

Significance
Bacterial pathogens are developing resistance to antibiotic compounds at an alarming rate. We use computer simulations to design inhibitors of the Escherichia coli multidrug resistance protein EmrE (efflux-multidrug resistance E) from the small multidrug family. Starting with low-resolution X-ray data, we obtain an atomic structure of EmrE using extensive molecular simulations. Based on the structure, we design hydrocarbon-stapled peptide inhibitors of EmrE, which are synthesized and shown to be effective in vivo. The rational drug design approach described here holds promise for combating efflux-mediated drug resistance in microbes and, more generally, in cancer.
Keywords: drug resistance, molecular dynamics, membrane proteins, structure refinement, stapled peptides
Abstract
Small multidrug resistance (SMR) pumps represent a minimal paradigm of proton-coupled membrane transport in bacteria, yet no high-resolution structure of an SMR protein is available. Here, atomic-resolution structures of the Escherichia coli efflux-multidrug resistance E (EmrE) multidrug transporter in ligand-bound form are refined using microsecond molecular dynamics simulations biased using low-resolution data from X-ray crystallography. The structures are compatible with existing mutagenesis data as well as NMR and biochemical experiments, including pKas of the catalytic glutamate residues and the dissociation constant () of the tetraphenylphosphonium+ cation. The refined structures show the arrangement of residue side chains in the EmrE active site occupied by two different ligands and in the absence of a ligand, illustrating how EmrE can adopt structurally diverse active site configurations. The structures also show a stable, well-packed binding interface between the helices H4 of the two monomers, which is believed to be crucial for EmrE dimerization. Guided by the atomic details of this interface, we design proteolysis-resistant stapled peptides that bind to helix H4 of an EmrE monomer. The peptides are expected to interfere with the dimerization and thereby inhibit drug transport. Optimal positions of the peptide staple were determined using free-energy simulations of peptide binding to monomeric EmrE. Three of the four top-scoring peptides selected for experimental testing resulted in significant inhibition of proton-driven ethidium efflux in live cells without nonspecific toxicity. The approach described here is expected to be of general use for the design of peptide therapeutics.
Multidrug resistance poses serious challenges for the treatment of many infectious diseases (1). A common mechanism by which multidrug resistance arises in bacteria involves active efflux of cytotoxic compounds by transmembrane pumps. The small multidrug resistance (SMR) family of membrane-bound transporters is ubiquitous in bacteria; they are present in Mycobacterium tuberculosis, Pseudomonas aeruginosa, Bordetella pertussis, Neissaria meningitis, Bacillus anthracis, and Staphylococcus aureus, among others (2–4). SMR proteins use the electrochemical potential of proton influx to pump toxic compounds from the cytoplasm into the periplasmic space where, in conjunction with the resistance/nodulation/cell division (RND) superfamily of proteins, substrates are then fully extruded from the cell (5). They confer resistance to a wide variety of quaternary ammonium compounds (QACs) (6), some of which are used as disinfectants in hospitals and in the food industry (e.g., benzalkonium chloride), and also promote resistance to antibiotics such as ampicillin, erythromycin, and tetracycline (7, 8). Although the efflux-multidrug resistance E (EmrE) transporter from Escherichia coli has been the subject of experimental studies (reviewed in ref. 9), atomic-level structural information that is needed for the understanding of the drug-pumping mechanism or rational design of inhibitors is not available. Recent cryo-EM and X-ray studies of EmrE have produced only low-resolution structures (10–12). Here, we use microsecond molecular dynamics (MD) simulations with restraints to the low-resolution X-ray density map to obtain all-atom structures of EmrE in ligand-free and ligand-bound states embedded in a lipid membrane. The structures are validated using available experimental data. We then use the refined structures and extensive free-energy MD simulations for the rational design of hydrocarbon-stapled peptides (13–15) that inhibit EmrE dimerization by binding to a monomer. Several of these peptides were synthesized and tested in live E. coli cells; they showed significant efflux inhibition at concentrations that did not cause nonspecific cytotoxicity. The drug design strategy used here could be applied to other types of efflux-mediated drug resistance in bacteria as well as to drug resistance in cancer.
Results
We begin with an analysis of the optimized structure and its use in the modeling of other structures—that is, EmrE with bound ethidium ligand, ligand-free EmrE, monomeric EmrE, as well as structures of proposed stapled peptide inhibitors—and conclude with the experimental testing of inhibitors in E. coli cells. Additional analysis, such as interpretation of mutagenesis data in light of the structures, is given in SI Appendix, SI Text.
Overall Structure of the EmrE Dimer.
The refined structure is superposed onto the Protein Data Bank (PDB) -only structure in Fig. 1 A–C. The main differences are in the loop regions, with the positions of the helices in good correspondence, consistent with the moderate root-mean-square distance (RMSD) between the simulation and the initial structure used for MD simulation shown in Fig. 1E. Further, root-mean-square fluctuations (RMSFs) computed from the simulation correlate with the data from B-factors with the Pearson coefficient of 0.7 (Fig. 1F). This value is comparable to those obtained for MD simulation starting from complete all-atom crystal structures (16). Furthermore, a strong correlation should not be expected, because the MD simulations are of a dimer in solution rather than in a crystal. The conformations of the intrahelical loops are somewhat more extended in the refined structure than in the PDB structure. This behavior is expected because loops are generally more confined by crystal packing and because they have more charged residues (9), which interact with the lipid head groups and water molecules. The loops fluctuate more than the helices, as can be seen from the RMSF and B-factor plot in Fig. 1F. The refined structure has a similar degree of compactness as the crystal structure (the radii of gyration of the X-ray and refined structures being 15.5 Å and 15.9 Å, respectively, when only the helices are included), which underscores that the structure is stable in the dimyristoylphosphatidylcholine (DMPC) bilayer. DMPC was chosen to approximate the experimental conditions used to measure the affinity of EmrE for tetraphenylphosphonium (TPP), which was done in DMPC:dihexanoyl phosphatidylcholine (DHPC) isotropic bicelles (17).
Fig. 1.

Refined structure of EmrE. (A and B) Side views showing monomer chains 1 (yellow) and 2 (blue), with helices H4 of both monomers drawn in purple. (C) Side view showing the dimer interface and TPP inside the binding pocket; the orientation in C is obtained from those (A and B) by and – rotations, respectively, about the vertical axis using the right-hand rule. The refined structure in ribbon representation is superimposed on the -only PDB structure, drawn as connected cylinders. (D) Side view showing the solvation environment of the dimer, with the lipids with any atom within 2.7 Å of the protein backbone outlined in dark blue and water oxygens within 5 Å of a protein atom drawn as small green spheres. (E) RMSD between the evolving simulation structure and the initial minimized structure. Only the helix backbones were used for the calculation. (F) RMSFs computed from MD with those obtained from B-factors in the PDB; for each residue, the RMSF shown represents an average over the coordinates of the residue heavy atoms; a 3-point smoothing filter was applied to the simulation and PDB data.
