

Abstract
Acromegaly is an uncommon, chronic disease, characterized by hypersecretion of a pituitary growth hormone by somatotroph adenomas, along with increased levels of insulin-like growth factor-1. Although acromegaly presents a wide array of clinical manifestations, the salient symptoms include acral and soft tissue enlargement, joint pain, heart and respiratory failure, diabetes mellitus, and hypertension, leading to increased morbidity and mortality. Hence, early diagnosis of the disease is critical to enhance life expectancy and quality of life. New approaches are being developed for diagnosis and surveillance (both screening and follow-up), including sensitive biochemical assays and the use of MRI to visualize extremely small tumors, and are helpful in the early diagnosis of acromegaly, subsequent treatment, and disease control. This mini-review summarizes the most common and effective tools used in the diagnosis of acromegaly.
Keywords: pituitary growth hormone, pituitary gland, diabetes mellitus, biochemical assays, acromegaly
Introduction
Acromegaly is a rare, chronic, endocrine disorder, usually caused by hypersecretion of growth hormone (GH) for a prolonged period from a somatotroph adenoma.1 The term “acromegaly” is derived from two Greek words: “akrom”, meaning extremity, and “megas”, meaning great; the meaning reflects one of the familiar symptoms of the disease, which is abnormal growth of hands and feet.2
Epidemiology of acromegaly
The exact prevalence of acromegaly is not quite clear; however, the estimated incidence is three to four cases per million per year, with an occurrence rate of ~40–125 people per million of the population.3,4 Even though the mean age of occurrence of the disease is 32 years, the mean age of diagnosis is 40–45 years, primarily owing to a delay in diagnosis.2,3 The incidence of acromegaly does not vary with gender, race, and ethnicity, although epidemiological studies conducted in Spain reported a predominance of acromegaly among women.5 A study in a highly polluted area in the Messina province of Italy revealed an increased prevalence of acromegaly, indicating the pathogenic role of environmental factors in the development of acromegaly.6
Causes of acromegaly
The pea-sized pituitary gland, located at the sella turcica, is responsible for secreting a multitude of hormones, including GH or somatotropin, the action of which is controlled by a complex feedback mechanism.7 The most common cause of acromegaly is the presence of a benign tumor or adenoma originating from pituitary somatotroph cells and secreting excess GH.1,7 This excessive secretion of GH leads to a persistent elevation of insulin-like growth factor-1 (IGF-1),8 which is produced by the liver, kidney, pituitary gland, muscle, and gastrointestinal tract; with liver being the primary source.9 IGF-1 facilitates the growth-promoting effects of GH.9 In 95% of acromegalic cases, enhanced levels of GH and serum IGF-1 are responsible for the distinctive features of the disease and numerous comorbidities. Alternatively, increased levels of growth hormone-releasing hormone (GHRH) produced in the hypothalamus account for 0.5% of acromegaly cases.10 Hence, serum IGF-1 concentration is considered as a sensitive measure of integrated GH levels in patients with acromegaly.9 Furthermore, some correlation between acromegaly and familial syndromes, such as McCune–Albright syndrome, multiple neoplasia type I, Carney complex, and isolated familial acromegaly, has been detected.5
Clinical manifestations and associated comorbidities of acromegaly
