

Abstract
BACKGROUND & AIMS:
Acute pancreatitis (AP) is characterized by severe inflammation and acinar cell death. Transmembrane protein 173 (TMEM173 or STING) is a DNA sensor adaptor protein on immune cells that recognizes cytosolic nucleic acids and transmits signals that activate production of interferons and the innate immune response. We investigated whether leukocyte STING signaling mediates inflammation in mice with AP.
METHODS:
We induced AP in C57BL/6J mice (control) and C57BL/6J-Tmem173gt/J mice (STING-knockout mice) by injection of cerulein or placement on choline-deficient DL-ethionine supplemented diet. In some mice, STING signaling was induced by administration of a pharmacologic agonist. AP was also induced in C57BL/6J mice with bone marrow transplants from control or STING-knockout mice and in mice with disruption of the cyclic GMP-AMP synthase (Cgas) gene. Pancreata were collected, analyzed by histology, and acini were isolated and analyzed by flow cytometry, quantitative polymerase chain reaction, immunoblots, and enzyme-linked immunosorbent assay. Bone-marrow–derived macrophages were collected from mice and tested for their ability to detect DNA from dying acinar cells in the presence and absence of deoxyribonuclease (DNaseI).
RESULTS:
STING signaling was activated in pancreata from mice with AP but not mice without AP. STING-knockout mice developed less severe AP (less edema, inflammation, and markers of pancreatic injury) than control mice, whereas mice given a STING agonist developed more severe AP than controls. In immune cells collected from pancreata, STING was expressed predominantly in macrophages. Levels of cGAS were increased in mice with vs without AP, and cGAS-knockout mice had decreased edema, inflammation, and other markers of pancreatic injury upon induction of AP than control mice. Wild-type mice given bone marrow transplants from STING-knockout mice had less pancreatic injury and lower serum levels of lipase and pancreatic trypsin activity following induction of AP than mice given wildtype bone marrow. DNA from dying acinar cells activated STING signaling in macrophages, which was inhibited by addition of DNaseI.
CONCLUSIONS:
In mice with AP, STING senses acinar cell death (by detecting DNA from dying acinar cells) and activates a signaling pathway that promotes inflammation. Macrophages express STING and activate pancreatic inflammation in AP.
Keywords: Mouse Model, Apoptosis, Signal Transduction, Innate Immunity
Graphical Abstract:
Acute pancreatitis (AP) is characterized by acute abdominal pain and increased circulating pancreatic enzymes due to pancreatic acinar cell death.1 In up to 20% cases, AP is severe with high morbidity and mortality.2 In severe AP, multiple organ dysfunction ensues following pancreatic necrosis.1 To date, there is still no approved active therapy for AP. Access to an acutely inflamed pancreas remains a challenge and animal models have been used to better understand disease mechanisms and find potential therapies.
Because acinar cell injury is central to AP,1 dying cells release damage-associated molecular patterns that activate and stimulate the immune system,3 and also induce inflammation.4 For example, damage-associated molecular patterns, such as extracellularly released high-mobility group box protein 1 (HMGB1), have been shown to increase pancreatic injury and proinflammatory cytokine release.5 During necrosis and apoptosis, DNA also can be released from the nucleus into the systemic circulation,6–8 and lead to the development of autoimmune disease.9 These findings led us to hypothesize that DNA sensors may recognize DNA released by dying acinar cells to promote inflammation in AP.
Stimulator of interferon genes (STING, encoded by TMEM173) is critical for controlling type I interferons (IFNs) and proinflammatory cytokines when it senses abnormal DNA or cyclic dinucleotides in the cytosol of the cell.10–12 STING also recognizes various bacteria-secreted DNA and virus DNA, such as HSV1, HBV, KSHV, and so on.13–16 Moreover, STING is reported to induce the autoimmune disease systemic lupus erythematosus by sensing apoptotic or necrotic DNA to trigger cytokine production.17 These discoveries related to STING led us to hypothesize that STING signaling may be involved in the development of AP, particularly because STING has been shown to sense self-DNA leaked from the nucleus of host cells.18
Here we investigated the role of STING signaling in AP using experimental models induced by cerulein injection and choline-deficient diet supplemented with DL-ethionine (CDE) feeding. We found that STING and its downstream signaling are activated in AP and that leukocyte STING signaling promotes inflammation associated with AP. Furthermore, using STING-deficient mice and pharmacologic activator of STING, we show disease amelioration and worsening, respectively. Interestingly, macrophages are the predominant leukocyte subset, which up-regulate STING during AP. Notably, our ex vivo studies demonstrate that acinar cell death released DNA activates STING signaling in macrophages. Taken together, these findings suggest that STING signaling plays an important role in the development of AP via sensing of DNA released by acinar cell death.
Material and Methods
Animals
C57BL/6J mice, and C57BL/6J-Tmem173gt/J mice (STINGknockout [KO] mice) were purchased from Jackson Laboratories (Bar Harbor, ME). Cyclic GMP-AMP synthase (cGAS) KO mice were kindly provided by Dr Lingyin Li (Stanford University, Stanford, CA). Mice were housed under pathogen-free conditions. Animal use was approved by Stanford University institutional animal care and use committees.
Induction of AP and Assessment of Pancreatic Severity
For the cerulein model of pancreatitis, female mice and age-matched mice (6–8 weeks old) were fasted for 12 to 16 hours, then mice received 7 hourly intraperitoneal injections of 50 mg/kg cerulein. Sera and pancreas tissue were harvested at 12 hours or the indicated time points. For the CDE model of pancreatitis, young female mice (16–20 g) were fasted for 12 to 16 hours, then fed a choline-deficient diet (Harlan Teklad, Madison, WI) supplemented with 0.5% DL-ethionine (Sigma-Aldrich, St Louis, MO). Mice were killed at 72 hours.
