

Abstract
The FKBP-12-rapamycin associated protein (FRAP, also known as mTOR and RAFT-1) is a member of the phosphoinositide kinase related kinase family. FRAP has serine/threonine kinase activity and mediates the cellular response to mitogens through signaling to p70s6 kinase (p70s6k) and 4E-BP1, resulting in an increase in translation of subsets of cellular mRNAs. Translational up-regulation is blocked by inactivation of FRAP signaling by rapamycin, resulting in G1 cell cycle arrest. Rapamycin is used as an immunosuppressant for kidney transplants and is currently under investigation as an antiproliferative agent in tumors because of its ability to block FRAP activity. Although the role of FRAP has been extensively studied in vitro, characterization of mammalian FRAP function in vivo has been limited to the immune system and tumor models. Here we report the identification of a loss-of-function mutation in the mouse FRAP gene, which illustrates a requirement for FRAP activity in embryonic development. Our studies also determined that rapamycin treatment of the early embryo results in a phenotype indistinguishable from the FRAP mutant, demonstrating that rapamycin has teratogenic activity.
The mammalian phosphoinositide kinase related kinase (PIKK) family of proteins is comprised of FKBP-12-rapamycin-associated protein (FRAP), ATM, ATR/FRP, and DNA-PKcs, all of which contain a C-terminal kinase domain related to the phosphoinositide kinase domain (1–5). However, the PIKK members are protein kinases and contain a short segment at their extreme C terminus not present in the PI3 and PI4 kinases. This C-terminal region is required for kinase activity and is not interchangeable within the family (6). Although the ATM, ATR/FRP, and DNA-PKcs proteins all respond to DNA damage in the cell, FRAP functions to regulate the rate of protein translation in response to mitogenic signals, allowing progression from the G1 to S phase of the cell cycle (7).
FRAP kinase activity promotes the translation of a subset of cellular mRNAs by phosphorylating its targets, p70s6k (S6K1 and S6K2) and 4E-BP1 and 4E-BP2. The phosphorylation of p70s6k promotes the phosphorylation of the s6 ribosomal subunit, leading to an increase in translation (8). FRAP kinase activity also phosphorylates the translation inhibitors 4E-BP1 and 4E-BP2, causing these proteins to dissociate from the translation initiation factor eIF4e, promoting translation (5, 9). One class of mRNA targets translated by the FRAP signaling pathway is the 5′ TOP messages, which contain a 5′ terminal oligopyrimidine tract (10). Additionally, FRAP activity increases the translation of mRNAs such as c-myc and cyclin D1 (11). The translation of these classes of mRNAs has been demonstrated to be blocked by rapamycin treatment, a potent inhibitor of FRAP activity (10, 11).
FRAP has been proposed to be a regulator of cell growth (12). In yeast, the FRAP homologs TOR1 and TOR2 are thought to act as nutrient sensors and promote translation initiation and cell cycle progression (13). In Drosophila, both the ds6k mutant and FRAP mutants demonstrate reduced cell size (14–16). Targeted deletion of the mouse S6K1 gene produces small mice as well, although the presence of a second s6 kinase gene in mice (S6K2) may compensate for some S6K1 functions and may mask a more severe phenotype (17). Additionally, the FRAP pathway has been implicated in the growth of mammalian tumors, and rapamycin is being investigated as an antitumor drug (18).
Although several roles of the FRAP signaling pathway have been identified, the function of FRAP in a mouse model has not been determined. Here we present the identification of an ethylnitrosourea-induced mutation in the mouse FRAP gene, which illustrates a critical requirement for FRAP in embryonic development. We demonstrate that the FRAP signaling pathway is affected in these mutants. Additionally, we show that cells in the mouse mutant are not reduced in cell size, unlike the Drosophila FRAP or dS6K mutants, or mouse S6K1 knockout. Finally, we also determine that rapamycin treatment of mouse embryos produces a phenocopy of the FRAP mouse mutant, indicating that rapamycin is a teratogen.
Methods
Physical Mapping.
Overlapping oligonucleotides were used to generate probes from unique sequences surrounding D4MIT206, D4MIT310, and D4 MIT129. These probes were used to screen the mouse RPCI-21 genomic bacterial artificial chromosome (BAC) library (http://www.chori.org/bacpac). End sequence from these clones was used to generate PCR primers to determine the order of clones as well as to perform a blast search to locate genes in the flat-top interval.
Mutation Analysis.
