

Abstract
The importance of interspecific introgression as a source of adaptive variation is increasingly recognized. Theory predicts that beneficial genetic variants cross species boundaries easily even when interspecific hybridization is rare and gene flow is strongly constrained throughout the genome. However, it remains unclear whether certain classes of genes are particularly prone to adaptive introgression. Genes affected by balancing selection (BS) may constitute such a class, because forms of BS that favour novel, initially rare alleles, should facilitate introgression. We tested this hypothesis in hybridizing newts by comparing 13 genes with signatures of BS, in particular an excess of common non-synonymous polymorphisms, to the genomic background (154 genes). Parapatric hybridizing taxa were less differentiated in BS candidate genes than more closely related allopatric lineages, while the opposite was observed in the control genes. Coalescent and forward simulations that explored neutral and BS scenarios under isolation and migration showed that processes other than differential gene flow are unlikely to account for this pattern. We conclude that BS, probably involving a form of novel allele advantage, promotes introgression. This mechanism may be a source of adaptively relevant variation in hybridizing species over prolonged periods.
Keywords: adaptive introgression, amphibians, balancing selection, hybridization
1. Introduction
Taxa capable of hybridization may benefit from genetic exchange, because they have access to a larger pool of genetic variation than would have been available if they had evolved independently. This effectively shared variation may provide the raw material for selection and thus accelerate adaptation [1]. In contrast to neutral variants or to alleles decreasing the fitness of hybrids, introgression of beneficial alleles is expected to proceed relatively unimpeded until reproductive isolation is complete or gene flow ceases owing to geographical isolation [2,3]. This view is supported by mounting empirical evidence for adaptive introgression between hybridizing species [4,5], including that from extinct hominins to our own species [6]. It remains unclear, however, whether certain classes of genes are particularly prone to adaptive introgression. Genes under balancing selection (BS) may constitute such a class.
BS is a general term that encompasses forms of selection that maintain variation within populations [7]. This definition, adopted here, does not include divergent selection causing local adaptation, which maintains variation at the species, but not at the population level. Several mechanisms of BS confer selective advantage to novel or rare alleles, which is expected to facilitate introgression, because alleles acquired via hybridization would less often be lost by chance and more likely to become established [8]. This effect may be further amplified by the mechanism of divergent allele advantage [9]. Contrary to universally beneficial variants under positive selection, introgressed variants under BS usually do not reach fixation, thereby increasing the amount of polymorphism shared between species. However, not all mechanisms of BS are equally likely to promote introgression. For example, while overdominance may do so [8], as introgressed alleles will initially be found predominantly in heterozygotes, the effect would diminish with increasing variation in the recipient species, because resident alleles will also occur mostly in heterozygotes. Other forms of BS, owing to intralocus sexual conflict, antagonistic pleiotropy or fluctuating selection pressures, may also affect the rate of introgression, but their effects are poorly understood.
Stronger introgression of genes affected by BS operating via negative frequency dependence (NFD), compared to neutral genes, is supported by both analytical theory and computer simulations [8,10,11], while scant empirical evidence comes from self-incompatibility (SI) genes in Arabidopsis [12] and major histocompatibility complex (MHC) genes in humans [13], alpine ibex [14] and newts [15]. However, a comprehensive test comparing introgression of multiple putative targets of BS to the genomic average is lacking. Here we present the results of such a test, employing BS candidates identified among immune genes. Genes involved in immune response are primary targets of BS because coevolution with pathogens favours novel variants and leads to the maintenance of numerous alleles [16].
Because BS slows down differentiation by opposing drift, ancestral variation in and around targets of BS is shared for a longer time than elsewhere in the genome even between isolated species [17], resulting sometimes in extensive trans-species polymorphism [18]. This complicates comparisons of the extent of introgression between targets of BS and other genes [4,7]. One way to disentangle the effects of introgression and BS is to compare taxa, in which patterns of relationships and gene flow allow these two sources of similarity to be distinguished between. European newts of the Lissotriton vulgaris species complex offer such opportunity. Here we consider six evolutionary lineages within this complex in three geographical groups: Mon, North and South (figure 1). Two groups, North and South, currently allopatric and with little evidence for historical gene flow [19] are more closely related, while the parapatric and more distantly related Mon and North hybridize, but genome-wide nuclear introgression between them has been limited [20,21]. Consequently, we expect (figure 1) that the pair North–South will exhibit the lowest genome-wide differentiation, followed by Mon–North and Mon–South. By contrast, if genes under BS introgress more easily, we expect the lowest differentiation between the groups exchanging genes (Mon–North). We test these predictions using two classes of genes: 13 BS candidates and 154 randomly selected control (CR) genes. The former comprise: (i) 11 single-copy genes identified as putative targets of recent BS (BS11) in a genomic scan involving more than 600 immune genes [22], (ii) multilocus MHC class I, evolving under strong, long-term BS, and (iii) TAP1, a gene that is tightly linked to MHC and shows signatures of BS.
Figure 1.

Sampling and predictions. (a) Sampling sites and distribution of six lineages of the Lissotriton vulgaris species complex used in the study: L. graecus (Lg), L. kosswigi (Lk), L. montandoni (Lm), L. schmidtleri (Ls), L. v. ampelensis (Lva), northern L. v. vulgaris (NLvv); the lineages are arranged into three geographical groups: North, South and Mon; the distribution of the L. vulgaris complex lineages not included in the study is in pale green; coloured dots—localities with data for both MHC and other genes, small black dots—localities where only MHC was investigated; (b) the TreeMix drift tree depicting relationships among the six lineages [19]; Mon and North, as well as lineages within North have been connected by historical gene flow (double-headed arrows); and (c) expected differentiation between the three groups for genes under balancing selection (BS, orange) and for control genes (CR, blue) under the assumption that BS facilitates introgression; the groups are at vertices and edge lengths reflect the relative differentiation in pairwise comparisons; differentiation is determined by retention of ancestral polymorphism (AP) caused by neutral or selective processes and introgression (I); arrow thicknesses show contributions of AP and I; inequalities show the expected FST rankings. (Online version in colour.)
