

Abstract
Background
Oral carcinoma, one of the most commonly diagnosed cancers, has a poor prognosis and low survival rate with treatment. In recent years, some studies reported the upregulation of miRNA-16 (miR-16) in the oral carcinoma, whereas some other studies confirmed the downregulation of miR-16. In the current study, we aimed to investigate the function of miR-16 in oral carcinoma.
Materials and methods
Cell proliferation assay was measured by MTT assay, quantitative real time polymerase chain reaction (qRT-PCR) was used to evaluate the expression of miR-16, and apoptosis was analyzed by flow cytometry. In addition, the expression of proteins was detected by Western blot. Moreover, xenograft tumor model was established to detect the effect of miR-16 in vivo.
Results
The results suggested that miR-16 was downregulated in the oral carcinoma tissues. Overexpression of miR-16 inhibited the growth and proliferation of oral squamous carcinoma cells (OSCCs) and induced apoptosis both in vitro and in vivo, which is due to the suppression of Wnt/β-catenin signaling pathway.
Conclusion
This study provides evidence that overexpression of miR-16 inhibits OSCC growth by regulating Wnt/β-catenin signaling. Our findings suggest that overexpression of miR-16 could be a potential approach for gene therapy of OSCC in future.
Keywords: oral carcinoma, miRNA-16, apoptosis, Wnt/β-catenin signaling
Introduction
Oral carcinoma, one of the most commonly diagnosed cancers, has a poor prognosis and low survival rate with treatment. Some oral cancer presents highly migrated feature and led to the emergence of second primary tumor, which are the core reason for poor prognosis.1 In recent years, there has been an increasing focus on gene therapy, which has been applied in primary immunodeficiencies,2 hemoglobin disorders,3 coagulation disorder hemophilia B,4 inherited neurological disorders, and cancer immunotherapy.5–7 However, gene therapy treatment for oral carcinoma has been seldom investigated.
MicroRNAs (miRNAs), a bunch of noncoding RNAs, are composed ~22 nucleotides, which play a role as posttranscriptional regulators of gene expression. miRNAs act as oncogenes or tumor suppressors to regulate pathogenesis of cancer.8–12 miRNA microarrays have demonstrated that abnormal miRNA expression may act as potential diagnostic and prognostic biomarkers for cancer. In addition, it is well known that miRNAs play critical role in many physiological processes by regulating the expression of apoptosis, differentiation, metabolism, and migration-related genes.13–15 More than 30 miRNAs have been reported which involved in apoptosis, among them, miR-16 is reported as proapoptosis miRNA.16 Some studies reported that the expression of miR-16 was upregulated in cancer, conversely, other studies reported that the expression of miR-16 was downregulated.36,37,43 Thereby, in the current study, we aimed to investigate the function of miR-16 in oral carcinoma.
The first gene that encoded a Wnt signaling component was identified from mouse tumor cells 30 years ago.17 The Wnt/β-catenin signaling directs fundamental processes, including metazoan development and tissue homeostasis, whereas deregulation of Wnt signaling underlies numerous congenital disorders and carcinomas.18 In addition, Wnt signaling plays an essential role in regulating cell fate, proliferation, survival, and migration.19–21 Increasingly, reports reveal that miRNA regulates cell differentiation, proliferation, and apoptosis. Many studies also reported that miRNA regulates the expression of Wnt signaling pathway. Zhu et al reported that upregulating miR-34a could decrease the expression level of Wnt/β-catenin to inhibit liver tumorigenesis.22–25 miR-218 is involved in cardiac stem cells (CSC) proliferation and differentiation via modulating Wnt signaling.23 Hence, in our study, we focused on the expression of Wnt/β-catenin signaling when regulated by miRNA in oral squamous carcinoma cells (OSCCs).
In the recent years, many studies investigated the expression of miR-16 in oral carcinoma, some reported that miR-16 was upregulated in some types of OSCCs, whereas, other studies reported that miR-16 was downregulated in OSCCs.26–28 To validate this discrepancy, we first detected the miR-16 expression level in 49 oral tissues and 44 normal tissues by quantitative real time polymerase chain reaction (qRT-PCR) assay.
Materials and methods
Clinical samples and cell culture
A total of 49 fresh tumor tissues and adjacent normal tissue samples were obtained from The First Affiliated Hospital of Xinjiang Medical University approved by the corresponding ethics committee. Written informed consent was obtained from patients whose tissue samples were used. The OSCC Cal-27 cell line was purchased from American Type Culture Collection (Manassas, VA, USA), which were maintained in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich Co., St Louis, MO, USA) and were incubated at 37°C with 5% CO2 in a humidified atmosphere.