Reverse transcription–PCR (RT-PCR) was performed on flat-top mutant, heterozygote, and wild-type samples by using FRAP primers that amplify across the exon7/exon 8 junction. The products were radioactively labeled and analyzed on a standard 6% acrylamide sequencing gel. A histogram analysis of the products was performed by using a Molecular Dynamics “Storm” PhosphorImager and analyzed by using NIH image software. The presence of intron 7 sequences in products A and B was confirmed by sequencing.
Rapamycin Injections.
Wild-type pregnant C57BL/6J females were treated with three daily doses of 3 mg/kg of rapamycin (Calbiochem) starting 5.5 days postcoitum (dpc). A stock solution of 1 mg/ml of rapamycin dissolved in methanol was diluted into 500 μl of Ringer's Saline immediately before i.p. injection. Control animals treated with carrier alone produced embryos indistinguishable from untreated wild type.
Enzyme Assays.
Western blots were performed as described in ref. 19. Blots were probed with anti-p70 (Santa Cruz Biotechnology, catalogue no. SC-230) or antiphospho-Thr-389 p70 (New England Biolabs, catalogue no. 9205s). Approximately 40 flat-top or wild-type embryos were resuspended in lysis buffer. Lysates were incubated with protein G agarose, cleared, and the supernatants were incubated with anti-FRAP or anti-p70s6k and fresh protein-G agarose. The samples were then washed as described in ref. 16. For the FRAP assay, p70s6k produced in Escherichia coli was used as the substrate. After the kinase reaction, the samples were analyzed on an 8% SDS acrylamide gel.
For the p70s6k assay, a peptide derived from the amino acid sequence of S6 ribosomal protein was used as the substrate. After the kinase reaction, the samples were transferred to phospho-Sepharose squares, washed, and the amount of label incorporated was determined with a scintillation counter.
4E-BP1 Phosphorylation Assay.
Plasmids and antibodies. pcDNA3-flag-FRAP wild-type, kinase dead (Asp-2357-Glu), and rapamycin-resistant (Ser-2035-Thr) constructs were kindly provided by Jie Chen (Department of Cell and Structural Biology, University of Illinois, Urbana–Champaign, IL) and were described previously (20). Flat-top insertion and deletion mutants were generated by PCR mutagenesis and cloned into the background of the rapamycin resistant flag-FRAP (Ser-2035-Thr) construct. The 4E-BP1 plasmid used for transfection (pACTAG-2–4E-BP1) was described previously (21). Anti-hemagglutinin (HA) and anti-flag monoclonal antibodies were purchased from Babco (Richmond, CA) and Sigma, respectively. The phosphospecific anti-Ser-65 and anti-Thr-70 antibodies were provided by Cell Signaling Technologies (Beverly, MA).
Expression and purification of a bacterially expressed glutathione S-transferase 4E-BP1 fusion protein (pGEX-6P1–4E-BP1 wild type) was described previously (9).
Cell Culture and Drug Treatment.
Human embryonic kidney (HEK)293T cells were maintained in DMEM containing 10% FBS. Cells were cotransfected with 1 μg of pACTAG-2–4E-BP1 and 10 μg of pcDNA3-flag-FRAP by using Lipofectamine Plus (Life Technologies, Rockville, MD). Transfected cells were split 1:2 the next day, and 16–24 h later, one set of cells was treated with rapamycin (100 ng/ml; Calbiochem) for 1 hour.
Protein Extract Preparation and Western Blotting.
After rapamycin treatment, cells were rinsed twice with PBS, scraped into 1.5-ml tubes, pelleted by centrifugation, and resuspended in lysis buffer (50 mM Tris, pH 7.4/100 mM NaCl/10% glycerol/0.2% Tween 20/1 mM DTT/50 mM β-glycerol phosphate, the phosphatase inhibitors microcystin, okadaic acid, and calyculin A from Calbiochem, and protease inhibitor mixture tablets from Roche, Mannheim, Germany). Cells were lysed by a series of five freeze-thaw cycles. For analysis of the phosphorylation state of HA-4E-BP1, 45 μg of protein extract was subjected to SDS/15% PAGE, separated proteins were transferred to nitrocellulose, and Western blotting was performed by using the phosphospecific antibody diluted 1:2,000 in PBS containing 0.1% Tween 20, 0.5% BSA 10 μg/ml, and recombinant glutathione S-transferase-4E-BP1 protein (to prevent interaction with the unphosphorylated 4E-BP1). All washes were conducted in PBS containing 0.1% Tween 20 and 0.5% BSA. The same membranes were reprobed with the anti-HA antibody diluted 1:5,000. The anti-flag antibody was diluted 1:5,000 and was used to probe a parallel blot. A peroxidase-coupled goat anti-rabbit secondary antibody was used for antiphospho-Ser-65 and antiphospho-Thr-70 blots [1:5,000, Amersham Pharmacia Pharmacia Biotech (APB)], whereas an anti-mouse secondary antibody (1:5,000, APB) was used for anti-HA and anti-flag blots. Proteins were detected by using chemiluminescence (Renaissance, APB)
Cell Size Measurements.