2. Methods
(a). Samples
A total of 28 (35 for MHC class I) populations (15–20 individuals per population) were sampled within the ranges of six evolutionary lineages in the L. vulgaris species complex (figure 1; electronic supplementary material, table S1). The lineages were assigned to three groups based on their geographical distribution: Mon, North and South (figure 1). Mon comprises a single lineage, Lissotriton montandoni (Lm, eight populations), North consists of two smooth newt lineages: northern Lissotriton vulgaris vulgaris (NLvv, 4) and Lissotriton vulgaris ampelensis (Lva, 4), whereas South includes three lineages: Lissotriton graecus (Lg, 4), Lissotriton kosswigi (Lk, 4) and Lissotriton schmidtleri (Ls, 4). Additionally, 13 Lissotriton helveticus (Lh) individuals served as an outgroup to estimate mutation rates and identify ancestral variants. Newts were sampled during the breeding season by dip-netting; tailtip biopsies were taken and the animals were released afterwards. Tissues were stored in 95% ethanol and DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega).
(b). Re-sequencing of balancing selection candidates and control genes
Eleven single-copy putative targets of BS, or BS candidates (BS11), were reported previously [22]. BS11 genes were identified using two composite tests looking for: (i) an excess of non-synonymous polymorphisms segregating at intermediate frequencies, and (ii) an elevated ratio of polymorphism to divergence and an excess of polymorphisms segregating at high frequencies. Nine out of eleven BS11 were identified using the former test, which compares non-synonymous and synonymous sites and should be thus resistant to the effect of introgression [7]. No targets of long-term BS were found among BS11 genes and the overall strength of evidence for BS was moderate. Most BS11 genes are thus probably targets of weak, transient or recent BS, and this set of candidates may also contain false positives, such as cases of relaxed purifying selection. Nevertheless, using this set of BS candidates to test the prediction of the introgression hypothesis is warranted, as the main effect of the complexities mentioned above would be a reduction of power, making the test more conservative. TAP1 was also identified in [22] as a putative target of BS, but was not analysed in detail owing to suspected gene duplication. Further analyses revealed that TAP1 is largely single-copy as haplotypes carrying two gene copies are rare (see the electronic supplementary material, results). We therefore included TAP1 here, but consider it separately from BS11 because tight linkage may have caused non-independence between TAP1 and MHC class I (see the electronic supplementary material, methods and results). The genomic background was represented by 178 CR genes randomly selected from the transcriptome (http://newtbase.eko.uj.edu.pl/ [22]). The BS11 and CR genes were re-sequenced on the Illumina platform using molecular inversion probes (MIPs) following an established protocol [23]. Each MIP targeted 112 bp of exon sequence; details of MIP design are given elsewhere [22]. For the BS11 genes we attempted to re-sequence as much coding sequence as possible (97 MIPs covering a total of 9.3 kbp), while for the CR genes one to three MIPs per gene were used (230 MIPs covering a total of 25.8 kbp). To estimate genotyping concordance 14 individuals were analysed in two replicates. Variation in TAP1 and MHC class I was examined with high-throughput amplicon sequencing (details in the electronic supplementary material, methods). To map MHC class I, we genotyped it in one of the families used for the construction of a linkage map [24].
BS11 and CR reads were mapped to reference transcripts using bwa-mips (https://github.com/brentp/bwa-mips) or bwa-mips combined with Bowtie 2 [25] (details in the electronic supplementary material, methods). Single nucleotide polymorphism (SNP) calling, quality filtering and tests of the Hardy–Weinberg expectations were performed in GATK [26] separately for each population. Genotyping concordance estimated from samples run in duplicates was 0.96. Twenty-four CR genes showing an excess of heterozygotes at the false discovery rate of 0.05 in any population were flagged as paralogues and excluded from further analyses, leaving 154 single-copy CR genes and all BS11 genes (electronic supplementary material, table S2). One hundred and fifty three out of 154 CR and 10 out of 11 BS11 genes as well as TAP1 were located on a linkage map [24] (electronic supplementary material, figure S1). The bioinformatics procedures are detailed in the electronic supplementary material, methods.
Mstatspop (https://bioinformatics.cragenomica.es/numgenomics/people/sebas/software/software.html) was used to calculate the following statistics: nucleotide diversity (π), number of segregating variants (S), shared variants (Ss), derived variants segregating in one but fixed in another population (Sxf), dxy, relative node depth (RND), Tajima's D, the Hudson et al. [27] estimate of FST and divergence (dxo) from the outgroup (Lh). Mutation rate was calculated for each gene using the average dxo between Lh and the six lineages within the L. vulgaris species complex, and assuming a split of Lh 4.6 × 106 generations ago [28].
MHC class I genes were genotyped using the adjustable clustering method [29,30] (see the electronic supplementary material, methods). MHC class I consists of multiple genes, which may share similar alleles and show copy number variation between haplotypes (see the electronic supplementary material, results). Therefore, to calculate FST we scored presence/absence of alleles within individuals, encoded each allele as a bi-allelic locus and calculated FST in Arlequin [31] using the number of pairwise differences between individuals as the measure of genetic distance. Such FST values are not directly comparable with those calculated for Mendelian markers. However, in this study we did not compare FST values but only rankings of FST Mon–North, FST Mon–South, FST North–South between categories of genes (figure 1). Pairwise FST matrices were calculated with all MHC class I alleles and also including only putative functional classical alleles (HEX, see the electronic supplementary material, Methods). TAP1 genotyping is described in the electronic supplementary material, methods. Because TAP1 is mostly a single-copy gene, FST was calculated as for BS11 and CR genes, excluding individuals with more than two alleles in any exon.