For treatment with agonists of STING, mice were intraper-itoneally injected with 10 mg/kg 5,6-dimethyllxanthenone-4-acetic acid (DMXAA; MedChem Express, Monmouth Junction, NJ) after the second injection of cerulein.19 For CDE model, mice were intraperitoneally injected with 10 mg/kg DMXAA after 2 hours and 24 hours of starting the CDE diet.
Histology and Immunofluorescence
Mice were killed by CO2 inhalation. Pancreas pieces were immediately taken and fixed in 10% formalin. Fixed tissues were sent to the Stanford Pathology Laboratory for staining and processing of hematoxylin-eosin slides. The severity of pancreatitis and the mean score of each histopathologic finding was analyzed as previously described.20,21
Bone Marrow Chimeric Mice
Bone marrow (BM) chimeric mice were generated as previously described.22 C57BL/6J wild-type (WT) recipient mice were subjected to irradiation with a dose of 9.5 Gy. BM cells were collected from WT or STING KO mice by flushing femur and tibia with Hanks’ balanced salt solution containing 2% bovine calf serum. Then 5 × 106 BM cells from donor WT (WT→WT) or STING KO (KO→WT) mice were transferred into each irradiated WT recipient mice by retro-orbital injection. Eight weeks later, the WT→WT and STING KO→WT mice were subjected to cerulein-induced AP.
BM-Derived Macrophage Preparation
Femur and tibia were flushed with complete RPMI 1640 containing 10% fetal bovine serum syringe and passed through a 100-mm filter. Then cells were collected in the 50-mL tube. After centrifugation and excluding red blood cells, BM cells were cultured with 20% L929 cell-cultured medium. On day 4, fresh medium containing 10% L929 cell-cultured medium was added. Cells were in M0 profile for use on day 6.23
Preparation of Pancreatic Acini
Pancreatic acini were obtained by collagenase digestion of mouse pancreas as previously described with minor modification.24 Fresh pancreas was infused with buffer A (140 mM NaCl, 4.7 mM KCl, 1.13 mM MgCl2, 1 mM CaCl2, 10 mM glucose, 10 mM HEPES, and 0.5 mg/mL soybean trypsin inhibitor [pH 7.3]) containing 1 mg/mL type IV collagenase, minced and digested (15 minutes,100 rpm/min shaking, 37°C). Then the digested tissue was passed through 100-μm nylon mesh filter to obtain dispersed acinar cells. Acini were cultured in Dulbecco’s modified Eagle’s medium/F12 for the indicated times.
Immunostaining for Flow Cytometry
For surface staining, cells were stained with antibody to the following markers: CD45.2, CD4, CD8, CD11b, F4/80, CD11c, Ly6C, and Ly6G (BioLegend, San Diego, CA). For intracellular STING staining, after surface staining, cells were fixed and permeabilized with a kit (eBioscience, San Diego, CA) according to the manufacturer’s guidelines. Rabbit unconjugated STING antibody and Rabbit immunoglobulin G isotype control (Invitrogen, Carlsbad, CA) were used as primary antibody, and AF488 conjugated goat anti-rabbit used as secondary antibody (Life Technologies, Carlsbad, CA). Dead cells were excluded from analysis with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (ThermoFisher Scientific, Waltham, MA). Cells were run on LSRII (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (Treestar Inc, Ashland, OR).
Quantitative Polymerase Chain Reaction
Mouse pancreas tissues and cells were homogenized in TRIZOL for RNA extraction. Then RNA was reverse-transcribed into cDNAs with GoScript reverse transcription system (Promega, Madison, WI). Quantitative polymerase chain reaction was performed on ABI-7900 sequence detection system (Applied Biosystems, Foster City, CA). Primers and probes were as follows: mouse cGAS forward, 5′-GCCGAGACGGTGAA TAAAGT-3′; mouse cGAS reverse, 5′- CATTAGGAGCAGAAATCT TCACA-3′; mouse cGAS probe, 5′-AACGCCTGCTGCGCAGAATG-3′; mouse STING forward, 5′-AGCGGAAGTCTCTGCAGTCT-3′; mouse STING reverse, 5′-GGAGCCCTGGTAAGATCAAC-3′; mouse STING probe, 5′-TATGACCAGGCCAGCCCGTG-3′; mouse Mx1 forward, 5′-CTCTGGGTGTGGAGCAGGAC-3′; mouse Mx1 reverse, 5′-GAGGGCCACTCCAGACAGTG-3′; mouse Mx1 probe, 5′-CTGGC CCTGCCTGCCATCGCTGT-3′; mouse IFNb forward, 5′-ACCAGCC TGGCTTCCATCAT-3′; mouse IFNb reverse, 5′-TGTGGTGGAGAA GCACAGCA-3′; mouse IFNb probe, 5′-ACAGGTGGATCCTCCACG CTGCGTTCC-3′; Results were normalized to glyceraldehyde-3phosphate dehydrogenase and shown as fold change to control.
Western Blot and Enzyme-Linked Immunosorbent Assay Analysis
Mouse pancreas tissue and acini were homogenized in RIPA buffer (Cell Signaling, Danvers, MA) containing protease inhibitor cocktail (Sigma, St Louis, MO). cGAS, STING, phosphor-interferon regulatory factor 3 (p-IRF3), cleaved-caspase3, p65, p-p65 (Cell Signaling), CD45, and IRF3 (Abcam, Cambridge, UK) antibodies were used for Western blot.
Enzyme-linked immunosorbent assay was performed for the measurement of tumor necrosis factor (TNF)α (BioLegend), IFNβ (R&D Systems, Minneapolis, MN) in pancreas tissue homogenates and cell culture supernatant according to the manufacturer’s instructions.
Biochemical Analysis
Blood was collected from mice by intracardiac puncture, and serum was isolated for detecting lipase. Lipase level was detected by diagnostic laboratory at Stanford University. Pancreas were homogenized for detection of trypsin activity and myeloperoxidase (MPO) activity with commercial kits (Biovision, Milpitas, CA) according to the manufacturer’s guidelines. Wet lung tissue was weighed and homogenized for MPO activity using the same method as for the pancreas.22 Pancreatic edema was determined by tissue water content and presented as the difference between the wet and dry tissue weight as a percentage of the wet tissue weight.