Flat-top or wild-type embryos at 9.5 dpc were dissected in PBS, 1 mM EDTA. Embryos were incubated on ice for 45 min, the midthoracic region was dissected and incubated in DMEM, and a single cell suspension was prepared by trituration. Only mesenchymal cells were liberated by this gentle preparation method, and the ectoderm, neuroectoderm, and endoderm remained largely intact. Digital images of the cells were captured by using DIC microscopy and cell size was measured by using NIH image. The single cell suspension prepared in this way consisted almost exclusively of spherical cells, so the cell diameter measurement provides a consistent measure of cell volume.
In Situ Hybridization.
A FRAP in situ probe was generated from a reverse RT-PCR product cloned into the TA cloning kit vector (Invitrogen). Primer sequences for RT-PCR are available on request. In situ hybridizations were carried out as described in ref. 22.
Results
The mouse flat-top mutant was isolated in an ethylnitrosourea screen designed to identify recessive forebrain defects (22). The mutants specifically lack the telencephalon, the anterior-most region of the forebrain. The flat-top phenotype has been described in detail (22) and includes a failure to up-regulate cell proliferation in the telencephalon and an inability to maintain gene expression in the prechordal region of the forebrain neuroectoderm. Mutants also fail to rotate around the embryonic body axis and die at 12.5 dpc at midgestation.
To identify the genetic basis of the developmental defects in the flat-top embryo, we mapped the mutation to high resolution. Approximately 3,000 meiotic events showed complete linkage of the flat-top locus with the marker D4MIT310, located between the markers D4MIT206 and D4MIT129, in a region that is homologous to human chromosome 1p35–36. More detailed mapping of the region was carried out by constructing a clone based physical map or contig across the flat-top region (Fig. 1A). Both BAC and PI artificial chromosome (PAC) libraries were screened to identify clones containing D4MIT206, 310, and 129. End sequences from these clones were then used to screen the same libraries until complete clone coverage of the region was obtained. We generated end sequence from BAC clones in the region and used blast searches of the nucleotide databases to align the flat-top region at very high resolution with human genomic sequence of the homologous interval.
Figure 1.

A mutation in the FRAP gene in flat-top mice and the predicted protein products. (A) Physical map of the flat-top region. BAC clones are shown in blue, PAC clones in orange. Genes identified in the flat-top interval from blast searches of clone end sequences are shown in boxes with the corresponding human PAC clone address in red. (B) RT-PCR by using primers that span the exon 7–8 splice junction produces products of 600 and 500 bp from flat-top RNA and one 500-bp product from wild-type RNA. flat, flat-top homozygote; wt, BTBR; neg, no template control. (C) The flat-top allele of the FRAP locus and the mRNAs it produces. The flat-top mutation is shown as a T to A base change in purple. The misspliced mRNA products are shown in red. mRNA A corresponds to the form that does not splice in this region, resulting in the inclusion of intron 7. Intron 7 has an in-frame stop codon (underlined nucleotides in intron) that would cause premature termination of translation. mRNA B is produced by an early splice in the intron, resulting in a 9-bp insert of intron sequence. Approximately 5% of the flat-top transcripts are wild type (wt). A similar proportion of the transcripts (mRNA C) splice into exon 8 resulting in a deletion of coding sequence. The genomic structure of the 5′ portion of the FRAP gene is shown in red, on the basis of the sequence of human PAC clone 647 M16 (GenBank accession no. AL049653). (D) Predicted protein products from the flat-top FRAP locus. The mRNA products shown in A are predicted to encode the proteins shown. mRNA A encodes an N-terminal fragment as a result of an in-frame stop codon in the intron. mRNA B encodes a three-amino acid insertion (in red in the one-letter amino acid code). mRNA C encodes a three-amino acid deletion (missing amino acids indicated by … ). (E) Western blot analysis of FRAP. An antibody recognizing the N terminus of the FRAP protein was used to probe a Western blot by using extracts from 293 cells, flat-top embryos, or wt control embryos. The position at which full length FRAP migrates is indicated by arrowheads. The blot has been overexposed in an attempt to detect the protein predicted by mRNA A. The expected mobility of this polypeptide is indicated by the arrow. The additional bands revealed by overexposure are caused by nonspecific binding by the antibody, as they are not blocked by an excess of soluble peptides used for the immunization that produced the antibody (data not shown). The identity of the FRAP band is known because it is competed by the peptide and it comigrates with a myc-tagged FRAP protein.