(c). Testing predictions of the introgression hypothesis and robustness of results
Pairwise FST values between groups (Mon, North, South) were calculated as the average of relevant entries in the matrix of pairwise FST between all populations, separately for BS11, MHC class I, TAP1 and CR. Significance of FST differences between group pairs was tested with 104 randomizations (reshuffling of population labels in the matrix of pairwise FST). The FST matrices were visualized using two-dimensional scaling with the cmdscale() function in R [32]. Percentage of variation explained by each principal coordinate was calculated by dividing the corresponding eigenvalue by the sum of the absolute values of all eigenvalues.
We then asked how likely is the reversal of FST ranking (expected under stronger introgression, figure 1) of the magnitude observed for BS11 under several historical scenarios that do or do not allow for BS that could cause differential introgression. The distributions of pairwise between-group FST under each scenario were obtained through simulations conditioned on the previously inferred [19,21] demographic history (divergence times between lineages, effective population sizes (Ne) and migration rates were estimated from sequences of multiple nuclear genes with approximate Bayesian computations) and tailored to the characteristics of BS11 and CR genes, such as mutation rate, sample size and the length of the assayed fragments. For each simulation we recorded the statistic diffFST = (FST Mon-North − FST North–South)/mean(FST Mon–North; FST North–South). Both isolation and gene flow assuming realistic migration parameters were modelled for the following scenarios: (i) neutral demography, (ii) neutral demography with Ne increased to obtain variation comparable to that in BS11 genes, (iii) BS operating via symmetric overdominance (SOD), and (iv) BS operating via NFD. Scenario (i) was explored with coalescent simulations in ms [33] using parameter values listed in the electronic supplementary material, table S3, including three migration rates that spanned the range estimated in a previous study [21]. Scenario (ii), which tested whether the reversal of FST ranking could be simply owing to higher variation in BS11 genes, was investigated by increasing the Ne of all extant and ancestral lineages (NeBS11/NeCR = 6.8 inferred from the observed nucleotide diversities). In scenario (iii) SOD was modelled with forward simulations using two strengths of heterozygote advantage, 0.05 and 0.02. Scenario (iv) modelled NFD with forward simulations using three strengths of selection (initial rare allele advantage 0.5, 0.05, 0.02). All forward simulations were performed in SLiM 2 [34], explored two migration rates and used demographic parameters rescaled by a factor of 100 to speed up computations (electronic supplementary material, table S3). We verified that the results for the neutral scenarios obtained using ms and SLiM 2 with rescaled parameters were virtually identical. Details of the simulations are given in the electronic supplementary material, methods.
3. Results
Variation of BS11, CR and TAP1 genes was examined in 28 populations and that of MHC class I in 35 populations (figure 1; electronic supplementary material, table S1). Both BS11 and CR genes are scattered over multiple chromosomes, while TAP1 and MHC class I are tightly linked (less than or equal to 0.5 cM) on linkage group 1 (electronic supplementary material, figure S1). In BS11 genes nucleotide diversity (π) was on average 6.8 times higher than in CR genes (table 1), despite only a slightly higher fraction of segregating sites and mutation rate (SBS11/SCR = 1.18, μBS11/μCR = 1.10). Higher π in BS11 genes results from polymorphisms segregating at higher frequencies, reflected in more positive values of Tajima's D compared to CR genes. The fraction of polymorphisms shared between the three geographical groups is similar for BS11 and CR genes (table 1).
Table 1.
Variation in 11 single-copy balancing selection candidates (BS11), TAP1, a target of BS tightly linked to MHC, and 154 control (CR) genes. (Means ± s.d. are given; L, re-sequenced length in bp; S, number of segregating sites per site in pooled sequences from all populations; π, nucleotide diversity per site, average of population means; μ, mutation rate per site per generation; Tajima's D, average of population means; Ss, number of shared polymorphisms per site in pairs of population groups; Sxf, fraction of sites with the derived variant fixed in one population group and polymorphic in another, summed for pairs of population groups.)
| BS11 | TAP1 | CR | |
|---|---|---|---|
| L | 672 ± 353 | 734 | 136 ± 65 |
| S | 0.13 ± 0.05 | 0.26 | 0.11 ± 0.05 |
| π (%) | 0.17 ± 0.07 | 1.46 | 0.02 ± 0.01 |
| μ | 2.6 × 10−9 ± 1.4 × 10−9 | 3.5 × 10−9 | 2.4 × 10−9 ± 1.5 × 10−9 |
| Tajima's D | 0.12 ± 0.42 | 0.38 | −0.01 ± 0.17 |
| Ss (%) North–South | 2.0 ± 2.0 | 6.5 | 2.0 ± 1.5 |
| Ss (%) Mon–North | 2.0 ± 1.9 | 12.3 | 2.1 ± 1.5 |
| Ss (%) Mon–South | 0.8 ± 0.9 | 6.2 | 1.0 ± 1.1 |
| Sxf (%) North–South | 0.25 ± 0.25 | 0.40 | 0.25 ± 0.47 |
| Sxf (%) Mon–North | 0.28 ± 0.22 | 0.13 | 0.20 ± 0.44 |
| Sxf (%) Mon–South | 0.39 ± 0.31 | 0.54 | 0.29 ± 0.46 |
MHC class I consists of several genes (the number differs between haplotypes), not all are expressed at high levels (see the electronic supplementary material, results). MHC class I is highly variable, with a total of 899 alleles and more than 100 alleles detected in some populations (electronic supplementary material, table S1 and figures S2, S3). A strong signal of positive selection (electronic supplementary material, table S4) combined with extreme allelic and sequence variation (electronic supplementary material, tables S1 and S5) indicate long-term BS on MHC class I. While less than 5% of alleles were shared in pairs Mon–South (4.0%) and North–South (4.3%), as much as 44.4% alleles were shared by Mon and North (see also the electronic supplementary material, table S6). Tightly linked to MHC, TAP1 is also highly polymorphic (table 1), with on average 73.8 alleles per exon. High polymorphism together with a signal of positive selection (see the electronic supplementary material, results) suggest the action of BS on TAP1. Similarly as in MHC, a much higher fraction of TAP1 alleles is shared by Mon and North (47.8%) than in the two remaining pairs (Mon–South: 10.0%, North–South: 12.4%; see also the electronic supplementary material, table S6).
