

Summary
An intronic GGGGCC repeat expansion in C9ORF72 is the most common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), but its pathogenic mechanism remains unclear. Here we use human induced motor neurons (iMNs) to show that repeat-expanded C9ORF72 is haploinsufficient in ALS. We show that C9ORF72 interacts with endosomes and is required for normal vesicle trafficking and lysosomal biogenesis in motor neurons. Repeat expansion reduces C9ORF72 expression, triggering neurodegeneration through two mechanisms: accumulation of glutamate receptors leading to excitotoxicity, and impaired clearance of neurotoxic dipeptide repeat proteins derived from the repeat expansion. Thus, cooperativity between gain- and loss-of-function mechanisms leads to neurodegeneration. Restoring C9ORF72 levels or augmenting its function with constitutively active RAB5 or chemical modulators of RAB5 effectors rescues patient neuron survival and ameliorates neurodegenerative processes in both gain- and loss-of function C9ORF72 mouse models. Thus, modulating vesicle trafficking can rescue neurodegeneration caused by the C9ORF72 repeat expansion. Coupled with rare mutations in ALS2, FIG4, CHMP2B, OPTN, and SQSTM1, our results reveal mechanistic convergence on vesicle trafficking in ALS/FTD.
The GGGGCC repeat expansion in C9ORF72 is the most common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), accounting for about 10% of each disease worldwide 1–4. In the central nervous system (CNS), neurons and microglia express the highest levels of C9ORF72 5, suggesting that C9ORF72 acts in part cell autonomously and effects in neurons are a key source of disease etiology. Studies showing that the repeat expansion generates neurotoxic species including nuclear RNA foci 6–8, RNA/DNA G-quadruplexes 9, and dipeptide repeat proteins (DPRs) 10–12 have oriented the field towards a therapeutic focus on blocking the toxicity of these products 6–8,13,14. However, these strategies have not fully rescued the degeneration of patient-derived neurons 7,13. Moreover, tandem GGGGCC repeats are transcribed from over 80 other genomic locations within human spinal motor neurons (Supplementary Tables 1 and 2), yet genetic studies have not linked repeat expansions in these regions to ALS/FTD. In addition, hexanucleotide repeat-mediated toxicity in mice requires supraphysiological expression levels or a specific genetic background 14–16. These observations suggest that there are additional pathogenic triggers caused by repeat expansion within C9ORF72.
The repeat expansion suppresses the production of C9ORF72 protein by inhibiting transcription 3,4,6,7,9,17, raising the possibility that haploinsufficiency for C9ORF72 activity triggers disease pathogenesis. Consistent with this hypothesis, elimination of C9orf72 activity alters myeloid cell behavior in mice 14,18,19 and in vitro studies suggest that C9ORF72 activity may enhance autophagy 20,21.
However, C9orf72-deficient mice do not display overt neurodegenerative phenotypes 14,18,19,22. Moreover, no studies have shown that reduced C9ORF72 activity leads to the degeneration of C9ORF72 ALS patient-derived motor neurons, nor have any provided direct evidence identifying a cellular pathway through which C9ORF72 activity modulates neuronal survival. Additionally, a patient homozygous for the C9ORF72 repeat expansion had clinical and pathological phenotypes that were severe but nonetheless did not fall outside the range of heterozygous patients, leaving it uncertain if reductions in C9ORF72 protein levels directly correlate with disease severity 23. Thus, the role of the C9ORF72 protein in C9ORF72 ALS/FTD disease pathogenesis remains unclear.
C9ORF72 patient iMNs recapitulate ALS disease processes
To study the pathogenic mechanism of the C9ORF72 repeat expansion in human motor neurons, we used the forced expression of the transcription factors Ngn2, Isl1, Lhx3, NeuroD1, Brn2, Ascl1, and Myt1l, to convert control and C9ORF72 ALS/FTD patient induced pluripotent stem cells (iPSCs)(for iPSC characterization, see Supplementary Fig. 1 and Supplementary Tables 3, 4) into iMNs (Supplementary Fig. 2a, b) 10,24. Control and patient iMNs labeled with an Hb9::RFP+ lentiviral reporter construct (Supplementary Fig. 2b-d)25 co-expressed spinal motor neuron markers including TUJ1, HB9, and VACHT; were produced at similar rates amongst different iPSC lines; and possessed electrophysiological properties of motor neurons (Supplementary Fig. 2c-i). Depolarizing voltage steps induced currents characteristic of sodium and potassium channels and iMNs fired single or repetitive action potentials (patient - 90%, n=10; control – 100%, n=10)(Supplementary Fig. 2g-i). When co-cultured with primary chick muscle, channel rhodopsin-expressing control and patient iMNs repeatedly induced myotube contraction upon depolarization with green light, indicating they formed neuromuscular junctions and actuated muscle contraction (Supplementary Fig. 2j and Supplementary Videos 1, 2).
To determine if C9ORF72 iMNs recapitulate neurodegenerative ALS processes, we examined their survival by performing longitudinal tracking of Hb9::RFP+ iMNs (Fig. 1a). This approach enabled us to distinguish differences in neurogenesis from differences in survival, which could not be addressed using previously-reported cross-sectional analyses6,7,10,26. In basal neuronal medium supplemented with neurotrophic factors, control and C9ORF72 patient iMNs survived equally well (Fig. 1b, Supplementary Fig. 3a, Supplementary Tables 5, 6). As human C9ORF72 ALS patients have elevated glutamate levels in their cerebrospinal fluid (possibly triggered by DPR-mediated aberrant splicing of the astrocytic excitatory amino acid transporter 2 EAAT2 4,27) we stimulated iMN cultures with a high glutamate pulse (12-hour treatment, 10 μM glutamate). This initiated a robust degenerative response in patient, but not control, iMNs (Fig. 1c-e and Supplementary Videos 3, 4) that was consistent across lines from multiple patients (n=6 patients) and controls (n=4 controls)(Fig. 1c, d and Supplementary Fig. 3d, e). While iMN survival varied slightly between live imaging systems, or between independent experiments due to the lengthy time course of neurodegeneration, the relative difference between control and C9-ALS patient iMNs was consistent (Fig. 1c - Nikon Biostation CT and Supplementary Fig. 3b - Molecular Devices ImageExpress). Moreover, iMNs from different iPSC lines derived from the same donor behaved similarly, suggesting genotypic differences accounted for these effects (Supplementary Fig. 3c). Treatment with glutamate receptor antagonists during glutamate administration prevented patient iMN degeneration (Fig. 1f). Alternatively, withdrawal of neurotrophic factors without glutamate stimulation also caused rapid degeneration of patient iMNs (n=3 patients, (Fig. 1g and Supplementary Fig. 3f).
Figure 1. C9ORF72 patient iMNs undergo rapid neurodegeneration.
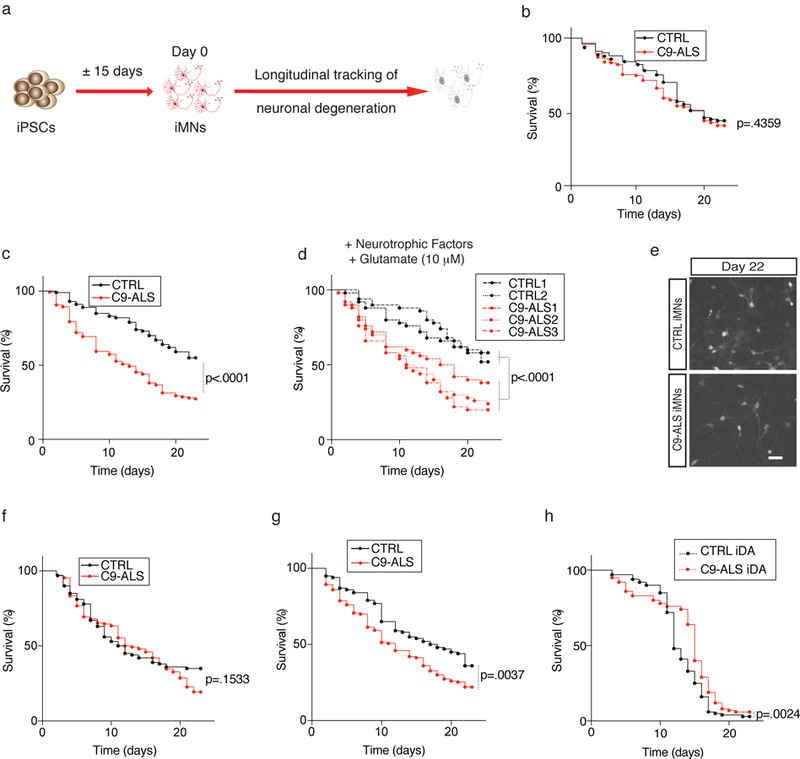
(a) Production of Hb9::RFP+ iMNs and survival tracking by time-lapse microscopy. (b-d) Survival of control (CTRL) and C9ORF72 patient (C9-ALS) iMNs with neurotrophic factors (b) or in excess glutamate (shown with iMNs from all lines in aggregate (b, c) or for each individual line separately (d)). For (b-d), n=50 iMNs per line for 2 control and 3 C9-ALS lines, iMNs quantified from 3 biologically independent iMN conversions per line. (e) iMNs at day 22 in excess glutamate. This experiment was repeated three times with similar results. (f-g) Survival of control and C9-ALS iMNs in excess glutamate with glutamate receptor antagonists (f) or without neurotrophic factors (g). For (f-g), n=50 iMNs per line for 2 control and 3 C9-ALS lines, iMNs quantified from 3 biologically independent iMN conversions per line. (h) Survival of induced dopaminergic (iDA) neurons in excess glutamate. n=50 iMNs per line for 2 control and 2 C9-ALS lines, iMNs quantified from 3 biologically independent iMN conversions per line. Except in (d), each trace includes neurons from at least 2 donors with the specified genotype; see full detail in Methods. Scale bar: 100 μm (e). All iMN survival experiments were analyzed by two-sided log-rank test, and statistical significance was calculated using the entire survival time course. iMN survival experiments in (b-g) were performed in a Nikon Biostation and experiments in (h) were performed in a Molecular Devices ImageExpress.
To determine if the survival difference between C9ORF72 patient iMNs and controls was specific to our transcription factor-based reprogramming approach, we also measured the survival of Hb9::RFP+ control and C9ORF72 patient motor neurons derived from iPSCs by small molecule activation of the Sonic Hedgehog and retinoic acid signaling pathways 28 (Supplementary Fig. 3g, h). Similarly to iMNs, morphogen-generated motor neurons showed a significant survival difference between C9ORF72 patients and controls (Supplementary Fig. 3i-l).
To determine if patient iMN degeneration resulted from bona fide ALS disease processes specific for motor neurons, we measured the survival of induced dopaminergic neurons (iDAs) generated by expression of FoxA2, Lmx1a, Brn2, Ascl1, and Myt1l 29. These neurons expressed high levels of tyrosine hydroxylase, indicating they had established a key aspect of the dopamine synthesis pathway and were distinct from iMNs, which do not express this enzyme 24 (Supplementary Fig. 3m, n). Unlike iMN cultures, iDA cultures from C9ORF72 patients (n=2 patients) did not show reduced survival compared to controls (n=2 controls) in either glutamate treatment and neurotrophic factor withdrawal conditions (Fig. 1h and Supplementary Fig. 3o), indicating that the in vitro neurodegenerative phenotype elicited by the C9ORF72 mutation is selective for motor neurons.
C9ORF72 is haploinsufficient in ALS
Consistent with previous studies 3,4,6–8, patient iMNs (n=5 patients) had reduced C9ORF72 expression compared to controls (n=3; Fig. 2a and Supplementary Fig. 4a, 5b). While previous studies have linked low C9ORF72 levels to changes in vesicle trafficking or autophagy 18,20,30–33, it remains unknown if loss of C9ORF72 protein directly contributes to degeneration. Thus, we re-expressed C9ORF72 (isoform A or B) in iMNs using a retroviral cassette (Supplementary Fig. 4b) and found that both isoforms rescued C9ORF72 patient iMN survival in response to glutamate treatment (n=3 patients Fig. 2b and Supplementary Fig. 4c). This effect was specific for C9ORF72 iMNs, as forced expression of C9ORF72 did not rescue SOD1A4V iMN survival (Fig. 2c), nor did it improve the survival of control iMNs (n=2 controls Fig. 2d and Supplementary Fig. 4d).
Figure 2. C9ORF72 protein levels determine iMN survival.
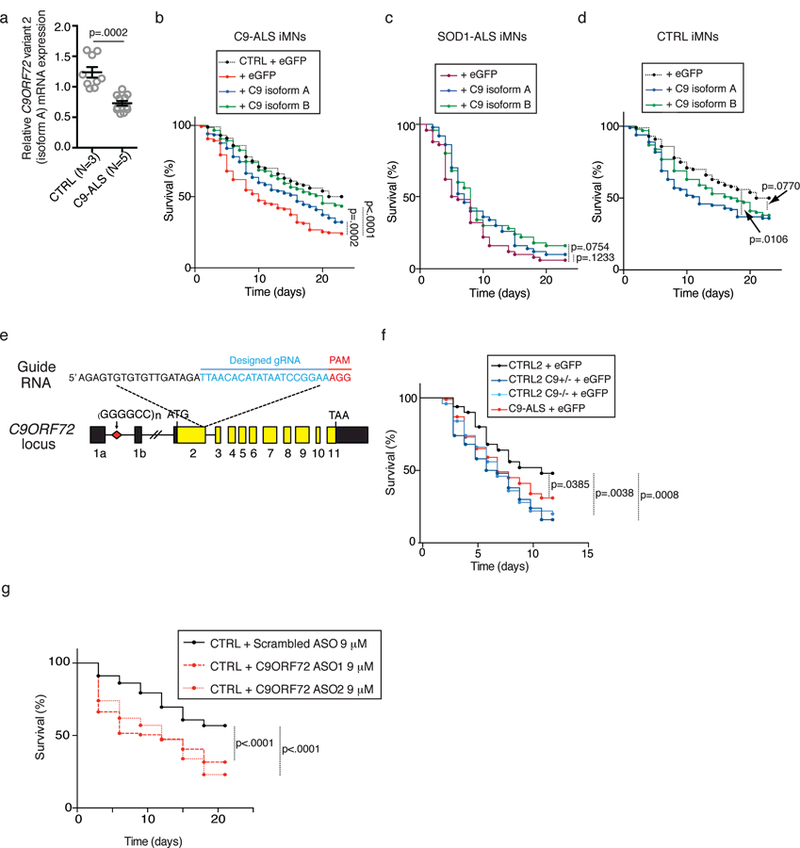
(a) The levels of C9ORF72 variant 2 mRNA transcript (encoding isoform A). Values are mean ± s.e.m., two-tailed t-test with Welch’s correction. t-value: 5.347, degrees of freedom: 11.08. n= 9 biologically independent iMN conversions from 3 control lines and 12 biologically independent iMN conversions from 5 C9-ALS lines. (b–d) iMN survival in excess glutamate following introduction of C9ORF72 (C9 isoform A or B) into C9ORF72 patient iMNs (b), but not control (b, d) or SOD1-ALS iMNs (c). For (b), n=50 iMNs per line for 2 control and 3 C9-ALS lines, iMNs quantified from 3 biologically independent iMN conversions per line. For (c), n=50 iMNs per condition, iMNs scored from 3 biologically independent iMN conversions. For (d), n=50 iMNs per line per condition for 2 control lines, iMNs quantified from 3 biologically independent iMN conversions. Each trace includes iMNs from 2–3 donors with the specified genotype (except SOD1-ALS (c)); see full details in Methods. (e) Strategy for knocking out C9ORF72 from control iPSCs using CRISPR/Cas9. (f) Survival of control (CTRL2) iMNs, the isogenic heterozygous (C9+/−) and homozygous (C9−/−) iMNs and C9ORF72 patient (C9-ALS) iMNs in excess glutamate. n=50 biologically independent iMNs per line per condition for one control and two C9-ALS lines, iMNs quantified from 3 biologically independent iMN conversions. (g) Control iMN survival in excess glutamate with scrambled or C9ORF72 antisense oligonucleotides (ASO). Each trace includes control iMNs from 2 donors. n=50 biologically independent iMNs per line per condition for 2 control lines, iMNs quantified from 3 biologically independent iMN conversions. All iMN survival experiments were analyzed by two-sided log-rank test, and statistical significance was calculated using the entire survival time course. iMN survival experiments in (b, d, and g) were performed in a Nikon Biostation, and (e and f) were performed in a Molecular Devices ImageExpress.