Among the genes identified by these approaches was FRAP (also known as mTOR and RAFT1). To determine whether flat-top mice carried a mutation in the FRAP gene, we generated overlapping RT-PCR products covering the entire coding sequence of ≈8 kb that were sequenced in their entirety. One of the primer sets generated two products from homozygous mutant embryo RNA and a single band from wild-type RNA (Fig. 1B). Sequence analysis showed that both PCR products from the flat-top embryos contained inserts relative to the wild-type product. Comparison of the cDNA sequence to the human genomic sequence indicated that the inserts corresponded to the location of an intron in the human genomic locus. Sequencing of both wild-type and mutant mouse genomic DNA corresponding to this intron revealed the existence of a single base change within the intron (Fig. 1C). The effects of the mutation on the splicing of the mRNA were examined by carrying out quantitative RT-PCR by using primers that spanned this intron. This analysis revealed the existence of two additional, but relatively rare, forms of mRNA in mutant embryos in contrast to the single form present in wild-type embryos (shown schematically in Fig. 1C). mRNA produced by a normal splice event comprised about 5% of the total FRAP message in mutant embryos, whereas the bulk of the mRNA was predicted to encode the altered proteins indicated in Fig. 1D.
The structure of the mutant mRNAs predicted that two species of altered protein would each make up about half of the total protein: an N-terminal fragment of about 385 amino acids and a full length protein with a three-amino acid insert at amino acid 372 (Fig. 1C). To examine this prediction, we performed Western blot analysis of embryo extracts by using an N-terminal FRAP antibody (Fig. 1E). The predicted N-terminal fragment was not detectable, suggesting that it was unstable or insoluble. The levels of the full-length form of FRAP were reduced to approximately one-half the level present in wild-type animals, consistent with the predictions of the mRNA analysis.
After confirming the mutation in the FRAP gene, we examined the activity of the FRAP signaling pathway in the flat-top mutants. We assayed the ability of FRAP immunoprecipitated from flat-top embryos to phosphorylate the S6 kinase p70s6k in vitro. Kinase activity of FRAP on this substrate is significantly reduced by the flat-top mutation (Fig. 2A). The extent of this defect on the overall phosphorylation level of p70s6k in vivo was assessed by Western blotting (Fig. 2B). p70s6k from flat-top embryos comigrates on SDS/PAGE with the hypophosphorylated form of p70s6k found in rapamycin-treated HEK293 cells, in contrast to the hyperphosphorylated state of the enzyme in wild-type embryos (Fig. 2B). The phosphorylation of a specific residue, Thr-389, is sensitive to rapamycin in vivo (8). Western blotting by using a phospho-specific antibody revealed a dramatic reduction in the phosphorylation of Thr-389 in flat-top embryos, similar to the results seen in rapamycin treated cells (Fig. 2B). Because phosphorylation of Thr-389 is thought to be an important means of regulating of p70s6k kinase activity (23), we assayed the ability of p70s6k to phosphorylate a peptide substrate derived from the S6 ribosomal subunit in vitro. This revealed that p70s6k activity in flat-top embryos is reduced to 17% of the level in wild-type embryos (Table 1).
Figure 2.

The flat-top mutation reduces FRAP activity significantly in homozygous embryos. (A) FRAP phosphorylation of p70s6k in vitro was analyzed by immunoprecipitation of FRAP from wild-type embryos (wt), flat-top mutant embryos (flat), and HEK293 cells, as described (16). There is ≈50% less labeled p70s6k substrate in flat-top embryos than in wild-type embryos or HEK293 controls. There was no activity in samples that were not incubated with substrate (−substrate). (B). Western blots showing the phosphorylation of p70s6k in flat-top mutant embryo extracts as compared with wild-type embryos and HEK293 cells. All of the signal represents S6 kinase. The slower migrating band in each lane is a splice variant of p70 generated by an alternative splice and sometimes designated p70a or p85. (Left) Anti-p70 antibody (Santa Cruz Biotechnology) was used to detect p70s6k on a Western blot. (Right Top) Santa Cruz p70s6k antibody on wild-type embryos, flat-top embryos, and 293 cells. (Center) NEB anti-p70-phospho-Thr-389 Western blot for the same samples as in Top. Note here that the flat-top embryos demonstrate little phosphorylation on Thr-389, a residue which is sensitive to FRAP activity. (Bottom) Double the amount of protein extracts was analyzed for Thr-389 phosphorylation. There is still little detectable Thr-389 phosophorylation in flat-top samples as compared with wild type.