DNaseI Treatment
A total of 100 μg/mL DNaseI (Worthington, Lakewood, NJ) was added to acinar cell supernatant and the reaction kept at 37°C for 1 hour. Then the samples were maintained at 75°C for 5 minutes to deactivate the DNaseI.
Apoptosis and Necrosis Assay
Apoptosis was detected with cleaved-caspase3 by Western blot analysis. Lactate dehydrogenase activity in acinar cell culture supernatant (ACS) was determined by the diagnostic laboratory at Stanford University.
Statistical Analysis
Unpaired Student t test was used to evaluate the significance between 2 groups. One-way analysis of variance was used to determine the differences among multiple groups. A value of P < .05 was regarded as statistically significant.
Results
STING and Its Downstream Effectors Are Activated in AP
Because acinar cell death occurs in AP, we sought to investigate the role of STING, a sensor of apoptotic and necrotic DNA. To address the role of STING signaling, we induced AP using the commonly used model of repeated injection of cerulein (50 μg/kg).25 As AP developed, cGAS and STING increased gradually at both the messenger RNA (mRNA) and protein level but decreased when assessed at 24 hours (Supplementary Figure 1A and B). For the subsequent experiments, the mice were killed at 12 hours after the first cerulein injection, when severity of pancreatic injury peaks. We found that STING, downstream genes M×1 and IFNβ mRNA level increased significantly in AP (Figure 1A). Consistent with STING activation, we detected downstream up-regulation of IRF3 (p-IRF3) and transcription of IFNβ (Figure 1A and B). STING is expressed highly in hematopoietic cells, such as macrophages and dendritic cells.10,11 Consistent with immune cell infiltration during AP, CD45 (pan-leukocyte marker) was also increased by Western blot (Figure 1B). We then analyzed STING expression in leukocytes. An increase in STING-positive leukocytes was observed in AP (Figure 1C and D) with macrophages accounting for most of the leukocyte STING expression but no significant difference was seen among CD11c+ (dendritic) cells (Figure 1E and F). To examine STING signaling in macrophages, we first sorted macrophages from control and AP mice pancreas and found that the absolute macrophage numbers increased more than 10-fold in AP (Figure 1G). STING mRNA level and protein were increased in the pancreatic macrophages (Figure 1H and I). Although cGAS was increased at mRNA and protein level in acinar cells of AP mice, STING protein level and downstream activation of p-IRF3 did not increase (Supplementary Figure 1C and D). Taken together, these results show that STING signaling is activated in AP, and leukocytes, particularly macrophage population, are the main leukocyte subset up-regulating STING in the pancreas.
Figure 1.

STING signaling is activated in AP. (A) Total tissue homogenates were obtained from pancreas of cerulein-induced AP and saline-treated (control [Con]) in C57BL/6J mice, and quantitative polymerase chain reaction (qPCR) was performed to determine STING signaling. Data are represented as mean ± SEM of n = 4 mice per group and per experiment. Representative data are shown from 3 independent experiments. (B) Total pancreas tissue was used to determine CD45, STING, and downstream proteins by Western blot. (C) C57BL/6J mice received 7 hourly cerulein injections (50 μg/kg) and were euthanized 12 hours after the first cerulein injection. Pancreas leukocytes were isolated for flow cytometry and scatter plots as well as those gated on live total leukocytes (CD45.2) are shown. (D) Percentage of STING+ cells in total pancreatic leukocytes (CD45+ cells). (E and F) STING+ leukocyte subsets in control and AP mice. Data are represented as mean ± SEM from at least 3 independent experiments (n = 4 mice per group and per experiment). (G) Pancreas absolute macrophage cell numbers from control and AP mice (6 mice were pooled for each group). Data shown are from 3 independent experiments. (H) Pancreas macrophages sorted from control and AP mice were used to analyze STING by qPCR and by (I) Western blot. Six mice were pooled for each group. Data shown are from 3 independent experiments.
STING Deficiency Attenuates AP
Because the above results showed that STING and its downstream signals are activated in AP, we speculated that STING signaling plays an important role in AP. To examine this, we induced AP with cerulein in STING-deficient mice and compared pancreas injury and inflammation with those in WT mice. Compared with WT mice, STING KO mice had less edema and inflammation in the pancreas (Figure 2A). Serum lipase, pancreas trypsin activity, pancreas MPO activity, and pancreas water content (edema) were also decreased in STING KO as compared with WT AP mice (Figure 2B and D). Importantly, the proinflammatory cytokine TNFα as well as IFNβ were decreased in the pancreas of STING KO mice (Figure 2E). Downstream activation of IRF3 (p-IRF3) and nuclear factor (NF)-kB (p-p65) were decreased in the pancreas of the STING KO AP mice (Figure 2F). Consistent with the decreased disease activity and inflammation, total leukocytes (CD45+) and neutrophils (Ly6G+) were decreased in STING KO mice (Figure 2G–I). However, there was no difference of AP-associated lung inflammation as measured by MPO between WT mice and STING KO mice (data not shown). These results suggest that STING is required for promoting pancreatic inflammation during AP.
Figure 2.

AP is attenuated in STING KO mice. (A) Histopathology and histology scores of C57BL/6J AP mice at 12 hours from first cerulein injection (50 μg/kg, 7 hourly injections) of WT and STING KO mice. Scale bar = 100 μm. (B) Serum levels of lipase in WT and STING KO in saline-treated (con) and cerulein-treated (AP) mice. (C) Pancreas trypsin and MPO activity from WT and STING KO in con and AP mice. (D) Pancreas water content from WT and STING KO in con and AP mice. (E) Bar graphs show pancreas TNFα and IFNβ from AP mice. (F) Total pancreas tissue was used to determine IRF3, p-IRF3, p65, and p-p65 by Western blot. (G) Pancreas leukocyte analyzed by flow cytometry. (H, I) Bar graph representing live total pancreas leukocytes (CD45+) and Ly6G+ neutrophils analyzed by fluorescence-activated cell sorter. All data are presented as mean ± SEM from at least 3 independent experiments (n = 5 mice per group and per experiment).