To confirm that reduced C9ORF72 protein levels are sufficient to cause neurodegeneration, we used CRISPR/Cas9-mediated genome editing to introduce a frameshift mutation into one or both alleles of C9ORF72 in control iPSCs (Fig. 2e and Supplementary Fig. 4e). qPCR showed that targeting one allele reduced C9ORF72 transcript levels due to nonsense-mediated decay and transcript levels were more severely reduced in homozygous mutant cells (Supplementary Fig. 4f). Frameshift mutations also decreased C9ORF72 protein expression (Supplementary Fig. 4g, 5c). RNA sequencing of flow-purified Hb9::RFP+ iMNs showed that targeting C9ORF72 did not significantly alter the expression of the top 10 genes with predicted off-target sites for the CRISPR guide RNA (Supplementary Fig. 4h and Supplementary Table 7). In addition, expression levels of the 20 genes nearest C9ORF72 on chromosome 9 were largely unperturbed in either the C9ORF72+/− and C9ORF72−/− iMNs, indicating that this approach specifically inactivated C9ORF72 (Supplementary Fig. 4i).
Eliminating C9ORF72 protein expression from one or both alleles reduced iMN survival to levels comparable to patient iMNs (Fig. 2f). Antisense oligonucleotide (ASO)-mediated suppression of C9ORF72 expression levels also reduced control iMN survival (Fig. 2g and Supplementary Fig. 4j), suggesting that reduced iMN survival was not due to an off-target effect of the CRISPR/Cas9 genome editing. Exogenously restoring C9ORF72 expression in C9ORF72+/− and C9ORF72−/− iMNs rescued survival (Supplementary Fig. 4k, l), verifying that depletion of C9ORF72 caused the observed neurodegeneration.
To determine if transcriptional changes in C9ORF72+/− and C9ORF72−/− iMNs also reflect the contribution of C9ORF72 protein levels to neurodegeneration, we performed RNA sequencing on flow-purified Hb9::RFP+ iMNs from C9ORF72+/−, C9ORF72−/−, and isogenic control iMNs, as well as C9ORF72 patient iMNs (Supplementary Table 7), and compared them to existing RNA-seq data from postmortem tissue 34,35. When examining consensus genes that were differentially expressed compared to controls in all C9ORF72 patient postmortem datasets (from GSE56504 and GSE67196)34,35, both C9ORF72+/− and C9ORF72 patient iMNs shared similar gene expression changes to the postmortem tissue (Supplementary Fig. 6). Thus, a reduction in C9ORF72 levels induces disease-associated transcriptional changes observed in C9ORF72 patient postmortem samples.
Reduced C9ORF72 activity disrupts lysosomal biogenesis in motor neurons
Determining the functional pathway in which C9ORF72 acts may enable design of function-directed therapies. C9ORF72 displays limited structural homology to Differentially Expressed in Normal and Neoplasia (DENN)-domain containing proteins36, many of which modulate RAB GTPase activity and vesicle trafficking 37.
There are conflicting reports as to whether C9ORF72 levels affect endosomes or lysosomes more significantly, and whether C9ORF72 levels affect vesicle trafficking in neurons in the same way as in cell lines 18,30–33,38,39. In addition, the functional role of C9ORF72 in patient motor neurons is unclear.
To examine C9ORF72 function, we determined its localization in iMNs. We first used an HA-tagged C9ORF72 construct to verify that the C9ORF72 antibody specifically recognizes C9ORF72 in cells (Supplementary Fig. 7a). In iMNs, C9ORF72 co-localized to cytoplasmic puncta and ASO-mediated knockdown of C9ORF72 expression reduced the number of antibody-detected cytoplasmic puncta in iMNs, indicating that the antibody specifically recognizes C9ORF72 in these puncta (Supplementary Fig. 7b, c). Super-resolution microscopy and z-stack imaging showed that about 80% of the C9ORF72+ vesicles also expressed the early endosomal proteins RAB5 and EEA1 (Fig. 3a and Supplementary 7d-h). Only rarely did C9ORF72 co-localize with the lysosomal marker LAMP1 (20%)(Supplementary Fig. 7e), and control and patient iMNs showed similar C9ORF72 localization (Supplementary Fig. 7h). We performed density gradient centrifugation on lysates from iPSC-derived motor neurons to separate light (endosomal) and heavy (lysosomal) membrane fractions. C9ORF72 co-segregated with EEA1 and not LAMP1, supporting the notion that C9ORF72 localizes predominantly in early endosomes (Fig. 3b, Supplementary Fig. 5d). In addition, we found that C9ORF72 isoform B bound strongly to an immobilized N-terminal fragment of EEA1 (Supplementary Fig. 7i). C9ORF72 isoform A did not interact as strongly with EEA1 (Supplementary Fig. 7i). The fact that not all EEA1+ vesicles contained high levels of C9ORF72 is consistent with this hypothesis and suggests that C9ORF72 may not localize to all types of EEA1+ vesicles (Fig. 3a).
Figure 3. Reduced C9ORF72 activity disrupts vesicle trafficking and lysosomal biogenesis in motor neurons.
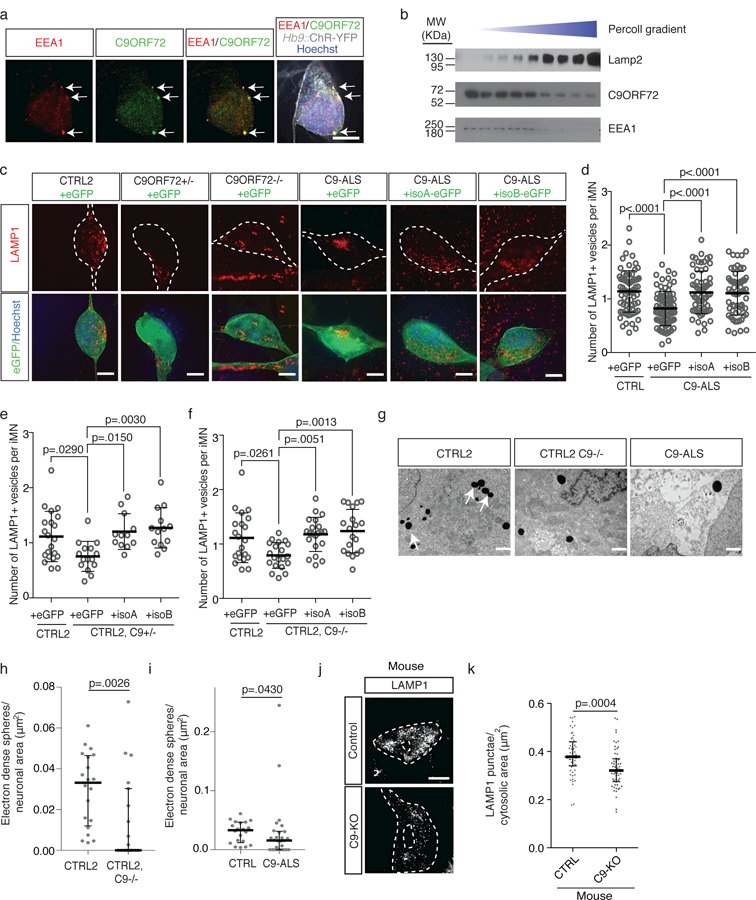
(a) Super-resolution microscopy images of control iMNs showing colocalization (arrows) of C9ORF72 (green) with EEA1 (red). Scale bar: 5 µm. This experiment was repeated 3 times with similar results. (b) Immunoblot against C9ORF72, EEA1, and LAMP1 on lysates from iPSC-derived motor neurons separated into light (endosomal) and heavy (lysosomal) membrane fractions using percoll gradient centrifugation. This experiment was repeated twice with similar results. (c) Super-resolution microscopy images of LAMP1 immunostaining in iMNs of specified genotypes expressing eGFP or C9ORF72 (isoform A or B)-eGFP. Scale bar: 5 µm. This experiment was repeated 3 times with similar results. (d-f) Number of LAMP1+ vesicles in control (d-f), patient (d), C9ORF72+/− (e), and C9ORF72−/− (f) iMNs overexpressing eGFP or C9ORF72 (isoform A or B)-eGFP. Each grey open circle represents a single iMN, Mean ± s.d. For (d), n=80 (CTRL + GFP), 80 (C9-ALS + GFP), 64 (C9-ALS + isoA), and 61 (C9-ALS + isoB) iMNs quantified from two biologically independent iMN conversions of 3 CTRL or 4 C9-ALS lines. For (e), n=20 (CTRL + GFP), 15 (C9ORF72+/− + GFP), 12 (C9ORF72+/− + isoA), and 13 (C9ORF72+/− + isoB) iMNs f quantified from two biologically independent iMN conversions per condition. For (f), n=20 iMNs quantified from two biologically independent iMN conversions per condition. One-way ANOVA with Tukey correction between CTRL2 and C9ORF72+/− and C9ORF72−/− (e, f), one-way ANOVA with Tukey correction between controls and patient conditions (d). F-value (DFn, DFd): (3, 273)=12.12 (d), (3, 57)=5.64 (e), (3, 77)=6.091 (f). Dotted lines outline iMNs. (g) Representative electron micrographs of control, C9ORF72−/−, and patient iMNs showing lysosomes as electron-dense spherical perinuclear structures (arrows). This experiment was repeated twice with similar results. Scale bar: 1 μm. (h-i) Number of electron-dense spheres per square micron of perinuclear cytosol in control (h-i), C9ORF72−/− (h), and patient iMNs (i) Median ± interquartile range, each data point represents a single cell, Two-sided Mann-Whitney test). For (h), n=20 (CTRL2) and 19 (C9ORF72+/−), and for (i) n=20 (CTRL2) and 26 (C9ORF72 patient) cells quantified from two biologically independent iMN conversions of one line per genotype. (j) Super-resolution microscopy images of Lamp1 immunoreactivity in control and C9-KO mouse spinal neurons. This experiment was repeated twice with similar results. Scale bar: 5 μm. (k) Number of Lamp1+ punctae in Chat+ mouse spinal neurons. Median ± interquartile range, two-tailed t-test. t-value: 3.681. Degrees of freedom: 113. n=59 (CTRL2) and 56 (C9ORF72−/−) cells quantified from sections of two mice per genotype.
To determine if a deletion of C9ORF72 or the C9ORF72 repeat expansion caused changes in endosomal trafficking in motor neurons, we examined the number of early endosomes (RAB5+, EEA1+), late endosomes (RAB7+), and lysosomes (LAMP1+, LAMP2+, LAMP3+) in control, C9ORF72 patient, C9ORF72+/−, and C9ORF72−/− iMNs. We observed the most significant difference in the lysosomal population, with C9ORF72 patient iMNs (n=4 patients) having fewer LAMP1+, LAMP2+, and LAMP3+ vesicles than control iMNs (n=4 controls)(Fig. 3c, d and Supplementary Fig. 8a-d). C9ORF72+/− and C9ORF72−/− also harbored fewer LAMP1+, LAMP2+, and LAMP3+ vesicles than isogenic control iMNs, indicating that reduced C9ORF72 levels alone leads to a loss of lysosomes (Fig. 3c, e, f and Supplementary Fig. 8a-d). ASO-mediated knockdown of C9ORF72 expression also decreased lysosome numbers in iMNs (Supplementary Fig. 8e). Although membrane fractionation showed that control and patient iMNs have similar amounts of LAMP2 in the lysosomal membrane fraction (Supplementary Fig. 8f), analysis of the immunofluorescence intensity of LAMP proteins suggests that this is likely due to the fact that C9ORF72 patient and C9ORF72+/− iMNs have a higher concentration of LAMP proteins in their lysosomal membranes, possibly as a result of fewer lysosomes being present (Supplementary Fig. 8g). Using electron microscopy to identify lysosomes by their high election density 40, we verified that the vesicles reduced in C9ORF72-deficient cells were lysosomes (Fig. 3g-i). Forced expression of either C9ORF72 isoform restored the number of LAMP1+, LAMP2+, and LAMP3+ lysosomes in patient (n=4 patients) and C9ORF72-deficient iMNs (Fig. 3c-f and Supplementary Fig. 8a-h). To determine if loss of C9ORF72 activity reduces lysosome numbers in motor neurons in vivo, we measured the number of lysosomes in spinal motor neurons in Nestin-Cre-Stop-Flox-C9orf72 mice 22. C9orf72−/− motor neurons contained significantly fewer Lamp1+ lysosomes than control motor neurons (Fig. 3j, k).
During lysosomal biogenesis, lysosomal proteins are transported in Mannose-6-Phosphate Receptor (M6PR)+ vesicles from the trans-Golgi Network to early and late endosomes for eventual incorporation into lysosomes 41. Disruption of M6PR+ vesicle trafficking can lead to a reduction in lysosome numbers 42 and altered localization of M6PR+ vesicles 43. In control iMNs (n=3 controls), M6PR+ vesicles were distributed loosely around the perinuclear region and to a lesser extent in the non-perinuclear cytosol (Supplementary Fig. 9a, b). In contrast, C9ORF72 patient (n=4 patients), C9ORF72+/−, and C9ORF72−/− iMNs frequently harbored densely-packed clusters of M6PR+ vesicles (Supplementary Fig. 9a, b). This was not due to a reduced number of M6PR+ vesicles in patient and C9ORF72-deficient iMNs (Supplementary Fig. 9c). Forced expression of C9ORF72 isoform B restored normal M6PR+ vesicle localization in patient (n=4 patients) and C9ORF72-deficient iMNs, confirming that a lack of C9ORF72 activity induced this phenotype (Supplementary Fig. 9a, b).