Acromegaly is an insidious disease, which typically takes about 10–12 years from the onset of symptoms to diagnosis.2 The exact reasons for the insidious nature of acromegaly remain unclear. It is likely that the gradual, progressive course of the disease leads to alterations ignored by the patient, family members, and even physicians. In addition, some of the comorbidities resemblance common disorders. As a result, before obtaining the correct diagnosis, patients usually have to visit primary care physicians and even other specialists several times.11 The clinical manifestations of acromegaly depend on several factors, including levels of GH and IGF-1, the sensitivity of different tissues and organs to increased levels of GH and IGF-1, age, and tumor size, as well as delays in diagnosis.12 In general, the clinical features of acromegaly include somatic effects due to excessive GH and IGF-1 levels and local effects caused by expansion of the pituitary tumor.2
Somatic effects
Somatic effects include increases in the thickness of the skin and connective tissue; uneven proliferation of cartilage, bone, and other epithelial tissues; and visceromegaly in the form of goiter, hepatomegaly, splenomegaly, and macroglossia.12 Acral enlargement, such as enlargement of hands, feet, and fingers, and typically coarse facial features, are the universal clinical manifestations of acromegaly. Facial changes include enlarged lips, tongue, and nose; deep nasolabial furrows; frontal skull bossing; mandibular prognathism; and separation of maxillary teeth.2 Changes in the skin and soft tissues involve thickening of the skin owing to glycosaminoglycan deposition, leading to hyperhidrosis or oily skin texture, acanthosis nigricans (dark skin patches), and skin tags.2,12
Local effects of tumor
In a 2015 demographic study conducted on 271 patients, 6.6% and 83% were found to have microadenoma and macroadenoma, respectively.24 According to another study, the prevalence of macroadenoma was found to be higher than that of microadenoma.25 The adenoma may compress local organs and even cause neurological symptoms along with visual disturbances, which include headaches, defects in the visual field, cranial nerve palsies, hypopituitarism, and hypothyroidism.2 The optic chiasm is an X-shaped structure formed by crossing of the optic nerves (CN II) in the brain. Upward growth of a pituitary adenoma leads to optic chiasm compression, thereby producing visual field disturbance, which starts in the mid-periphery of the superior temporal sectors and finally leads to bitemporal hemianopsia. The cavernous sinus is an important structure in the brain which is formed by a large channel of venous blood forming a cavity surrounded by sphenoid and temporal bones. This cavity is significant because of its location and content. It contains the third (oculomotor) and fourth (trochlear) cranial nerves. It also contain parts one (the ophthalmic nerve) and two (the maxillary nerve) of the fifth cranial (trigeminal) nerve, and the sixth cranial (abducens) nerve. Cavernous sinus invasion by a pituitary adenoma is of clinical significance it makes surgery more difficult and less efficient.31
Associated comorbidities
Manifestations of acromegaly include soft and acral tissue overgrowth, joint pain, hypertension, and heart and respiratory failure.12 Many studies have provided diagnostic algorithms from the associated comorbidities to identify acromegaly.8 A scoring system called “ACROSCORE” has been used to investigate and confirm acromegaly in the very early stages (Figure 1).28
Figure 1.
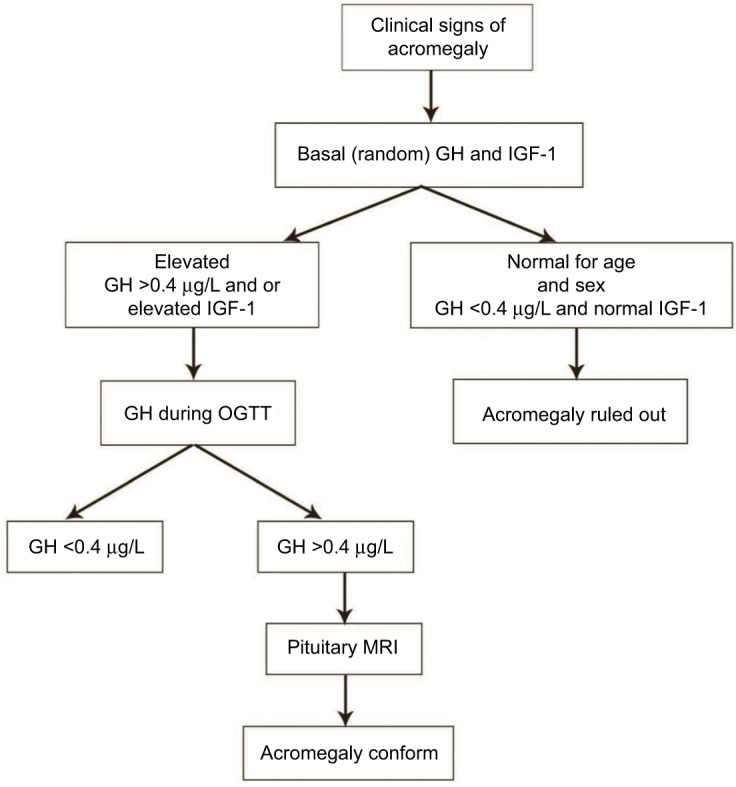
Diagnostic flow of acromegaly.