Treatment With STING Agonist Worsens AP
To further confirm the role of STING in AP, we used pharmacologic agent to induce STING activation (Figure 3A). Previous reports show that DMXAA as a STING agonist with ability to induce IFNβ and mediate antiviral activity.26,27 In contrast to STING deficiency, activation of STING with DMXAA worsened disease and led to more severe pancreas pathology (Figure 3B). Lipase in the serum, pancreas trypsin activity, pancreas MPO activity, and pancreas water content increased with DMXAA treatment (Figure 3C–E). The proinflammatory cytokine TNFα, and IFNβ, which is downstream of STING activation, increased in the pancreas of DMXAA-treated mice (Figure 3F). STING activation by DMXAA treatment is further shown by the downstream activation of IRF3 (p-IRF3) and NF-kB (p-p65) in the pancreas of the AP mice (Figure 3G). Disease worsening was also associated with increase in total leukocyte (CD45+) and neutrophil (Ly6G+) infiltration in the pancreas of mice treated with DMXAA (Figure 3H–J). Consistent with the results in which absence of STING led to amelioration of disease, activation of STING with DMXAA led to worsening of experimental AP.
Figure 3.

Treatment with STING agonist worsens AP. (A) DMXAA treatment of C57BL/6J AP mice induced as in Figure 1 with 7 hourly injections of cerulein (50 μg/kg). DMXAA (10 mg/kg) or vehicle control (VE) were administered intraperitoneally 30 minutes after second cerulein injection and mice were killed at 12 hours post first cerulein injection. (B) Representative pancreas hematoxylin-eosin images and histology scores. Scale bar = 100 μm. (C) Serum levels of lipase in VE and DMXA-Atreated con and AP mice. (D) Pancreas trypsin and MPO activity from VE and DMXAA-treated saline control (con) and AP mice. (E) Pancreatic water content from VE and DMXAA-treated con and AP mice. (F) Bar graphs show pancreas TNFα and IFNβ from AP mice. (G) Expression of IRF3, p-IRF3, p65, and p-p65 in whole pancreatic extract of AP mice shown by Western blot. (H) Pancreas leukocyte analyzed by flow cytometry. (I, J) Bar graph representing live total pancreas leukocytes (CD45+) and Ly6G+ neutrophils analyzed by fluorescence-activated cell sorter. All data are presented as mean ± SEM from at least 3 independent experiments (n = 5 mice per group and per experiment).
STING Upstream DNA-binding Protein cGAS Is Up-regulated in AP
Upstream DNA-binding proteins such as cGAS enable STING signaling and activate innate immune responses.28 Thus, to confirm that cGAS-STING pathway of the innate immune system is involved in AP, we tested whether cGAS is up-regulated and its absence leads to protection from AP. We found that cGAS was up-regulated in total pancreas tissue (Figure 4A) and in sorted pancreatic macrophages (Figure 4B and C) in AP. Similar to STING KO findings, cGAS KO mice had decreased pancreatic injury as compared with WT mice with less edema and inflammation (Figure 4D). Other markers of pancreatic injury (serum lipase, pancreas trypsin activity, pancreas MPO activity, and pancreas water content) were also decreased in the cGAS KO mice (Figure 4E–G). Proinflammatory cytokine TNFa decreased in the pancreas of cGAS KO mice relative to WT mice. Moreover, inhibition of downstream signals in the pancreas of the cGAS KO mice was demonstrated by the decrease in IFNb (Figure 4H). Total leukocytes (CD45+) and neutrophils (Ly6G+) were also decreased in the cGAS KO pancreas (Figure 4I and J). These results suggest that cGAS, which is upstream of STING, plays an important role in mediating inflammation during AP.
Figure 4.

cGAS is upregulated in AP and worsens AP. (A) Pancreas homogenates were obtained from AP and control (con) C57BL/6J mice (same as in Figure 1A), and mRNA was extracted to determine cGAS expression by quantitative polymerase chain reaction (qPCR). (B, C) Pancreas macrophages sorted from con and AP mice were used to analyze cGAS by qPCR and by Western blot (same as in Figure 1H). (D) Representative pancreas hematoxylin-eosin images and histology scores of AP mice at 12 hours following first cerulein injection (50 μg/kg, 7 hourly injections). Scale bar = 100 μm. (E) Serum levels of lipase from WT and cGAS KO in con and AP mice. (F) Pancreas trypsin and MPO activity from WT and cGAS KO in con and AP mice. (G) Pancreatic water content from WT and cGAS KO in con and AP mice. (H) Bar graphs show pancreas TNFα and IFNβ from AP mice. (I, J) Bar graph representing live total pancreas leukocytes (CD45+) and Ly6G+ neutrophils analyzed by fluorescence-activated cell sorter. All data are presented as mean ± SEM from at least 3 independent experiments (n = 5 mice per group and per experiment).
Leukocyte STING Signaling Plays an Important Role in Promoting Inflammation in AP
Considering that STING is expressed in hematopoietic cells, we sought to investigate whether STING in leukocytes plays a role in AP. To test this, we generated chimeric mice by engrafting WT mice with either WT (WT-WT) or STING KO (KO-WT) BM and induced AP with cerulein injections (Figure 5A). Similar to STING KO mice, WT mice engrafted with STING KO BM had less pancreatic injury as compared with WT chimeras (Figure 5B). Serum lipase and pancreas trypsin activity were lower in STING KO-WT as compared with WT-WT chimeric mice (Figure 5C and D). TNFα also decreased in the pancreas of STING KO-WT mice as compared with WT-WT mice (Figure 5E). IFNβ decreased in the pancreas of STING KO-WT mice (Figure 5F) highlighting contribution of leukocyte STING activity in AP. Consistent with the decrease in inflammation, total leukocytes (CD45+) and neutrophils (Ly6G+) infiltrating the pancreas were reduced in STING KO engrafted mice (Figure 5G–I). These studies show that STING signaling in leukocytes plays a critical role in experimental AP.
