induced protein denaturation is sufficient for HS-induced HSR (Freeman et al. 1995) even though we cannot exclude a role of local oxidation of key Cys residues in HSF1 proteins to regulate its activity (Lu et al. 2008).
Of all the types of oxidative stress examined during this work, none was able at non-toxic doses to induce a level of HSR superior or similar to the one observed after HS. Indeed, all types of oxidative stress were much less tolerated than HS. Actually, earlier work (Jacquier-Sarlin and Polla 1996) had suggested from in vitro experiments that the low level of peroxide-induced HSR observed in some cases may be explained by its double action: simultaneously to its activation due to denaturation of cellular proteins, HSF1 activity can be diminished by oxidation of some of its Cys residues. One hypothesis to consider is therefore the possibility that the absence of oxidative stress symptoms after HS warrants a high HSR level triggered by protein denaturation because oxidative stress interferes with HSR signaling.
Strikingly, the treatments leading to the highest HSR level (MN, DA, and HS) in bovine cells displayed the lowest increase in reactive oxygen species detected by utilizing the two ROS probes, CM-H2DCFDA and DHR123, while the treatments leading to the lowest HSR level (UVA radiation and H2O2) displayed the highest increase in reactive oxygen species. Furthermore, in HeLa cells, depletion of GSH by BSO treatment resulted in the formation of deleterious intramolecular HSF1 disulfide bonds after HS that block the heat-induced HSF1 DNA binding activity (Manalo and Liu 2001). These data support the view that intracellular ROS production may prevent efficient HSR. However, Yu et al. have demonstrated that the expression of heat-shock protein induced by metal ions was linearly related to the cellular superoxide anion level (Yu et al. 2006). In the present work, we showed that BSO treatment in the primary bovine cells had no consequences on HS-induced HSR and increased (UVA, H2O2, DA) or maintained (MN) HSR after the four types of oxidative stress. These data therefore do not fit with the first hypothesis since they strongly suggest that increasing intracellular ROS production or decreasing the cell content of a major anti-oxidant and redox regulator like GSH exacerbated HSR instead of reducing it.
An alternative hypothesis to consider is therefore that the HSR level is in relation with the level of protein denaturation. Differences in redox regulation might translate into a different level and nature of protein oxidation-induced denaturation. It has been recently demonstrated that iHSPA proteins participate in proteasome-mediated elimination of H2O2-induced carbonylated protein (Reeg et al. 2016). In bovine cells, the level of total carbonylated proteins was found significantly but transiently higher early after peroxide and UVA treatments, which led to the highest ROS production but the weakest HSR and protein polyubiquitination levels. These results fit with what is generally admitted, i.e., that limited oxidized proteins are degraded by 20S proteasome independently of ubiquitination (Jung and Grune 2008; Shringarpure et al. 2003). The large difference in HSR level between H2O2 and UVA treatments might be due to quantitative differences in protein carbonylation that will require more sensitive methods or to another regulation (see below). In contrast, a significant increased level of protein polyubiquitination was observed 4 h after MN and DA treatment, both treatments leading to a lower or barely detectable level of ROS production, the weakest level of carbonylation but the highest oxidative stress-induced HSR. Moreover, the kinetics observed suggest that the denatured proteins are polyubiquitinated during the recovery period following these treatments to be targeted to the ubiquitin proteasome system (UPS). Strikingly, the moderate increase in protein polyubiquitination observed immediately after heat stress in bovine cells may be rather due to transient proteasome inhibition (Pajonk et al. 2005). Our results therefore indicated that HSR and recovery-associated protein ubiquitination are correlated after some types of oxidative stress. Importantly, this is not true after HS. Different protein species are targeted by heat stress and oxidative stress as shown earlier (Freeman et al. 1995). It will be crucial to determine whether it is the rate or the nature of proteins irreversibly damaged after some types of oxidative stress that overwhelm—or inhibit—the capacity of UPS to degrade them. Clearly, other covalent modifications of proteins (Hnat et al. 2005; Jacobs and Marnett 2007) that have not been studied in this work deserve to be examined in protein extracts and in situ since they could identify key elements to better understand the relationship between HSR, unfolded and denatured proteins, and their processing.