Fig. 1D shows the lipids that have at least one atom within 2.7 Å of the protein backbone. Most of these lipids have their tails running alongside the transmembrane helices, as expected in view of their hydrophobicity. A notable lipid molecule is marked with an asterisk in Fig. 1D near TPP. It has one aliphatic chain protruding into the ligand binding site from the open side of the dimer and interacting with TPP, as well as with the residues Y40[2] and W63[1]. This lipid penetration formed spontaneously during the simulation, as the initial placement of all lipid molecules was several angstroms away from the protein structure (see Materials and Methods). To investigate whether the lipid penetration into the binding site was a reproducible event, we performed an additional 0.65 s MD simulation in which the above lipid molecule was deleted and the simulation box re-equilibrated. Further, to investigate whether the observed lipid penetration is dependent on the lipid composition, we transferred the EmrE dimer into a POPC:POPG (3:1) membrane patch, re-equilibrated the simulation structure, and performed a 0.7 s MD simulation. In both MD simulations, a lipid molecule penetrated the binding site (see SI Appendix, Fig. S1) in similar locations, suggesting that it contributes to stabilizing the EmrE dimer structure. The closed side of EmrE remained compact throughout the simulation, with no lipid penetration.
EmrE Active Site.
The TPP-bound EmrE is described first because the position of TPP is identifiable from the electron density (ED) of the structure used for the refinement (12). Because TPP was bound to the dimer and biochemical studies indicate that the addition of substrate to detergent-solubilized EmrE releases about one proton per monomer (18), the active site glutamates were modeled in the deprotonated state.
EmrE with TPP.
To validate the binding pose and conformation of TPP in the optimized structure, we computed the standard free energy of TPP binding to EmrE (see Materials and Methods) and compared the result with experimental measurements; excellent agreement is observed (Table 1) (17, 18).
Table 1.
The standard free-energy of TPP binding to EmrE
| Experimental values | Ref. | ||
| −10.3 0.7 | −10.1 | DDM micelles at C, pH 7.0 | 17 |
| −10.0 | DMPC:DHPC 1:2 isotropic bicelles at C, pH 7.0 | 17 | |
| −9.38 | DDM micelles at C, pH 7.0 | 18 |
Three experimental values are given (computed from KDs).
Fig. 2A illustrates the geometry of the active site with bound TPP. It is known from biochemical studies that residues E14, W63, and Y60 are important for substrate binding and transport, while Y40 modifies substrate specificity (19, 20). The main differences in the interactions between the two E14 residues and nearby active site residues are summarized in Table 2. Both of the E14 residue side chains interact with TPP, but the orientation of the residues in the two monomers is somewhat different; both carboxy oxygens of E14[1] are oriented toward TPP, compared with only one of E14[2] (Fig. 2A); here and in the following, the monomer is indicated in square brackets. TPP is displaced slightly toward the E14[2], with the distance between the TPP phosphorous and the closest carboxy oxygen in E14 being 3.5 Å and 5 Å for monomers 2 and 1, respectively. Consistent with the asymmetric displacement of TPP toward E14[2], examination of the MD trajectory revealed that a water molecule was present between the phosphorous of TPP and the carboxyl of E14[1] (Fig. 2A) 77% of the time; about 200 distinct water molecules occupied this position in a 500 ns trajectory segment, with the average residence time of 1 ns (see SI Appendix, SI Text and SI Methods). Other persistent water-mediated interactions were not observed. The conformations of both E14s are stabilized by electrostatic interactions with the nitrogen on the W63 ring of the corresponding monomer. Further, the position of E14[1] is stabilized by a hydrogen bond with Y60[2] on the opposite monomer [d(E14OE, Y60OH) 3 Å] and, to a lesser extent, by interactions with Y40[1] in the same monomer [d(E14OE, Y40OH) 5 Å]. Further, Y60[2]HO interacts with W63[1]NE, and thus, the residue triplet E14[1]/W63[1]/Y60[2] forms an H-bond network on the left side of the active site (Fig. 2A). The additional stabilization of E14[1] compared with E14[2] appears to be due in part to the asymmetric position of TPP in the binding pocket, which does not allow Y60[1] to be sufficiently close to E14[2] to form a hydrogen bond. Another cause for the asymmetry is the binding pose of TPP, which does not have a plane of symmetry that aligns with the dimer interface. The E14[2] carboxyl is in closer contact with the W63 side chain nitrogen than the E14[1] carboxyl by about 1 Å. However, the E14[1] carboxyl, unlike the E14[2] carboxyl, is able to transiently H-bond to Y60[2] on the opposite monomer (see Table 2). The difference in the E14/Y60 interactions exists because the active site is closed on the side on which Y60[2] is located, bringing Y60[2] into contact with E14[1], but open on the side of Y60[1] (see Fig. 2A).
Fig. 2.

Stereoviews of the active site conformations of EmrE. (A) With ligand TPP. (B) With ligand ethidium. (C) Ligand-free. EmrE monomers 1 and 2 are shown in yellow and in blue, respectively. The active site is visualized through the open side of EmrE; the closed side is thus farther from the reader in the direction perpendicular to the page. In this, as in all stereo figures that follow, side-by-side wall-eyed arrangement is used. In A, the red asterisk indicates the average position of a water molecule that mediates the interaction between E14[1] and TPP.
Table 2.
Average distances (mean SD in Ångstroms) between E14 carboxyls and three stabilizing atoms in the active site
| EmrE-TPP | apoEmrE | |
| E14[1]CD/W63[1]NE | 4.3 1.4 | 5.3 1.1 |
| E14[1]CD/Y40[1]OH | 7.0 1.3 | 6.9 1.9 |
| E14[1]CD/Y60[1]OH | 4.5 1.2 | 5.3 1.3 |
| E14[2]CD/W63[2]NE | 3.5 0.3 | 4.3 1.2 |
| E14[2]CD/Y40[2]OH | 6.4 0.4 | 6.2 0.8 |
| E14[2]CD/Y60[1]OH | 7.4 1.0 | 10.1 1.5 |
EmrE with Ethidium.