Figure 5.

Leukocytes play an important role in STING signaling and inflammation during AP. (A) BM chimeras were generated by transplanting WT or STING KO BM into irradiated WT mice. AP was induced following 8 weeks of BM engraftment as in the preceding figures using cerulein injection. (B) Representative hematoxylin-eosin pancreas staining and histology scores are shown from mice engrafted with WT (WT-WT) and STING KO (KO-WT) BM following AP induction. Scale bar = 100 μm. (C) Serum levels of lipase in WT-WT and STING KO-WT mice. (D) Pancreas trypsin from WT-WT and STING KO-WT mice. (E, F) Bar graphs show pancreas TNFα and IFNβ from AP mice. (G) Pancreas leukocytes analyzed by flow cytometry. (H, I) Bar graph representing live total pancreas leukocytes (CD45+) and Ly6G+ neutrophils analyzed by fluorescence-activated cell sorter. All data are presented as mean ± SEM from 2 independent experiments (n = 6 mice per group and per experiment).
Acinar Cell Death–Derived DNA Activates STING Signaling in Macrophages
Previous reports have shown that as a DNA sensor, STING can be activated by virus DNA,16 intrinsic self-DNA,29 as well as apoptotic and necrotic DNA.17 Acinar cell death is well described in AP and, based on our finding that STING is important in mediating inflammation in AP and macrophages are the predominant leukocyte population up-regulating STING, we sought to investigate the relationship between dying acinar cells and macrophage STING signaling. We took advantage of the fact that acinar cells die spontaneously post-isolation and cultured them over time as shown by the increase in cleaved-caspase3 (Figure 6A and B) as well as lactate dehydrogenase activity (Figure 6C). These findings are consistent with acinar cells undergoing apoptosis and necrosis gradually after isolation. We then collected the supernatant from the dying acinar cell cultures and exposed BM-derived macrophages (BMDM) to investigate STING activation. STING downstream genes Mx1 and IFNβ were increased in the BMDM cultured in the presence of the acinar cell culture supernatant and were inhibited in the BMDM derived from STING KO mice (Figure 6D and E). IFNβ in the WT macrophage supernatant was also increased at a protein level and diminished in the STING KO BMDM cultures (Figure 6F). STING activation as shown by overexpression of downstream p-IRF3 was present in ACS-treated BMDM (Figure 6G). These results show that acinar cell death can release molecules to activate STING signaling in macrophages. Because STING is a DNA sensor, we postulated that DNA in the ACS was the mediator for macrophage STING activation. To test this, we added DNaseI to degrade DNA present in the ACS and inhibit macrophage STING activity. Consistent with the known function of STING in sensing DNA, DNaseI treatment led to inhibition of STING activity as transcription of downstream genes (IFNβ and Mx1) and secretion of IFNβ were abolished (Figure 6H–J). These results underscore the role of leukocyte STING and cross talk between immune cells (macrophages) and dying acinar cells.
Figure 6.

Acinar cell death–derived DNA-activated STING signaling in macrophages. (A) Acinar cells were isolated from WT mice and cultured over time. Acinar cells (Acini) and supernatant (ACS) were collected at indicated times. Then BMDMs from WT and STING KO mice were cultured with the ACS with lipofectamine 2000 (0.75 mL/well) (Invitrogen). (B) Expression of cleaved-caspase3 (C-casp3) and tubulin in acinar cells at indicated time as determined by Western blot. (C) Lactate dehydrogenase (LDH) activity was tested at indicated time of acinar cell culture. (D, E) ACS was added to BMDM from WT and STING KO mice and the BMDM collected at indicated times for mRNA isolation, which was used to analyze IFNβ and Mx1 by quantitative polymerase chain reaction. (F) ACS was added to BMDM from WT and STING KO mice and cultured over time. The culture media was then collected to determine protein expression of IFNβ by enzyme-linked immunosorbent assay (ELISA). (G) ACS from acini cultured at 0 hour and 24 hours was added to stimulate BMDMs from WT and STING KO mice for 45 minutes and then the BMDM collected to determine the expression of STING, IRF3, p-IRF3, and Actin by Western blot. (H, I) ACS from acini cultured for 24 hours was collected and subjected to DNaseI treatment. BMDMs from WT mice were stimulated with ACS with or without DNaseI treatment. The BMDMs were then collected for mRNA isolation to determine expression of Mx1 and IFNβ by quantitative polymerase chain reaction and the culture supernatant was used to determine (J) IFNb by ELISA. All data are presented as mean ± SEM from 3 independent experiments (n = 3 repeats per group and per experiment).
STING Signaling Also Plays an Important Role in AP Induced With CDE Diet Feeding
As shown previously, STING plays an important role in cerulein-induced AP. To ensure that these findings are not model or strain dependent, we investigated whether STING also plays an important role in the CDE diet–induced model of AP.30 Similar to the cerulein model, the severity of AP was much reduced in the CDE-fed STING KO mice as shown by the pancreas histology (Supplementary Figure 2A), serum lipase, and pancreas trypsin activity (Supplementary Figure 2B and C). TNFα cytokine and IFNβ also decreased in the pancreas of the STING KO mice (Supplementary Figure 2D and E). Similar to cerulein model findings, DMXAA treatment worsened the pancreas injury (Supplementary Figure 2G). Serum lipase and pancreatic trypsin also increased with DMXAA treatment (Supplementary Figure 2H and I). As expected, pancreas TNFα and IFNβ levels increased in the pancreas of DMXA-Atreated CDE-fed mice (Supplementary Figure 2J and K). Because the STING KO mice are in C57BL/6J background, the CDE-fed mice here have less severe disease as previously reported and compared with our and other reports using FVB/n or Swiss Webster mice.30,31 All together, these experiments prove that STING plays a significant role in experimental models of AP.