The extent of PSSG was neither a good predictor of HSR level in bovine cells. Indeed, a review of the literature fails to show a clear, generalizable correlation between HSR level and PSSG level as postulated earlier (Dandrea et al. 2002). In HeLa cells, peroxide treatment yielded to the highest level of protein glutathionylation among the different stresses studied (Zou et al. 1998) while it was very limited in primary bovine cells (this study) and cells of the human type II lung epithelial A549 and endothelial ECV304 cell lines (Dandrea et al. 2002). In bovine cells (this study) and in HeLa cells (Zou et al. 1998), DA-induced HSR level is high but PSSG level is low. In the A549 and ECV304 human cell lines, subtoxic DA treatment—although at doses superior to the one used in bovine cells—promotes HSR at a level that correlates with PSSG level and a decrease in reduced GSH (Dandrea et al. 2002). DA-induced protein glutathionylation is, however, often observed at a concentration superior to 0.5 mM (Girard et al. 2013) or in relation to cell death (Hill et al. 2010). In addition, in bovine cells, UVA radiation gave rise to the highest extent of proteins S-glutathionylated followed by MN, which agrees with data published in CHO cells (Girard et al., 2013), while the HSR was on average 10 times more efficient after MN than UVA radiation (this study). The mechanisms of protein S-glutathionylation are not fully established (Girard unpublished results, Sanchez-Gomez et al. 2013) but it is generally considered as a mechanism of protection of protein thiols since it is rapidly reversed after moderate oxidative attack (Sanchez-Gomez et al. 2013). If PSSG is primarily a mechanism of protection that participates in reduction of non-native protein disulfide bonds formed after oxidative stress (Dalle-Donne et al. 2003), it is expected to prevent protein denaturation. This may explain why its yield of formation is not a good predictor of oxidative stress-induced HSR, at least in some cellular situations.
To understand the differences in the extent of oxidative stress-induced HSR, an alternative hypothesis is to explore the role of redox regulation. The observation that the level of cellular oxidized GSH—and to a less extent oxidized NADPH—was positively correlated with the increase in HSR level following several types of oxidative stress in bovine cells brought some support to this hypothesis. However, it cannot be the sole redox response to consider insofar as HSR in response to oxidative stress is not decreased in GSH-depleted cells, as discussed above. We cannot, however, exclude a specific glutathionylation-mediated (or thioredoxin-mediated) redox regulation of a member of the HSR pathway. Forward and reverse protein glutathionylation has been demonstrated to be catalyzed by glutaredoxins and possibly glutathione S-transferase (Sanchez-Gomez et al. 2013). PSSG may have negative or positive consequences due to inhibition of specific protein activities (Aesif et al. 2011; Chrestensen et al. 2000; Kuipers et al. 2012). The role of PSSG in enzyme regulation and DNA binding activity is now well established (Pastore and Piemonte 2012; Sanchez-Gomez et al. 2013). In addition, several examples of the role for PSSG in redox signaling have also been reported (Zhang and Forman 2012). Accordingly, the role of Cys residues in HSF1 activity that has been demonstrated after HS in HeLa cells (Ahn and Thiele 2003; Lu et al. 2008; Manalo et al. 2002; Manalo and Liu 2001) may actually be more relevant after oxidative stress in most cells.