The asymmetry in the EmrE binding site reflects its plasticity—that is, ability to bind a diverse set of ligands through rearrangements in the active site geometry. To illustrate this point concretely, we examined the active site geometry of EmrE bound to ethidium. Ethidium binds to EmrE with submicromolar affinity (12), although no structural details of the binding pose are available. Starting from the equilibrated structure of EmrE with TPP, we replaced TPP with ethidium and performed a 0.5 s of MD simulation. The simulation structure was stable, with the backbone RMSD from the initial conformation of about 1.5 Å. The insertion of ethidium was performed using different ethidium orientations, all of which resulted in essentially the same binding mode within several nanoseconds. The final structure of the active site is shown in Fig. 2B. The active site conformation is similar to that of EmrE with TPP, with the most important difference involving the positions of residues E14[1] and W63[1]. The differences can be explained by the planar geometry of ethidium, with the exception of the phenyl and methyl groups bound to the central aromatic ring, whereas TPP is tetrahedral. Ethidium occupies less volume in the active site cavity than TPP, providing space for W63 to rotate its side chain closer to ethidium to increase nonpolar interactions. The importance of W63 for the active site plasticity is further discussed below in the context of ligand-free EmrE. Unlike in the TPP-ligated case, we did not observe stable water-mediated interactions between E14[2] and ethidium, possibly because the positive charge is more localized in the case of TPP than ethidium. To examine the possible origins of the lower binding affinity of EmrE for ethidium versus TPP, we computed interaction energies between seven residues in the active site whose side chains were found to be in the closest proximity to the ligand (see SI Appendix, Fig. S4 and SI Text for details). For both ligands, the dominant electrostatic and van der Waals (vdW) interactions involved residues E14 and W63, respectively; TPP had stronger electrostatic interactions with the E14s than did ethidium but weaker interactions with the W63s than did ethidium. However, increased vdW interactions with ethidium were insufficient to offset the decreased electrostatic interactions, relative to TPP, resulting in more favorable interaction energies between EmrE and TPP. The energy analysis underscores that both electrostatic and vdW interactions are important to binding, with the corresponding contributions dependent on the ligand and the particular importance of residues E14 and W63.
EmrE in the Absence of Ligand.
Because EmrE spends a part of its transport cycle in ligand-free form, it is instructive to examine the possible ligand-free structures in different protonation states of the two E14s. For this analysis, we consider four simulations, initiated from the structure equilibrated in the presence of TPP, but now with TPP deleted, with both E14 deprotonated, with both E14 protonated, and with either E14[1] or E14[2] protonated.
After the TPP is removed without protonation of either E14, EmrE undergoes a conformational change that corresponds to a backbone RMSD of about 2 Å from the structure equilibrated with TPP. The simulation trajectory in the first 600 ns shows an increase in the separation between the dimers from 16 Å to 19 Å, followed by a decrease to 15.5 Å (see Fig. 3A). An examination of the active site residue motions suggests that the initial separation phase is caused by the elimination of the positively charged TPP, which can no longer stabilize the negatively charged E14 carboxyls (see Fig. 2). Because the active site contains only a few water molecules (see Fig. 3B), the removal of TPP creates a void that is partially filled by an inward rotation of W63[2] 10 ns into the simulation and by a subsequent inward rotation of W63[1] 200 ns into the simulation (see Fig. 3 B and C). The repositioning of W63s is consistent with the active site structure of EmrE bound to ethidium discussed above. The aromatic side chains provide a low-dielectric environment, which does not shield effectively the electrostatic repulsion between the E14 side chains, and the dimer separates slightly. Over the course of the simulation, water molecules enter the active site from the open side of EmrE, hydrating the E14 carboxyls and providing dielectric screening (Fig. 3D). It is noteworthy that, despite the increased number of water molecules in the active site, the closed side of the active site remains essentially “water-proof,” preventing the formation of a water channel or a proton wire that could compromise the proton gradient present in an actual E. coli cell. The reversal of the dimer separation occurs after the ring of Y40[1] rotates away from the active site to interact with residues Y40[2], F44[2], and E14[2] (see Fig. 3D). The first interaction involves the hydroxyl of Y40[1] and the backbone carbonyl of Y40[2], the second mimics a stacking interaction between the corresponding aromatic rings, and the third involves the Y40[1] hydroxyl and the E14 carboxyl. [We use the term “mimic” to emphasize that in classical potential functions such as the CHARMM forcefield, nonbonded interactions are modeled as radial functions (i.e., Coulomb, van der Waals) and do not include explicit representations for orbitals.] The last interaction is transiently mediated by active site water molecules, as the distance between the hydroxyl and carbonyl oxygens is 6.5 Å. In contrast, in the three simulations involving a protonated E14, the dimers have moved closer together in the first 300 ns and without significant conformational changes, as the hydrophobic residues Y60 and W63 in the active site move closer together to strengthen their interactions (see SI Appendix, Fig. S6).
Fig. 3.

Evolution of EmrE dimer after removal of TPP. (A) Distance between the centers-of-mass (COMs) of helices H1–H3 of EmrE monomers 1 and 2; the legend entries indicate the protonation states of E14 residues in monomer 1/monomer 2, respectively. B–D correspond to the structure of the doubly deprotonated EmrE near the beginning, middle, and end of the 900 ns MD trajectory, respectively. Black spheres represent water oxygens within 5 Å of an E14 residue. The closed side of EmrE is at the top and away from the reader in the direction perpendicular to the page.
The ligand-free simulations illustrate how the EmrE dimer can retain stability over a broad range of pH, including basic conditions under which both E14s are deprotonated. The fact that the conformations protonated on E14 do not differ significantly from the ligand-bound ones (SI Appendix, Fig. S6) rationalizes the NMR observations that EmrE is capable of undergoing the inward–outward transition in the absence of ligand (21).
Finally, the EmrE dimer stability and active site plasticity observed in the simulations is consistent with recent experimental evidence that, at low pH, EmrE can bind TPP and a proton simultaneously (22).
pKa of Active Site Glutamates.
pKa values of the active site glutamates have been estimated to be in the range of 7.3–8.5 (18, 24); both estimates provide a single effective pKa value. In recent solution NMR experiments, Morrison et al. (25) studied the pKas of the two E14 residues. Because the interpretation was limited by a macroscopic model of deprotonation (26), it was not possible to determine the pKa values of the individual E14s (discussed further in SI Appendix, SI Text). While Morrison et al. (25) did not assign specific pKa values to the monomers, they hypothesized on the basis of the asymmetry in the NMR spectra and the low-resolution X-ray structure (12) that the E14s occupy different environments but also protonate anticooperatively.
The asymmetry observed in the present structures (see Fig. 2 and Table 2) also indicates that the environments of the E14s are somewhat different. This was proposed earlier by Lehner et al. (27) on the basis of solid-state NMR experiments with 13C-labeled E14 residues; two distinct chemical shifts for the E14 side chain atoms were observed, both in the ligand-free and in the ethidium-bound dimer (27). Further, the E14 pKas computed from the present MD simulation trajectories using Poisson–Boltzmann (PB) theory (23) show that both pKas are not very sensitive to the protonation state of the other glutamate (see Table 3). The computed difference is 0.5–1.0 pKa units, within the margin of error of the calculation of 1 unit. This finding should not seem surprising, given that the E14 carboxyls are separated by 7 Å in ligand-free EmrE. If the active site cavity in the absence of ligands is accessible to a sufficient number of water molecules to hydrate the E14 side chains, as we observed in the ligand-free EmrE simulations, the distance between the E14 is comparable to a typical Debye length of a charge in an aqueous solution (28), beyond which electrostatic forces are effectively screened by the medium. We caution, however, that continuum electrostatics models of dielectric screening, such as PB theory, have limited accuracy in partially hydrated environments, in which the effect of individual discrete solvent molecules cannot be captured by a continuum.