Table 1.
p70s6 kinase activity
| Sample | Counts | % of 293 | % of wild type |
|---|---|---|---|
| 293 + rap | 8,461 | 1.1 | |
| 293 | 800,675 | 100 | |
| 293, no Ab | 0 | 0 | |
| Flat-top | 30,409 | 17.3 | |
| Wild-type | 175,939 | 100 |
The ability of p70S6k to phosphorylate S6 was assessed in extracts from flat-top mutants, wild-type embryos, and HEK293 cells was analyzed as described (16). No activity was seen in samples that were not incubated with antibody for immunoprecipitation (293, no Ab). The results of a representative experiment are shown. The experiment was repeated four times with similar results.
The reduction in FRAP activity could be the result of a 50% in the levels of “full length” protein product or might also reflect reduced activity of the altered proteins encoded by mRNAs C and B. Activity against p70s6k is reduced more than the predicted 50%, suggesting that the altered proteins have reduced activity. To examine this, we produced plasmids that express each of the flat-top mRNAs separately (wild type, mRNA B, and mRNA C; see Fig. 1 C and D). A FLAG epitope and a mutation conferring rapamycin resistance were added to allow FRAP activity to be measured in transfected cells against the background of endogenous FRAP. The activity of the mutant proteins was assessed against a second major target of FRAP kinase activity, 4E-BP1 (Fig. 3). HEK293 cells were cotransfected with an individual FRAP expression plasmid and with a HA-tagged 4E-BP1 construct. Each combination was assessed in the presence or absence of rapamycin. 4E-BP1 was immunoprecipitated from the cells, and phosphorylation of Ser-65 and Thr-70, residues shown to be FRAP targets, were analyzed by Western blot with an antiphospho Ser-65 antibody (Fig. 3A) and antiphospho Thr-70 antibody (Fig. 3B). The results show that HEK293 cells transfected with the wild-type FRAP protein efficiently phosphorylate 4E-BP1 at Ser-65 and Thr-70. In contrast, both the protein containing a three-amino acid insertion (mRNA B) and that with a three-amino acid deletion (mRNA C) have drastically reduced activity, similar to that produced by a mutation in the active site of the kinase (kinase dead in Fig. 3.) These results demonstrate that the altered proteins produced by the flat-top allele of FRAP have drastically reduced activity, despite the fact that the lesions are far from the active site.
Figure 3.

FRAP flat-top mutants do not effect phosphorylation of 4E-BP1 at Ser-65 or Thr-70. 293T cells were cotransfected with pACTAG-2–4E-BP1 (HA-4E-BP1) and pcDNA3-flag-FRAP constructs. Cells were either treated with 100 ng/ml of rapamycin (+) or untreated (−). (A) Western blot analyses of whole cell extracts were performed with (Top) a phosphospecific antibody directed against 4E-BP1 serine 65 (α-P-Ser65), (Middle) an α-HA antibody to compare HA-4E-BP1 expression levels, and (Bottom) an α-flag antibody for detection of the flag-FRAP protein. All FRAP constructs were generated from a rapamycin-resistant form of the protein, Ser-2035-Thr. The experiment was carried out in duplicate with each of the transfected forms of FRAP. (B) The same cell extracts were used for Western blot analysis with (Top) a phosphospecific antibody directed against threonine 70 (α-P-thr-70); (Bottom) an α-HA antibody to examine HA-4E-BP1 expression. (C) The results of the serine 65 phosphorylation experiment were quantitated and expressed as a percent relative to the wild type without rapamycin treatment. The standard error is indicated.