Cell stress response is a key element in cell survival and functions, which has led to the proposal that HSPA genes belong to a group of “Vitagenes” (Calabrese et al. 2012). In this work, we present data in favor of a regulation of “oxidative stress-induced” HSR by a global change in thiol redox state since the better correlation we observed was between the oxidative state of the key cellular GSH/GSSG redox couple and HSR levels. Interestingly, the GSH redox state has been shown to be modified in other cases of HSR (Calabrese et al. 2007; Davis et al. 2014). The comparison of the effect of different quinones (DMNQ, MN, BQ) brought further support to the role of GSH. MN is unique among these molecules since it is able to both conjugate directly with glutathione and protein thiols and generate superoxide through a one-electron redox cycle when oxidized by cytochromes P450 and a few other oxidases (Giulivi and Cadenas 1994; Watanabe et al. 2004). The weak effect of DMNQ, which can only redox cycle, and BQ, which can only conjugate, is strongly in favor of a dual role of both thiol oxidation and protein denaturation to mediate the effect of oxidative stress on HSR. Strong HSR inducer reagents like DA and other natural molecules with strong sulfhydryl activity (Zhang et al. 2011) may directly react with both GSH and sensitive protein thiols, disturbing the folding of numerous proteins (Freeman et al. 1995). However, contrary to the situation observed by Freeman et al., the absence of a measurable delay between kinetics in HSR induced by HS or oxidative stress in primary bovine cells suggests strongly that transcriptional HSR response followed similar kinetics after the different types of stress, possibly because the balance between free and chaperone-associated HSF1 molecules (Jacobs and Marnett 2007) or other HSF1-associated signaling events were also rapidly altered. HSPA proteins themselves are likely targets of redox regulation (West et al. 2012). The extent of the response should most likely depend on the different redox networks and their dynamics in any given cell. In some cases, heat stress itself may translate into a redox stress (Liu et al. 2016). Some of the assays presented here could be systematically performed to be able to compare the response of different cell types and different HSR inducers. Additionally, we provided data showing that fluorescent probes largely used to establish the presence of ROS in relation with HSR are inappropriate. Emerging methods (Kalyanaraman et al. 2012) will have to be utilized to identify the primary or secondary reactive species that could be key mediators of redox changes leading to HSR. In this way, one will be able to identify the precise redox regulation active or not in the cell and its relationship with the mode of action of different HSR inducers.
Electronic supplementary material
(PPTX 697 kb)
Acknowledgements
We are indebted to Drs. Hervé Blottière (Micalis Institute, INRA) and Marie Dutreix (CNRS, Institut Curie) who brought us their support to complete this study. We also thank Jean-François Oudin and Sylvie Ruffini (BDR INRA) for part of the protein dosages and some initial tests of antibodies in western analysis respectively. We are grateful to Dr. Pascal Rigolet (Université Paris-Saclay, Institut Curie) for his help in statistical analysis.
Funding information