RNA isolation and qRT-PCR
The total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instruction. Expressions of miR-16 in cell lines and patient tissue samples were detected using All-in-One™ miRNA qRT-PCR Detection kit following the manufacturer’s protocol (Cat. No: AOMD-Q020; GeneCopoeia™, Rockville, MD, USA). Polymerase chain reactions for each gene were performed three times separately.
Transient and stable transfection
To establish transient and stable transfection, Cal-27 cells were incubated at a concentration of 1×105/mL for a day before transfection. Cal-27 cells were transfected with miR-16, inhibitor or controls (Genepharma, Guangzhou, China) using the Lipofectamine 2000 reagent (Thermo Fisher Scientific) according to manufacturer’s protocol. The efficiency of transfection was evaluated to be >80%. The mature types of miR-16 in the study were 3′GCGGUUAUAAAUGCACGACGAU5′. After transfection, the cells were harvested for assays in 48 hours. Lenti-miR-16 was constructed by pSin-EF2-puromcin lentiviral plasmid (Addgene, Cambridge, MA, USA) which can synthesize and clone the precursor sequence of miR-16. The empty pSin-EF2 vector (lenti-vector) was used as a control.
Observation of morphologic changes
The cells were seeded into six-well culture (Corning Incorporated, Corning, NY, USA) plates and transferred with miR-16 or miR-Ctrl. After incubation for 24 hours, the cellular morphology was observed by phase contrast microscope (Olympus America Inc., Center Valley, PA, USA).
Cell viability assay
The cells were seeded into 96-well culture plates (Corning Incorporated) and cultured for 24 hours at 5% CO2 atmosphere. Then miR-16 or miR-Ctrl was transfected into the cells. After incubation for another 24 hours, the cells were rinsed twice with ice-cold PBS and cultured with 100 mL of 0.5 mg/mL MTT (Thermo Fisher Scientific) solution. After 3 hours, the crystal was dissolved in 150 μL dimethyl sulfoxide and measured by a mirco-plate reader (Thermo Fisher Scientific), and the inhibition rate was calculated by the following formula:
Cell apoptosis assays
Cell apoptosis was performed by Annexin V- Fluorescein isothiocyanate (FITC)/Propidiumiodide (PI) Apoptosis Detection kit (Pierce Biotechnology, Rockford, IL, USA). The cells that were cultured in six-well transfected plates with miR-16 or miR-Ctrl were detected with the kit. The cells were washed with PBS and collected after being labeled with Annexin-V-FITC and PI (BD Biosciences, San Jose, CA, USA). Finally, the cells were analyzed by a flow cytometer (BD Biosciences).
In vivo xenograft tumor model
BALB/c null mice about 4–6 weeks old were obtained from Vital River (Beijing, China) and housed in barrier facilities on a 12 hour light and dark cycle. All animal experiments were approved by the First Affiliated Hospital of Xinjiang Medical University Institutional Animal Care and Use Committee. NIH Guidelines and Animal Welfare were strictly followed. For establishing xenograft tumor model, the cells transfected with lenti-miR-16 or lenti-vector were subcutaneously inoculated in the right flank of the mice (3×106 cells/mouse). Every 5 days, tumor size was measured, and volumes were calculated. In another sister experiment, the mice were subcutaneously injected with lenti-vector transfected cells and divided into two groups. One group was orally daily dosed with 10 mg/kg XAV-939, and the other group was dosed with vehicle. The mice were sacrificed after 25 days treatment, and the tumors were excised and collected.
Immunohistochemistry
Immunohistochemistry was handled as described previously with slight modification.29,30 The tissues were incubated overnight with anti-Wnt3a primary antibody (Abcam). After incubation with secondary antibodies, the sections were washed and mounted.
Western blotting assay
Some Cal-27 cells were transferred with miR-16 and then treated with R-spondin1; inhibition of Wnt signaling in the other cells was achieved by the addition of 5 mM tankyrase inhibitor XAV939. After incubated for 24 hours, both adherent and floating cells were collected and lysed with Radio-Immunoprecipitation Assay (RIPA) lysis buffer (Beyotime, Haimen, China) supplemented with Phenylmethanesulfonyl fluoride (PMSF) (1 mM) for 30 minutes. Then, the cells were centrifuged at 12,000× g for 10 minutes, and the supernatant was collected. The protein concentration was detected by the Bio-Rad protein assay reagent (Bio-Rad Laboratories Inc., Hercules, CA, USA). Equal amounts of the total protein were separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Millipore Immobilon®-P Transfer Membrane (EMD Millipore, Billerica, MA, USA). The antibody of β-catenin, β-actin, APC, Axin-1, and horseradish peroxidase-conjugated secondary antibodies were obtained from Sigma Chemical (Perth, Australia). The expression levels of proteins were visualized by electro-chemiluminescence (Thermo Fisher Scientific).