We next examined the developmental expression pattern of FRAP, p70s6k, and 4E-BP1 to determine whether the genes were expressed in tissues that are abnormal in the flat-top embryo. At 8.5 dpc, prominent FRAP mRNA expression in the midthoracic region of the embryo is apparent (Fig. 4A). Expression spreads quickly and is quite widespread by 9.5 dpc (Fig. 4B). Some regional variation in expression was apparent; for example, expression is higher in the neuroectoderm, higher yet in rhombomeres 3 and 5, and absent or at low levels in the heart. This widespread pattern suggested that the expression of downstream targets might be a more important determinant of the specific defects seen in flat-top mice. We also examined the expression pattern of 4E-BP1 and p70s6k in wild-type embryos. The expression patterns of p70s6k and 4E-BP1 are more restricted, consistent with the onset of developmental defects in flat-top embryos. High levels of expression are found in the neuroectoderm of the developing telencephalon and the surrounding mesenchyme (shown for 4E-BP1 in Fig. 4). The highest levels of 4E-BP1 expression at 8.5 dpc are also in the presumptive telencephalon, suggesting that defects in 4E-BP1 and p70s6k activity result in the specific proliferation defects in the telencephalic neuroectoderm of flat-top mutants.
Figure 4.
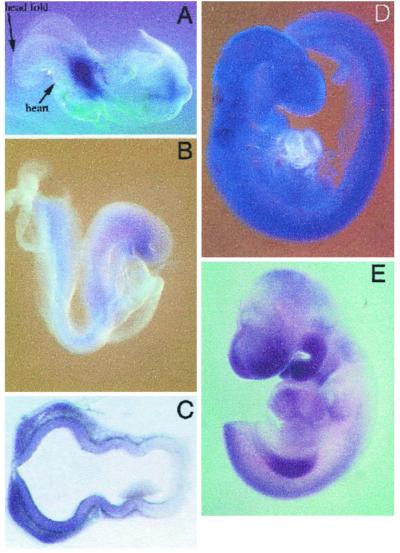
FRAP and 4E-BP1 expression in wild-type embryos. (A) FRAP expression in a wild-type embryo at 8.5 dpc. The expression is most prominent in the midthoracic region of the embryo. (D) FRAP expression in a wild-type embryo at 9.5 dpc. The expression domain has expanded from that seen at 8.5 dpc. However, there appears to be little or no FRAP expression in the heart. Also, hindbrain rhombomeres 3 and 5 have higher levels of FRAP expression than surrounding tissues. (B) 4E-BP1 expression at 8.5 dpc. The expression is relatively low overall with the highest level of expression in the ventral portion of the forebrain and the surrounding mesenchyme. (E) Overall 4E-BP1 expression at 9.5 dpc. A section through the head (C) shows high levels of expression in the ventral telencephalic neuroectoderm and surrounding mesenchyme but not in the surface ectoderm.
Our previous analysis of the flat-top phenotype showed that the telencephalic defect was because of a failure to up-regulate the rate of proliferation in the neuroectoderm (22). In Drosophila, both FRAP and S6 kinase activities are necessary to maintain a consistent cell size during development, and a 50% reduction in S6 kinase activity has a discernible effect (15–16). Mice have two S6 kinase genes, and the activity of both is strongly inhibited by rapamycin (17). Thus it seemed possible that an effect on cell size might also play a role in the developmental defects in flat-top mice. We examined the size of cells in flat-top mice by two different means. The size of dissociated cells from whole, mutant, or unaffected littermate controls was compared by using forward light scatter in a fluorescence activated cell sorter. This analysis revealed the same distribution of cell sizes in mutant and wild-type embryos but approximately one-third fewer cells in mutant embryos. To examine cell size in a more direct fashion, we recovered mesenchymal cells from the midthoracic region of embryos, the earliest site of FRAP expression, and measured cell diameters (Fig. 5). This analysis did not detect an effect of the mutation on cell size.
Figure 5.
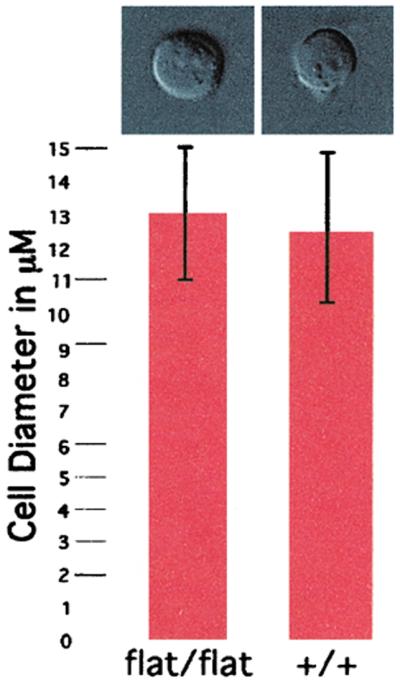
Loss of FRAP activity does not affect regulation of cell size. Cell size comparisons of flat-top embryos and wild-type embryos. A single cell suspension was generated from the midthoracic region of either wild-type or flat-top mutant embryos at 9.5 dpc, and cells were measured to determine size. There is no significant difference between flat-top (Left) and wild-type (Right) cell diameters.