Live imaging of iMNs expressing a M6PR-GFP fusion protein that localizes to M6PR+ vesicles 44 confirmed that C9ORF72 patient and C9ORF72-deficient iMNs possess increased numbers of M6PR+ vesicle clusters, and that overexpression of C9ORF72 isoform A or B rescues this phenotype (Supplementary Fig. 9c-g and Supplementary Videos 5-9). Clusters did not disperse over the time course of the assay, suggesting that they are relatively stable and not in rapid flux (Supplementary Videos 5-9). In addition, M6PR+ puncta moved with a slower average speed in C9ORF72 patient and C9ORF72+/− iMNs than controls (Supplementary Fig. 9h, i). Thus, reduced C9ORF72 levels lead to fewer lysosomes in motor neurons in vitro and in vivo, and this may be due in part to altered trafficking of M6PR+ vesicles.
Low C9ORF72 activity sensitizes iMNs to glutamate
Our iMN survival results (Fig. 1c-e) suggest that the repeat expansion alters iMN glutamate sensing. In cortical neurons, homeostatic synaptic plasticity is maintained through endocytosis and subsequent lysosomal degradation of glutamate receptors in response to chronic glutamate signaling 45,46. Defects in this process lead to the accumulation of glutamate receptors on the cell surface 45,46.
Immunostaining revealed that C9ORF72+/− and C9ORF72−/− iMNs contained elevated levels of NMDA (NR1) and AMPA (GLUR1) receptors on neurites and dendritic spines compared to control iMNs under basal conditions (Fig. 4a, c, d and Supplementary Fig. 5b and 10a, c-e, g, h, j, k). In addition, control iMNs treated with C9ORF72-specific ASOs displayed increased numbers of NMDA and AMPA receptors in their neurites (Supplementary Fig. 10l, m). C9ORF72 patient iMNs (n=3 patients) also showed elevated NR1 and GLUR1 levels compared to controls (n=3 controls), and forced expression of C9ORF72 isoform B reduced glutamate receptor levels in patient iMNs (n=3 patients) to that of controls (n=3 controls) (Fig. 4a-c and Supplementary Fig. 10a-h). mRNA levels of NR1 (GRIN1) and GLUR1 (GRIA1) were not elevated in flow-purified C9ORF72+/− iMNs, indicating that increased transcription could not explain the increased glutamate receptor levels (Supplementary Fig. 10n).
Figure 4. Low C9ORF72 activity sensitizes neurons to glutamate.
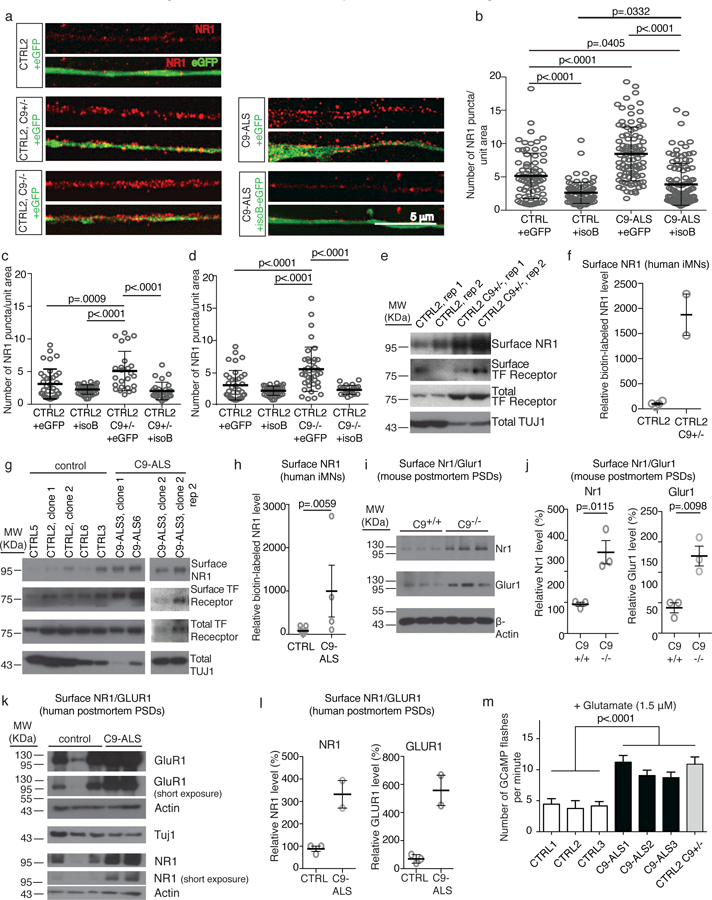
(a) Super-resolution microscopy images of immunofluorescence shows NR1+ puncta on neurites of iMNs overexpressing eGFP or C9ORF72 isoform B-eGFP. Scale bar: 5 µm. This experiment was repeated 3 times with similar results. (b-d) Number of NR1+ puncta per unit area in control (b-d), patient (b), C9ORF72+/− (c), and C9ORF72+/− (d) iMNs. Mean ± s.d. Each grey open circle represents the number of NR1+ puncta per area unit on a single neurite (one neurite quantified per iMN). For (b), n=75 (CTRL + GFP), 84 (C9-ALS + GFP), 95 (C9-ALS + isoA), and 111 (C9-ALS + isoB) iMNs quantified from two biologically independent iMN conversions of 3 CTRL or 4 C9-ALS lines. For (c), n=37 (CTRL + GFP), 37 (C9ORF72+/− + GFP), 25 (C9ORF72+/− + isoA), and 27 (C9ORF72+/− + isoB) iMNs quantified from two biologically independent iMN conversions per condition. For (d), n=37 (CTRL + GFP), 37 (C9ORF72−/− + GFP), 38 (C9ORF72−/− + isoA), and 23 (C9ORF72−/− + isoB) iMNs quantified from two biologically independent iMN conversions per condition. One-way ANOVA with Tukey correction for all comparisons. F-value (DFn, DFd): (3, 360) = 56.63 (b), (3, 122) = 13.42 (c), (3, 131) = 17.11 (d). (e-h) Immunoblotting analysis of surface NR1 after surface protein biotinylation in control (e-h), C9ORF72+/− (e-f), and C9-ALS patient (g-h) iMNs generated with 3 factors (NGN2, ISL1, and LHX3). In (f), n=4 biologically independent iMN conversions from CTRL2 and 2 biologically independent iMN conversions from the C9ORF72+/− line. Mean +/− s.d. In (h), two-tailed Mann-Whitney test. n=11 biologically independent motor neuron cultures from 11 independent control lines and 4 biologically independent motor neuron cultures from 4 independent C9-ALS patient lines. Experiments in (e-h) were repeated twice with similar results. Mean +/− s.e.m. (i-j) Immunoblotting analysis of surface Nr1 and Glur1 in post-synaptic densities (PSDs) from C9orf72 control and knockout mice, two-tailed t-test. t-value: 4.424 (Nr1), 4.632 (Glur1), degrees of freedom: 4 (Nr1), 4 (Glur1). n= 3 control PSD preparations isolated from 3 control mice and 3 C9orf72−/− PSD preparations isolated from 3 C9orf72−/− mice. This experiment was repeated twice with similar results. Mean +/− s.e.m. (k-l) Immunoblotting analysis of surface NR1 and GLUR1 in post-synaptic densities (PSDs) from post mortem control and C9-ALS patient motor cortices, n=3 control and 2 C9-ALS patient PSD preparations isolated from 3 control and 2 C9-ALS patients. This experiment was repeated twice with similar results. Mean +/− s.d. (m) Average Ca2+ flux in the presence of glutamate per minute. n=28 (CTRL1), 15 (CTRL2), 15 (CTRL3), 26 C9-ALS1), 20 (C9-ALS2), 24 (C9-ALS3), and 15 (C9ORF72+/−) iMNs analyzed from two biologically independent iMN conversions for each line. Mean ± s.e.m. One-way ANOVA with Tukey correction between all controls and all patients and C9ORF72+/−. F-value (DFn, DFd): (6, 136) = 11.21.
To confirm that glutamate receptor levels were increased on the surface of C9ORF72+/− and C9ORF72 patient iMNs, we used CRISPR/Cas9 editing to introduce a Dox-inducible polycistronic cassette containing NGN2, ISL1, and LHX3 into the AAVS1 safe-harbor locus of control, C9ORF72+/− and C9ORF72 patient iPSCs. This enabled large-scale production of iMNs that expressed motor neuron markers and had transcriptional profiles similar to 7F iMNs (Supplementary Fig. 11). Using this approach, we quantified the amount of surface-bound NR1 by immunoblotting after using surface protein biotinylation to isolate membrane-bound proteins. This confirmed that surface NR1 levels were higher on C9ORF72+/− and C9ORF72 patient iMNs (n=2 patients) than controls (n=3 controls)(Fig. 4e-h, Supplementary Fig. 5g, h).
To determine if reduced C9orf72 levels leads to glutamate receptor accumulation in vivo, we examined spinal motor neurons deleted of C9orf72 in Nestin-Cre-Stop-Flox-C9orf72 mice 22. Immunofluorescence analysis indicated that Nr1 (NMDA) and GluR1 (AMPA) levels were elevated in C9orf72-null motor neurons (Supplementary Fig. 12a, b). To confirm these findings, we isolated post-synaptic densities from the spinal cords of control and C9orf72 knockout mice. Post-synaptic density fractions contained glutamate receptors and PSD-95, but not p53 or synaptophysin, indicating they were enriched for post-synaptic density proteins (Supplementary Fig. 12c, 5i). Immunoblotting showed that post-synaptic densities in C9orf72 knockout mice contained significantly higher levels of Nr1 and Glur1 than in control mice (Fig. 4i, j and Supplementary Fig. 5j).
To determine if glutamate receptor accumulation occurs on C9ORF72 patient motor neurons in vivo, we measured glutamate receptor expression in ventral horn neurons in lumbar spinal cord samples from 3 C9ORF72 ALS patients and 3 unaffected controls. We identified motor neurons by size and confirmed that most neurons selected in this manner were CHAT+ and SMI-32+ (Supplementary Fig. 12d). Spinal motor neurons from the C9ORF72 ALS patients displayed higher NR1 levels than control neurons (Supplementary Fig. 12e). In addition, post-synaptic densities isolated from the motor corticesof C9ORF72 patients had higher levels of NR1 and GLUR1 than controls (Fig. 4k, l and Supplementary Fig. 5k).
To determine if the extra glutamate receptors were functional, we used Gcamp647 to measure calcium influx into iMNs in response to glutamate. Glutamate triggered more frequent calcium influxes in C9ORF72 patient (n=3 patients) and C9ORF72+/− iMNs than controls (n=3 controls)(Fig. 4m)(Supplementary Videos 10-12
Since glutamate receptor activation and neuronal firing both induce calcium influx, we determined their relative contributions to the increased Gcamp6 activation by. using the ion channel inhibitors TTX and TEA to block neuronal firing. C9ORF72+/− iMNs still displayed more frequent Gcamp6 activation than C9ORF72+/+ iMNs (Supplementary Fig. 13a), indicating that part of the hyperexcitability is due to increased glutamate receptor activation. To determine which receptors were responsible for the increased glutamate response, we tested small molecule agonists of specific glutamate receptor subtypes. Activation of NMDA, AMPA, and kainate receptors was higher in C9ORF72+/− iMNs than controls (Supplementary Fig. 13a).
Removal of TTX and TEA during glutamate receptor agonist treatment revealed additional increases in Gcamp6 activation in C9ORF72+/− iMNs compared to controls, suggesting that C9ORF72+/− iMNs also fire action potentials more frequently than controls (Supplementary Fig. 13a), although we did not detect large changes in sodium or potassium current amplitudes in C9ORF72+/− iMNs (Supplementary Fig. 13b, c). To determine if increased neuronal activity due in part to elevated glutamate receptor levels contributes to neurodegeneration in C9ORF72 patient and C9ORF72+/− iMNs, we measured iMN survival in the presence or absence of retigabine. Retigabine is approved by the U.S. Food and Drug Administration for the treatment of epilepsy and reduces neuronal excitability by activating Kv7 potassium channels 48. In the glutamate treatment assay, retigabine increased the survival of C9ORF72 patient (n=2 patients) and C9ORF72-deficient iMNs, but not controls (n=2 controls)(Supplementary Fig. 13d-g).
Although C9orf72 knockout mice do not show overt neurodegeneration, gain-of-function disease processes may trigger neurodegeneration through mechanisms induced by reduced C9ORF72 levels. For example, DPRs cause mis-splicing of the EAAT2 glutamate transporter in astrocytes, which couldincrease excitotoxicity in neurons with elevated glutamate receptor levels 12. To determine if DPRs alter glutamate uptake by astrocytes, we compared glutamate uptake in human primary astrocytes expressing either GFP or GR50 –GFP. Indeed, GR50 –GFP significantly impaired glutamate uptake by astrocytes (Supplementary Fig. 13h).
Low C9ORF72 activity sensitizes iMNs to DPR toxicity
We wondered if reduced C9ORF72 activity renders motor neurons vulnerable to other insults that might be exacerbated by impaired endosomal-lysosomal system function, such as DPR toxicity. Without excess glutamate treatment, C9ORF72+/−, C9ORF72−/−, and C9ORF72 patient iMN survival was similar to control iMNs (Fig. 5a-d and Supplementary Fig. 14a, b). Exogenous expression of PR50 or GR50 DPRs using cassettes that do not contain the GGGGCC repeat expansion induced control iMN degeneration (Fig. 5a, b). Interestingly, C9ORF72+/−, C9ORF72−/−, and C9ORF72 patient iMNs (n=2 patients) degenerated significantly faster than controls in response to PR50 or GR50 expression (Fig. 5a-d and Supplementary Fig. 14a, b).
Figure 5. C9ORF72 levels determine dipeptide repeat turnover.