Table 3.
pKas of active site glutamates
| Residue | E | E | E | E |
| E14[1] | 11.38 | 10.45 | n/a | n/a |
| E14[2] | n/a | n/a | 8.80 | 9.32 |
The values are computed using PB theory implemented in Adaptive Poisson–Boltzmann Solver (APBS) (23) (see Materials and Methods). For each of the two glutamates, two pKa values are given, which correspond to the protonation state of the other glutamate, as indicated by the residue charge. The SE in the values is ∼1 pKa per unit.
Monomeric EmrE with TPP.
Because an objective of this study was to design dimerization inhibitors, it is instructive to examine the behavior of monomeric EmrE with TPP bound. To this end, we deleted monomer 1 from the structure equilibrated with TPP, performed an equilibration simulation that allowed the lipid membrane to fill the space occupied by the deleted monomer, and simulated the resulting structure for 1 s. Monomer 2 undergoes conformational changes in the first 100 ns (Fig. 4); thereafter, the backbone RMSD from the initial dimeric conformation is at a plateau of 4 Å. The largest observed conformational change involves an unkinking of helix H3 (Fig. 4), accompanied by a displacement of TPP closer toward this helix, and farther away from the catalytic residue E14. In the simulations with stapled peptides, in which one of the monomers was replaced with a stapled peptide (discussed below), we observed unkinking in the majority of cases and displacements of TPP of similar magnitudes but in various directions. The unkinking of H3 suggests that the kinked helix in the dimer is stabilized by interactions with the second monomer. Stabilizing interactions could be provided by nonpolar residues such as I71 (Fig. 4). On the basis of changes in NMR chemical shifts upon pH changes, Gayen et al. (29) proposed an allosteric pathway that couples the protonation state of Glu14 to large motions of H3 (figure 3c in ref. 29). In view of the present simulations, this pathway could involve unkinking of H3.
Fig. 4.

Monomeric EmrE. Shown is a stereoview of equilibrated EmrE monomer bound to TPP (colors) overlaid on the conformation of the monomer in the dimer (gray scale). Before deletion of monomer 1, the closed side of the EmrE dimer was at the top.
The displacements of TPP observed in the monomeric and stapled peptide simulations indicate that TPP is no longer bound to a particular site on the monomer, and the variability of displacements from the active site suggests that the binding is substantially weaker than in dimeric EmrE. It is noteworthy that the downward displacement of TPP is accompanied by an outward rotation of W63, in accord with what was observed for the dimer simulations of ligand-free EmrE (see Fig. 2 B and C and SI Appendix, Fig. S6), suggesting that this is the preferred conformation of W63 in the absence of other constraints.
Structure of H4 Dimerization Helices.
In the low-resolution structures of EmrE, helix 4 does not form part of the binding pocket; it is believed to serve as a dimerization motif (30, 31). The interface between the H4s in the EmrE dimer was found to be very stable in the present simulations (see Fig. 5). In particular, nonpolar residues I89, L93, I94, I100, I101, and L104 have their side chains in close proximity to the partner helix (Fig. 5A). The distance between the COMs of the two H4s is 10.3 0.24 Å in the final microsecond of the equilibration simulation, and the average backbone RMSD with respect to the first structure in the final microsecond is 1.3 Å. The low SD of the interhelix distance suggests that the attractive interactions between the two helices are strong enough to prevent significant distance fluctuations as a result of thermal motion. The role of H4 as dimerization motifs is further supported by our previous experiments, in which structural peptide analogs of H4 were found to inhibit the efflux activity of EmrE homolog Hsmr (32, 33), and the fact that a truncated construct lacking H4 was prone to aggregation (34).
Fig. 5.

Helices H4. (A) Side view and (B) top view show the close packing of hydrophobic side chains. In A, side chains of hydrophobic residues that are in contact with the opposite H4 helix (and residue C95, which faces the lipid) are drawn in thick lines; the remaining residues are drawn in thin lines. In B, side chains are drawn as black lines, and side chain atoms are also drawn as a transparent surface formed by the union of spheres with the corresponding vdW radii. Hydrogen atoms are not shown.
To determine the strength of the interactions quantitatively, we performed simulations using a modified system, in which monomer 1 was deleted, except for H4. First, the system was simulated by MD for 1.4 s. The average RMSD of the backbone atoms of H4 helices from the initial structure was 0.97 Å, indicating a stable interface. Next, we carried out a free-energy simulation to determine the affinity between the H4 helix and the monomer. The separation between the H4s in the modified system was increased gradually from the initial value of 10 Å by 12 Å, and the forces required to achieve the separation were integrated to compute the association Potential of Mean Force (PMF), as described in SI Appendix, SI Methods. The PMF at the final separation was 20 1 kcal/mol. Using the rigid rotor formula for the rotational partition function of the free H4 helix (35) and a standard concentration of 1M, we obtain standard state corrections of −13.1 kcal/mol and −2.9 kcal/mol for the two contributions, respectively (see SI Appendix, SI Methods). The result is a standard binding free energy of 4 kcal/mol (1 mM). However, the above estimate assumes (i) that the free H4 peptide is in the lipid phase and (ii) that it explores all possible orientations. If the peptide were transferred to aqueous solution, the free-energy contribution from the PMF would most likely increase because of its hydrophobic residue content. Further, if not all peptide orientations in the medium were equally likely, as would be the case for a nonisotropic medium such as a membrane, the rotational free-energy correction would be smaller in magnitude. Both of these factors would increase the binding free energy. Overall, the calculated free energy and dimer structures are consistent with the identification of H4 as the dimerization element.
Design of Stapled Peptide Inhibitors.
The starting structure for the inhibitor design was obtained by deleting one EmrE monomer, except for the H4 helix involved in the dimerization (see SI Appendix, SI Methods for full details). The +7 hydrocarbon staple was chosen for incorporation into the peptide-based inhibitors. The purpose of the staple is to shield a portion of the peptide backbone from protease degradation and to increase favorable hydrophobic contacts with lipids in the bilayer and, possibly, with EmrE helix H4 (13–15, 36).