Note: Data from Rosario et al26 and Cordido et al.27
Abbreviations: GH, growth hormone; IGF-1, insulin-like growth factor-1; OGTT, oral glucose tolerance test; MRI, magnetic resonance imaging.
Cardiovascular complications
One of the most prevalent comorbidities related to acromegaly is cardiovascular disorder,8 occurring in 60% of patients with acromegaly.12 Cardiovascular disorders act as negative determinants for life expectancy in acromegaly patients. Acromegaly is associated with a typical cardiomyopathy, characterized by biventricular hypertrophy, and hypertension is one of the most common cardiovascular comorbidities in acromegaly, with an average prevalence of ~35%. In addition, congestive heart failure, cardiomyopathy, arrhythmias, coronary disease, and hypertension commonly occur.
Pulmonary and respiratory complications
Patients suffering from acromegaly have altered structures of the respiratory apparatus, which lead to severe respiratory problems.2,8 Respiratory complications associated with acromegaly include sleep breathing disorders, generally represented by sleep apnea, which affects up to 80% of patients, and respiratory insufficiency. The anatomical changes in respiratory structures affect lung volumes, respiratory mucosa/cartilage, lung elasticity, chest muscle structure, and respiratory muscle activities. Some patients develop a barrel chest as a result of the changes in vertebral and costal morphology. Other complications include hyperventilation, and hypertrophy of the laryngeal mucosa and cartilage, causing sleep apnea and excessive snoring.8,12
Gastrointestinal complications
Development of colon carcinoma, adenomatous polyps, and dolichocolon are some of the common gastrointestinal problems detected in acromegaly patients.12 It is accepted that the risk of colon cancer is higher in acromegalic patients than in the general population.32
Orthopedic comorbidities
Arthropathy widely occurs in patients with acromegaly, along with vertebral fractures, joint disease, and loss of weight-bearing ability. Early diagnosis of acromegaly may decrease the risk of bone and joint disease, resulting in improved quality of life.
Metabolic disorders
Increased secretion of GH and IGF-1 leads to abnormal glucose regulation, causing diabetes mellitus. A defect in lipid metabolism is also observed in some patients with acromegaly.13
Cancer
The high level of IGF-1 in acromegaly is related to an increased risk of some cancers, particularly colon cancer and thyroid cancer.8 An increased incidence of micronucleus (MN) and oxidative DNA damage in lymphocytes of patients with acromegaly was found by Hamurcu et al.22 An increased MN frequency (chromosomal DNA damage) in peripheral blood lymphocytes is predictive of an increased risk of cancer in humans. Thus, the increase in chromosomal/oxidative DNA damage and the positive association between MN frequency and serum IGF-1 levels may predict an increased risk of malignancy in acromegalic patients.23
Other complications
Women suffering from acromegaly have higher probabilities of menstrual irregularities and infertility. Hyperprolactinemia was also found to develop in 30% of patients.12
Mortality
The mortality rate in patients with active acromegaly is two to four times higher than in the general population.3 This is mostly due to cardiovascular disorders, pulmonary complications, and diabetes mellitus. Cardiovascular disease has historically been reported to represent the primary cause of death in these patients, contributing to nearly 50% of increased mortality. However, according to recent studies, the most common cause of mortality in acromegaly was malignancy, followed by cardiovascular diseases,29,30 then respiratory problems and neoplasias, accounting for ~25% and 15% of mortality, respectively.14 There is a direct correlation between the mortality rate and excess GH and IGF-1 secretion. Normalization of GH and IGF-1 has been found to reduce the mortality rate and disease-related morbidity.3 A positive correlation also exists between mortality in acromegaly patients and colorectal cancer.12 However, with advances in the management strategies of acromegaly and its associated comorbidities, there have been marked improvements in the survival rate and quality of life of patients with acromegaly.13
Diagnosis of acromegaly
Acromegaly is a chronic, debilitating disorder with slow disease onset, which impedes accurate diagnosis in the early stages.15 None of the signs and symptoms is sensitive enough to be detected at the beginning of the disease. Failure to recognize appropriate symptoms causes a delay in disease diagnosis, leading to a high incidence of acromegaly.4 According to a study by Reid et al, involving 324 acromegaly patients from the periods 1981–1994 and 1995–2006, the majority of patients showed many clinical symptoms and complications consistent with the disease being at an advanced stage at diagnosis, indicating that there is a high prevalence of underdiagnosed acromegaly.11,13 In general, primary care physicians diagnose ~40% of patients with acromegaly.33 Nevertheless, patients often consult other specialists and dentists, besides primary care physicians, before obtaining the correct diagnosis, and are then treated according to the presenting symptoms, instead of being treated for acromegaly.16
The phenotype of acromegaly develops gradually over several years; usually, the first clinical manifestation of the disease includes the growth of acral parts and physiognomic alterations.12 Thus, comparison of new and old facial photographs was conventionally used in early detection of the disease. The main drawback in considering phenotypic alterations for the initial recognition of acromegaly is that only 50% of patients with somatotroph adenomas reveal distinct phenotypic changes, whereas the other 50% either show insignificant symptoms or are completely asymptomatic, even having elevated levels of GH and IGF-1, and even after several years of observation.13 Another possible method for early diagnosis of acromegaly is to use software to depict the features that have developed as a result of acromegaly in a single photograph of the patient’s face.12 However, the sensitivity and specificity of the computer modeling system in early acromegaly are not yet clear. Once there is a clinical suspicion of acromegaly, biochemical tests should be performed for every patient to confirm the diagnosis.