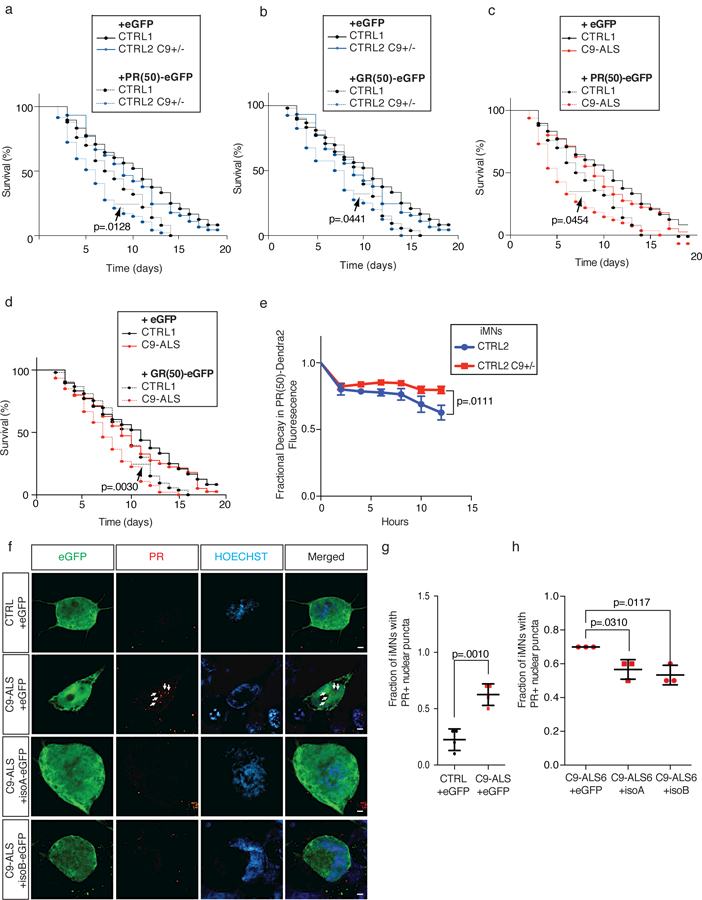
(a-b) Survival of control and CRISPR-mutant iMNs without excess glutamate with overexpression of eGFP or PR(50)-eGFP (a) or GR(50)-eGFP (b). (c-d) Survival of control and C9-ALS iMNs without excess glutamate with overexpression of eGFP or PR(50)-eGFP (c) or GR(50)-eGFP (d). For (a), n=50 (CTRL1 + GFP AND CTRL1 + PR(50)), 49 (C9ORF72+/− + GFP), and 47 (C9ORF72+/− + PR(50)) iMNs per line, iMNs quantified from 3 biologically independent iMN conversions per line. For (b), n=50 (CTRL1 + GFP AND CTRL1 + GR(50)), 49 (C9ORF72+/− + GFP), and 40 (C9ORF72+/− + GR(50)) iMNs per line, iMNs quantified from 3 biologically independent iMN conversions per line. For (c), n=50 (CTRL1 + GFP AND CTRL1 + PR(50)), 50 (from each of two C9-ALS lines + GFP), and 41 (from each of two C9-ALS lines + PR(50)) iMNs per line, iMNs quantified from 3 biologically independent iMN conversions per line per condition. For (d), n=50 (CTRL1 + GFP AND CTRL1 + GR(50)), 50 (from each of two C9-ALS lines + GFP), and 46 and 47 (from two C9-ALS lines + GR(50)) iMNs per line, iMNs quantified from 3 biologically independent iMN conversions per line per condition. All iMN survival experiments in (a-d) were analyzed by two-sided log-rank test, and statistical significance was calculated using the entire survival time course. Survival curves for the “+GFP” condition were included as a reference, but were not used in statistical analyses. (e) Relative decay in Dendra2 fluorescence over 12 hours in iMNs of specified genotypes. Mean +/− s.e.m. n = 18 (control) and 24 (C9ORF72+/−) iMNs quantified from two biologically independent iMN conversions each, two-tailed t-test with Welch’s correction between data points at each time point, t-value: 2.739, degrees of freedom: 25.62). (f-h) Immunostaining to determine endogenous PR+ puncta in control or C9-ALS iMNs with or without overexpression of C9ORF72 isoform A or B. Scale bar = 2 μm. This experiment was repeated twice with similar results. (g) Mean +/− s.d. n= 4 biologically independent iMN conversions generated from two different iPSC lines per genotype. Quantified values represent the average number of PR+ puncta in 40 iMNs from a single iMN conversion. Two-tailed t-test, t-value: 5.908, degrees of freedom: 6. (h) Mean +/− s.e.m. n= 3 biologically independent iMN conversions per condition. Quantified values represent the average number of PR+ puncta in 40 iMNs from a single iMN conversion. One-way ANOVA with Tukey correction, F-value (DFn, DFd): (2, 6)=10.5. iMN survival experiments in (a-d) were performed in a Molecular Devices ImageExpress.
Because C9ORF72 activity is required to maintain normal lysosomal function, we measured the effect of C9ORF72 activity on PR50 clearance by monitoring the clearance of PR50-Dendra2 fusion proteins in C9ORF72−/− iPSC-derived fibroblasts with or without exogenous C9ORF72. Dendra2 is a green fluorescent protein that irreversibly converts to red fluorescence when exposed to blue light, enabling quantification of its degradation 49. PR50-Dendra2 formed discrete punctae within cells, indicating that Dendra2 did not prevent intracellular aggregation of PR50 (Supplementary Fig. 14c). Expression of C9ORF72-T2A-GFP in C9ORF72−/− iPSC-derived fibroblasts significantly enhanced the decay of PR50-Dendra2 fluorescence over GFP alone (Supplementary Fig. 14d). To determine if C9ORF72 activity modulates DPR aggregate clearance in human motor neurons, we compared the decay of PR50-Dendra2 in C9ORF72+/+ and C9ORF72+/− iMNs (Fig. 5e and Suppementary Fig. 14e). Consistent with the hypothesis that C9ORF72 activity promotes DPR aggregate clearance, PR50-Dendra2 decayed significantly slower in C9ORF72+/− iMNs (Fig. 5e).
To measure the effect of C9ORF72 activity on endogenous DPR levels in human motor neurons, we quantified endogenous PR+ puncta in C9-ALS iMNs with or without C9ORF72 overexpression. Using a validated anti-PR antibody 10,50, we found that the majority of PR+ punctae were localized in the nucleus (Fig. 5f), although we also detected cytoplasmic PR+ punctae to a larger extent than we had previously observed with exogenous PR(50) 10. C9-ALS iMNs (n=2 patients) had higher levels of nuclear PR+ puncta than controls (n=2 controls)(Fig. 5f, g) and overexpression of C9ORF72 isoform B significantly reduced the number of PR+ puncta in C9-ALS iMNs (Fig. 5f, h).
Given our observation that iMNs with reduced C9ORF72 levels are hypersensitive to DPR toxicity, we wondered if this might be due to a general disruption of protein turnover by DPRsHowever, PR50-GFP expression did not impair turnover of APP or Tau (Supplementary Fig. 14f, g and Supplementary Fig. 5l). Thus the neurotoxicity caused by DPRs that accumulate rapidly in C9-ALS motor neurons due to reduced C9ORF72 levels is not due to global disruption of protein turnover.
Small molecule and genetic regulators of endosomal trafficking rescue patient iMN survival
Therapeutic strategies in development for C9ORF72 ALS/FTD target gain-of-function mechanisms. These include ASOs 6–8 and small molecules 13 that disrupt RNA foci formation. However, these approaches have not fully rescued neurodegeneration in human patient-derived neurons 6–8,13, indicating that replacing C9ORF72 function or new therapeutic targets may be required.
To this end, we performed a phenotypic screen to identify small molecules that rescue C9ORF72 patient iMN survival (Fig. 6a). To enrich for chemicals that might replace C9ORF72 function, we screened 800 bioannotated compounds targeting diverse cellular processes including vesicle trafficking.
Figure 6. Small molecule modulators of vesicle trafficking rescue neurodegeneration in vitro and in vivo.
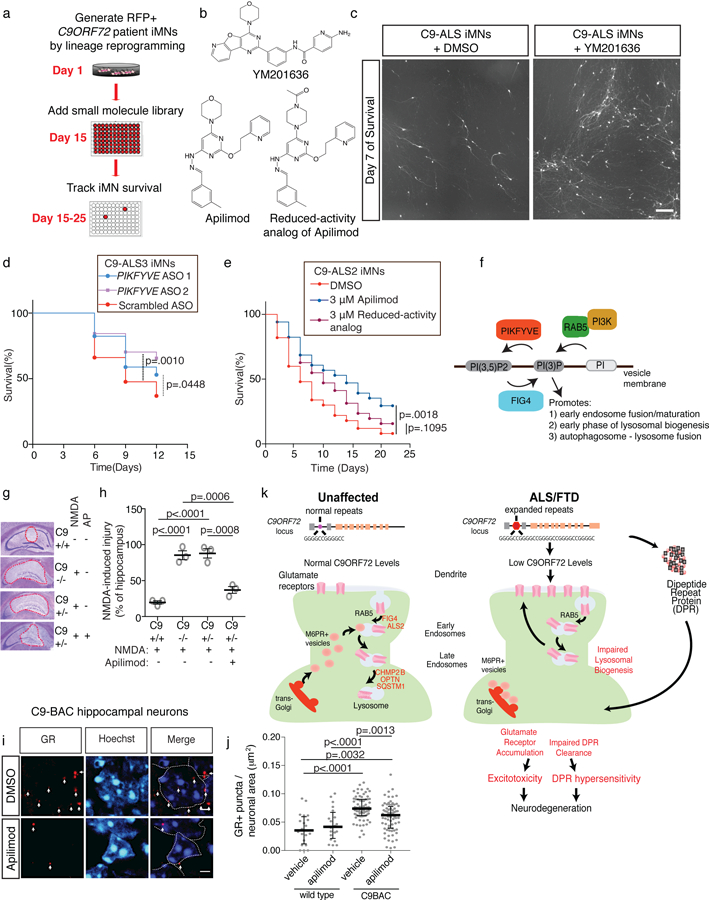
(a) Phenotypic screening for small molecules that enhance the survival of C9-ALS iMNs. (b) Chemical structure of the PIKFYVE inhibitors YM201636 and Apilimod, and a reduced-activity analog of Apilimod. (c) Live cell images of iMNs at day 7 of treatment with DMSO or YM201636 (scale bar: 1 mm). This experiment was performed 3 times with similar results. (d) Survival effect of scrambled or PIFKVYE ASOs on C9-ALS iMNs in excess glutamate. n=50 iMNs per condition, iMNs quantified from 3 biologically independent iMN conversions per condition. (e) Survival effect of Apilimod and the reduced-activity analog on C9-ALS patient iMNs with neurotrophic factor withdrawal. n=50 iMNs per condition, iMNs quantified from 3 biologically independent iMN conversions per condition. All iMN survival experiments in (d, e) were analyzed by two-sided log-rank test, and statistical significance was calculated using the entire survival time course. (f) Activities of therapeutic targets in C9ORF72 ALS. (g, h) The effect of 3 μM Apilimod on NMDA-induced hippocampal injury in control, C9orf72+/−, or C9orf72−/− mice. (Mean +/− s.e.m. of n=3 mice per condition, one-way ANOVA with Tukey correction across all comparisons, F-value (DFn, DFd): (3, 8)=43.55, AP = Apilimod, red dashed lines outline the injury sites). (i, j) The effect of 3 μM Apilimod on the level of GR+ puncta in the dentate gyrus of control or C9-BAC mice. Mean +/− s.d. of the number of GR+ puncta per cell, each data point represents a single cell. n=20 (wild-type + DMSO), 20 (wild-type + Apilimod), 87 (C9-BAC + DMSO), and 87 (C9-BAC + Apilimod) cells quantified from 3 mice per condition, one-way ANOVA with Tukey correction for all comparisons, F-value (DFn, DFd): (3, 180) = 16.29. Scale bars = 2 μm, dotted lines outline nuclei, and white arrows denote GR+ punctae (i). (k) Model for the mechanisms that cooperate to cause neurodegeneration in C9ORF72 ALS/FTD. Proteins in red are known to be mutated in ALS or FTD. iMN survival experiments in (d, e) were performed in a Molecular Devices ImageExpress.
Amongst four reproducible hit compounds, we identified a PIKFYVE kinase inhibitor (YM201636) that significantly increased C9ORF72 patient iMN survival (n=2 patients) (Fig. 6b, c and Supplementary Fig. 15a). PIKFYVE is a lipid kinase that converts phosphatidylinositol 3-phosphate (PI3P) into phosphtidylinositol (3,5)-bisphosphate (PI(3,5)P2)51(Fig. 6f). PI3P is primarily generated by PI3-kinases recruited to early endosomes by RAB5, and PI3P anchors EEA1 to early endosomes to drive endosomal maturation 52(Fig 6f). Following endosomal maturation into lysosomes, PI3P drives fusion of lysosomes with autophagosomes 53. PIKFYVE regulates PI3P levels by converting PI3P into PI(3,5)P2 52, which disfavors lysosomal fusion with endosomes and autophagosomes 53,54. Therefore, inhibition of PIKFYVEincreases autophagosome-lysosome fusion 53 and may compensate for reduced C9ORF72 activity and other disease processes by increasing PI3P levels to facilitate removal of glutamate receptors or DPRs (Fig. 6f). Interestingly, FIG4 is a phosphatase that opposes PIKFYVE kinase by converting PI(3,5)P2 to PI3P and loss-of-function mutations in FIG4 cause ALS 55. Thus, genetic evidence suggests that PIKFYVE inhibition may be capable of modulating ALS disease processes in humans.
To verify that PIKFYVE is the functional target of the inhibitor, we first confirmed PIKFYVE expression by qPCR in control and patient (n=3 patients) iMNs (Supplementary Fig. 15b). Next, we verified that YM201636 rescued C9ORF72 patient iMN survival in a dose-dependent manner (Supplementary Fig. 15c). We then asked if Apilimod, a structurally distinct PIKFYVE inhibitor, could rescue patient iMN survival 51(Fig. 6b). To verify target engagement by Apilimod in iPSC-derived motor neurons, we administered Apilimod for three hours and measured EEA1+ early endosome size. PIKFYVE inhibition increases PI3P levels, leading to increased recruitment of EEA1 to early endosomes, more homotypic early endosomal fusion, and larger EEA1+ early endosomes 54. As expected, Apilimod treatment increased EEA1+ endosome size in a dose-dependent manner, verifying target engagement in motor neurons (Supplementary Fig. 15d, e).
Consistent with PIKFYVE being the relevant target in the iMN survival assay, Apilimod increased C9ORF72 patient, but not control, iMN survival in either neurotrophic withdrawal conditions (Fig. 6d) or excess glutamate (n=4 patients, Supplementary Fig. 15f (n=3 controls, Supplementary Fig. 15g). Automated neuron tracking software independently verified Apilimod efficacy on C9ORF72 patient iMNs (Supplementary Fig. 15h). As further confirmation that PIKFYVE was the active target, ASO-mediated suppression of PIKFYVE also rescued C9ORF72 patient iMN survival (Fig. 6d and Supplementary Fig. 15i). In addition, we synthesized a structural analog of Apilimod with a reduced ability to inhibit PIKFYVE kinase activity in a biochemical assay using purified PIKFYVE protein (Fig. 6b and Supplementary Fig. 15j, 16). The reduced activity analog was significantly less effective at rescuing C9ORF72 patient iMN survival (Fig. 6e). Thus, small molecule inhibition of PIKFYVE can rescue patient motor neuron survival.
To verify that PIKFYVE-dependent modulation of vesicle trafficking was responsible for rescuing C9ORF72 patient iMN survival, we tested the ability of a constitutively active RAB5 mutant to block C9ORF72 patient iMN degeneration. Active RAB5 recruits PI3-kinase to synthesize PI3P from PI and therefore, like PIKFYVE inhibition, increases PI3P levels 56. Constitutively active RAB5 did not improve control iMN survival (n=2 controls)(Supplementary Fig. 15k), but successfully rescued C9ORF72 patient iMN survival (n=3 patients)(Supplementary Fig. 15l). In constrast, dominant negative RAB5, wild-type RAB5, or constitutively active RAB7 did not rescue C9ORF72 patient iMN survival (n=1, 3, 3 patients, respectively)(Supplementary Fig. 14m-o).