The close packing of the EmrE dimerization interface (Fig. 5) suggested that it has been optimized by natural selection. Therefore, we did not consider inhibitor peptides with sequences different from wild-type H4 and instead focused on optimizing the location of the staples. Following Verdine and Hilinski (13) and Guo et al. (38), proteolysis-resistant peptides were designed by replacing selected residue side chains by all-hydrocarbon staples (see Fig. 6A). Twelve stapled peptides were considered, in which the N-terminal position of the staple was one of the residues in the H4 sequence 86PAIIGMMLICAG97 and the C-terminal position of the staple was placed seven residues downstream in the sequence, corresponding to two turns of the helix.
Fig. 6.

Design of stapled peptides. (A) Illustration of a hydrocarbon staple (Top); a stable EmrE-stapled peptide complex stapled at positions M92–L99 (Bottom). (B) Profiles of the free energy as a function of monomer–peptide displacement. (C) Monomer–peptide interaction of vdW energy (blue) computed using the Generalized Born membrane model (37) and monomer–peptide interaction of free energy (red); the correlation coefficient between the interaction energy and the free energy is 0.74.
The 12 EmrE–peptide complexes were simulated by MD for 100 ns each, and interaction energies were computed between the peptides and EmrE using a Generalized Born membrane model (37). For validation, free energies of interaction were computed by reversibly separating the stapled peptides from the EmrE monomer in an explicit lipid environment, as described above and in Materials and Methods. The simulation results are summarized in Fig. 6 B and C, which shows that staple placement has a significant effect on the predicted binding affinity. With the exception of the peptide stapled at M92, peptides with the highest computed affinity for the EmrE monomer correspond to staple locations near the N terminus of the helix and low-affinity peptides tend to have staples near the C terminus. The binding free energy was not computed for staple G90 because it spontaneously dissociated from the monomer. The binding interfaces between EmrE and four representative stapled peptides are discussed in SI Appendix, SI Text and shown in SI Appendix, Fig. S9.
Guided by the computational predictions, we selected four peptides with the highest computed affinity for EmrE, with staples at positions 87 to 89 and 92 for experimental synthesis and testing. The staple locations for these peptides are shown in Table 4, and the coordinates of all staples in complex with EmrE are given in a public dataset available at dx.doi.org/10.17632/3pvz4hytfd.2.
Table 4.
EmrE inhibitor peptides designed on the basis of the H4 -helix
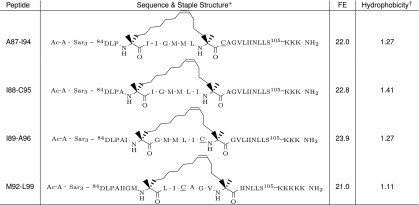 |
The staple structures are adapted from Guo et al. (38). The free energy of separation from EmrE (FE) is expressed in kcal/mol and has an SE of ∼1 kcal/mol.
*Peptide sequences are shown with attached sarcosine (Sar) and lysine (K) tags. Ac and NH2 denote acetylation and amidation of N and C termini, respectively. Alanine-sarcosine and lysine tags were present in experiments only; acetylated and amidated termini were used both in experiment and in simulation. In the experiments, the wild-type C95 in peptides A87–I94, I89–A96, and M92–L99 was replaced with a serine (underlined symbol) to prevent disulfide cross-linking.
†Core peptide hydrophobicity was calculated by the Liu–Deber hydrophobicity scale (39) for each residue and averaged over the sequence (tags and staples were excluded).
Although the hydrophobicity imparted by the staple increases peptide solubility in the lipid membrane, it made peptide synthesis, purification, and handling more challenging. To mitigate these challenges, we added solubility tags to the and termini of all peptides and substituted the native cysteine (C95) in EmrE H4 with serine (see Table 4). The C95S substitution was intended to preserve the steric volume of the side chain, while eliminating the possibility of disulfide bond formation between peptides and increasing peptide solubility for in vitro handling and addition to cells. Lysine residues were added to the terminus of each peptide to promote ease of handling and to aid in targeting the peptides to the anionic bacterial membrane (32, 40). -terminal acetylation removes a positive charge, while the addition of the peptoid sarcosine (N-methyl glycine)-containing tag directs this terminus of the peptide to insert into the membrane (32, 33).
Peptide Helicity in Membrane Mimetics.
First, peptide structure in anionic detergent micelles was assessed using circular dichroism (CD) spectroscopy. All peptides had very similar CD spectra and displayed high helicity (Fig. 7A). To investigate the peptide secondary structure in a more realistic bacterial membrane mimetic, CD spectra were also collected in anionic lipid bilayers (POPC:POPG, 3:1 M ratio). All four peptides adopted -helical structures in the bilayer but with wide variations in the degree of helicity (Fig. 7B). Peptide I88-C95 showed the least helical structure of the four peptides, and peptides I89-A96 and M92-L99 showed moderate helicity ranging between I88-C95 and A87-I94.
Fig. 7.

Experimental tests of inhibitors. (A and B) CD spectra of hydrocarbon-stapled EmrE TM4 peptides in bacterial membrane mimetics: (A) 20 M peptides in buffer (10 mM Tris HCl, 10 mM NaCl, pH 7.4) and 140 mM SDS; (B) 20 M peptides in 2.5 mM POPC:POPG (3:1 M ratio). Spectra shown represent the average of three independent samples. (C) Inhibition of ethidium efflux by stapled peptides. E. coli cells were grown in minimal media and incubated with the ionophore CCCP, EtBr, and either DMSO or peptide (4 M) in DMSO. Cells were placed in fresh media (lacking CCCP) to observe fluorescence decay of ethidium as it is pumped from the cells. Fluorescence intensity is normalized to the initial value. Spectra shown represent the average of three independent experiments; dashed lines represent computed fits to the exponential decay model with initial plateau (see Materials and Methods). (D) Cell toxicity assay. E. coli cells were grown in minimal media in the presence or absence of peptide (4 ). was measured over 1 h in 15-min intervals. All time points are normalized to the starting time point and cell growth in the absence of peptide. S-CAP-2G is included as a positive control for cell toxicity [sequence: KKKKKK-AGFAAWAAFGA-; hydrocarbon-stapled positions indicated by A; the shorter + 4 hydrocarbon staple was used (41)]. Each curve represents the average of two independent experiments. Error is indicated as SEM.
Peptide Inhibition of Ethidium Efflux.
To quantify the inhibitory activity of the designed peptides, E. coli cells were initially treated with the ionophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP) to dissipate the proton gradient and subsequently loaded with the fluorescent toxin EtBr; ethidium efflux was then monitored over time by fluorescence spectroscopy (see Materials and Methods) in the presence and in the absence of the peptides.
Upon removal of the ionophore, a period is needed to re-establish the proton gradient across the cell membrane, during which EmrE lacks the energy to pump out ethidium and ethidium fluorescence is at an initial plateau (Fig. 7C). As EmrE begins to expel ethidium from the cells, fluorescence decays to a final equilibrium plateau, at which point efflux of ethidium by EmrE is exactly balanced by passive influx from the extracellular medium. Inhibition of EmrE dimerization results in a higher level of final fluorescence plateau, due to a reduction in the availability of functional EmrE and, therefore, in ethidium efflux.