Because reduced FRAP kinase activity causes abnormal development in flat-top embryos, we examined whether rapamycin treatment of normal embryos could cause similar defects in embryonic development. We injected wild-type pregnant mice with a dose regimen of rapamycin comparable to the therapeutic dose given to humans and analyzed the embryos at 9.5 dpc. One hundred percent of embryos (n = 33) from these females phenocopied the flat-top homozygous mutant embryo (Fig. 6B). Western blot analysis with p70s6k Thr-398 phospho-specific antibody demonstrated that phosphorylation of p70s6k is reduced in rapamycin treated embryos (data not shown), consistent with the idea that the effects of rapamycin on embryogenesis were because of the known targets of FRAP. These results provide further evidence that the FRAP mutation is the cause of abnormal development in the flat-top mutant embryo.
Figure 6.

The flat-top phenotype and a rapamycin-induced phenocopy. (A) Wild-type (Left) and flat-top embryos at 9.5 days of gestation (dpc). The flat-top embryo is slightly developmentally delayed and smaller than somite matched wild-type embryos. Mutant embryos fail to turn, fail to form the telencephalic vesicles, and growth arrest by about 10.5 dpc. (B) Rapamycin treatment of pregnant females produces a phenocopy of the flat-top phenotype in wild-type embryos. Treatment of pregnant females with vehicle had no discernable effects on embryonic development.
Discussion
We present, to our knowledge for the first time, a mouse with a mutation in the FRAP gene. The flat-top mouse mutant has many patterning defects, especially in the developing telencephalon, demonstrating that FRAP activity is necessary for proper embryonic forebrain development. The targets of the FRAP kinase, p70s6k, and 4E-BP1 are not phosphorylated in the flat-top mutants, demonstrating that the mutation affects FRAP kinase activity. Although the flat-top mutant allele of FRAP has a three-amino acid insertion into a region of the FRAP protein that has not been previously characterized to perform a specific function, our analysis of the flat-top FRAP allele in transfection assays demonstrates that this mutation blocks FRAP kinase activity toward 4E-BP1.
The mouse FRAP mutation does not affect cell size, which is an interesting difference from the described Drosophila FRAP mutants (15, 16) and the mouse and Drosophila p70s6k mutants (14, 17). We propose that, whereas in Drosophila FRAP regulates cell growth, in the mouse this function is controlled by downstream targets of FRAP, such as p70s6k. However, in the flat-top mutant cell proliferation is abnormal, suggesting that in the mouse, FRAP does integrate mitogenic signals to up-regulate cell proliferation in development. Tissues that require a rapid increase in cell proliferation, such as the telencephalon, ventral body walls, and limb buds, are all abnormal in flat-top mutants (22). FRAP activity may also be required for a shortening of the G1 phase of the cell cycle during embryonic development, thus allowing more rapid cell proliferation in certain embryonic regions. The embryo does not have a G1 phase of the cell cycle until after gastrulation, indicating that FRAP activity does not function in the regulation of development until after gastrulation.
The rapamycin-induced phenocopy of the flat-top mutant provides strong evidence that the mutation in FRAP is causative of the flat-top phenotype. There are no other known rapamycin sensitive genes in the flat-top critical region determined by meiotic mapping of the mutation, and characterization of the region from the human genome sequencing project has not identified any other genes in the region involved in the FRAP sequencing pathway. The phenocopy of the flat-top mutation by injection of rapamycin into wild-type pregnant mice also demonstrates that rapamycin is a teratogen. As its use increases as an immunosuppressant and a possible anticancer agent, its teratogenic potential should be considered.
Further studies are necessary to determine the mechanism of the flat-top mutation. However, new studies on FRAP signaling show that FRAP must shuttle between the nucleus and cytoplasm to have signaling activity (24). We can speculate that perhaps the flat-top FRAP allele with the three-amino acid insertion disrupts the ability of FRAP either to enter or to leave the nucleus. It has also been demonstrated that FRAP kinase activity is required for the expression of cyclin D1 protein in pancreatic cancer cells (11). It is possible that the flat-top FRAP mutation blocks the translation of cyclin D1 in the developing embryo, resulting in a loss of cell proliferation in the telencephalon and other areas affected by the flat-top mutation. Further study of the flat-top mutant will validate in vitro results regarding the FRAP signaling pathway and will seek to determine the role of FRAP activity in embryonic development and cell proliferation.