To determine if PIKFYVE inhibition rescued patient iMN survival by reversing phenotypic changes caused by C9ORF72 haploinsufficiency, we measured glutamate receptor levels with and without PIKFYVE inhibitor treatment. PIKFYVE inhibition significantly lowered NR1 (NMDA receptor) and GLUR1 (AMPA receptor) levels in patient (n=4 patients) and C9ORF72+/− iMNs (Supplementary Fig. 15p-s). PIKFYVE inhibition also reduced electrophysiological activity in patient motor neurons (C9-ALS1) during glutamate treatment (Supplementary Fig. 15t). To determine if small molecule inhibition of Pikfyve rescues C9ORF72 disease processes in vivo, we first established an NMDA-induced hippocampal injury model in C9orf72-deficient mice. In control mice, hippocampal injection of NMDA caused neurodegeneration after 48 hrs as we have shown previously 57 (Supplementary Fig. 17a, b). Consistent with C9orf72-deficient mice having elevated NMDA receptor levels (Fig. 4h, i and Supplementary Fig. 11a-d), injection of NMDA caused significantly greater neurodegeneration in C9orf72+/− and C9orf72−/− mice than in controls (Fig. 6g, h). Importantly, co-administration of Apilimod rescued the NMDA-induced neurodegeneration in C9orf72-deficient mice (Fig. 6g, h).
To determine if Pikfyve inhibition rescues gain-of-function processes in vivo, we measured DPR levels in C9-BAC transgenic mice 58 with or without Apilimod treatment. Although it was not previously reported 58, we observed significantly higher levels of GR+ punctae in hippocampal neurons in C9-BAC mice than controls (Fig. 6j) using a previously-validated poly(GR) antibody 11. These data are consistent with findings in another published C9-BAC mouse model 14, suggesting that poly(GR) may be a common feature of C9-BAC mice. We also detected a low level of poly(GR) in neurons from control mice (Fig. 6j), which may be derived from other repeat regions or proteins with short poly(GR) sequences. Nevertheless, GR+ punctae levels were significantly higher in C9-BAC mouse neurons than in controls (Fig. 6j). Importantly, Apilimod treatment significantly reduced the number of GR+ punctae in hippocampal neurons in C9-BAC mice after 48 hrs (Fig. 6i, j). Therefore, small molecule inhibition of Pikfyve rescues both gain- and loss-of-function disease processes induced by C9ORF72 repeat expansion in vivo.
Discussion
Our results indicate that haploinsufficiency for C9ORF72 activity triggers neurodegeneration in C9ORF72 ALS, and this occurs by at least two mechanisms. First, reduced C9ORF72 activity causes the accumulation of glutamate receptors and excitotoxicity in response to glutamate. Although C9orf72 knockout mice do not display overt neurodegeneration14,18,22, these mice may be protected from excitotoxicity because they lack gain-of-function disease processes such as DPRs, which induce aberrant splicing and dysfunction of the EAAT2 glutamate transporter in astrocytes in vitro 12 and in C9ORF72 ALS patients 4,27. EAAT2 dysfunction causes glutamate accumulation in the cerebrospinal fluid of ALS patients 27, and consistent with this notion, we found that poly(PR) expression in human astrocytes reduced their rate of glutamate uptake. By using human iMNs, mice, and human post mortem tissue, we show for the first time that reduced C9ORF72 activity modulates the vulnerability of human motor neurons to degenerative stimuli and establish a mechanistic link between the C9ORF72 repeat expansion and glutamate-induced excitotoxicity
We also found that Reduced C9ORF72 activity also induces iMN hypersensitivity to DPRs by impairing their clearance. This uncovers a more direct form of cooperative pathogenesis between gain- and loss-of-function mechanisms in C9ORF72 ALS/FTD. Through a potentially similar mechanism, reduced C9orf72 levels can also facilitate cytoplasmic TDP-43 accumulation in mouse neurons 20.
Although studies in mice have indicated that C9orf72 activity is important for myeloid cell function 14,18, a report in mouse neurons and zebrafish suggests that C9orf72 isoform A may modulate poly-Q Ataxin-2 toxicity 20, and studies have implicated C9ORF72 in regulating autophagy 20,31,32,38, our study provides the first direct evidence showing that gain- and loss-of-function C9ORF72 mechanisms cooperate to cause the degeneration of human motor neurons. Recent studies have shown that C9ORF72 isoform A, but not isoform B, can form a functional complex with SMCR8 and WDR41 20. However, since both C9ORF72 isoforms rescue C9-ALS iMN degeneration in our assay, other mechanisms or protein interactions may underlie the rescue of patient iMNs.
Importantly, our work establishes a new approach for suppressing DPR protein toxicity and blocking C9ORF72 pathogenesis: restoring or replacing C9ORF72 activity. Although high levels of C9ORF72 isoform A may have slightly detrimental effects on control motor neuron survival, we have only observed this in neurons without C9ORF72 repeat expansion. Thus, we would not anticipate a harmful effect of forced C9ORF72 expression in C9ORF72 patients. In addition, a better understanding of the effects of forced C9ORF72 expression could inform safe development of this therapeutic strategy. For example, determining if C9ORF72 accelerates turnover of DPR aggregates by stimulating autophagy could lead help to identify new therapeutic targets.
We show for the first time that chemical or genetic modulators of vesicle trafficking can fully rescue iMN degeneration caused by the C9ORF72 repeat expansion. Previous studies have implicated several rare ALS or FTD mutations linked to these vesicle trafficking pathways, but by showing that C9ORF72 is haploinsufficient in ALS/FTD and demonstrating that perturbation of vesicle trafficking rescues C9ORF72 neurodegeneration, our findings highlight mechanistic convergence in a large portion of ALS.
Our results highlight the importance of C9ORF72 protein function, RAB5 activity, PI3P levels, and lysosomeal function as key therapeutic targets for C9ORF72 ALS/FTD. By generating PI3P, RAB5 drives early endosomal maturation and the initial stages of lysosomal biogenesis (Fig. 6f)59. PI3P also plays important roles in autophagosome formation and autophagsome-lysosome fusion. Indeed, a previous study suggests that PIKFYVE inhibition may increase autophagic flux 53, and this should be investigated in the context of motor neurons. Loss-of-function mutations in two other genes whose proteins function to increase PI3P levels, ALS2 and FIG4, also cause ALS 1. ALS2 encodes the RAB5 guanine exchange factor ALSIN 60, while FIG4 converts PI(3,5)P2 into PI3P 55(Fig. 6f). In addition, proteins encoded by several other ALS genes play key roles in lysosomal biogenesis, including CHMP2B, OPTN, and SQSTM1 1. The fact that FIG4 and ALS2 loss-of-function mutations can cause ALS suggests that PIKFYVE inhibition or RAB5 activation may be capable of modulating ALS disease processes in humans.
In myeloid cells, endosomal-lysosomal trafficking regulates inflammatory cytokine release 51 and indeed, C9orf72-deficient macrophages release inflammatory cytokines 18. Interestingly, the PIKFYVE inhibitor Apilimod inhibits the release of pro-inflammatory cytokines IL-12 and IL-23 from human and mouse peripheral blood mononuclear cells 51. If impaired endosomal and lysosomal trafficking in C9ORF72 patients increases the production of pro-inflammatory cytokines that accelerate disease progression 18, PIKFYVE inhibitors or other modulators of this pathway may provide therapeutic benefit by lowering cytokine release.
We anticipate three key implications of our findings: 1) ALS/FTD caused by the C9ORF72 repeat expansion requires both gain- and loss-of-function mechanisms, 2) increasing C9ORF72 activity in motor neurons should mitigate disease and provides a new therapeutic target, and 3) PIKFYVE inhibition and other approaches that modulate vesicle trafficking may ameliorate C9ORF72 disease processes in both neurons and myeloid cells. The fact that mutations in FIG4 cause ALS, epilepsy, and Charcot-Marie-Tooth 55 illustrates the broad implications of impaired vesicle trafficking within the CNS. The identification of targets that effectively modulate vesicle trafficking in neurons, glia, and myeloid cells could hold tremendous therapeutic value for C9ORF72 ALS/FTD and other CNS disorders.
Methods
iPSC reprogramming
Human lymphocytes from healthy subjects and ALS patients were obtained from the NINDS Biorepository at the Coriell Institute for Medical Research and reprogrammed into iPSCs as previously described using episomal vectors61. Briefly, mammalian expression vectors containing Oct4, Sox2, Klf4, L-Myc, Lin28, and a p53 shRNA were introduced into the lymphocytes using the Adult Dermal Fibroblast Nucleofector™ Kit and Nucleofector™ 2b Device (Lonza) according to the manufacturer’s protocol. The cells were then cultured on mouse feeders until iPSC colonies appeared. The colonies were then expanded and maintained on Matrigel (BD) in mTeSR1 medium (Stem Cell Technologies).
Repeat Primed PCR (RP-PCR)
To provide a quantitative measure of (GGGGCC)n hexanuceotide expansion in C9ORF72, 100 ng of genomic DNA was amplified by touchdown PCR using primers shown in Supplementary Data Table 4, in a 28-µl PCR reaction consisting of 0.2 mM each of 7-deaza-2-deoxyguanine triphosphate (deaza-dGTP) (NEB), dATP, dCTP and dTTP, 7% DMSO, 1X Q-Solution, 1X Taq PCR buffer (Roche), 0.9 mM MgCl2, 0.7 µM reverse primer (four GGGGCC repeats with an anchor tail), 1.4 µM 6FAM-fluorescently labeled forward primer, and 1.4 µM anchor primer corresponding to the anchor tail of reverse primer (Supplementary Data Table 4). During the PCR, the annealing temperature was gradually decreased from 70 ºC and 56 ºC in 2 ºC increments with a 3 min extension time for each cycle. The PCR products were purified using the QiaQuick PCR purification kit (Qiagen) and analyzed using an ABI3730 DNA Analyzer and Peak Scanner™ Software v1.0 (Life Technologies).
C9ORF72 Southern Blotting
A 241-bp digoxigenin (DIG)-labeled probe was generated from 100 ng control genomic DNA (gDNA) by PCR reaction using Q5® High-Fidelity DNA Polymerase (NEB) with primers shown in Supplementary Data Table 4. Genomic DNA was harvested from control and patient iPSCs using cell lysis buffer (100 mM Tris-HCl pH 8.0, 50 mM EDTA, 1% w/v sodium dodecyl sulfate (SDS)) at 55ºC overnight and performing phenol:chloroform extraction. A total of 25 µg of gDNA was digested with XbaI at 37 ºC overnight, run on a 0.8% agarose gel, then transferred to a positive charged nylon membrane (Roche) using suction by vacuum and UV-crosslinked at 120 mJ. The membrane was pre-hybridized in 25 ml DIG EasyHyb solution (Roche) for 3 h at 47 ºC then hybridized at 47 ºC overnight in a shaking incubator, followed by two 5-min washes each in 2X Standard Sodium Citrate (SSC) and in 0.1% SDS at room temperature, and two 15-min washes in 0.1x SSC and in 0.1% SDS at 68 ºC. Detection of the hybridized probe DNA was carried out as described in DIG System User’s Guide. CDP-Star® Chemilumnescent Substrate (Sigma-Aldrich) was used for detection and the signal was developed on X-ray film (Genesee Scientific) after 20 to 40 min.
Molecular cloning and viral production
Complementary DNAs (cDNAs) for the iMN factors (Ngn2, Lhx3, Isl1, NeuroD1, Ascl1, Myt1l, and Brn2) and iDA neuron factors (Ascl1, Brn2, Myt1l, Lmx1a, and Foxa2), were purchased from Addgene. cDNA for C9ORF72 was purchased from Thermo Scientific. Each cDNA was cloned into the pMXs retroviral expression vector using Gateway cloning technology (Invitrogen). The Hb9::RFP lentiviral vector was also purchased from Addgene (ID: 37081). Viruses were produced as follows. HEK293 cells were transfected at 80–90% confluency with viral vectors containing genes of interest and viral packaging plasmids (PIK-MLV-gp and pHDM for retrovirus; pPAX2 and VSVG for lentivirus) using polyethylenimine (PEI)(Sigma-Aldrich). The medium was changed 24h after transfection. Viruses were harvested at 48h and 72 h after transfection. Viral supernatants were filtered with 0.45 µM filters, incubated with Lenti-X concentrator (Clontech) for 24 h at 4 ºC, and centrifuged at 1,500 x g at 4ºC for 45 min. The pellets were resuspended in 300 µl DMEM + 10% FBS and stored at −80 ºC.
Conversion of iPSCs into induced motor neurons and dopaminergic neurons
Reprogramming was performed in 96-well plates (8 × 103 cells/well) or 13mm plastic coverslips (3.2 × 104 cells/coverslip) that were sequentially coated with gelatin (0.1%, 1 hour) and laminin (2–4 hours) at room temperature. To enable efficient expression of the transgenic reprogramming factors, iPSCs were cultured in fibroblast medium (DMEM + 10% FBS) for at least 48 hours and either used directly for retroviral transduction or passaged before transduction for each experiment. 7 iMN factors or 5 iDA factors were added in 100–200 µl fibroblast medium per 96-well well with 5 μg/ml polybrene. For iMNs, cultures were transduced with lentivirus encoding the Hb9::RFP reporter 48 hours after transduction with transcription factor-encoding retroviruses. On day 5, primary mouse cortical glial cells from P1 ICR pups (male and female) were added to the transduced cultures in glia medium containing MEM (Life Technologies), 10% donor equine serum (HyClone), 20% glucose (Sigma-Aldrich), and 1% penicillin/streptomycin. On day 6, cultures were switched to N3 medium containing DMEM/F12 (Life Technologies), 2% FBS, 1% penicillin/streptomycin, N2 and B27 supplements (Life Technologies), 7.5 µM RepSox (Selleck), and 10 ng/ml each of GDNF, BDNF, and CNTF (R&D). The iMN and iDA neuron cultures were maintained in N3 medium, changed every other day, unless otherwise noted.
Immunocytochemistry
iMNs were fixed in 4% paraformaldehyde (PFA) for 1h at 4 ºC, permeabilized with 0.5% PBS-T overnight at 4 ºC, blocked with 10% FBS in 0.1% PBS-T at room temperature for 2 h, and incubated with primary antibodies at 4 ºC overnight. Cells were then washed with 0.1% PBS-T and incubated with Alexa Fluor® secondary antibodies (Life Technologies) in blocking buffer for 2 h at room temperature. To visualize nuclei, cells were stained with DAPI (Life Technologies) then mounted on slides with Vectashield® (Vector Labs). Images were acquired on an LSM 780 confocal microcope (Zeiss). The following primary antibodies were used: mouse anti-HB9 (Developmental Studies Hybridoma Bank); mouse anti-TUJ1 (EMD Millipore); rabbit anti-VACHT (Sigma); rabbit anti-C9ORF72 (Sigma-Aldrich); mouse anti-EEA1 (BD Biosciences); mouse anti-RAB5 (BD Biosciences); mouse anti-RAB7 (GeneTex); mouse anti-LAMP1 (Abcam); mouse anti-LAMP3 (DSHB, cat. no. H5C6); rabbit anti-LAMP3 (Proteintech, cat. no. 12632); mouse anti-LAMP2 (DSHB, cat. no. H4B4); mouse anti-M6PR (Abcam, cat. no. Ab2733); rabbit anti-GluR1 (EMD Millipore, cat. no. pc246); mouse anti-GluR1 (Santa Cruz); rabbit anti-NR1 (EMD Millipore); mouse anti-NR1 (EMD Millipore, cat. no. MAB363); chicken anti-GFP (GeneTex).