This work was financially supported by INRA, CNRS, and Institut Curie.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Adachi M, Liu Y, Fujii K, Calderwood SK, Nakai A, Imai K, Shinomura Y. Oxidative stress impairs the heat stress response and delays unfolded protein recovery. PLoS ONE. 2009;4:e7719. doi: 10.1371/journal.pone.0007719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aesif SW, Anathy V, Kuipers I, Guala AS, Reiss JN, Ho YS, Janssen-Heininger YM. Ablation of glutaredoxin-1 attenuates lipopolysaccharide-induced lung inflammation and alveolar macrophage activation. Am J Respir Cell Mol Biol. 2011;44:491–499. doi: 10.1165/rcmb.2009-0136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierkamp C, Luxey M, Metchat A, Audouard C, Dumollard R, Christians E. Lack of maternal Heat Shock Factor 1 results in multiple cellular and developmental defects, including mitochondrial damage and altered redox homeostasis, and leads to reduced survival of mammalian oocytes and embryos. Dev Biol. 2010;339:338–353. doi: 10.1016/j.ydbio.2009.12.037. [DOI] [PubMed] [Google Scholar]
- Bui LC et al (2009) Retrotransposon expression as a defining event of genome reprogramming in fertilized and cloned bovine embryos. Reproduction (138):289–299 [DOI] [PubMed]
- Calabrese V, et al. In vivo induction of heat shock proteins in the substantia nigra following L-DOPA administration is associated with increased activity of mitochondrial complex I and nitrosative stress in rats: regulation by glutathione redox state. J Neurochem. 2007;101:709–717. doi: 10.1111/j.1471-4159.2006.04367.x. [DOI] [PubMed] [Google Scholar]
- Calabrese V, et al. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim Biophys Acta. 2012;1822:753–783. doi: 10.1016/j.bbadis.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Chrestensen CA, Starke DW, Mieyal JJ. Acute cadmium exposure inactivates thioltransferase (glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J Biol Chem. 2000;275:26556–26565. doi: 10.1074/jbc.M004097200. [DOI] [PubMed] [Google Scholar]
- Christians E, Michel E, Adenot P, Mezger V, Rallu M, Morange M, Renard JP. Evidence for the involvement of mouse heat shock factor 1 in the atypical expression of the HSP70.1 heat shock gene during mouse zygotic genome activation. Mol Cell Biol. 1997;17:778–788. doi: 10.1128/MCB.17.2.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza A, et al. Simultaneous analysis of reactive oxygen species and reduced glutathione content in living cells by polychromatic flow cytometry. Nat Protoc. 2009;4:1790–1797. doi: 10.1038/nprot.2009.189. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. Actin S-glutathionylation: evidence against a thiol-disulphide exchange mechanism. Free Radic Biol Med. 2003;35:1185–1193. doi: 10.1016/S0891-5849(03)00504-5. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandrea T, Bajak E, Warngard L, Cotgreave IA. Protein S-glutathionylation correlates to selective stress gene expression and cytoprotection. Archives Biochemistry Biophysics. 2002;406:241–252. doi: 10.1016/S0003-9861(02)00462-9. [DOI] [PubMed] [Google Scholar]
- Davis AL et al (2014) The quinone methide aurin is a heat shock response inducer that causes proteotoxic stress and Noxa-dependent apoptosis in malignant melanoma cells. J Biological Chemistry. 10.1074/jbc.M114.592626 [DOI] [PMC free article] [PubMed]
- Dinkova-Kostova AT. The role of sulfhydryl reactivity of small molecules for the activation of the KEAP1/NRF2 pathway and the heat shock response. Scientifica. 2012;2012:606104. doi: 10.6064/2012/606104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulias P-T, et al. Involvement of heat shock protein-70 in the mechanism of hydrogen peroxide-induced DNA damage: the role of lysosomes and iron free radical. Biology Medicine. 2007;42:567–577. doi: 10.1016/j.freeradbiomed.2006.11.022. [DOI] [PubMed] [Google Scholar]