Biochemical tests
GH level
The biochemical diagnosis of acromegaly is traditionally based on the levels of oversecreted GH and IGF-1. However, in normal healthy individuals the levels of circulating GH secreted from the pituitary fluctuate greatly throughout the day owing to the pulsatile nature of GH production.17 Maximum secretion of GH occurs at night in accordance with sleep stages. The value of GH usually ranges between 0.1–0.2 µg/L and 5–30 µg/L during the secretory bursts, and these values overlap with the values observed in acromegaly patients.12 A random GH value <0.04 µg/L with a normal level of IGF-1 (matched for age and gender) excludes the diagnosis. Thus, determination of the GH level is of minimal diagnostic utility in acromegaly. Instead, a GH level <0.4 ng/mL in an oral glucose tolerance test (OGTT) is the gold standard for diagnosis.13 In normal individuals, the GH nadir value during an OGTT is undetectable as secretion is suppressed, but the value is very high in acromegaly patients owing to the lack of suppression.2
IGF-1 level
IGF-1, the level of which is correlated with mean GH level, has been used as a biomarker of acromegaly since the turn of the millennium.5 Measurement of circulating IGF-1 is considered a vital biochemical tool because of its long half-life of 18–20 hours and stability throughout the day.5 An elevated IGF-1 level along with inability to reduce GH levels <1 µg/L in an OGTT is considered an important criterion for acromegaly detection.1 Although a correlation exists between the levels of serum IGF-1 and GH, some acromegaly patients show high IGF-1 levels in spite of a normal daily GH concentration, indicating a complex correlation between them, which cannot be simply explained by GH elevation.13 Regardless of the substantial advantages of IGF-1 as a biochemical tool, there are certain limitations to its use. The IGF-1 level is affected by multiple physiological factors such as age, gender, and body mass index, and these should be considered during data interpretation. The IGF-1 level is raised during normal pregnancy and puberty and rarely represents a clinical problem. In contrast, malnutrition, liver and renal failure, hypothyroidism, and insulin-dependent diabetes reduce the level of IGF-1. Major technical problems of IGF-1 assay include interference caused by endogenous IGF-1-binding protein and the tendency of IGF-1 to plateau at mean GH levels.5 All these factors lead to variability in IGF-1 assay performance. Therefore, technical refinements of IGF-1 biochemical assays, validation of assay performance, and assay-specific reference data are essential to detect specific clinical conditions related to acromegaly.18
Immunoassays
For accurate diagnosis of acromegaly, it is imperative to have a highly sensitive GH assay, with the ability to detect GH levels <1.0 ng/mL. The development of a radioimmunoassay (RIA) to measure serum GH and IGF-1 levels presented a biochemical device to diagnose acromegaly. RIA has several limitations as a result of which more sensitive immunoassays, such as immunoluminometric and immunoradiometric assays based on the use of monoclonal antibodies, have been developed to measure plasma GH and IGF-1 levels precisely in patients with suspected acromegaly. These assays can determine GH concentrations <0.05 µg/mL;19 the sensitivity of these immunoassays is up to 100 times greater than RIA.20 However, one limitation of these assays is proper reproducibility. Variability of measured GH values occurs owing to the lack of universal standards, recognition of non-uniform antibodies by GH isoforms, and the presence of circulating GH-binding protein. Hence, more authentic and reliable ultrasensitive GH assays based on strong reference standards are required.19
Imaging studies