Discussion
In severe AP, pancreatic necrosis and acinar cell death play a critical role in the development and progression of the disease. DNA leaked from necrotic pancreatic cells into circulation has been reported as a marker of disease.32 In clinical studies, plasma level of HMGB1, soluble receptor for advanced glycation end-products, and circulating DNA are regarded as markers of cell death and severity of AP.32 In mice, intracellular HMGB1 was found to inhibit inflammatory nucleosome release and reduce AP.33 However, extracellular HMGB1 promotes pancreatic injury and proinflammatory cytokine release.5 Unlike the HMGB1, presence of cytosolic DNA and cell-free DNA in circulation are perceived as a danger signal.18,32 Moreover, TLR9, which senses genomic DNA to produce pro-interleukin 1β can further activate the NLRP3 inflammasome and promote inflammation in AP.34 However, the mechanisms for acinar cell death in promoting inflammation and/or activating leukocytes remains poorly understood.
Cytosolic nucleic acids derived from self-DNA activate STING and play an important role in certain autoimmune diseases.35 STING also senses apoptotic or necrotic DNA to trigger the secretion of cytokines and IFNs that promote inflammatory diseases.17 Herein, we explored the role of STING in AP and the ability of leukocyte STING in sensing DNA from dying acinar cells. STING was significantly up-regulated in 2 independent experimental models of AP. Interestingly, macrophages in the pancreas were the predominant source for leukocyte STING expression in AP. Although STING signaling in the literature has been reported and far more studied in dendritic cells, this subset of leukocytes did not have significant alteration in STING expression during AP. Thus, our findings suggest that macrophages play an important role in sensing nucleic acids and promoting innate immune responses in AP.
To better understand the function of STING in AP, we modulated STING activation using KO mice and widely used STING agonist DMXAA.26,27,36 Consistent with previous reports that STING plays a central role in DNA recognition to drive proinflammatory cytokine and type I IFN generation, our studies led to similar findings in which absence of STING diminished AP-associated inflammation, whereas its activation led to worsening of disease. Although in development, currently pharmacologic inhibitors of STING are nonexistent, and such studies will be important in targeting proinflammatory responses in AP. In addition, and consistent with previously reports,37 STING activation in AP also led to activation of NF-kB.
Cytosolic DNA activates cGAS and generates the second messenger cyclic GMP-AMP, which activates STING and ultimately lead to production of type I IFN. Previous reports have shown that activation of the cGAS-STING pathway by self-DNA can lead to autoimmune and inflammatory disease.28 Given these findings, we tested the functional role of cGAS-STING pathway in AP using cGAS-deficient mice. In agreement with the results of the STING-deficient mice, absence of cGAS led to a decrease in IFNβ release and also had a protective role in AP. These findings demonstrate that the DNA sensing cGAS-STING pathway is active in AP and mediates inflammation.
Given the preceding findings and that STING has been well described in immune cells to mediate host innate immune responses, we performed BM chimeras to determine contribution of leukocyte STING signaling in the promotion of inflammation in AP. In support of our observation that macrophage STING is highly up-regulated in the inflamed pancreas, chimeric mice generated with adoptive transfer of STING KO BM were less susceptible to AP and had diminished IFNβ response. These results were similar to the global STING-deficient mice, indicating that leukocyte STING signaling plays an important role in AP.
Granted that acinar cell death is central to AP and our results show that macrophages are the predominant leukocyte subset that overexpresses STING, we established ex vivo cultures to study acinar cell–derived DNA sensing ability by macrophages. In fact, we found that macrophages mount a strong IFNb response to dying acinar cells and this response is abolished in the absence of STING. Because there are many factors released by dying acinar cells in the culture, we confirmed that dying cells released DNA is necessary for STING signaling in the macrophages using the DNA digesting enzyme DNaseI. Our findings highlight novel mechanism for a crosstalk between dying acinar cells and leukocytes leading to innate immune cell responses.
In our in vitro study, the released DNA from dying acinar cells can activate STING signaling in macrophages, which is consistent with the finding that adding apoptotic DNA to BMDM will cause activation of cGAS-STING signaling in vitro.17 In vivo, STING is responsible for sensing phagocytosed DNA from apoptotic cells that cannot be adequately digested.17 Besides this, tumor-derived DNA also induces IFNb production by a cGAS-STING dependent pathway in vitro and in vivo.38 Our study enriched another disease model that cGAS-STING signaling plays a role to sense DNA to release inflammatory cytokines.
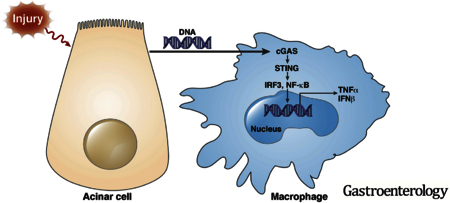
Type I IFN signaling induced by nucleotide-binding oligomerization domain-containing protein 1 (NOD1) sensing of commensal organisms has been shown in the development of pancreatitis.39 Here, we show that the DNA sensor cGAS-STING pathway mediates inflammation in AP and sensing of acinar cell death activates IFN signaling by STING in macrophages, demonstrating that IFNs play an important role in AP via various innate immune sensing pathways. In summary, we demonstrate that leukocyte STING signaling plays an important role in AP and that acinar cell death released necrotic or apoptotic DNA can activate STING signaling in macrophages and promote inflammatory cytokine and IFN production, which likely mediates AP-related inflammatory responses in the pancreas (Figure 7). Moreover, the results indicate that STING signaling–activated macrophages cause further acinar cell injury as represented by increased trypsin activation in the pancreatic tissue, a finding consistent with previous work showing the role of leukocytes in acinar injury and trypsin activation.40 These discoveries provide new ways to target inflammation and treat AP.