- Figueiredo-Pereira ME, Yakushin S, Cohen G. Disruption of the intracellular sulfhydryl homeostasis by cadmium-induced oxidative stress leads to protein thiolation and ubiquitination in neuronal cells. J Biological Chemistry. 1998;273:12703–12709. doi: 10.1074/jbc.273.21.12703. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman ML, Borrelli MJ, Syed K, Senisterra G, Stafford DM, Lepock JR. Characterization of a signal generated by oxidation of protein thiols that activates the heat shock transcription factor. J Cell Physiol. 1995;164:356–366. doi: 10.1002/jcp.1041640216. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Prahlad V, Morimoto RI. The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol. 2011;3:3. doi: 10.1101/cshperspect.a009704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilda JE, Gomes AV. Stain-free total protein staining is a superior loading control to β-actin for Western blots. Analytical Biochemistry. 2013;440:186–188. doi: 10.1016/j.ab.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- Girard PM, Graindorge D, Smirnova V, Rigolet P, Francesconi S, Scanlon S, Sage E. Oxidative stress in mammalian cells impinges on the cysteines redox state of human XRCC3 protein and on its cellular localization. PLoS One. 2013;8:e75751. doi: 10.1371/journal.pone.0075751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard PM, Arbabian A, Fleury M, Bauville G, Puech V, Dutreix M, Sousa JS. Synergistic effect of H2O2 and NO2 in cell death induced by cold atmospheric He plasma. Scientific Reports. 2016;6:29098. doi: 10.1038/srep29098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulivi C, Cadenas E. One- and two-electron reduction of 2-methyl-1,4-naphthoquinone bioreductive alkylating agents: kinetic studies, free-radical production, thiol oxidation and DNA-strand-break formation. Biochem J. 1994;301(Pt 1):21–30. doi: 10.1042/bj3010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go YM, Gipp JJ, Mulcahy RT, Jones DP. H2O2-dependent activation of GCLC-ARE4 reporter occurs by mitogen-activated protein kinase pathways without oxidation of cellular glutathione or thioredoxin-1. J Biol Chem. 2004;279:5837–5845. doi: 10.1074/jbc.M307547200. [DOI] [PubMed] [Google Scholar]
- Goossens K, Van Poucke M, Van Soom A, Vandesompele J, Van Zeveren A, Peelman LJ. Selection of reference genes for quantitative real-time PCR in bovine preimplantation embryos. BMC Dev Biol. 2005;5:27. doi: 10.1186/1471-213X-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nature Rev Drug Discovery. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Gosslau A, Ruoff P, Mohsenzadeh S, Hobohm U, Rensing L (2001) Heat shock and oxidative stress-induced exposure of hydrophobic protein domains as common signal in the induction of hsp68. J Biol Chem 276:1814-1821 doi:10.1074/jbc.M008280200 [DOI] [PubMed]
- Graindorge D, Martineau S, Machon C, Arnoux P, Guitton J, Francesconi S, Frochot C, Sage E, Girard PM. Singlet oxygen-mediated oxidation during UVA radiation alters the dynamic of genomic DNA replication. PLoS One. 2015;10:e0140645. doi: 10.1371/journal.pone.0140645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith OW. Depletion of glutathione by inhibition of biosynthesis. Methods Enzymol. 1981;77:59–63. doi: 10.1016/S0076-6879(81)77011-3. [DOI] [PubMed] [Google Scholar]
- Guettouche T, Boellmann F, Lane WS, Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC biochemistry. 2005;6:4. doi: 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8(2):R19 [DOI] [PMC free article] [PubMed]
- Hill BG, Higdon AN, Dranka BP, Darley-Usmar VM. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim Biophys Acta. 2010;1797:285–295. doi: 10.1016/j.bbabio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnat MD, Meadows JW, Brockman DE, Pitzer B, Lyall F, Myatt L. Heat shock protein-70 and 4-hydroxy-2-nonenal adducts in human placental villous tissue of normotensive, preeclamptic and intrauterine growth restricted pregnancies. Am J Obstetrics Gynecology. 2005;193:836–840. doi: 10.1016/j.ajog.2005.01.059. [DOI] [PubMed] [Google Scholar]
- Horowitz M, Assadi H. Heat acclimation-mediated cross-tolerance in cardioprotection: do HSP70 and HIF-1alpha play a role? Ann N Y Acad Sci. 2010;1188:199–206. doi: 10.1111/j.1749-6632.2009.05101.x. [DOI] [PubMed] [Google Scholar]