We tested the predictions outlined in figure 1 by comparing genetic differentiation between population groups using FST. The ranking of FST values differs between the BS candidates and CR genes (figure 2; electronic supplementary material, figure S4 and table S7) in a way consistent with easier introgression of BS candidates (figure 1), i.e. in the BS candidates FST is the lowest between hybridizing groups, while in the CR genes FST is the lowest between more closely related groups. In BS11 genes, Mon–North are the least differentiated, although the difference between FST Mon–North and FST North–South is not significant. Nevertheless, this pattern supports easier introgression of BS11 genes, because it contrasts with that seen in the genomic background (CR genes, figure 2; electronic supplementary material, table S8), where FST North–South is the lowest and differences between all pairs are significant (randomization test, p < 0.01). The ranking of FST values in the genomic background can be explained by relationships between lineages as depicted by the drift tree in figure 1b obtained from an independent, genome-wide dataset [19]. North and South are most closely related and the latter has experienced more drift, which results in higher FST Mon–South relative to FST Mon–North (but FST North–South is still the lowest). Apparently genome-wide gene flow between Mon and North has been too weak to increase similarity between these taxa above that observed for more closely related North and South. Reduction of FST Mon–North relative to FST North–South in BS11 genes is clearly visible in multidimensional scaling plots and is even more pronounced for MHC class I and TAP1 (figure 2; electronic supplementary material, figure S5). Consequently, differentiation between Mon and North in MHC class I and TAP1 genes is the lowest (randomization test, p < 0.001, electronic supplementary material, tables S7, S9, S10). Results for TAP1 and MHC class I may be correlated owing to the tight linkage of these genes, but even then TAP1 is useful, because it demonstrates that the binary encoding of multilocus MHC class I genotypes (see Methods) did not affect the outcome of the tests. The FST values are generally lower in BS genes (figure 2), which is expected because FST, a relative measure of differentiation, is affected by levels of diversity, which differ between the BS and CR genes. However, the differences in variation between the BS and CR genes per se should not change the ranking of pairwise FST values, as we show below using simulations. The measures of absolute sequence divergence (dxy and RND) in CR were substantially lower between North and South, consistent with their closer relationship, while absolute divergence in BS11 was similar between all group pairs (electronic supplementary material, table S11).
Figure 2.

Differentiation between groups and populations calculated for 154 control genes (CR), 11 single-copy putative targets of balancing selection (BS11), TAP1 and MHC class I. (a) Triangles: FST between groups, calculated as the average of pairwise FST between populations from the respective groups or (in brackets) as the average of pairwise FST between lineages from the respective groups (populations within lineages were pooled). Significant differences (randomization test) between FST values on the edges adjacent to a vertex are designated with asterisks (** p < 0.01, *** p < 0.001). (b) Scatterplots: first two principal coordinate axes of multidimensional scaling ordination of populations based on the matrix of pairwise FST; percentages of variation explained by each coordinate are also shown. (Online version in colour.)
To see whether the reversal of FST ranking of the magnitude observed for BS11 could be caused by factors other than differential gene flow, we performed a series of simulations conditioned on the previously inferred demographic history (figure 3; electronic supplementary material, table S3). Both neutrality and BS were simulated under isolation and migration. Under isolation the reversed ranking as observed for BS11 (diffFST = −0.289) was unlikely for genes of the characteristics of BS11 evolving neutrally (p < 10−4 and 0.005 for scenarios (i) and (ii), respectively), under SOD (p ≤ 0.004) or NFD (p ≤ 0.004) BS (table 2; electronic supplementary material, table S12). Hence, BS would not reverse the FST ranking in isolation. The probability of obtaining the reversed ranking under neutrality increased with increasing migration (table 2; electronic supplementary material, table S12), but even for the highest evaluated rate it was still small (p = 0.005 and 0.038 for scenarios (i) and (ii), respectively). Note that the highest migration rate used in the simulations is probably an overestimate, as simulations for genes of the CR characteristics suggest (table 2; electronic supplementary material, table S12). As expected, both SOD and NFD caused the reversal of FST ranking even under low migration, but the effect depended on the strength of selection (table 2; electronic supplementary material, table S12). Thus, the simulations confirmed that processes other than differential gene flow are unlikely to account for the differences in the FST ranking observed between the BS11 and CR genes.
Figure 3.
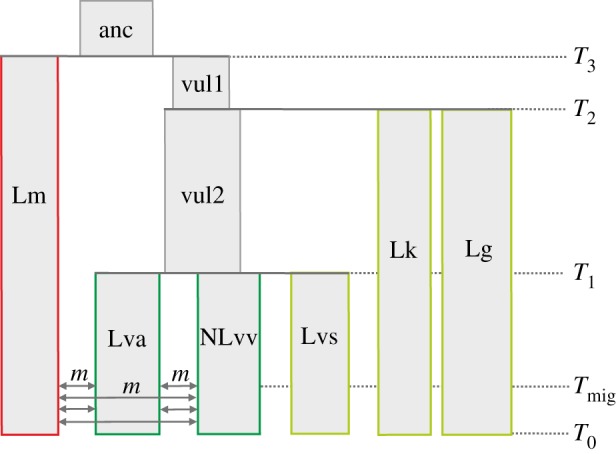
The demographic model used to test whether processes other than differential gene flow could account for the observed results. Parameter values used in simulations, taken from previous studies [19,21], are given in the electronic supplementary material, table S3. (Online version in colour.)
Table 2.