MRI with contrast administration is considered to be the most effective imaging technique to locate the pituitary source of excess GH.12 MRI is a sensitive and reliable technique, which enables the identification of very small tumors, even smaller than 2 mm, tumor invasiveness, proximity to the optic chiasm, and compression of surrounding structures by the tumor which previously remained undetected.5 The majority of somatotroph adenomas (75%–85%) are macroadenomas (>10 mm in diameter) at the time of diagnosis, which rarely grow into the cavernous sinus.12 MRI helps to identify a pituitary macroadenoma in acromegaly patients.2 Advanced MRI techniques such as post-contrast, volumetric interpolated breath-hold MRI can detect acromegaly in patients with pituitary tumors, which was otherwise unidentified by traditional spin-technique T1-weighted MRI. Hence, improved MRI techniques have advantages over conventional methods for the diagnosis of acromegaly.13
Rarely, acromegaly occurs as a result of ectopic tumors producing GH or GHRH. When a biochemical diagnosis has been established for acromegaly without any pituitary tumor or diffused pituitary enlargement is detected, physicians should suspect ectopic sources.12 Measurement of the plasma GHRH level is beneficial in identifying ectopic sources of tumor.5
Treatment of acromegaly
Improved understanding of the disease mechanism has led to the development of successful treatment modalities of acromegaly. Novel surgical techniques are used to operate on pituitary tumors.
The majority of pituitary tumor operations in patients with acromegaly are performed by transsphenoidal surgery.30 Some surgeons use an image intensifier for navigation while others choose neuronavigation. A submucosal tunnel is dissected, usually followed by a medial nasal incision. The mucosal tunnel is kept open by a nasal speculum. Then the operating microscope is introduced. Sphenoidotomy is performed using the vomer as a midline orientation. A direct perinasal intervention to the sphenoid sinus is usually chosen by endoscopic transsphenoidal surgeons. The transnasal/transsphenoidal technique seems to be accurate and safe, with good surgical results. Based on the preoperative MRI, the decision is made to carry out a curative or debulking procedure. The resectioning of the adenoma depends on the size and invasive character of the tumor. For patients who are not cured by surgery, or for those whose health status prohibits surgery, medical therapy using somatostatin analogs and GH-receptor antagonists is considered to be the principal therapy.1,13 For those patients in whom both surgical and medical therapy fails to provide biochemical control of acromegaly, radiation therapy is the best curative technique. Sometimes, more than one treatment procedure is required. In almost all cases, long-term treatment and monitoring are essential.1 With progress in disease management, the prognosis of the disease has improved, and in the majority of the cases adequate hormonal disease control is attained, resulting in life expectancy comparable to the general population.21
Conclusion
Acromegaly is a rare, devastating disorder associated with multiple comorbidities, poor quality of life, and increased mortality. Timely diagnosis and subsequent therapies are vital for successful prevention of disease and premature death. To mitigate the problems related to the delay in diagnosis of acromegaly, it is essential to develop awareness about the early signs and symptoms of the disease among primary care physicians, specialists, dentists, and other health care professionals. In addition, better communication and collaboration are necessary between patients and health care providers for disease diagnosis as well as long-term care. Recently, several techniques have been developed to aid in the early identification and monitoring of acromegaly and its associated complications. These advances in disease identification have led to a rise in the number of patients diagnosed with the disease and the development of better treatment modalities. This, in turn, will result in increased cure and decreased mortality rates in acromegaly patients.
Acknowledgments
The author is thankful to www.manuscriptedit.com for providing English language editing and proofreading services for this manuscript.
Footnotes
Disclosure
The author reports no conflicts of interest in this work.