Live Imaging of M6PR-GFP+ Vesicles in iMNs
iMNs were transduced with a lentvirus encoding M6PR-GFP and imaged at > 10 frames per second over the course of 60 seconds in a Zeiss LSM780 confocal microscope with environmental control. Quantification of vesicle movement was performed using Imaris, using automated detection and tracking of vesicles.
Super-resolution Imaging of iMNs
Structured illumination microscopy (SIM) images were acquired using a Zeiss Elyra PS.1 system equipped with a 100X 1.46 NA or 63X 1.4NA objective. Acquisition was performed with PCO edge sCMOS camera and image reconstruction was done with built-in structured illumination model. Confocal microscopy images were acquired using Zeiss LSM800 microcopy with 63X 1.4NA objective or Zeiss LSM780 microcopy with 40X 1.1NA objective. Z stack images were done with a step size of 2.5 um. Further image process was done with Fiji.
Quantification of Glutamate Receptors
For neurites, Image J was used to automate detection of NR1+ and GLUR1+ puncta in a given are using a thresold of 30. For dendritic spines, Image J was used to quantify the Signal Intensity on dendritic spines, which were selected manually. Neurons from at least two biological replicates were quantified for every sample condition.
Whole Cell Patch Clamp Electrophysiology
Whole cell membrane potential and current recordings in voltage- and current-clamp configurations were made using an EPC9 patch clamp amplifier controlled with PatchMaster software (HEKA Electronics). Voltage- and current-clamp data was acquired at 50 kHz and 20 kHz, respectively, with a 2.9 kHz low-pass Bessel filter, while spontaneous action potential recordings were acquired at 1 kHz sampling frequency. For experiments, culture media was exchanged with warm extracellular solution consisting of (in mM): 140 NaCl, 2.8 KCl, 10 HEPES, 1 MgCl2, 2 CaCl2, and 10 glucose, with pH adjusted to 7.3 and osmolarity adjusted to 305 mOsm. Glass patch pipettes were pulled on a Narishige PC-10 puller and polished to 5–7 MΩ resistance. Pipettes were also coated with Sylgard 184 (Dow Corning) to reduce pipette capacitance. The pipette solution consisted of (in mM): 130 K-gluconate, 2 KCl, 1CaCl2, 4 MgATP, 0.3 GTP, 8 phosphocreatine, 10 HEPES, 11 EGTA, adjusted to pH 7.25 and 290 mOsm. Pipettes were sealed to cells in GΩ-resistance whole cell configuration, with access resistances typically between 10–20 MΩ, and leakage currents less than 50 pA. Capacitance transients were compensated automatically through software control. For voltage clamp, cells were held at −70 mV. For Current-voltage traces, a P/4 algorithm was used to subtract leakage currents from the traces. Measurements were taken at room temperature (approximately 20–25 °C). Data was analyzed and plotted in Igor Pro 6 (WaveMetrics) using Patcher’s Power Tools plug-in and custom programmed routines. Current density was obtained by dividing the measured ion channel current by the cell capacitance. For control iMNs, 10/10 tested fired action potentials. For C9-ALS iMNs, 9/10 tested fired action potentials.
Multielectrode array recordings
Local field potentials (LFPs) were recorded from iPSC-derived motor neurons on days 17–21 in culture in 6-well multielectrode chips (9 electrodes and 1 ground per well) using a MultiChannel Systems MEA-2100 multielectrode array (MEA) amplifier (ALA Scientific) with built-in heating elements set to 37°C. Cells were allowed to acclimate for 5 minutes after chips were placed into the MEA amplifier, and after glutamate addition (10 μM final concentration). For 1 μM Apilimod treatments, chips were incubated for 35 min in a humidified incubator in the presence of the particular drug, then returned to the MEA amplifier and acclimated for 5 min before beginning recordings. For each condition, recordings (5 min baseline, 10 min glutamate and/or drug, 40 kHz sampling rate) were filtered between 1–500 Hz, and average LFP frequency per well was determined using the accompanying MC Rack software.
Neuromuscular junction assay
Primary chick myoblasts were dissected from D11 chick embryos and plated onto plastic dishes pre-coated with 0.1% gelatin. After 3 days of culture in muscle medium containing F10 (Life Technologies), 10% horse serum, 5% chicken serum (Life Technologies), 0.145 mg/ml CaCl2 (Sigma), and 2% Penicillin/Streptomycin, myoblasts were trypsinized and replated onto iMNs which were at days 15–18 post-transduction. The co-culture was maintained in neuronal medium containing DMEM/F12, 2% B27, 1% GlutaMax and 1% Penicillin/Streptomycin, supplemented with 10ng/ml BDNF, GDNF, and CNTF for 7 days in order to allow neuromuscular junctions to form. Videos were taken using Nikon Eclipse Tis microscope with NIS Element AR software. Light-stimulated contraction shown in Supplementary Figure 2j are representative of contraction observed in 2 biological replicates, with 5 contractile sites per replicate.
Induced neuron survival assay
Hb9::RFP+ iMNs appeared between days 13–16 after retroviral transduction. RepSox was removed at day 17 and the survival assay was initiated. For the glutamate treatment condition, 10 µM glutamate was added to the culture medium on day 17 and removed after 12 hours. Cells were then maintained in N3 medium with neurotrophic factors without RepSox. For the glutamate treatment condition with glutamate receptor antagonists, cultures were co-treated with 10 μM MK801 and CNQX, and 2 μM Nimodipine during the 12 hour glutamate treatment. The antagonists were maintained for the remainder of the experiment. For the neurotrophic factor withdrawal condition, BDNF, GDNF, and CNTF were removed from the culture medium starting at day 17. Longitudinal tracking was performed by imaging neuronal cultures in a Nikon Biostation CT or Molecular Devices ImageExpress once every 24–72 hours starting at day 17. Tracking of neuronal survival was performed using SVcell 3.0 (DRVision Technologies). Neurons were scored as dead when their soma was no longer detectable by RFP fluorescence. All neuron survival assays were performed at least twice, with equal numbers of neurons from three individual replicates from one of the trials being used for the quantification shown. All trials quantified were representative of other trials of the same experiment. When iMNs from multiple independent donors are combined into one survival trace in the Kaplan-Meier plots for clarity, the number of iMNs tracked from each line can be found in Supplementary Table 5.
CRISPR/Cas9 genome editing of iPSCs
CRISPR/Cas9-mediated genome editing was performed in human iPSCs as previously described, using Cas9 nuclease62. To generate loss-of-function alleles of C9ORF72, control iPSCs were transfected with a sgRNA targeting exon 2 of the C9ORF72 gene. Colonies were picked on day 7 after transfection and genotyped by PCR amplification and sequencing of exon 2. Colonies containing a frameshift mutation were clonally purified on MEF feeders and the resulting clones were re-sequenced to verify the loss-of-function mutation in C9ORF72.
Quantitative Real Time PCR (qRT-PCR)
Total RNA was extracted from sorted iMNs at day 21 post-transduction with Trizol RNA Extraction Kit (Life Technologies) and reverse transcribed with an Oligo dT primer using ProtoScript® II First Strand Synthesis Kit (NEB). RNA integrity was checked using the Experion system (Bio-Rad). Real-time PCR was performed with iTaq Universal SYBR Green Supermix (Bio-Rad) using primers shown in Supplementary Data Table 4.
Western Blotting
iMNs from healthy controls and ALS patients were collected on day 21 post-transduction in RIPA buffer (Sigma-Aldrich) with a protease inhibitor cocktail (Roche). Protein quantity was measured by the BCA assay (Pierce) and samples were run on a 10% SDS gel at 4 °C. The gel was transferred onto an Immobilon membrane (Millipore). The membrane was blocked with 5% milk in 0.1% PBS-Tween 20 (PBS-T)(Sigma-Aldrich), incubated with primary antibodies overnight at 4 °C, washed three times with 0.1% PBS-T, then incubated with horseradish peroxidase (HRP)-conjugated (Santa Cruz). After three washes with 0.1% PBS-T, blots were visualized using an Amersham ECL Western Blotting Detection Kit (GE) or the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo) and developed on X-ray film (Genesee). The following primary antibodies were used: rabbit anti-C9ORF72 (Proteintech, cat. no. 22637–1-AP), mouse anti-GAPDH (Santa Cruz, cat. no. sc-32233), chicken anti-MAP2 (Abcam, cat. no. ab11267), mouse anti-FLAG (Sigma, cat. no. F1804), rabbit anti-GLUR1 (Millipore, cat. no. 1504), mouse anti-NR1 (Novus, cat. no. NB300118), mouse anti-Transferrin receptor (Thermo, cat. no. 136800), mouse anti-LAMP3 (DSHB, cat. no. H5C6), rabbit anti-LAMP3 (Proteintech, cat. no. 12632), mouse anti-LAMP2 (DSHB, cat. no. H4B4), mouse anti-LAMP1 (Abcam, cat. no. Ab25630), goat anti-HRP (Santa Cruz, cat. no. sc-47778 HRP), mouse anti-EEA1 (BD Biosciences, cat. no. BD610457), mouse anti-TUJ1 (Biolegend, cat. no. MMS-435P), rabbit anti-APP (Abcam, cat. no. ab32136), mouse anti-Tau5 (Thermo, cat. no. AHB0042), mouse anti-PSD-95 (Thermo, cat. no. MA1–045) , mouse anti-p53 (Cell Signaling, cat. no. 2524S), anti-mouse HRP (Cell Signaling, cat. no. 7076S), anti-rabbit HRP (Cell Signaling, cat. no. 7074S). For C9ORF72 western blots, to generate enough motor neurons for C9ORF72 protein detection, we used a directed differentiation method described previously 28.
Post-synaptic Density Extraction
Postsynaptic density extraction was done following a protocol published previously 63. Briefly, mouse spinal cord tissue or human cortical tissue was homogenized in cold Sucrose Buffer (320 mM Sucrose, 10 mM HEPES pH 7.4, 2 mM EDTA, 30 mM NaF, 40 mM β-Glycerophosphate, 10 mM Na3VO4, and protease inhibitor cocktail (Roche)) using a tissue grinder and then spun down at 500 g for 6 min at 4℃. The supernatant was re-centrifuged at 10,000 g for 10 min at 4℃. The supernatant was collected as the “Total” fraction, and the pellet was resuspended in cold Triton buffer (50 mM HEPES pH 7.4, 2 mM EDTA, 50 mM NaF, 40 mM β-Glycerophosphate, 10 mM Na3VO4, 1% Triton X-100 and protease inhibitor cocktail (Roche)) and then spun down at 30,000 RPM using a Beckman rotor MLA-130 for 40 min at 4℃. The supernantant was collected as the “Triton” fraction and the pellet was resuspended in DOC buffer (50 mM HEPES pH 9.0, 50 mM NaF, 40 mM β-Glycerophosphate, 10 mM Na3VO4, 20 uM ZnCl2, 1% Sodium Deoxycholate and protease inhibitor cocktail (Roche)) and collected as the “DOC”, PSD-enriched fraction. Collected samples were boiled with SDS-PAGE sample buffer and analyzed by western blot. Purity of the PSD preps was analyzed by immunoblotting for PSD-95 (PSD), p53 (non-PSD), and synaptophysin (non-PSD).
Generation of Dox-NIL iMNs and Biotinylation of Surface-bound Glutamate Receptors
To generate Dox-NIL iMNs, the Dox-NIL construct was integrated into the AAVS1 safe harbor locus of the control, C9+/−, and C9-ALS patient iPSC lines using CRISPR/Cas9 editing (gRNA sequence shown in Supplementary Table 4). Dox-NIL iMNs were generated by plating at ~25% confluency on matrigel coated plates and adding 1 μg/ml of doxycylin in N3 media +7.5 μM RepSox 1 day after plating. Mouse primary mixed glia were added to the cultures at day 6, and doxycyline was maintained throughout conversion. iMN cultures were harvested at day 17.
Biotinylation of plasma membrane localized glutamate receptors was performed using the Piece™ Cell Surface Protein Isolation Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Briefly, Dox-NIL iMNs were incubated with 0.25mg/ml Sulfo-NHS-SS-Biotin in cold room for 1~2 hrs with end-to-end shaking. After quenching, cells were harvested by scraping and lysed with lysis buffer from the Piece™ Cell Surface Protein Isolation Kit or the M-PER™ mammilian protein extraction buffer (Thermo Fisher Scientific). Cell lysate was incubated with High Capacity NeutrAvidin™ agorase beads (Thermo Fisher Scientific), and the bound protein was eluted in 2X SDS-PAGE sample buffer supplemented with 50mM DTT for 1 hr at room temperature with end-to-end rotation, and further analyzed by western blot.
Human Astrocyte Glutamate Uptake Assay
Human primary astrocytes from Lonza (cat. no. CC-2565) were cultured in Lonza Astrocyte Growth Medium (cat. no. CC-3186) according to the manufacturer’s instructions. To force expression of dipeptide repeat proteins in the astrocytes, lentiviruses were produced in HEK 293T cells and concentrated ~80-fold using Lenti-X Concentrator (Clontech). When astrocytes reached 90% confluency, they were transduced with lentiviruses encoding either GFP or GR(50)-GFP. Fluorescence microscopy analysis was used to confirm that 60–80% of astrocytes were transduced in both conditions. To measure glutamate uptake, GFP- or GR(50)-GFP-expressing astrocytes were incubated with 200 mM glutamate in Astrocyte Growth Medium for 2 hours at 37 degrees Celsius in biological quadruplicate. Media samples were collected after 2 hours and glutamate levels were quantified using the BioVision Glutamate Colorimetric Assay Kit (cat. no. K629) according to the manufacturer’s instructions. Colorimetric substrate levels were quantified at 450 nm using a Molecular Devices SpectraMax i3x Multi-Mode Microplate Reader.
GST pull-down
Human EEA1 (1–209) with an N-terminal GST tag in pGEX-6P-1 vector or GST only were expressed in E. Coli BL21 (DE3) cells (Thermo Fisher Scientific) for 12 hr at 18℃. Harvested cells were lysed by sonication in cold GST Purification Buffer (50 mM Tris pH 8.0, 200 mM NaCl, 2 mM DTT, 0.5 mg/ml Lysozyme, 0.2% Triton X-100 and protease inhibitor cocktail (Roche)). After centrifugation at 15,000 g for 30 min at 4℃, clarified lysate was incubated with Glutathione Sepharose 4B beads (GE Healthcare Life Science) for 3 hours to purify GST-EEA1 or GST. HEK cells were transfected with C-terminal 3XFLAG tagged C9ORF72 isoform A or B, or eGFP constructs and harvested 36–48 hr post-transfection in cold Lysis Buffer (25 mM HEPES pH 7.4, 100 mM NACl, 5 mM MgCl2, 1 mM DTT, 10% Glycerol, 0.1% Triton X-100 and protease inhibitor cocktail (Roche)).After centrifugation at 8,000 g for 10 min at 4℃, the clarified supernatant was incubated with washed GST-EEA1 or GST beads for 2 hr at 4℃ with end-to-end rotation. Beads were then boiled in 2X SDS-PAGE sample buffer and pulled-down protein was analyzed by western blot.