Simulations evaluating probability of the reversal of FST ranking of the magnitude observed for BS11 genes under various evolutionary scenarios. (Both neutral scenarios were explored with 10 000 coalescent simulations, while BS scenarios were evaluated with 500 forward simulations; for details see text. Simulations were performed according to the model in figure 3 with parameter values from the electronic supplementary material, table S3. Neutrality, Ne × 6.8—neutral demography with the effective population size increased to obtain variation comparable to that in BS11 genes; BS11-like and CR-like—genes of the characteristics (length, mutation rate) of balancing selection candidates (BS11) and control (CR) genes. SOD, symmetric overdominance, NFD, negative frequency dependence, diffFST, difference in FST between Mon–North and North–South divided by the average value; s, selective advantage of a heterozygote (SOD), or the initial selective advantage of a new allele (NFD). p-values—proportions of simulations with diffFST lower than observed diffFST in BS11-like genes or higher than observed diffFST in CR-like genes, significant values are in italics.)
| scenario | genes | migration rate | observed diffFST | simulated diffFST |
p-value | |
|---|---|---|---|---|---|---|
| percentile | ||||||
| 5th | 95th | |||||
| neutrality | CR-like | 0 | 0.268 | 0.104 | 0.299 | 0.13 |
| neutrality | CR-like | m1 | 0.268 | 0.079 | 0.276 | 0.0701 |
| neutrality | CR-like | m2 | 0.268 | 0.013 | 0.222 | 0.0086 |
| neutrality | CR-like | m3 | 0.268 | −0.017 | 0.192 | 0.0024 |
| neutrality | BS11-like | 0 | −0.289 | −0.019 | 0.310 | <0.0001 |
| neutrality | BS11-like | m1 | −0.289 | −0.050 | 0.285 | 0.0001 |
| neutrality | BS11-like | m2 | −0.289 | −0.133 | 0.227 | 0.0023 |
| neutrality | BS11-like | m3 | −0.289 | −0.166 | 0.195 | 0.0050 |
| neutrality, Ne × 6.8 | BS11-like | 0 | −0.289 | −0.142 | 0.262 | 0.0047 |
| neutrality, Ne × 6.8 | BS11-like | m1 | −0.289 | −0.166 | 0.240 | 0.0046 |
| neutrality, Ne × 6.8 | BS11-like | m2 | −0.289 | −0.240 | 0.179 | 0.0249 |
| neutrality, Ne × 6.8 | BS11-like | m3 | −0.289 | −0.270 | 0.155 | 0.0380 |
| BS: moderate SOD, s = 0.05 | BS11-like | 0 | −0.289 | −0.112 | 0.298 | 0.004 |
| BS: moderate SOD, s = 0.05 | BS11-like | m1 | −0.289 | −0.755 | −0.031 | 0.618 |
| BS: moderate SOD, s = 0.05 | BS11-like | m3 | −0.289 | −1.250 | −0.415 | 0.986 |
| BS: weak SOD, s = 0.02 | BS11-like | 0 | −0.289 | −0.045 | 0.322 | <0.002 |
| BS: weak SOD, s = 0.02 | BS11-like | m1 | −0.289 | −0.125 | 0.276 | 0.010 |
| BS: weak SOD, s = 0.02 | BS11-like | m3 | −0.289 | −0.356 | 0.121 | 0.100 |
| BS: strong NFD, s = 0.5 | BS11-like | 0 | −0.289 | −0.182 | 0.185 | 0.004 |
| BS: strong NFD, s = 0.5 | BS11-like | m1 | −0.289 | −1.603 | −0.680 | 0.998 |
| BS: strong NFD, s = 0.5 | BS11-like | m3 | −0.289 | −1.656 | −0.775 | >0.998 |
| BS: moderate NFD, s = 0.05 | BS11-like | 0 | −0.289 | −0.112 | 0.303 | 0.002 |
| BS: moderate NFD, s = 0.05 | BS11-like | m1 | −0.289 | −0.749 | −0.066 | 0.692 |
| BS: moderate NFD, s = 0.05 | BS11-like | m3 | −0.289 | −1.255 | −0.462 | 0.996 |
| BS: weak NFD, s = 0.02 | BS11-like | 0 | −0.289 | −0.084 | 0.331 | <0.002 |
| BS: weak NFD, s = 0.02 | BS11-like | m1 | −0.289 | −0.124 | 0.296 | 0.004 |
| BS: weak NFD, s = 0.02 | BS11-like | m3 | −0.289 | −0.341 | 0.141 | 0.106 |
4. Discussion
Our study demonstrates stronger interspecific introgression of BS candidates identified among newt immune genes, compared to CR. Higher effective migration rate of genes affected by the negative frequency-dependent BS, resulting in easier introgression, has been predicted by theory and observed in simulations [8,10]. Stronger introgression of BS targets has also been detected in empirical studies, but these have been limited to single cases of long-term BS [12,14,15]. Here we show, for the first time, to our knowledge, increased introgression of BS candidates as a group, which probably includes targets of long-term as well as recent or transient BS. This supports the hypothesis that BS, most probably involving a mechanism of novel allele advantage, facilitates introgression of its targets in general.
How robust are these results? Our test of differential introgression compared the ranking of pairwise FST between three geographical groups for two predefined classes of genes: putative targets of BS and CR genes. Specifically, we expected that pervasive introgression would change the ranking of FST values, reducing markedly FST between more distantly related groups for BS candidates but not for CR genes. The tests did not rely on a direct comparison of FST values between the two classes of genes, so they were not affected by the issues which prompted criticism of FST as a tool to detect differential introgression [35]. We also demonstrate with simulations that higher genetic variation alone does not affect ranking of FST values observed for the candidate genes. Variation in recombination rate across the genome affects the intensity of background selection and, in consequence, the level of genetic variation [36]. This in turn, even in the absence of introgression, generates genomic heterogeneity of FST, which as a relative measure of differentiation is sensitive to variation within populations. If genomic variation in recombination rate were to affect our results, individual genes would have to experience different recombination environments in various evolutionary lineages within the L. vulgaris species complex. This is unlikely for two reasons: (i) both BS candidates and CR genes are scattered in the newt genome, so a rapid, massive reorganization of the recombination landscape would be required; and (ii) regions of increased differentiation as detected by elevated FST are relatively stable across related taxa, implying an overall stability of recombination landscapes over shorter evolutionary scales [37,38]. The robustness of our results was evaluated against several types of simulations conditioned on the properties of the investigated genes and demographic history of the taxa. Simulations showed that BS acting under isolation did not produce the reversed FST order as observed for BS candidates; under realistic migration rates and neutrality the differences in variation between BS11 and CR genes did not cause the observed pattern either. By contrast, the reversed ranking was easily obtained in BS scenarios when migration was allowed. The reversal of the magnitude similar to that observed for BS11 was produced by relatively weak BS. We thus conclude that increased introgression of the BS candidates is well supported, and suggest it has been driven by BS involving novel allele advantage.