References
- 1.Adelman DT, Liebert KJ, Nachtigall LB, Lamerson M, Bakker B. Acromegaly: the disease, its impact on patients, and managing the burden of long-term treatment. Int J Gen Med. 2013;6:31–38. doi: 10.2147/IJGM.S38594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banerjee A, Patel K, Wren AM. Acromegaly- clinical manifestations and diagnosis. Pharm J. 2003;13:273–278. [Google Scholar]
- 3.Nachtigall L, Delgado A, Swearingen B, Lee H, Zerikly R, Klibanski A. Extensive clinical experience: changing patterns in diagnosis and therapy of acromegaly over two decades. J Clin Endocrinol Metab. 2008;93:2035–2041. doi: 10.1210/jc.2007-2149. [DOI] [PubMed] [Google Scholar]
- 4.Schneider HJ, Sievers C, Saller B, Wittchen HU, Stalla GK. High prevalence of biochemical acromegaly in primary care patients with elevated IGF-1 levels. Clin Endocrinol. 2008;69(3):432–435. doi: 10.1111/j.1365-2265.2008.03221.x. [DOI] [PubMed] [Google Scholar]
- 5.Cordero RA, Barkan AL. Current diagnosis of acromegaly. Rev Endocr Metab Disord. 2008;9(1):13–19. doi: 10.1007/s11154-007-9060-2. [DOI] [PubMed] [Google Scholar]
- 6.Cannavò S, Ferraù F, Ragonese M, et al. Increased prevalence of acromegaly in a highly polluted area. Eur J Endocrinol. 2010;163(4):509–513. doi: 10.1530/EJE-10-0465. [DOI] [PubMed] [Google Scholar]
- 7.Melmed S. Medical progress: Acromegaly. N Engl J Med. 2006;355(24):2558–2573. doi: 10.1056/NEJMra062453. [DOI] [PubMed] [Google Scholar]
- 8.Abreu A, Tovar AP, Castellanos R, et al. Challenges in the diagnosis and management of acromegaly: a focus on comorbidities. Pituitary. 2016;19(4):448–457. doi: 10.1007/s11102-016-0725-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brabant G. Insulin-like growth factor-I: marker for diagnosis of acromegaly and monitoring the efficacy of treatment. Eur J Endocrinol. 2003;148(Suppl 2):S15–S20. doi: 10.1530/eje.0.148s015. [DOI] [PubMed] [Google Scholar]
- 10.Isidro ML, Iglesias Díaz P, Matías-Guiu X, Cordido F. Acromegaly due to a growth hormone-releasing hormone-secreting intracranial gangliocytoma. J Endocrinol Invest. 2005;28(2):162–165. doi: 10.1007/BF03345360. [DOI] [PubMed] [Google Scholar]
- 11.Reid TJ, Post KD, Bruce JN, Nabi Kanibir M, Reyes-Vidal CM, Freda PU. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol. 2010;72(2):203–208. doi: 10.1111/j.1365-2265.2009.03626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lugo G, Pena L, Cordido F. Clinical manifestations and diagnosis of acromegaly. Int J Endocrinol. 2012;2012:540398. doi: 10.1155/2012/540398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribeiro-Oliveira A, Barkan A. The changing face of acromegaly--advances in diagnosis and treatment. Nat Rev Endocrinol. 2012;8(10):605–611. doi: 10.1038/nrendo.2012.101. [DOI] [PubMed] [Google Scholar]
- 14.Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab. 2004;89(2):667–674. doi: 10.1210/jc.2003-031199. [DOI] [PubMed] [Google Scholar]
- 15.Ben-Shlomo A, Sheppard MC, Stephens JM, Pulgar S, Melmed S. Clinical, quality of life, and economic value of acromegaly disease control. Pituitary. 2011;14(3):284–294. doi: 10.1007/s11102-011-0310-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kreitschmann-Andermahr I, Siegel S, Kleist B, et al. Diagnosis and management of acromegaly: the patient’s perspective. Pituitary. 2016;19(3):268–276. doi: 10.1007/s11102-015-0702-1. [DOI] [PubMed] [Google Scholar]
- 17.Melarvie S, Jeevanandam M, Holaday NJ, Petersen SR. Pulsatile nature of growth hormone levels in critically ill trauma victims. Surgery. 1995;117(4):402–408. doi: 10.1016/s0039-6060(05)80060-1. [DOI] [PubMed] [Google Scholar]
- 18.Clemmons DR. Consensus statement on the standardization and evaluation of growth hormone and insulin-like growth factor assays. Clin Chem. 2011;57(4):555–559. doi: 10.1373/clinchem.2010.150631. [DOI] [PubMed] [Google Scholar]