Figure 7.

Schematic diagram of acinar cell death–mediated DNA sensing by macrophage STING during AP.
Supplementary Material
Supplementary Figure 1. cGAS-STING expression and signaling over time in total and acinar cells in AP. (A) Total pancreas homogenates were obtained from 7 hourly cerulein-injected C57BL/6J AP mice euthanized at the indicated time points (0–24 hours). cGAS and STING mRNA as determined by quantitative polymerase chain reaction (qPCR). Data are presented as mean ± SEM from 2 independent experiments (n ¼ 5 mice per group and per experiment). (B) Total pancreas tissue was used to determine cGAS, STING, and downstream proteins by Western blot. (C) Pancreas acinar cells were obtained from pancreas of control (saline-treated) and cerulein-treated C57BL/6J AP mice at 12 hours following first cerulein injection (50 mg/kg, 7 hourly injections). cGAS and STING mRNA as determined by qPCR. Data are represented as mean ± SEM from 2 independent experiments (n ¼ 5 mice per group per experiment). (D) Acinar cells from experiments in (C) were used to determine cGAS, STING, and downstream proteins by Western blot.
Supplementary Figure 2. STING signaling also plays an important role in AP induced with CDE diet feeding. (A) Representative hematoxylin-eosin (H&E) pancreas staining and histology scores of C57BL/6J WT and STING KO mice euthanized 72 hours following CDE diet feeding. (B) Serum levels of lipase in WT and STING KO mice. (C) Pancreas trypsin activity from WT and STING KO mice. (D, E) Bar graphs show pancreas TNFa and IFNb from WT and STING KO mice. (F) DMXAA treatment of AP mice induced with 72 hours of CDE diet feeding. DMXAA (10 mg/kg) or vehicle control (VE) was administered intraperitoneally at 6 hours and 36 hours post initiation of CDE diet feeding. (G) Representative H&E pancreas staining and histology scores of mice at 72 hours following CDE diet feeding. (H) Serum levels of lipase in VE and DMXAA-treated mice. (I) Pancreas trypsin activity from VE and DMXAA-treated mice. (J, K) Bar graphs show pancreas TNFa and IFNb from VE and DMXAAtreated mice. All data are presented as mean ± SEM of 3 independent experiments (n ¼ 5 mice per group and per experiment).
EDITOR’S NOTES
BACKGROUND AND CONTEXT
Acute pancreatitis is associated with significant acinar cell damage and death, but the mechanism via which acinar cell death results in inflammation and activation of immune cells remains poorly understood.
NEW FINDINGS
The researchers show that DNA released from dying acinar cells is sensed by STING in macrophages and leads to activation of downstream pro-inflammatory cytokines. Moreover, deletion of STING resulted in amelioration of acute pancreatitis, whereas its activation worsened the disease.
LIMITATIONS
This Study involves experimental models and needs to be validated in clinical studies.
IMPACT
The study improves our understanding of the inflammatory signals involved in acute pancreatitis and currently there are no active therapies for this disease and the study might lead to novel therapies.
Acknowledgments
Funding
This work was supported by the National Institutes of Health grant DK092421 (to Aida Habtezion), and P01 DK098108 and R01 AA024464 (to Stephen J.Pandol).
Abbreviations used in this paper:
- ACS
acinar cell culture supernatant
- AP
acute pancreatitis
- BM
bone marrow
- BMDM
BM-derived macrophages
- CDE
choline-deficient diet supplemented with DL-ethionine
- cGAS
cyclic GMP-AMP synthase
- DMXAA
5,6-dimethyllxanthenone-4-acetic acid
- HMGB1
high-mobility group box protein 1
- IFN
interferon
- IRF3
interferon regulatory factor 3
- KO
knockout
- MPO
myeloperoxidase
- mRNA
messenger RNA
- NF-kB
nuclear factor–kB
- STING
stimulator of interferon genes
- TNF
tumor necrosis factor
- WT
wild-type
Footnotes
Conflicts of interest
The authors disclose no conflicts.
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2018.01.065.
Author names in bold designate shared co-first authorship.