The strength of evidence for BS varies among the candidate genes. While the signal is strong for MHC class I and TAP1, it is moderate for BS11. This is not surprising, as long-term BS, generating strong signatures, is rare in vertebrate genomes outside the MHC [39]. The BS11 genes may thus include mostly cases of recent/transient BS, probably weaker than that acting on MHC. Moreover, even in MHC genes strong BS does not operate via pure NFD or SOD. Although a contribution of novel allele advantage has been demonstrated experimentally [40], empirical patterns reported in the ample MHC literature point to a complex mixture of selective and demographic mechanisms operating at various spatial and temporal scales [41,42]. These complexities do not erase the signal of BS, but make the patterns idiosyncratic and generalizations difficult [42,43]. Combined with probably weaker BS, it is not surprising that the observed diffFST for BS11 was not as extreme as observed for MHC and TAP1 or in simulations assuming strong NFD. In general, distinguishing between specific mechanisms of BS is extremely difficult [42,44], and these can be intricately linked, for example there is an inherent frequency-dependent component in the heterozygote advantage model [42]. At the same time, without BS, the reversal of the ranking did not occur in simulations. Therefore, even if BS11 include false positives, for example cases of relaxed purifying selection, they apparently did not dominate the signal—such genes should introgress neutrally and then the reversal is not expected.
This finding has implications for understanding the impact of hybridization on the long-term maintenance of variation relevant for adaptation. If incipient species retain the ability to hybridize for prolonged periods and experience multiple cycles of isolation and contact, they could effectively share a pool of adaptive variation in BS targets. These genes also are usually affected by drift and other forms of selection, including possible episodes of directional selective pressure, which may cause loss of variation [42,43,45]. Hybridization would then provide a potent mechanism for restoring variation, boosting the long-term adaptive potential of the hybridizing lineages. Alleles of genes under BS are often old and differ by many mutations [7], hence recovery of adaptive variation through hybridization would be more rapid and efficient than via de novo mutation and recombination. The importance of introgression as a mechanism maintaining adaptive variation in BS genes may vary among taxa. It should be most relevant in organisms characterized by deep phylogeographic structure, ecological niche conservatism and prolonged evolution of reproductive isolation, such as amphibians, freshwater fishes and many invertebrates. In such taxa evolutionary lineages capable of hybridization may persist for a long time with extended periods of geographical isolation, allowing accumulation of adaptive variation that may introgress adaptively during episodes of secondary contact. Interspecific introgression of BS targets may also be important in parasites [46]. In plants, while adaptive introgression of SI genes is well documented, introgression of immune genes may be less likely. Although these highly dynamic gene families are often affected by BS, they are also hotspots for genetic incompatibilities between plant populations and species, so selection may prevent their introgression [47,48].
The results of our study may be of relevance for conservation and management of threatened taxa. Interspecific hybridization has been advocated as a radical yet potentially efficient tool for practical conservation [49]. The idea remains controversial [50] because of the risks of outbreeding depression, swamping adaptation through hybridization and inadvertent transfer of pathogens. However, as restoration of variation in BS targets is likely to increase fitness and co-introgressed variants causing outbreeding depression are efficiently eliminated in a few generations [51], the benefits may outweigh costs in many cases. Therefore, interspecific introgression may hold conservation potential if genetic rescue [52] through intraspecific translocations is not a viable option.
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We are grateful to P. Arntzen, S. Bury, D. Cogălniceanu, M. Herdegen-Radwan, K. Olgun, M. Liana, K. Nadachowska-Brzyska, M. Pabijan, J. Radwan, M. Stuglik, B. Wielstra and P. Zieliński for help in sampling. M. Herdegen-Radwan, M. Konczal, M. Pabijan, J. Radwan and members of the Molecular and Behavioral Ecology Group IES JU provided valuable comments. We thank M. Konczal for computational assistance. Part of the simulations were run in the Poznań Supercomputing and Networking Center. The work was funded by the Polish National Science Centre (2012/04/A/NZ8/00662 to W.B.) and the Jagiellonian University (DS/WBiNoZ/INoS/762/17).
Ethics
Samples were collected for the purpose of the present study according to the protocols conforming to the animal ethics approval (numbers 101/2009 and 28/2011), issued by the First Local Ethical Committee on Animal Testing at the Jagiellonian University in Krakow.
Data accessibility
Raw reads from sequencing BS11 and CR genes and corresponding bam files are deposited in NCBI SRA under accession SRP150740 (BioProject number PRJNA476511, BioSample numbers SAMN09435882-SAMN09436459). Genotype data and reference sequence data are available in the Dryad Digital Repository http://dx.doi.org/10.5061/dryad.1h48b38 [53].
Authors' contributions
W.B. and A.F. conceived and designed the study. A.F., K.D., M.N. and W.B. collected and analysed the data. A.F. and W.B. prepared the manuscript, with comments from the remaining authors.
Competing interests
We have no competing interests.