- Jacobs AT, Marnett LJ. Heat shock factor 1 attenuates 4-hydroxynonenal-mediated apoptosis: critical role for heat shock protein 70 induction and stabilization of Bcl-XL. J Biol Chem. 2007;282:33412–33420. doi: 10.1074/jbc.M706799200. [DOI] [PubMed] [Google Scholar]
- Jacquier-Sarlin MR, Polla BS. Dual regulation of heat-shock transcription factor (HSF) activation and DNA-binding activity by H2O2: role of thioredoxin. Biochem J. 1996;318(Pt 1):187–193. doi: 10.1042/bj3180187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger AM, Makley LN, Gestwicki JE, Thiele DJ. Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J Biol Chem. 2014;289:30459–30469. doi: 10.1074/jbc.M114.591578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP, Sies H. The Redox Code Antioxid Redox Signal. 2015;23:734–746. doi: 10.1089/ars.2015.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP, Go YM, Anderson CL, Ziegler TR, Kinkade JM, Jr, Kirlin WG. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. Faseb J. 2004;18:1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- Jung T, Grune T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life. 2008;60:743–752. doi: 10.1002/iub.114. [DOI] [PubMed] [Google Scholar]
- Kalyanaraman B, et al. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic biol Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Klotz LO, Hou X, Jacob C. 1,4-Naphthoquinones: from oxidative damage to cellular and inter-cellular signaling. Molecules. 2014;19:14902–14918. doi: 10.3390/molecules190914902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosower NS, Kosower EM. Diamide: an oxidant probe for thiols. Methods Enzymol. 1995;251:123–133. doi: 10.1016/0076-6879(95)51116-4. [DOI] [PubMed] [Google Scholar]
- Kuipers I, Bracke KR, Brusselle GG, Wouters EF, Reynaert NL. Smoke decreases reversible oxidations S-glutathionylation and S-nitrosylation in mice. Free Radic Res. 2012;46:164–173. doi: 10.3109/10715762.2011.647011. [DOI] [PubMed] [Google Scholar]
- Ladner CL, Yang J, Turner RJ, Edwards RA. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal Biochem. 2004;326:13–20. doi: 10.1016/j.ab.2003.10.047. [DOI] [PubMed] [Google Scholar]
- Lelievre JM, Le Bourhis D, Breton A, Hayes H, Servely JL, Vignon X. Heat-induced and spontaneous expression of Hsp70.1Luciferase transgene copies localized on Xp22 in female bovine cells. BMC Res Notes. 2010;3:17. doi: 10.1186/1756-0500-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelièvre JM, Peynot N, Ruffini S, Laffont L, Le Bourhis D, Girard PM, Duranthon V (2016) Regulation of heat-inducible HSPA1A gene expression during maternal-to-embryo transition and in response to heat in in vitro-produced bovine embryos. Reprod Fertil Dev doi:10.1071/RD15504 [DOI] [PubMed]
- Liu H, Lightfoot R, Stevens JL. Activation of heat shock factor by alkylating agents is triggered by glutathione depletion and oxidation of protein thiols. J Biol Chem. 1996;271:4805–4812. doi: 10.1074/jbc.271.9.4805. [DOI] [PubMed] [Google Scholar]
- Liu F, et al. Selenium and vitamin E together improve intestinal epithelial barrier function and alleviate oxidative stress in heat-stressed pigs. Exp Physiol. 2016;101:801–810. doi: 10.1113/EP085746. [DOI] [PubMed] [Google Scholar]
- Lu M et al. (2008) Two distinct disulfide bonds formed in human heat shock transcription factor 1 act in opposition to regulate its DNA binding activity. Biochemistry 47:6007-6015 doi:10.1021/bi702185u [DOI] [PubMed]
- Mahmood Q, Irshad M, Hussain J, Dinkova-Kostova AT (2012) The role of sulfhydryl reactivity of small molecules for the activation of the KEAP1/NRF2 pathway and the heat shock response. BioMed Research International 2012:606104 doi:10.1155/2014/56413610.6064/2012/606104 [DOI] [PMC free article] [PubMed]
- Manalo DJ, Liu AY. Resolution, detection, and characterization of redox conformers of human HSF1. J Biol Chem. 2001;276:23554–23561. doi: 10.1074/jbc.M011300200. [DOI] [PubMed] [Google Scholar]