References
- 1.Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. Lancet 2008;371:143–152. [DOI] [PubMed] [Google Scholar]
- 2.Lund H, Tonnesen H, Tonnesen MH, et al. Long-term recurrence and death rates after acute pancreatitis. Scand J Gastroenterol 2006;41:234–238. [DOI] [PubMed] [Google Scholar]
- 3.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008;8:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majno G, La Gattuta M, Thompson TE. Cellular death and necrosis: chemical, physical and morphologic changes in rat liver. Virchows Arch Pathol Anat Physiol Klin Med 1960;333:421–465. [DOI] [PubMed] [Google Scholar]
- 5.Li G, Wu X, Yang L, et al. TLR4-mediated NF-kappaB signaling pathway mediates HMGB1-induced pancreatic injury in mice with severe acute pancreatitis. Int J Mol Med 2016;37:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med 1999; 5:1249–1255. [DOI] [PubMed] [Google Scholar]
- 7.Sauter B, Albert ML, Francisco L, et al. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med 2000;191:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barker RN, Erwig L, Pearce WP, et al. Differential effects of necrotic or apoptotic cell uptake on antigen presentation by macrophages. Pathobiology 1999;67:302–305. [DOI] [PubMed] [Google Scholar]
- 9.Napirei M, Karsunky H, Zevnik B, et al. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 2000;25:177–181. [DOI] [PubMed] [Google Scholar]
- 10.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008;455:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009;461:788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burdette DL, Monroe KM, Sotelo-Troha K, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011;478:515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauer JD, Sotelo-Troha K, von Moltke J, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 2011;79:688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su C, Zheng C. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J Virol 2017;91(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Zhao K, Su X, et al. MITA/STING and its alternative splicing isoform MRP restrict hepatitis B virus replication. PLoS One 2017;12:e0169701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Z, Jacobs SR, West JA, et al. Modulation of the cGAS-STING DNA sensing pathway by gamma-herpesviruses. Proc Natl Acad Sci U S A 2015; 112:E4306–E4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahn J, Gutman D, Saijo S, et al. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A 2012;109:19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn J, Barber GN. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr Opin Immunol 2014;31:121–126. [DOI] [PubMed] [Google Scholar]
- 19.Zhao L, Ching LM, Kestell P, et al. Improvement of the antitumor activity of intraperitoneally and orally administered 5,6-dimethylxanthenone-4-acetic acid by optimal scheduling. Clin Cancer Res 2003;9:6545–6550. [PubMed] [Google Scholar]
- 20.Spormann H, Sokolowski A, Letko G. Experimental acute pancreatitis—a quantification of dynamics at enzymic and histomorphologic levels. Pathol Res Pract 1989; 185:358–362. [DOI] [PubMed] [Google Scholar]
- 21.Spormann H, Sokolowski A, Letko G. Effect of temporary ischemia upon development and histological patterns of acute pancreatitis in the rat. Pathol Res Pract 1989; 184:507–513. [DOI] [PubMed] [Google Scholar]
- 22.Xue J, Habtezion A. Carbon monoxide-based therapy ameliorates acute pancreatitis via TLR4 inhibition. J Clin Invest 2014;124:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc 2008;2008:pdb prot5080. [DOI] [PubMed] [Google Scholar]
- 24.Menozzi D, Jensen RT, Gardner JD. Dispersed pancreatic acinar cells and pancreatic acini. Methods Enzymol 1990;192:271–279. [DOI] [PubMed] [Google Scholar]
- 25.Baumann B, Wagner M, Aleksic T, et al. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest 2007;117: 1502–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shirey KA, Nhu QM, Yim KC, et al. The anti-tumor agent, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), induces IFN-beta-mediated antiviral activity in vitro and in vivo. J Leukoc Biol 2011;89:351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo F, Han Y, Zhao X, et al. STING agonists induce an innate antiviral immune response against hepatitis B virus. Antimicrob Agents Chemother 2015;59: 1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 2016;17:1142–1149. [DOI] [PubMed] [Google Scholar]
- 29.Ahn J, Ruiz P, Barber GN. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J Immunol 2014;193:4634–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Habtezion A, Kwan R, Akhtar E, et al. Panhematin provides a therapeutic benefit in experimental pancreatitis. Gut 2011;60:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lombardi B, Rao NK. Acute hemorrhagic pancreatic necrosis in mice. Influence of the age and sex of the animals and of dietary ethionine, choline, methionine, and adenine sulfate. Am J Pathol 1975;81:87–100. [PMC free article] [PubMed] [Google Scholar]
- 32.Kocsis AK, Szabolcs A, Hofner P, et al. Plasma concentrations of high-mobility group box protein 1, soluble receptor for advanced glycation end-products and circulating DNA in patients with acute pancreatitis. Pancreatology 2009;9:383–391. [DOI] [PubMed] [Google Scholar]
- 33.Kang R, Zhang Q, Hou W, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 2014;146: 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoque R, Sohail M, Malik A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011;141: 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gall A, Treuting P, Elkon KB, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 2012;36:120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Curran E, Chen X, Corrales L, et al. STING pathway activation stimulates potent immunity against acute myeloid leukemia. Cell Rep 2016;15:2357–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessi-tates canonical NF-kappaB activation through TBK1. J Virol 2014;88:5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41:830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuji Y, Watanabe T, Kudo M, et al. Sensing of commensal organisms by the intracellular sensor NOD1 mediates experimental pancreatitis. Immunity 2012; 37:326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gukovskaya AS, Vaquero E, Zaninovic V, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 2002;122:974–984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. cGAS-STING expression and signaling over time in total and acinar cells in AP. (A) Total pancreas homogenates were obtained from 7 hourly cerulein-injected C57BL/6J AP mice euthanized at the indicated time points (0–24 hours). cGAS and STING mRNA as determined by quantitative polymerase chain reaction (qPCR). Data are presented as mean ± SEM from 2 independent experiments (n ¼ 5 mice per group and per experiment). (B) Total pancreas tissue was used to determine cGAS, STING, and downstream proteins by Western blot. (C) Pancreas acinar cells were obtained from pancreas of control (saline-treated) and cerulein-treated C57BL/6J AP mice at 12 hours following first cerulein injection (50 mg/kg, 7 hourly injections). cGAS and STING mRNA as determined by qPCR. Data are represented as mean ± SEM from 2 independent experiments (n ¼ 5 mice per group per experiment). (D) Acinar cells from experiments in (C) were used to determine cGAS, STING, and downstream proteins by Western blot.
Supplementary Figure 2. STING signaling also plays an important role in AP induced with CDE diet feeding. (A) Representative hematoxylin-eosin (H&E) pancreas staining and histology scores of C57BL/6J WT and STING KO mice euthanized 72 hours following CDE diet feeding. (B) Serum levels of lipase in WT and STING KO mice. (C) Pancreas trypsin activity from WT and STING KO mice. (D, E) Bar graphs show pancreas TNFa and IFNb from WT and STING KO mice. (F) DMXAA treatment of AP mice induced with 72 hours of CDE diet feeding. DMXAA (10 mg/kg) or vehicle control (VE) was administered intraperitoneally at 6 hours and 36 hours post initiation of CDE diet feeding. (G) Representative H&E pancreas staining and histology scores of mice at 72 hours following CDE diet feeding. (H) Serum levels of lipase in VE and DMXAA-treated mice. (I) Pancreas trypsin activity from VE and DMXAA-treated mice. (J, K) Bar graphs show pancreas TNFa and IFNb from VE and DMXAAtreated mice. All data are presented as mean ± SEM of 3 independent experiments (n ¼ 5 mice per group and per experiment).