References
- 1.Abbott R, et al. 2013. Hybridization and speciation. J. Evol. Biol. 26, 229–246. ( 10.1111/j.1420-9101.2012.02599.x) [DOI] [PubMed] [Google Scholar]
- 2.Barton N. 1979. Gene flow past a cline. Heredity 43, 333–339. ( 10.1038/hdy.1979.86) [DOI] [Google Scholar]
- 3.Piálek J, Barton NH. 1997. The spread of an advantageous allele across a barrier: the effects of random drift and selection against heterozygotes. Genetics 145, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hedrick PW. 2013. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 22, 4606–4618. ( 10.1111/mec.12415) [DOI] [PubMed] [Google Scholar]
- 5.Liu KJ, Steinberg E, Yozzo A, Song Y, Kohn MH, Nakhleh L. 2015. Interspecific introgressive origin of genomic diversity in the house mouse. Proc. Natl Acad. Sci. USA 112, 196–201. ( 10.1073/pnas.1406298111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gittelman RM, Schraiber JG, Vernot B, Mikacenic C, Wurfel MM, Akey JM. 2016. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr. Biol. 26, 3375–3382. ( 10.1016/j.cub.2016.10.041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fijarczyk A, Babik W. 2015. Detecting balancing selection in genomes: limits and prospects. Mol. Ecol. 24, 3529–3545. ( 10.1111/mec.13226) [DOI] [PubMed] [Google Scholar]
- 8.Schierup MH, Vekemans X, Charlesworth D. 2000. The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet. Res. 76, 51–62. ( 10.1017/S0016672300004535) [DOI] [PubMed] [Google Scholar]
- 9.Pierini F, Lenz TL. In press Divergent allele advantage at human MHC genes: signatures of past and ongoing selection. Mol. Biol. Evol. ( 10.1093/molbev/msy116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muirhead CA. 2001. Consequences of population structure on genes under balancing selection. Evolution 55, 1532–1541. ( 10.1111/j.0014-3820.2001.tb00673.x) [DOI] [PubMed] [Google Scholar]
- 11.Leducq J, Llaurens V, Castric V, Saumitou-Laprade P, Hardy OJ, Vekemans X. 2011. Effect of balancing selection on spatial genetic structure within populations: theoretical investigations on the self-incompatibility locus and empirical studies in Arabidopsis halleri. Heredity 106, 319–329. ( 10.1038/hdy.2010.68) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castric V, Bechsgaard J, Schierup MH, Vekemans X. 2008. Repeated adaptive introgression at a gene under multiallelic balancing selection. PLoS Genet. 4, e1000168 ( 10.1371/journal.pgen.1000168) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abi-Rached L, et al. 2011. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334, 89–94. ( 10.1126/science.1209202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grossen C, Keller L, Biebach I, Croll D, International Goat Genome Consortium. 2014. Introgression from domestic goat generated variation at the major histocompatibility complex of alpine ibex. PLoS Genet. 10, e1004438 ( 10.1371/journal.pgen.1004438) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nadachowska-Brzyska K, Zielinski P, Radwan J, Babik W. 2012. Interspecific hybridization increases MHC class II diversity in two sister species of newts. Mol. Ecol. 21, 887–906. ( 10.1111/j.1365-294X.2011.05347.x) [DOI] [PubMed] [Google Scholar]
- 16.Ejsmond MJ, Radwan J. 2015. Red Queen processes drive positive selection on major histocompatibility complex (MHC) genes. PLoS Comput. Biol. 11, e1004627 ( 10.1371/journal.pcbi.1004627) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charlesworth D. 2006. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2, e64 ( 10.1371/journal.pgen.0020064) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein J, Sato A, Nagl S, O'hUigín C. 1998. Molecular trans-species polymorphism. Annu. Rev. Ecol. Syst. 29, 1–21. ( 10.1146/annurev.ecolsys.29.1.1) [DOI] [Google Scholar]
- 19.Pabijan M, Zielinski P, Dudek K, Stuglik M, Babik W. 2017. Isolation and gene flow in a speciation continuum in newts. Mol. Phyl. Evol. 116, 1–12. ( 10.1016/j.ympev.2017.08.003) [DOI] [PubMed] [Google Scholar]
- 20.Zieliński P, Dudek K, Stuglik MT, Liana M, Babik W. 2014. Single nucleotide polymorphisms reveal genetic structuring of the Carpathian newt and provide evidence of interspecific gene flow in the nuclear genome. PLoS ONE 9, e97431 ( 10.1371/journal.pone.0097431) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zieliński P, Nadachowska-Brzyska K, Dudek K, Babik W. 2016. Divergence history of the Carpathian and smooth newts modelled in space and time. Mol. Ecol. 25, 3912–3928. ( 10.1111/mec.13724) [DOI] [PubMed] [Google Scholar]
- 22.Fijarczyk A, Dudek K, Babik W. 2016. Selective landscapes in newt immune genes inferred from patterns of nucleotide variation. Genome Biol. Evol. 8, 3417–3432. ( 10.1093/gbe/evw236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niedzicka M, Fijarczyk A, Dudek K, Stuglik M, Babik W. 2016. Molecular inversion probes for targeted resequencing in non-model organisms. Sci. Rep. 6, 24051 ( 10.1038/srep24051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niedzicka M, Dudek K, Fijarczyk A, Zieliński P, Babik W. 2017. Linkage map of Lissotriton newts provides insight into the genetic basis of reproductive isolation. G3 7, 2115–2124. ( 10.1534/g3.117.041178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. ( 10.1038/nmeth.1923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. ( 10.1038/ng.806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudson RR, Slatkin M, Maddison W. 1992. Estimation of levels of gene flow from DNA sequence data. Genetics 132, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pabijan M, Zieliński P, Dudek K, Chloupek M, Sotiropoulos K, Liana M, Babik W. 2015. The dissection of a Pleistocene refugium: phylogeography of the smooth newt, Lissotriton vulgaris, in the Balkans. J. Biogeogr. 42, 671–683. ( 10.1111/jbi.12449) [DOI] [Google Scholar]