In the absence of peptide, E. coli cells remove 90% of the intracellular ethidium within 30 min (Fig. 7C and Table 5). In contrast, cells treated with peptide show varying differences in the amount of ethidium remaining within the cells as well as rate of efflux activity. Addition of peptides M92-L99 or A87-I94 results in markedly elevated final fluorescence intensity, relative to that in the absence of peptide (0.72 and 0.64 vs. 0.13; levels normalized to the initial intensity). This corresponds to a 5-fold increase in the intracellular ethidium or to the retention of 60% to 68% of the ethidium expelled in the peptide-free experiment (Fig. 7C). Peptide I89-A96 has a smaller but significant effect compared with A87-I94 and M92-L99, resulting in more than doubling of the intracellular ethidium (0.35 vs. 0.13), corresponding to the retention of 26% of the ethidium expelled in the peptide-free assay. Finally, addition of peptide I88-C95 does not lead to inhibition of ethidium efflux.
Table 5.
Ethidium fluorescence plateau and efflux rates (see Materials and Methods)
| Peptide | Ft→∞/F0* | EmrE efflux rate, † | Rel. EmrE efflux rate, %‡ | Inhibition, %§ |
| No peptide | 0.16 (0.02) | 2.47 (7.3 ) | 100.0 | 0 |
| A87-I94 | 0.67 (0.05) | 1.03 (8.3 ) | 42 | 58 |
| I88-C95 | 0.16 (0.04) | 2.66 (1.2 ) | 108 | 0 |
| I89-A96 | 0.38 (0.05) | 1.98 (1.0 ) | 80 | 20 |
| M92-L99 | 0.77 (0.07) | 0.60 (5.0 ) | 24 | 76 |
Final normalized ethidium fluorescence plateau.
EtBr efflux rates are corrected for the passive background influx of ethidium from the extracellular medium (see Materials and Methods).
Relative to cells without peptide.
Computed as 100% – EmrE efflux rate (%). Parentheses report 95% confidence limits.
Variations in inhibitory action likely arise from the varying solubility of the highly hydrophobic peptides. Although the stapled EmrE peptides only differ in the positioning of the hydrocarbon staple, each staple replaces a different amino acid pair along the TM4 sequence with a nonpolar hydrocarbon link, which implies that the peptides have different hydrophobicities (Table 4). The replacement of C95 (S95 in the other three peptides) with the hydrophobic staple precursor amino acid renders I88-C95 the most hydrophobic peptide (Table 4). This heightened hydrophobicity makes I88-C95 more susceptible to aggregation, a feature that is consistent with a decreased helical structure in the presence of anionic lipid bilayers (Fig. 7B). Thus, the I88-C95 peptide may not insert as stably into lipid bilayers as the other peptides, resulting in diminished ability to disrupt intramembrane protein–protein interactions within the target protein.
Peptides Show Minimal Intrinsic Toxicity to Cells.
The four synthetic peptides were tested for nonspecific cytotoxicity against E. coli cells in the absence of toxins EtBr and CCCP. The stapled selective cationic antimicrobial peptide (SCAP)2G, used as a positive control for toxicity, showed a 40% decrease in cell growth, compared with cells treated with DMSO alone (Fig. 7D). In contrast, cells treated with 4 M peptide showed minimal decreases in cell growth after 60 min (Fig. 7D). This demonstrates that, at the tested concentration, the stapled peptides have minimal intrinsic cytotoxicity against E. coli and therefore that the efflux inhibition observed above is not due to nonspecific toxicity.
Concluding Discussion
Starting from a low-resolution -only X-ray crystal structure of the EmrE transporter (12), we used MD simulations biased to the corresponding X-ray ED map to obtain an atomistically detailed structure. The modeled structure was stable for more than 1 s of MD simulation in the lipid membrane and was validated against experimental data using free-energy simulations. Using the refined structure, we created and evaluated models of stapled peptides, designed to inhibit EmrE dimerization, as a strategy for interfering with drug resistance. The four best models proposed on the basis of the simulations were synthesized and tested experimentally for efflux inhibition in live E. coli cells. Three of the four peptides caused significant inhibition, with two- to fivefold higher intracellular ethidium accumulation relative to a control without peptide (Fig. 7C). Because these peptides did not cause nonspecific cytotoxicity at the concentrations tested, they are promising candidates for therapeutic development. MD simulations have been used previously to fit structures into EDs obtained from cryo-EM and X-ray diffraction (42–45), and the use of peptides to disrupt protein–protein interactions is known (14, 46–48). The strategy described here successfully combines structure optimization by state-of-the-art simulations with rational structure-based design.
The structures obtained here can be used as a starting point for refining homology models of SMR proteins in other organisms (e.g. M. tuberculosis, Klebsiella pneumoniae, S. aureus) to which EmrE has a high degree of sequence homology (49). In light of recent evidence suggesting that EmrE can also form parallel dimers (50), it could be instructive to search for a stable structure of the parallel dimer using the present methods, possibly starting from the model of Gottschalk et al. (51), and the monomeric structure obtained here.
The strategy of disrupting subunit interactions within EmrE is applicable to other multidomain membrane proteins of therapeutic significance, such as G protein-coupled receptors involved in cell signaling, ion channels essential for viral replication (52), or ABC cassette proteins involved in drug-resistant cancers (53), especially for cases in which small-molecule drugs are not available.
Finally, the refined structure of EmrE presented here is expected to be of broad interest, because SMR pumps are believed to be the ancestors of most membrane transporters and could in fact be the minimal biological paradigm for the conversion of electrochemical gradients into motion.
Materials and Methods
Computer Modeling of Initial Structure.
-only structure of EmrE (3B5D) was obtained from the PDB. The protein backbone was generated using CHARMM (54). Several different algorithms were used to construct side-chain conformations for subsequent refinements (see SI Appendix, SI Methods). The models were energy-minimized using the implicit membrane model 1 (IMM1) (55) and used to start MD simulations.
MD Simulations.
To improve the starting structures for the explicit-solvent simulations, an intermediate refinement step was added. The ED map corresponding to the 3B5D X-ray crystal structure was obtained from Geoffrey Chang, Department of Pharmacology, University of California, San Diego. Restraints to a smoothed ED were incorporated into MD simulations using the enhanced sampling method self-guided Langevin dynamics (43). These calculations were performed in IMM1 implicit solvent (55) with CHARMM. For explicit solvent MD, patches of DMPC lipid bilayers were obtained from www.charmm-gui.org (56), and the protein structures were inserted into the membrane by deleting the lipids that overlapped with the protein. Structures were immersed in TIP3P water and equilibrated for 100 ns at standard pressure and temperature with weak harmonic restraints applied to the carbons of EmrE, using the program ACEMD (57). Detailed parameters of the various MD simulations performed after this stage are given in SI Appendix, SI Methods. Simulation parameters and coordinates are available in an online dataset (58).
pKa Shift Calculations.