- Manalo DJ, Lin Z, Liu AY. Redox-dependent regulation of the conformation and function of human heat shock factor 1. Biochemistry. 2002;41:2580–2588. doi: 10.1021/bi0159682. [DOI] [PubMed] [Google Scholar]
- McDuffee AT, et al. Proteins containing non-native disulfide bonds generated by oxidative stress can act as signals for the induction of the heat shock response. J Cell Physiol. 1997;171:143–151. doi: 10.1002/(SICI)1097-4652(199705)171:2<143::AID-JCP4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Menck M, Mercier Y, Campion E, Lobo RB, Heyman Y, Renard JP, Thompson EM. Prediction of transgene integration by noninvasive bioluminescent screening of microinjected bovine embryos. Transgenic Res. 1998;7:331–341. doi: 10.1023/A:1008841222138. [DOI] [PubMed] [Google Scholar]
- Nivon M, Abou-Samra M, Richet E, Guyot B, Arrigo AP, Kretz-Remy C (2012) NF-kappaB regulates protein quality control after heat stress through modulation of the BAG3-HspB8 complex, J Cell Sci. 125:1141–1151. 10.1242/jcs.091041 [DOI] [PubMed]
- Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24:1311–1317. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajonk F, van Ophoven A, WH MB. Hyperthermia-induced proteasome inhibition and loss of androgen receptor expression in human prostate cancer cells. Cancer Res. 2005;65:4836–4843. doi: 10.1158/0008-5472.CAN-03-2749. [DOI] [PubMed] [Google Scholar]
- Park J, Liu AY. Pervanadate induces the hyperphosphorylation but not the activation of human heat shock factor 1. J Cell Physiol. 2000;185:348–357. doi: 10.1002/1097-4652(200012)185:3<348::AID-JCP5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Pastore A, Piemonte F. S-Glutathionylation signaling in cell biology: progress and prospects. Eur J Pharm Sci. 2012;46:279–292. doi: 10.1016/j.ejps.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Pias EK, Aw TY. Apoptosis in mitotic competent undifferentiated cells is induced by cellular redox imbalance independent of reactive oxygen species production. Faseb J. 2002;16:781–790. doi: 10.1096/fj.01-0784com. [DOI] [PubMed] [Google Scholar]
- Reeg S, Jung T, Castro JP, Davies KJ, Henze A, Grune T. The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic Biol Med. 2016;99:153–166. doi: 10.1016/j.freeradbiomed.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokutan K, Hirakawa T, Teshima S, Honda S, Kishi K. Glutathione depletion impairs transcriptional activation of heat shock genes in primary cultures of Guinea pig gastric mucosal cells. J Clin Invest. 1996;97:2242–2250. doi: 10.1172/JCI118665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph TK, Freeman BA (2009) Transduction of redox signaling by electrophile-protein reactions. Sci Signal 2:re7. 10.1074/jbc.M110.152686 10.1126/scisignal.290re7 [DOI] [PMC free article] [PubMed]
- Sagirkaya H, Misirlioglu M, Kaya A, First NL, Parrish JJ, Memili E. Developmental and molecular correlates of bovine preimplantation embryos. Reproduction. 2006;131:895–904. doi: 10.1530/rep.1.01021. [DOI] [PubMed] [Google Scholar]
- Sanchez-Gomez FJ, Espinosa-Diez C, Dubey M, Dikshit M, Lamas S. S-glutathionylation: relevance in diabetes and potential role as a biomarker. Biol Chem. 2013;394:1263–1280. doi: 10.1515/hsz-2013-0150. [DOI] [PubMed] [Google Scholar]
- Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol. 1993;13:1392–1407. doi: 10.1128/MCB.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senisterra GA, Huntley SA, Escaravage M, Sekhar KR, Freeman ML, Borrelli M, Lepock JR. Destabilization of the Ca2+-ATPase of sarcoplasmic reticulum by thiol-specific, heat shock inducers results in thermal denaturation at 37 degrees C. Biochemistry. 1997;36:11002–11011. doi: 10.1021/bi9711590. [DOI] [PubMed] [Google Scholar]