- 19.Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119(11):3189–3202. doi: 10.1172/JCI39375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costa AC, Rossi A, Martinelli CE, Machado HR, Moreira AC. Assessment of disease activity in treated acromegalic patients using a sensitive GH assay: should we achieve strict normal GH levels for a biochemical cure? J Clin Endocrinol Metab. 2002;87(7):3142–3147. doi: 10.1210/jcem.87.7.8631. [DOI] [PubMed] [Google Scholar]
- 21.Giustina A, Chanson P, Bronstein MD, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95(7):3141–3148. doi: 10.1210/jc.2009-2670. [DOI] [PubMed] [Google Scholar]
- 22.Hamurcu Z, Cakir I, Donmez-Altuntas H. Micronucleus evaluation in mitogen-stimulated lymphocytes of patients with acromegaly. Metabolism. 2011;60:1620–1626. doi: 10.1016/j.metabol.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 23.Bayram F, Bitgen N, Donmez-Altuntas H, et al. Increased genome instability and oxidative DNA damage and their association with IGF-1 levels in patients with active acromegaly. Growth Horm IGF Res. 2014;24(1):29–34. doi: 10.1016/j.ghir.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Dutta P, Hajela A, Pathak A, et al. Clinical profile and outcome of patients with acromegaly according to the 2014 consensus guidelines: Impact of a multi-disciplinary team. Neurol India. 2015;63(3):360–368. doi: 10.4103/0028-3886.158210. [DOI] [PubMed] [Google Scholar]
- 25.Day PF, Loto MG, Glerean M, Picasso MF, Lovazzano S, Giunta DH. Incidence and prevalence of clinically relevant pituitary adenomas: retrospective cohort study in a Health Management Organization in Buenos Aires, Argentina. Arch Endocrinol Metab. 2016;60(6):554–561. doi: 10.1590/2359-3997000000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosario PW, Calsolari MR. Laboratory investigation of acromegaly: Is basal or random GH > 0.4 µg/L in the presence of normal serum IGF-1 an important result? Arch Endocrinol Metab. 2015;59(1):54–58. doi: 10.1590/2359-3997000000010. [DOI] [PubMed] [Google Scholar]
- 27.Cordido F, García Arnés JA, Marazuela Aspiroz M, Torres Vela E. Practical guidelines for diagnosis and treatment of acromegaly. Endocrinología y Nutrición. 2013;60(8):457.e1–457. doi: 10.1016/j.endonu.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Prencipe N, Floriani I, Guaraldi F, et al. ACROSCORE: a new and simple tool for the diagnosis of acromegaly, a rare and underdiagnosed disease. Clin Endocrinol. 2016;84(3):380–385. doi: 10.1111/cen.12959. [DOI] [PubMed] [Google Scholar]
- 29.Öberg K, Lamberts SW. Somatostatin analogues in acromegaly and gastroenteropancreatic neuroendocrine tumours: past, present and future. Endocr Relat Cancer. 2016;23(12):R551–R566. doi: 10.1530/ERC-16-0151. [DOI] [PubMed] [Google Scholar]
- 30.Buchfelder M, Schlaffer S. Surgical treatment of pituitary tumours. Best practice & research. Clin Endocrinol Metab. 2009;23:677–692. doi: 10.1016/j.beem.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Cottier JP, Destrieux C, Brunereau L, et al. Cavernous sinus invasion by pituitary adenoma: MR imaging. Radiology. 2000;215(2):463–469. doi: 10.1148/radiology.215.2.r00ap18463. [DOI] [PubMed] [Google Scholar]
- 32.Tirosh A, Shimon I. Complications of acromegaly: thyroid and colon. Pituitary. 2017;20(1):70–75. doi: 10.1007/s11102-016-0744-z. [DOI] [PubMed] [Google Scholar]
- 33.Chiaghana CO, Bauerfeind JM, Sulek CA, Goldstein JC, Awoniyi CA. Acromegaly discovered during a routine out-patient surgical procedure: a case report. J Med Case Rep. 2017;11(1):169. doi: 10.1186/s13256-017-1338-8. [DOI] [PMC free article] [PubMed] [Google Scholar]