- 29.Biedrzycka A, Sebastian A, Migalska M, Westerdahl H, Radwan J. 2016. Testing genotyping strategies for ultra-deep sequencing of a co-amplifying gene family: MHC class I in a passerine bird. Mol. Ecol. Res. 17, 642–655. ( 10.1111/1755-0998.12612) [DOI] [PubMed] [Google Scholar]
- 30.Sebastian A, Herdegen M, Migalska M, Radwan J. 2016. Amplisas: a web server for multilocus genotyping using next-generation amplicon sequencing data. Mol. Ecol. Res. 16, 498–510. ( 10.1111/1755-0998.12453) [DOI] [PubMed] [Google Scholar]
- 31.Excoffier L, Lischer HE. 2010. Arlequin suite ver. 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 10, 564–567. ( 10.1111/j.1755-0998.2010.02847.x) [DOI] [PubMed] [Google Scholar]
- 32.Team RC. 2017. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See https://www.R-project.org/ [Google Scholar]
- 33.Hudson RR. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18, 337–338. ( 10.1093/bioinformatics/18.2.337) [DOI] [PubMed] [Google Scholar]
- 34.Haller BC, Messer PW. 2016. SLiM 2: flexible, interactive forward genetic simulations. Mol. Biol. Evol. 34, 230–240. ( 10.1093/molbev/msw211) [DOI] [PubMed] [Google Scholar]
- 35.Cruickshank TE, Hahn MW. 2014. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23, 3133–3157. ( 10.1111/mec.12796) [DOI] [PubMed] [Google Scholar]
- 36.Ellegren H, Galtier N. 2016. Determinants of genetic diversity. Nat. Rev. Genet. 17, 422–433. ( 10.1038/nrg.2016.58) [DOI] [PubMed] [Google Scholar]
- 37.Burri R, et al. 2015. Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 25, 1656–1665. ( 10.1101/gr.196485.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vijay N, Bossu CM, Poelstra JW, Weissensteiner MH, Suh A, Kryukov AP, Wolf JB. 2016. Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat. Commun. 7, 13195 ( 10.1038/ncomms13195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quintana-Murci L, Clark AG. 2013. Population genetic tools for dissecting innate immunity in humans. Nat. Rev. Immunol. 13, 280–293. ( 10.1038/nri3421) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phillips KP, Cable J, Mohammed RS, Herdegen-Radwan M, Raubic J, Przesmycka KJ, van Oosterhout C, Radwan J. 2018. Immunogenetic novelty confers a selective advantage in host–pathogen coevolution. Proc. Natl Acad. Sci. USA 115, 1552–1557. ( 10.1073/pnas.1708597115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herdegen M, Babik W, Radwan J. 2014. Selective pressures on MHC class II genes in the guppy (Poecilia reticulata) as inferred by hierarchical analysis of population structure. J. Evol. Biol. 27, 2347–2359. ( 10.1111/jeb.12476) [DOI] [PubMed] [Google Scholar]
- 42.Spurgin LG, Richardson DS. 2010. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc. R. Soc. B 277, 979–988. ( 10.1098/rspb.2009.2084) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radwan J, Biedrzycka A, Babik W. 2010. Does reduced MHC diversity decrease viability of vertebrate populations. Biol. Conserv. 143, 537–544. ( 10.1016/j.biocon.2009.07.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hedrick PW. 2012. What is the evidence for heterozygote advantage selection? Trends Ecol. Evol. 27, 698–704. ( 10.1016/j.tree.2012.08.012) [DOI] [PubMed] [Google Scholar]
- 45.Ejsmond MJ, Radwan J. 2011. MHC diversity in bottlenecked populations: a simulation model. Cons. Genet. 12, 129–137. ( 10.1007/s10592-009-9998-6) [DOI] [Google Scholar]
- 46.King KC, Stelkens RB, Webster JP, Smith DF, Brockhurst MA. 2015. Hybridization in parasites: consequences for adaptive evolution, pathogenesis, and public health in a changing world. PLoS Pathog. 11, e1005098 ( 10.1371/journal.ppat.1005098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chae E, et al. 2014. Species-wide genetic incompatibility analysis identifies immune genes as hot spots of deleterious epistasis. Cell 159, 1341–1351. ( 10.1016/j.cell.2014.10.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sicard A, Kappel C, Josephs EB, Lee YW, Marona C, Stinchcombe JR, Wright SI, Lenhard M. 2015. Divergent sorting of a balanced ancestral polymorphism underlies the establishment of gene-flow barriers in Capsella. Nat. Commun. 6, 7960 ( 10.1038/ncomms8960) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamilton JA, Miller JM. 2016. Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv. Biol. 30, 33–41. ( 10.1111/cobi.12574) [DOI] [PubMed] [Google Scholar]
- 50.Kovach RP, Luikart G, Lowe WH, Boyer MC, Muhlfeld CC. 2016. Risk and efficacy of human-enabled interspecific hybridization for climate-change adaptation: response to Hamilton and Miller (2016). Conserv. Biol. 30, 428–430. ( 10.1111/cobi.12678) [DOI] [PubMed] [Google Scholar]
- 51.Aitken SN, Whitlock MC. 2013. Assisted gene flow to facilitate local adaptation to climate change. Annu. Rev. Ecol. Evol. Syst. 44, 367–388. ( 10.1146/annurev-ecolsys-110512-135747) [DOI] [Google Scholar]
- 52.Whiteley AR, Fitzpatrick SW, Funk WC, Tallmon DA. 2015. Genetic rescue to the rescue. Trends Ecol. Evol. 30, 42–49. ( 10.1016/j.tree.2014.10.009) [DOI] [PubMed] [Google Scholar]
- 53.Fijarczyk A, Dudek K, Niedzicka M, Babik W. 2018. Data from: Balancing selection and introgression of newt immune-response genes Dryad Digital Repository. ( 10.5061/dryad.1h48b38) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Fijarczyk A, Dudek K, Niedzicka M, Babik W. 2018. Data from: Balancing selection and introgression of newt immune-response genes Dryad Digital Repository. ( 10.5061/dryad.1h48b38) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
Raw reads from sequencing BS11 and CR genes and corresponding bam files are deposited in NCBI SRA under accession SRP150740 (BioProject number PRJNA476511, BioSample numbers SAMN09435882-SAMN09436459). Genotype data and reference sequence data are available in the Dryad Digital Repository http://dx.doi.org/10.5061/dryad.1h48b38 [53].