- Shang F, Taylor A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic Biol Med. 2011;51:5–16. doi: 10.1016/j.freeradbiomed.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shringarpure R, Grune T, Mehlhase J, Davies KJ. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem. 2003;278:311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- Steels EL, Watson K, Parsons PG. Relationships between thermotolerance, oxidative stress responses and induction of stress proteins in human tumour cell lines. Biochemical Pharmacology. 1992;44:2123–2129. doi: 10.1016/0006-2952(92)90338-J. [DOI] [PubMed] [Google Scholar]
- Taylor A, Shang F, Nowell T, Galanty Y, Shiloh Y. Ubiquitination capabilities in response to neocarzinostatin and H(2)O(2) stress in cell lines from patients with ataxia-telangiectasia. Oncogene. 2002;21:4363–4373. doi: 10.1038/sj.onc.1205557. [DOI] [PubMed] [Google Scholar]
- Tulapurkar ME, Asiegbu BE, Singh IS, Hasday JD. Hyperthermia in the febrile range induces HSP72 expression proportional to exposure temperature but not to HSF-1 DNA-binding activity in human lung epithelial A549 cells. Cell Stress Chaperones. 2009;14:499–508. doi: 10.1007/s12192-009-0103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol. 2010;2:a004390. doi: 10.1101/cshperspect.a004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallen ES, Buettner GR, Moseley PL. Oxidants differentially regulate the heat shock response. Int J Hyperth. 1997;13:517–524. doi: 10.3109/02656739709023550. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Dickinson DA, Liu RM, Forman HJ. Quinones and glutathione metabolism. Methods Enzymol. 2004;378:319–340. doi: 10.1016/S0076-6879(04)78024-6. [DOI] [PubMed] [Google Scholar]
- West JD, Wang Y, Morano KA (2012) Small molecule activators of the heat shock response: chemical properties, molecular targets, and therapeutic promise. Chem Res Toxicol 25:2036–2053. 10.1007/s12192-012-0375-x 10.1021/tx300264x [DOI] [PMC free article] [PubMed]
- Westerheide SD, Raynes R, Powell C, Xue B, Uversky VN. HSF transcription factor family, heat shock response, and protein intrinsic disorder. Curr Protein Pept Sci. 2012;13:86–103. doi: 10.2174/138920312799277956. [DOI] [PubMed] [Google Scholar]
- Wirth D, Christians E, Munaut C, Dessy C, Foidart JM, Gustin P. Differential heat shock gene hsp70-1 response to toxicants revealed by in vivo study of lungs in transgenic mice. Cell Stress Chaperones. 2002;7:387–395. doi: 10.1379/1466-1268(2002)007<0387:DHSGHR>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002;21:5164–5172. doi: 10.1093/emboj/cdf528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, Rajasekaran NS, Sathyanarayanan S, Benjamin IJ. Mouse HSF1 disruption perturbs redox state and increases mitochondrial oxidative stress in kidney. Antioxid Redox Signal. 2005;7:465–471. doi: 10.1089/ars.2005.7.465. [DOI] [PubMed] [Google Scholar]
- Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- Yu Z, Yang X, Wang K. Metal ions induced heat shock protein response by elevating superoxide anion level in HeLa cells transformed by HSE-SEAP reporter gene. Toxicology. 2006;223:1–8. doi: 10.1016/j.tox.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Zhang H, Forman HJ. Glutathione synthesis and its role in redox signaling. Semin Cell Dev Biol. 2012;23:722–728. doi: 10.1016/j.semcdb.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ahn YH, Benjamin IJ, Honda T, Hicks RJ, Calabrese V, Cole PA, Dinkova-Kostova AT. HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem Biol. 2011;18:1355–1361. doi: 10.1016/j.chembiol.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Salminen WF, Roberts SM, Voellmy R. Correlation between glutathione oxidation and trimerization of heat shock factor 1, an early step in stress induction of the Hsp response. Cell Stress Chaperones. 1998;3:130–141. doi: 10.1379/1466-1268(1998)003<0130:CBGOAT>2.3.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PPTX 697 kb)