Percoll Density Gradient Centrifugation
IPSC-MNs at differentiation D35 were harvested in cold Hypotonic buffer (20 mM HEPES pH 7.4, 10 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1mM EGTA, 1 mM DTT and protease inhibitor cocktail (Roche)) and lysed by passing through G25 needles 25 times and then spun down at 700 x g for 10min at 4℃. The Supernatant was loaded onto pre-made 30% Percoll solution and re-centrifuged at 33,000 RPM using Beckman rotor SWI55 for 50min at 4℃. 300 ul aliquots were taken from top to bottom as fractions and all the collected samples were boiled with SDS-PAGE sample buffer and analyzed by western blot.
Membrane Fraction Isolation for Immunoblot Analysis of Lysosomal Protein Levels
iMN cultures were harvested with the Plasma Membrane Isolation Kit (Abcam) according to the manufacturer’s instructions.
Fluorescent In Situ Hybridization (FISH) for RNA foci detection
Samples were first fixed in 4% PFA (1x PBS) overnight at 4°C and were subsequently washed three times with 1x PBS. Next, cells were permeabilized with 0.3% Triton X-100 (1x PBS) for 10 min at room temperature, followed by three washes with 1x PBS for 10 min each. After permeabilization, the samples were equilibrated in 1x SSC buffer for 10 min at room temperature and then transferred into 50% formamide (2x SSC) for 10 min at 60°C. The repeat expansion-targeting probe and the negative control probe were ordered from Exiqon 58. During this step, the probe mixture (1µl salmon sperm (10 µg/µl), 0.5 µl E. coli tRNA (20 µg/µl), 0.4 µl probe (25 µM), 25 µl 80% formamide/per sample) was made and placed at 95°C for at least 10 min. The samples were submerged in 200 µl of hybridization buffer (4ml 100% formamide, 0.5 ml 20x SSC, 1 ml BSA fraction V, 0.5ml RVC (20 mM), 1ml NaPO4 (0.1 M), 3 ml nuclease-free water) and 27 µl of the probe mixture was added to each sample and incubated for 1 hour at 60°C. After probe hybridization, the samples were washed twice with 50% formamide (2x SSC) for 20 min each at 65°C and once more with 40% formamide (1x SSC) for 10 min at 60°C. The remaining formamide was removed by briefly washing with 1x SSC three times. A final crosslinking step was performed by first incubating the samples with 1x Tris-Glycine for 5 minutes followed by a 5 min incubation in 4% PFA. Samples were washed three times with 1x PBS, stained with DAPI, and imaged using a Zeiss LSM 800 confocal microscope.
GCaMP6 calcium influx assay
GCaMP6 was cloned into the pMXs-Dest-WRE retroviral vector and transduced into reprogramming cultures concurrently with the motor neuron factors. To assess GCaMP6 activity, 1.5 μm glutamate was added to iMN cultures and cells were imaged continuously for 2 minutes at 24 frames per second. GFP flashes were scored manually using the video recording. At least 3 different fields of view from three independent cultures, totalling 50–100 iMNs, were scored per condition.
Dipeptide repeat protein expression in iMNs and PR50-Dendra2 turnover analysis
To measure the effect of dipeptide repeat protein expression on iMN survival, PR50 and GR50 were cloned into the pHAGE lentiviral vector as fusions with GFP to allow tracking of protein expression. iMN cultures were transduced with PR50 and GR50 lentiviruses at day 17 of reprogramming and longitudinal survival analysis was started the same day. 10 ng/ml of GDNF, BDNF, and CNTF was maintained throughout the experiment, and glutamate treatment was not performed. To measure PR50 turnover, PR50 was cloned into the pHAGE lentiviral vector as a fusion with Dendra2 (Addgene). iPSC-derived fibroblasts were generated according to Daley and colleagues64. Briefly, when C9ORF72−/− iPSC cultures reached 80% confluence, the medium was switched from mTeSR1 (Stem Cell Technologies) to human fibroblast medium containing DMEM (Life Technologies), 10% fetal bovine serum (FBS)(Thermo Fisher Scientific), and 1% penicillin/streptomycin (Life Technologies). Cells were passaged 2 to 3 times using Accutase (Life Technologies) before use in experiments. iPSC-derived fibroblasts were transduced with either pMXs-eGFP or pMXs-C9ORF72 isoform B-T2A-eGFP retrovirus and treated with 10 μg/ml mitomycin C for 3 hrs to inhibit cell proliferation. The cells were then transduced with the PR50–Dendra2 lentivirus and exposed to blue light for 1.5 sec using a lumencor LED light source to initiate photoconversion. The amount of decay (as a fraction of the starting level) of the red fluorescent punctae was monitored by longitudinal time lapse imaging in a Molecular Devices ImageExpress and analyzed using SVCell 2.0 (DRVision Technologies). Fluorescence was quantified at t = 0 and 12 hours after photoconversion. Distinct photoconverted punctae were treated as discrete objects for analysis (n = 20 each for +eGFP and +C9ORF72-T2A-eGFP). For each object, background fluorescence was subtracted and fluorescence was normalized according to object size. The fractional decay was statistically analyzed by two-tailed Student’s t-test. ** - p<.01.
For iMN Dendra2 experiments, the same procedure was used except that PR50–Dendra2 lentivirus was transduced into iMN cultures at day 14 of conversion.
Small Molecule Screen and PIKFYVE inhibitor assays
Hb9::RFP+ C9ORF72 ALS/FTD iMNs were generated in 96-well plates. On Day 15 post transduction, neurotrophic factors and RepSox were withdrawn and the small molecule library was added (EMD Millipore kinase collection and Stemselect library, 3.3 µM final concentration) and added fresh every other day until the screen was terminated on Day 25 post-transduction. Identification of neuroprotective compounds was identified using SVcell 3.0 (DRVision Technologies) and further verification by manual iMN tracking.
Synthesis and activity assays of Apilimod and the reduced activity analog
For experiments other than the comparison of Apilimod and the reduced-activity analog, Apilimod was purchased from Axon Medchem (cat. no. 1369). For the reduced-activity analog assays, Apilimod and the reduced activity analog were synthesized at Icagen, Inc. according to the schemes shown in Supplementary Fig. 16. PIKFYVE kinase inhibition was measured using the ADP-Glo kinase assay from SignalChem according to the manufacturer’s instructions, using purified PIKFYVE kinase (SignalChem cat. no. P17–11BG-05).
RNA Sequencing
Libraries were prepared from total RNA using Clontech SMARTer Stranded RNA-Seq kit, with Clonetech RiboGone ribodepletion performed ahead of cDNA generation. Amounts of input RNA were estimated using the Bioanalyzer and libraries produced according to Clontech’s protocol. Library generation and sequencing were performed at the Norris Cancer Center Sequencing Core at USC. All FASTQ files were analyzed using FastQC (version 0.10.1), trimmed using the FASTQ Toolkit (v 1.0), aligned to the GRCh37/hg19 reference genome using Tophat (version 2), and transcripts assembled and tested for differential expression using Cufflinks (version 2.1.1). Raw data is available for public download in the NCBI database under accession code PRJNA296854.
Bioinformatics analysis of gene expression.
RNA sequencing output was aligned to the GRCh38 Reference Genome and quantified using the STAR aligner.65 Genes were annotated against the GENCODE version 23 Comprehensive Gene Annotation. Quality control was performed using Picard Tools AlignmentSummaryMetrics. Samples passing quality control and having RNA Integrity Number (RIN) > 5 were used in downstream analysis. To identify differentially expressed genes, the R package DESeq2 was used as previously described.66 The function DESeq was used to estimate size factors, estimate dispersion, fit the data to a negative binomial generalized linear model, and generate differential expression statistics using the Wald test. KEGG enrichment analysis was performed for internal analysis using the R package clusterProfiler.67
We compared the differential expression results from our data to other transcriptomic datasets in ALS, obtained from the Gene Expression Omnibus (GEO). Raw Affymetrix array data (.CEL files) were downloaded for dataset GSE56504, and preprocessed using a standard exon array pipeline implemented using the R Bioconductor package oligo. For GSE56504, only the laser-capture microdissection samples were included/ Differential expression was calculated using the R Bioconductor package limma. RNA-seq counts data was obtained for dataset GSE67196. For GSE67196, only the frontal cortex samples were included. Normalization and differential expression analysis were performed using DESeq2.
Retinoic acid/purmorphamine protocol for iPSC-motor neuron differentiation
iPSC motor neurons were generated as described previously28, with slight modifications. On day 0, iPSCs were dissociated with Accutase (Life Technologies) and 300,000 iPSCs were seeded into one Matrigel (Corning)-coated well of a 6-well plate in mTeSR medium (Stem Cell Technologies) with 10 μM Rock Inhibitor (Selleck). On day 1, the medium was changed to Neural Differentiation Medium (NDM) consisting of a 1:1 ratio of DMEM/F12 (Genesee Scientific) and Neurobasal medium (Life Technologies), 0.5x N2 (Life Technologies), 0.5x B27 (Life Technologies), 0.1 mM ascorbic acid (Sigma), 1x Glutamax (Life Technologies). 3 μM CHIR99021 (Cayman), 2 μM DMH1 (Selleck), and 2 μM SB431542 (Cayman) were also added. On day 7, cells were dissociated with Accutase and 4.5 million cells were seeded into Matrigel coated 10cm dishes in NDM plus 1 μM CHIR99021, 2 μM DMH1, 2 μM SB431542, 0.1 μM RA (Sigma), 0.5 μM Purmorphamine (Cayman), and 10 μM Rock Inhibitor. Rock inhibitor was removed on day 9. On day 13, cells were dissociated with Accutase and seeded at a density of 40 million cells per well in a non-adhesive 6 well plate (Corning) in NDM plus 0.5 μM RA, 0.1 μM Purmorphamine, and 10 μM Rock Inhibitor. On day 19, the media was changed to NDM plus 1 μM RA, 1 μM Purmorphamine, 0.1 μM Compound E (Cayman), and 5 ng/ml each of BDNF, GDNF and CNTF (R&D Systems). Cells were used for experiments between days 25–35 of differentiation.
Electron microscopy
Cells were fixed in 6-well culture plates in 2.5 % glutaraldehyde in 0.1M cacodylate buffer, post-fixed in 1% osmium tetroxide for 1 hour and block stained in 1% uranyl acetate in 0.1M acetate buffer pH 4.4 overnight at 4 ˚C. Dehydration was performed in increasing concentrations of ethanol (10%/25%/50%/75%/90%/100%/100%/100%) for 15 minutes each and infiltrated with increasing concentrations of Eponate12 (Ted Pella Inc., Redding, CA, USA), 25% Eponate12 (no catalyst) in ethanol for 3 hours, 50% overnight, 100% for 5 hours, 100% overnight, and polymerized in fresh Eponate12 with DMP-30 for 48 hours at 60 ˚C. Previously marked areas were sawed out, the tissue culture plastic was removed and the selected area sectioned parallel to the substrate at a thickness of 70 nm. Sections at a depth of 3–5 µm were collected on formvar-filmed 50 mesh copper grids and imaged at 80 kV in an FEI 208 Morgagni (FEI is in Hillsboro, OR, USA). Per micrograph, cytosol was used to quantify the number of electron dense spheres that were defined as lysosomes 40.
Mouse immunostaining and quantification
All animal use and care were in accordance with local institution guidelines of the University Medical Center Utrecht (Utrecht, the Netherlands) and approved by the Dierexperimenten Ethische Commissie Utrecht with the protocol number DEC 2013.I.09.069. Wild-type C57BL6/J (strain: 000664), C9ORF72 KO (C57BL/6J-3110043021Rikem5Lutzy/J, strain: 027068), and C9-BAC (C57BL/6J-Tg(C9orf72_i3)112Lutzy/J, strain: 023099) were purchased from Jackson Laboratories (The Jackson Laboratory, Bar Harbor, USA). Mice were housed in standard conditions with food and water ad libitum in the conventional vivarium at the University of Southern California. All animal use and care were in accordance with local institution guidelines of the University of Southern California and the IACUC board of the University of Southern California (Los Angeles, USA) with the protocol numbers 20546 and 11938.
Previously described Nestin‐Cre+/− C9orf72loxP/loxP mice22 (18 months old) and age-matched controls (n=2 per genotype, 1 male and 1 female for C9orf72−/− and 2 males for C9orf72+/+) were transcardially perfused with phosphate buffered saline (PBS) and subsequently with 4% formaldehyde. Cryoprotection occurred in 30% sucrose. Additionally, 6 month old C9-BAC and wildtype controls were transcardially perfused with phosphate buffered saline (PBS) and subsequently with 4% formaldehyde. Cryoprotection occurred in 20% sucrose. After snap freezing, tissue was sectioned by cryostat at 20 µm thickness and stained with the following primary antibodies: NMDAR1, GluR1, GluR6/7, Lamp1, NeuN, Chat, and GR. Antigen retrieval by sodium citrate occurred prior to GR staining. Images were collected using a Zeiss LSM780 or LSM800 confocal microscope. Glutamate subunit intensity measurements occurred with ImageJ where the mean cytosolic intensity was divided by a background measurement collected near to the measured neuron. Lamp1 and GR quantification occurred with the help of ImageJ. The scientist performing the glutamate receptor subunit intensity, LAMP1 vesicle, and GR quantification was blinded to the genotypes or treatment of the samples.
NMDA/Apilimod hippocampal administration and NMDA-lesion site analysis
Mice were anesthetized with i.p. ketamine (100 mg ⁄ kg) and xylazine (10 mg ⁄ kg), and body temperature kept at 36.9 ± 0.1°C with a thermostatic heating pad. Mice were placed in a stereotactic apparatus (ASI Instruments, USA) and the head is fixed accordingly. A burr hole was drilled, and an injection needle (33 gauge) was lowered into the hippocampus between CA1 and the dentate gyrus (AP −2.0, ML +1.5, DV −1.8). NMDA (20 nmol in 0.3 μl of phosphate-buffered saline, pH 7.4) was infused over 2 min using a micro-injection system (World Precision Instruments, Sarasota, FL, USA). Simultaneously, or independently, Apilimod (0.3 μl of 20 μM in phosphate-buffered saline, pH 7.4) was infused over 2 min using a micro-injection system (World Precision Instruments, Sarasota, FL, USA). The needle was left in place for an additional 8 min after the injection. Animals were euthanized 48 h later. Brains were quickly removed, frozen on dry ice, and stored at −80°C until processing. Thirty-micrometer-thick coronal sections were prepared using a cryostat. Every fifth section 1 mm anterior and posterior to the site of injection was stained with cresyl violet. The lesion area was identified by the loss of staining, measured by NIH ImageJ software and integrated to obtain the volume of injury.