To compute the pKa of active-site residues E14 in ligand-free EmrE, we used PB solvation theory, as implemented in the program Adaptive Poisson–Boltzmann Solver (APBS) (23). To account for the conformation flexibility of EmrE, structures were taken from the equilibrium MD trajectories of EmrE without ligand, in 20 ns increments.
Peptide Synthesis.
Peptides were synthesized on an automated PS3 peptide synthesizer (Protein Technologies Inc.) using standard solid-state N-(9-fluorenyl)methoxycarbonyl (Fmoc) and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (Novabiochem) chemistry on a low-load PAL-PEG resin (Applied Biosystems) that produced an amidated C terminus after cleavage. The incorporation of staple precursor amino acids, (S)-N-Fmoc-2-(4′-pentenyl)alanine (Fmoc-S5Ala-OH) and (R)-N-Fmoc-2-(7′-octenyl) alanine (Okeanos Technology Co.), was performed as previously described (33).
Growth Inhibition Assay.
E. coli K12 cells were grown overnight to saturation in LB (Luria broth). Cells were harvested and resuspended in fresh media to a final of 0.1. Cells were then grown in the presence of DMSO alone or in DMSO-solubilized peptide (4 ) over 1 h, while was recorded in 15-min intervals. E. coli growth curves were normalized to the starting .
Ethidium Efflux Assay.
EtBr efflux assays were performed as described previously (5, 32). Complete method details are given in SI Appendix, SI Methods.
Supplementary Material
Acknowledgments
We thank Prof. Geoffrey Chang for providing the X-ray density maps. This work was supported by NIH Grant 5R03AI111416 (to V.O. and M.K.) and Natural Science and Engineering Research Council of Canada (NSERC) Discovery Grant RGPIN-2016-05577 and Canadian Institutes of Health Research (CIHR) Grant 376666 (to C.M.D.). Computer resources were provided by National Energy Resource Scientific Computing Center Contract DE-AC02-05CH11231.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Simulation parameters and coordinates are provided as a Mendeley Dataset available at dx.doi.org/10.17632/3pvz4hytfd.2.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1802177115/-/DCSupplemental.
References
- 1.Nikaido H. Multidrug resistance in bacteria. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paulsen I, Brown M, Skurray R. Proton-dependent multidrug efflux systems. Microbiol Rev. 1996;60:575–608. doi: 10.1128/mr.60.4.575-608.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bay D, Rommens K, Turner R. Small multidrug resistance proteins: A multidrug transporter family that continues to grow. Biochim Biophys Acta. 2008;1778:1814–1838. doi: 10.1016/j.bbamem.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Bay DC, Turner RJ. Diversity and evolution of the small multidrug resistance protein family. BMC Evol Biol. 2009;9:140. doi: 10.1186/1471-2148-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tal N, Schuldiner S. A coordinated network of transporters with overlapping specificities provides a robust survival strategy. Proc Natl Acad Sci USA. 2009;106:9051–9056. doi: 10.1073/pnas.0902400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heir E, Sundheim G, Holck A. Identification and characterization of quaternary ammonium compound resistant Staphylococci from the food industry. Int J Food Microbiol. 1999;48:211–219. doi: 10.1016/s0168-1605(99)00044-6. [DOI] [PubMed] [Google Scholar]
- 7.Fuentes D, et al. The product of the QacC gene of Staphylococcus epidermidis CH mediates resistance to beta-lactam antibiotics in gram-positive and gram-negative bacteria. Res Microbiol. 2005;156:472–477. doi: 10.1016/j.resmic.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Nishino K, Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia Coli. J Bacteriol. 2001;183:5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuldiner S. EmrE, a model for studying evolution and mechanism of ion-coupled transporters. Biochim Biophys Acta. 2009;1794:748–762. doi: 10.1016/j.bbapap.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 10.Ubarretxena-Belandia I, Baldwin JM, Schuldiner S, Tate CG. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. Embo J. 2003;22:6175–6181. doi: 10.1093/emboj/cdg611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleishman SJ, et al. Quasi-symmetry in the cryo-EM structure of EmrE provides the key to modeling its transmembrane domain. J Mol Biol. 2006;364:54–67. doi: 10.1016/j.jmb.2006.08.072. [DOI] [PubMed] [Google Scholar]
- 12.Chen YJ, et al. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci USA. 2007;104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 14.Baek S, et al. Structure of the stapled P53 peptide bound to Mdm2. J Am Chem Soc. 2012;134:103–106. doi: 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]
- 15.Brown CJ, et al. Stapled peptides with improved potency and specificity that activate P53. ACS Chem Biol. 2013;8:506–512. doi: 10.1021/cb3005148. [DOI] [PubMed] [Google Scholar]
- 16.Li DW, Brusächweiler R. All-atom contact model for understanding protein dynamics from crystallographic B-factors. Biophys J. 2009;96:3074–3081. doi: 10.1016/j.bpj.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison EA, et al. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature. 2012;481:45–52. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adam Y, Tayer N, Rotem D, Schreiber G, Schuldiner S. The fast release of sticky protons: Kinetics of substrate binding and proton release in a multidrug transporter. Proc Natl Acad Sci USA. 2007;104:17989–17994. doi: 10.1073/pnas.0704425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elbaz Y, Salomon T, Schuldiner S. Identification of a glycine motif required for packing in EmrE, a multidrug transporter from Escherichia Coli. J Biol Chem. 2008;283:12276–12283. doi: 10.1074/jbc.M710338200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rotem D, Steiner-Mordoch S, Schuldiner S. Identification of tyrosine residues critical for the function of an ion-coupled multidrug transporter. J Biol Chem. 2006;281:18715–18722. doi: 10.1074/jbc.M602088200. [DOI] [PubMed] [Google Scholar]
- 21.Cho M, Gayen A, Banigan J, Leninger M, Traaseth N. Intrinsic conformational plasticity of native EmrE provides a pathway for multidrug resistance. J Am Chem Soc. 2014;136:8072–8080. doi: 10.1021/ja503145x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson A, Thomas N, Morrison E, Balthazor B, Henzler-Wildman K. New free-exchange model of EmrE transport. Proc Natl Acad Sci USA. 2017;114:E10083–E10091. doi: 10.1073/pnas.1708671114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker N, Sept D, Joseph S, Holst M, McCammon J. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soskine M, Adam Y, Schuldiner S. Direct evidence for substrate induced proton release in detergent solubilized EmrE, a multidrug transporter. J Biol Chem. 2004;279:9951–9955. doi: 10.1074/jbc.M312853200. [DOI] [PubMed] [Google Scholar]