Abbreviations
- PIKK
phosphoinositide kinase related kinase
- BAC
bacterial artificial chromosome
- PAC
PI artificial chromosome
- dpc
days postcoitum
- HEK
human embryonic kidney
- RT-PCR
reverse transcription–PCR
- HA
hemagglutinin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley J N, Ried T, Tagle D, Wynshaw-Boris A. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 2.Cliby W A, Roberts C J, Cimprich K A, Stringer C M, Lamb J R, Schreiber S L, Friend S H. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartley K O, Gell D, Smith G C, Zhang H, Divecha N, Connelly M A, Admon A, Lees-Miller S P, Anderson C W, Jackson S P. Cell. 1995;82:849–856. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- 4.Hoekstra M F. Curr Opin Genet Dev. 1997;7:170–175. doi: 10.1016/s0959-437x(97)80125-6. [DOI] [PubMed] [Google Scholar]
- 5.Gingras A-C, Raught B, Sonnenberg N. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi T, Hara K, Inoue H, Kawa Y, Tokunaga C, Hidayat S, Yoshino K, Kuroda Y, Yorezawa K. Genes Cells. 2000;5:765–775. doi: 10.1046/j.1365-2443.2000.00365.x. [DOI] [PubMed] [Google Scholar]
- 7.Kuruvilla F, Schrieber S L. Chem Biol. 1999;6:R129–R136. doi: 10.1016/S1074-5521(99)80070-2. [DOI] [PubMed] [Google Scholar]
- 8.Dennis P B, Pullen N, Kozma S C, Thomas G. Mol Cell Biol. 1996;16:6242–6251. doi: 10.1128/mcb.16.11.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gingras A C, Gygi S P, Raught B, Polakiewicz R D, Abraham R T, Hoekstra M F, Aebersold R, Sonenberg N. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loreni F, Thomas G, Amaldi F. Eur J Biochem. 2000;267:6594–6601. doi: 10.1046/j.1432-1327.2000.01753.x. [DOI] [PubMed] [Google Scholar]
- 11.Grewe M, Gansauge F, Schmid R, Alder G, Seufferlein T. Cancer Res. 1999;59:3581–3587. [PubMed] [Google Scholar]
- 12.Schmelzle T, Hall M N. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 13.Barbet N C, Schneider U, Helliwell S B, Stanfield I, Tuite U F, Hall M N. Mol Biol Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montagne J, Stewart M J, Stocker H, Hafen E, Kozma S C, Thomas G. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- 15.Zheng H, Stallock J P, Ng J C, Reinhard C, Neufield J P. Genes Dev. 2000;14:2717–2724. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E. Genes Dev. 2000;14:2689–2694. doi: 10.1101/gad.845700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma S C. EMBO J. 1998;17:6649–6659. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudkin L, Dilling M, Cheshire R J, Harwood F C, Hollingshead M, Arbuck S, Travis K, Janesville E A, Houghton P J. Clin Cancer Res. 2001;7:1758–1764. [PubMed] [Google Scholar]
- 19.Burnett P E, Barrow R K, Cohen N A, Snyder S H, Sabatini D M. Proc Natl Acad Sci USA. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vilella-Bach M, Nuzzi P, Fang Y, Chen J. J Biol Chem. 1999;274:4266–4272. doi: 10.1074/jbc.274.7.4266. [DOI] [PubMed] [Google Scholar]
- 21.Gingras A C, Kennedy S G, O'Leary M A, Sonenberg N, Hay N. Genes Dev. 1998;12:502–513. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hentges K, Thompson K, Peterson A. Development (Cambridge, UK) 1999;126:1601–1609. doi: 10.1242/dev.126.8.1601. [DOI] [PubMed] [Google Scholar]
- 23.Hara K, Yonezawa K, Weng Q P, Kozlowski M T, Belham C, Avruch J. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- 24.Kim J E, Chen J. Proc Natl Acad Sci USA. 2000;97:14340–14345. doi: 10.1073/pnas.011511898. . (First Published December 12, 2000; 10.1073/pnas.011511898) [DOI] [PMC free article] [PubMed] [Google Scholar]