Immunohistochemistry of human tissue
Post mortem tissues were kindly provided by Neil Shneider (Columbia) and were collected from the following individuals: Sample 1 – age: 64, diagnosis: ALS, genotype: positive for C9ORF72 repeat expansion, Sample 2 – age: 55, diagnosis: ALS, genotype: positive for C9ORF72 repeat expansion, Sample 3 – age: 65, diagnosis: ALS, genotype: positive for C9ORF72 repeat expansion, Sample 4 – age: 65, diagnosis: control, genotype: negative for C9ORF72 repeat expansion, Sample 5 – age: 50, diagnosis: control, genotype: negative for C9ORF72 repeat expansion, Sample 6 – age: 50, diagnosis: control, genotype: negative for C9ORF72 repeat expansion, Sample 7 – age: 53, diagnosis: ALS, genotype: negative for C9ORF72 repeat expansion, Sample 8 - age: 64, diagnosis: ALS, genotype: negative for C9ORF72 repeat expansion. All donors except donor 7 (sample 7) were female. For immunofluorescence, 10 µm sections were sliced from flash frozen lumbar spinal cord tissues. Sections were then air dried and fixed with ice cold acetone for 10 minutes, and blocked with 10% normal goat serum/1% BSA/0.3% Triton-X/PBS at room temperature for 1 hour followed by incubation with NR1 antibody (1:200, BD Bioscience) in blocking buffer overnight at 4 ºC. Sections subsequently were blocked using avidin/biotin kit (Vector Lab), and washed with PBS. Then, sections were incubated with goat anti-rabbit IgG Biotin conjugate secondary antibody (1:750, Invitrogen) or with goat anti-mouse IgG Biotin conjugate secondary antibody (1:750, Invitrogen) for 1 hour at room temperature, washed and incubated with streptavidin-Alexa Fluor 488 conjugate (1:500, Invitrogen) in dark for 1 hour at room temperature. Sections were washed and blocked again in blocking buffer for 1 hour at room temperature. For neuronal marker staining, sections were incubated with Tu-20 antibody (1:1000, Abcam) or NeuN antibody (1:500, Abcam) at 37 ºC for 1 hour. Sections were washed with PBS and incubated with goat anti-mouse Alexa Fluor 546 (1:500, Invitrogen) or goat anti-rabbit Alexa Fluor 546 (1:500, Invitrogen) for 1 hour at room temperature. Lipofuscin autofluorescence was quenched by immersing sections in autofluorescence eliminator reagent (Millipore) for 4 minutes following manufacture’s instruction. Sections were then counterstained and mounted with Prolong Gold antifade mounting medium with DAPI (Invitrogen).
The following information can also be found in the Life Sciences Reporting Summary:
Statistical Analysis
Analysis was performed with the statistical software package Prism Origin (GraphPad Software, La Jolla, USA). Statistical analysis of iMN survival experiments was performed using a two-sided log-rank test to account for events that did not occur (i.e. iMNs that did not degenerate before the end of the experiment). For each line, the survival data from 50 iMNs were selected randomly using Microsoft Excel, and these data were used to generate the survival curve. If all iMNs degenerated in a given experiment, statistical significance was calculated using a two-tailed Student’s t-test. For all other experiments, differences between two groups were analyzed using a two-tailed Student’s t-test, unless the data was non-normally distributed for which two-sided Mann-Whitney testing was used. Differences between more than two groups were analyzed by one way-ANOVA with Tukey correction for multiple testing. Significance was assumed at p < 0.05. Error bars represent the standard deviation unless otherwise stated.
For all experiments, sample size was chosen using a power analysis based on pilot experiments that provided an estimate of effect size (http://ww.stat.ubc.ca/~rollin/stats/ssize/n2.html). Mice used for immunohistochemical analysis were selected randomly from a set of genotyped animals (genotypes were known to investigators). Mouse and human tissue sections used for immunohistochemical analysis were selected randomly. For mouse tissues, sections were prepared using an approximately equal representation of all levels of the spinal cord, and of those, all were imaged and quantified. The sections were only not used if NeuN or Chat immunostaining failed. For iMN survival assays, assays were repeated at least twice, with each round containing 3 biologically independent iMN conversions. iMNs from the 3 biologically independent iMN conversions in one representative round was used to generate the Kaplan-Meier plot shown. iMN survival times were confirmed by manual longitudinal tracking by an individual who was blinded to the identity of the genotype and condition of each sample. To select 50 iMNs per condition for analysis, >50 neurons were selected for tracking randomly at day 1 of the assay. Subsequently, the survival values for 50 cells were selected at random using the RAND function in Microsoft Excel. For quantification of immunofluorescence, samples were quantified by an individual who was blinded to the identity of the genotype of each sample.
iMN survival times were confirmed by manual longitudinal tracking by an individual who was blinded to the identity of the genotype and condition of each sample. All other quantification was performed by individuals blinded to the identity of each sample.
Additional Reagent Information
The following antibodies were used in this manuscript: mouse anti-HB9 (Developmental Studies Hybridoma Bank); 81.5C10. chicken anti-TUJ1 (EMD Millipore); AB9354. rabbit anti-VACHT (Sigma); SAB4200559. rabbit anti-C9ORF72 (Sigma-Aldrich); HPA023873. rabbit anti-C9ORF72 (Proteintech); 25757–1-AP. mouse anti-EEA1 (BD Biosciences); 610457. mouse antiRAB5 (BD Biosciences); 610281. mouse anti-RAB7 (GeneTex); GTX16196. mouse anti-LAMP1 (Abcam); ab25630. mouse anti-M6PR (Abcam); ab2733. rabbit anti-GluR1 (EMD Millipore); pc246. mouse anti-NR1 (EMD Millipore); MAB363. chicken anti-GFP (GeneTex); GTX13970. rabbit anti-Glur6/7 (EMD Millipore); 04–921. mouse anti-FLAG (Sigma); F1804. mouse anti-GAPDH (Santa Cruz); sc-32233. chicken anti-MAP2 (Abcam); ab11267, rabbit anti-GLUR1 (Millipore, cat. no. 1504), mouse anti-NR1 (Novus, cat. no. NB300118), mouse anti-Transferrin receptor (Thermo, cat. no. 136800), mouse anti-LAMP3 (DSHB, cat. no. H5C6), rabbit anti-LAMP3 (Proteintech, cat. no. 12632), mouse anti-LAMP2 (DSHB, cat. no. H4B4), goat anti-HRP (Santa Cruz, cat. no. sc-47778 HRP), mouse anti-TUJ1 (Biolegend, cat. no. MMS-435P), rabbit anti-APP (Abcam, cat. no. ab32136), mouse anti-Tau5 (Thermo, cat. no. AHB0042), mouse anti-PSD-95 (Thermo, cat. no. MA1–045), mouse anti-p53 (Cell Signaling, cat. no. 2524S), anti-mouse HRP (Cell Signaling, cat. no. 7076S), anti-rabbit HRP (Cell Signaling, cat. no. 7074S).
HEK 293T cells were used to produce retrovirus, lentivirus, and C9ORF72 protein. HEK cells were used for these purposes based on previous published studies using HEK cells in order to produce viral particles and mammalian proteins. HEK cells were obtained from American Type Culture Collection, catalog number CRL-11268. HEK and iPS cells were tested for mycoplasma before, during, and after the study and were negative.
All experiments involving live vertebrates (cortical glial isolation) performed at USC were done in compliance with ethical regulations approved by the USC IACUC committee. All animal use and care at the University Medical Center Utrecht were in accordance with local institution guidelines of the University Medical Center Utrecht (Utrecht, the Netherlands) and approved by the Dierexperimenten Ethische Commissie Utrecht with the protocol number DEC 2013.I.09.069.
Supplementary Material
Acknowledgments
We thank the NINDS Biorepository at Coriell Institute for providing the following cell lines for this study: ND12133, ND03231, ND01751, ND11976, ND03719, ND00184, ND5280, ND06769, ND10689, ND12099, ND14954, ND08957, ND12100, and ND014587. We thank Helena Chui and Carol Miller at the University of Southern California Alzheimer’s Disease Research Center and Neil Shneider at the Columbia University Medical Center for control and C9ORF72 patient tissue. We thank the Choi Family Therapeutic Screening Facility for chemical screening support and the Translational Imaging Center at USC for imaging support. We thank Max Koppers, Youri Adolfs, Christiaan van der Meer, and Mark Broekhoven for help with mouse breeding and kainate injection experiments. We thank Prof. Satoshi Waguri for providing the M6PR-GFP construct. We thank Christopher Buser for assistance with electron microscopy. We also thank Sam Alworth (DRVision Technologies, LLC), Katja Hebestreit, and Raj Bhatnagar (Verge Genomics), Bob Baloh, Jacqueline O’Rourke, Christopher Donnelly, Chang Tong, Andrew McMahon and Qing Liu-Michael for reagents, technical support, and discussions. E.Y.S. is a Walter V. and Idun Berry Postdoctoral Fellow. K.A.S. was supported in part by a Muscular Dystrophy Association Development Grant. L.M. was supported by NIH grant T32DC009975–04. This work was supported by NIH grants AG039452, AG023084, and NS034467 to B.V.Z. R.J.P. was supported by grants from ALS Foundation Netherlands (TOTALS), Epilepsiefonds (12–08, 15–05), and VICI grant Netherlands Organisation for Scientific Research (NWO). This work was also supported by NIH grants R00NS077435 and R01NS097850, U.S. Department of Defense grant W81XWH-15–1-0187, and grants from the Donald E. and Delia B. Baxter Foundation, the Tau Consortium, the Frick Foundation for ALS Research, the Muscular Dystrophy Association, the New York Stem Cell Foundation, the USC Keck School of Medicine Regenerative Medicine Initiative, the USC Broad Innovation Award, and the Southern California Clinical and Translational Science Institute to J.K.I. J.K.I. is a New York Stem Cell Foundation-Robertson Investigator.
Footnotes
Competing financial interests
J.K.I. and P.A. are co-founders of Acurastem, Inc. P.A. is an employee of Icagen Corporation. J.K.I. and P.A. declare that they are bound by confidentiality agreements that prevent them from disclosing details of their financial interests in this work. S-J.L. is a founder of DRVision Technologies and T-Y.C. is an employee of DRVision Technologies. A.Z. and J.A.C. are co-founders of Verge Genomics and V.H-S., N.W., and T.G.B. are employees of Verge Genomics.
Code availability
Code for RNA sequencing analysis can be obtained from Verge Genomics by emailing alice@vergegenomics.com.
Data Availability Statement
Code for RNA sequencing analysis (Supplementary Fig. 6) can be obtained from Verge Genomics by emailing alice@vergegenomics.com.
References
- 1.Renton AE, Chio A & Traynor BJ State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majounie E, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11, 323–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5, 208ra149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donnelly CJ, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 110, E4530–4539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haeusler AR, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen X, et al. Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates that Initiate In Vitro and In Vivo Neuronal Death. Neuron 84, 1213–1225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mori K, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Kwon I, et al. Poly-dipeptides encoded by the C9ORF72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science (2014). [DOI] [PMC free article] [PubMed]
- 13.Su Z, et al. Discovery of a Biomarker and Lead Small Molecules to Target r(GGGGCC)-Associated Defects in c9FTD/ALS. Neuron ePub Aug(2014). [DOI] [PMC free article] [PubMed]
- 14.Jiang J, et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron (2016). [DOI] [PMC free article] [PubMed]
- 15.Chew J, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, et al. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron (2016). [DOI] [PubMed]
- 17.Liu EY, et al. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol 128, 525–541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Rourke JG, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burberry A, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8, 347ra393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sellier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J (2016). [DOI] [PMC free article] [PubMed]
- 21.Webster CP, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J (2016). [DOI] [PMC free article] [PubMed]
- 22.Koppers M, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol (2015). [DOI] [PMC free article] [PubMed]
- 23.Fratta P, et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol 126, 401–409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Son EY, et al. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchetto MC, et al. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3, 649–657 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Kiskinis E, et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin CL, et al. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20, 589–602 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Du ZW, et al. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat Commun 6, 6626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfisterer U, et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A 108, 10343–10348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aoki Y, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Sullivan PM, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta neuropathologica communications 4, 51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webster CP, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 35, 1656–1676 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farg MA, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23, 3579–3595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prudencio M, et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci 18, 1175–1182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Highley JR, et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathology and applied neurobiology 40, 670–685 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Levine TP, Daniels RD, Gatta AT, Wong LH & Hayes MJ The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marat AL, Dokainish H & McPherson PS DENN domain proteins: regulators of Rab GTPases. J Biol Chem 286, 13791–13800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amick J, Roczniak-Ferguson A & Ferguson SM C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell 27, 3040–3051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sellier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J 35, 1276–1297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neiss WF The electron density of light and dark lysosomes in the proximal convoluted tubule of the rat kidney. Histochemistry 77, 63–77 (1983). [DOI] [PubMed] [Google Scholar]
- 41.Saftig P & Klumperman J Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10, 623–635 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Ganley IG, Carroll K, Bittova L & Pfeffer S Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol Biol Cell 15, 5420–5430 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez-Gabin AG, Yin X, Si Q & Larocca JN Transport of mannose-6-phosphate receptors from the trans-Golgi network to endosomes requires Rab31. Exp Cell Res 315, 2215–2230 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waguri S, et al. Visualization of TGN to endosome trafficking through fluorescently labeled MPR and AP-1 in living cells. Mol Biol Cell 14, 142–155 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang G, Gilbert J & Man HY AMPA receptor trafficking in homeostatic synaptic plasticity: functional molecules and signaling cascades. Neural plasticity 2012, 825364 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen P, Gu Z, Liu W & Yan Z Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol Pharmacol 72, 40–51 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wainger BJ, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 7, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gurskaya NG, et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol 24, 461–465 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Mackenzie IR, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol 130, 845–861 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Cai X, et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chemistry & biology 20, 912–921 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lemmon MA Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol 9, 99–111 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Martin S, et al. Inhibition of PIKfyve by YM-201636 dysregulates autophagy and leads to apoptosis-independent neuronal cell death. PLoS One 8, e60152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikonomov OC, Sbrissa D & Shisheva A Localized PtdIns 3,5-P2 synthesis to regulate early endosome dynamics and fusion. American journal of physiology. Cell physiology 291, C393–404 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Chow CY, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet 84, 85–88 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lodhi IJ, et al. Insulin stimulates phosphatidylinositol 3-phosphate production via the activation of Rab5. Mol Biol Cell 19, 2718–2728 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong Z, et al. Protein S protects neurons from excitotoxic injury by activating the TAM receptor Tyro3-phosphatidylinositol 3-kinase-Akt pathway through its sex hormone-binding globulin-like region. J Neurosci 30, 15521–15534 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Rourke JG, et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 88, 892–901 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeigerer A, et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature 485, 465–470 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Topp JD, Gray NW, Gerard RD & Horazdovsky BF Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J Biol Chem 279, 24612–24623 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods 8, 409–412 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, et al. Long-term potentiation modulates synaptic phosphorylation networks and reshapes the structure of the postsynaptic interactome. Sci Signal 9, rs8 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anders S, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protocols 8, 1765–1786 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Yu G, Wang L-G, Han Y & He Q-Y clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A Journal of Integrative Biology 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.