- 25.Morrison E, Robinson A, Liu Y, Henzler-Wildman K. Asymmetric protonation of EmrE. J Gen Physiol. 2015;146:445–461. doi: 10.1085/jgp.201511404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McIntosh L, et al. Dissecting electrostatic interactions in Bacillus circulans Xylanase through NMR-monitored PH titrations. J Biomol NMR. 2011;51:5–19. doi: 10.1007/s10858-011-9537-x. [DOI] [PubMed] [Google Scholar]
- 27.Lehner I, et al. The key residue for substrate transport (Glu14) in the EmrE dimer is asymmetric. J Biol Chem. 2008;283:3281–3288. doi: 10.1074/jbc.M707899200. [DOI] [PubMed] [Google Scholar]
- 28.Russel W, Saville DA, Schowalter W. Colloidal Dispersions. Cambridge Univ Press; Cambridge, UK: 1989. [Google Scholar]
- 29.Gayen A, Leninger M, Traaseth N. Protonation of a glutamate residue modulates the dynamics of the drug transporter EmrE. Nat Chem Biol. 2016;12:141–145. doi: 10.1038/nchembio.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poulsen B, Rath A, Deber C. The assembly motif of a bacterial small multidrug resistance protein. J Biol Chem. 2009;284:9870–9875. doi: 10.1074/jbc.M900182200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poulsen BE, Cunningham F, Lee KKY, Deber CM. Modulation of substrate efflux in bacterial small multidrug resistance proteins by mutations at the dimer interface. J Bacteriol. 2011;193:5929–5935. doi: 10.1128/JB.05846-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poulsen BE, Deber CM. Drug efflux by a small multidrug resistance protein is inhibited by a transmembrane peptide. Antimicrob Agents Chemother. 2012;56:3911–3916. doi: 10.1128/AAC.00158-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellmann-Sickert K, Stone T, Poulsen B, Deber C. Efflux by small multidrug resistance proteins is inhibited by membrane-interactive helix-stapled peptides. J Biol Chem. 2015;290:1752–1759. doi: 10.1074/jbc.M114.616185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banigan J, Gayen A, Cho M, Traaseth N. A structured loop modulates coupling between the substrate-binding and dimerization domains in the multidrug resistance transporter EmrE. J Biol Chem. 2015;290:805–814. doi: 10.1074/jbc.M114.601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill TL. An Introduction to Statistical Thermodynamics. Dover; New York: 1986. [Google Scholar]
- 36.Joseph T, Lane D, Verma C. Stapled BH3 peptides against MCL-1: Mechanism and design using atomistic simulations. PLoS One. 2012;7:e43985. doi: 10.1371/journal.pone.0043985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanizaki S, Feig M. A generalized born formalism for heterogeneous electric environments: Application to the implicit modeling of biological membranes. J Chem Phys. 2005;122:124706. doi: 10.1063/1.1865992. [DOI] [PubMed] [Google Scholar]
- 38.Guo Z, et al. Probing the alpha-helical structural stability of stapled P53 peptides: Molecular dynamics simulations and analysis. Chem Biol Drug Des. 2010;75:348–359. doi: 10.1111/j.1747-0285.2010.00951.x. [DOI] [PubMed] [Google Scholar]
- 39.Liu L, Deber C. Combining hydrophobicity and helicity: A novel approach to membrane protein structure prediction. Bioorg Med Chem. 1999;7:1–7. doi: 10.1016/s0968-0896(98)00233-8. [DOI] [PubMed] [Google Scholar]
- 40.Melnyk R, et al. Polar residue tagging of transmembrane peptides. Biopolymers. 2003;71:675–685. doi: 10.1002/bip.10595. [DOI] [PubMed] [Google Scholar]
- 41.Stone TA, Cole GB, Nguyen HQ, Sharpe S, Deber CM. Influence of hydrocarbon-Stapling on membrane interactions of synthetic antimicrobial peptides. Bioorg Med Chem. 2018;26:1189–1196. doi: 10.1016/j.bmc.2017.10.020. [DOI] [PubMed] [Google Scholar]
- 42.Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. Molecular dynamics flexible fitting: A practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu X, Subramaniam S, Case D, Wu K, Brooks B. Targeted conformational search with map-restrained self-guided Langevin dynamics: Application to flexible fitting into electron microscopic density maps. J Struc Biol. 2013;183:429–440. doi: 10.1016/j.jsb.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGreevy R, et al. xMDFF: Molecular dynamics flexible fitting of low-resolution X-ray structures. Acta Crystallog D Biol Crystallogr. 2014;70:2344–2355. doi: 10.1107/S1399004714013856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vitalis A, Caflisch A. Equilibrium sampling approach to the interpretation of electron density maps. Structure. 2014;22:156–167. doi: 10.1016/j.str.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 46.Yin H, et al. Computational design of peptides that target transmembrane helices. Science. 2007;315:1817–1822. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 47.Muppidi A, et al. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J Am Chem Soc. 2012;134:14734–14737. doi: 10.1021/ja306864v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stone T, Deber C. Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochim Biophys Acta. 2017;1859:577–585. doi: 10.1016/j.bbamem.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 49.Rossi ED, et al. Mmr, a mycobacterium tuberculosis gene conferring resistance to small cationic dyes and inhibitors. J Bacteriol. 1998;188:6068–6071. doi: 10.1128/jb.180.22.6068-6071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lloris-Garcerá P, et al. Antiparallel dimers of the small multidrug resistance protein EmrE are more stable than parallel dimers. J Biol Chem. 2012;287:26052–26059. doi: 10.1074/jbc.M112.357590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gottschalk K, Soskine M, Schuldiner S, Kessler H. A structural model of EmrE, a multi-drug transporter from Escherichia coli. Biophys J. 2004;86:3335–3348. doi: 10.1529/biophysj.103.034546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, et al. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza a virus. Proc Natl Acad Sci USA. 2013;110:1315–1320. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tarasova NI, et al. Transmembrane inhibitors of P-Glycoprotein, an ABC transporter. J Med Chem. 2005;48:3768–3775. doi: 10.1021/jm049065t. [DOI] [PubMed] [Google Scholar]
- 54.Brooks B, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins Struct Func Genet. 2003;52:176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- 56.Jo S, Lim J, Klauda J, Im W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harvey M, Giupponi G, Fabritiis GD. ACEMD: Accelerated molecular dynamics simulations in the microseconds timescale. J Chem Theor Comput. 2009;5:1632–1639. doi: 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- 58.Ovchinnikov V. 2018 EmrE MD simulation coordinates and simulation parameters. Mendeley Data, v2. dx.doi.org/10.17632/3pvz4hytfd.2. Accessed July 20, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.