Statistical analysis
To determine the statistical significance between groups, all results and data were detected by at least three separate experiments. All data were analyzed by analysis of variance by using SPSS software (version 13.0; SPSS, Chicago, IL, USA). *p<0.05 and **p<0.01 were considered statistically different or significantly different, respectively. All data are expressed as mean ± SD.
Results
miR-16 expression was downregulated in oral carcinoma tissues
The patient information was summarized in Table 1. The results showed that the average expression level of miR-16 in oral carcinoma tissues was decreased compared with the normal tissues samples (Figure 1). These results indicated that miR-16 is downregulated in oral carcinoma and may be involved in oral carcinoma tumorigenesis and progression. The relative expression of miR-16 in Cal-27 cells (0.68) was also lower than normal human tissues.
Table 1.
Patient information
| Patient no | Gender | Age (years) | Disease stage |
|---|---|---|---|
| 1 | M | 39 | II |
| 2 | F | 41 | III |
| 3 | F | 78 | IV |
| 4 | F | 67 | III |
| 5 | M | 56 | II |
| 6 | M | 81 | II |
| 7 | F | 45 | 0 |
| 8 | M | 44 | I |
| 9 | M | 78 | III |
| 10 | F | 61 | IV |
| 11 | M | 55 | III |
| 12 | F | 51 | II |
| 13 | F | 56 | IV |
| 14 | F | 38 | I |
| 15 | M | 32 | II |
| 16 | M | 67 | III |
| 17 | F | 68 | IV |
| 18 | M | 45 | III |
| 19 | M | 48 | II |
| 20 | M | 56 | IV |
| 21 | F | 54 | III |
| 22 | F | 53 | II |
| 23 | M | 41 | I |
| 24 | M | 43 | III |
| 25 | M | 78 | III |
| 26 | F | 45 | II |
| 27 | F | 51 | I |
| 28 | M | 58 | III |
| 29 | M | 66 | III |
| 30 | F | 61 | II |
| 31 | F | 41 | III |
| 32 | F | 44 | III |
| 33 | M | 65 | II |
| 34 | F | 41 | III |
| 35 | M | 56 | II |
| 36 | F | 67 | II |
| 37 | F | 31 | III |
| 38 | M | 44 | II |
| 39 | M | 33 | II |
| 40 | F | 56 | IV |
| 41 | F | 67 | II |
| 42 | F | 41 | III |
| 43 | M | 78 | II |
| 44 | M | 51 | II |
| 45 | F | 43 | III |
| 46 | M | 39 | IV |
| 47 | M | 51 | III |
| 48 | F | 57 | IV |
| 49 | M | 58 | III |
Abbreviations: M, male; F, female.
Figure 1.
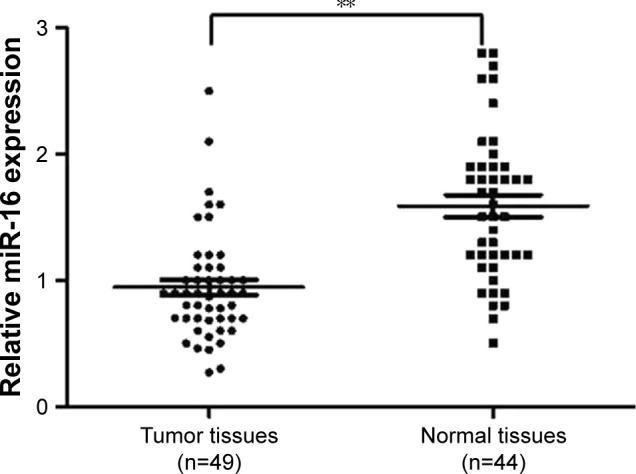
miR-16 is decreased in oral carcinoma tissues. The total RNA was isolated using TRI zol reagent from oral carcinoma tissues (n=49) and adjacent normal tissues (n=44). Relative miR-16 expression was detected using All-in-One™ miRNA qRT-PCR Detection kit. **p<0.01, normal tissues compared with tumor tissues
miR-16 inhibited oral carcinoma cell growth via inducing apoptosis
The MTT and apoptotic assays were performed in order to evaluate the functions of miR-16 in oral carcinoma. The observation of morphology showed that the cells transfected with miR-16 had a low viability and a remarkable nuclear fragmentation compared with that transfected with miR-Ctrl (Figure 2A). In addition, the results of MTT demonstrated the suppressive effect of miR-16 on cell proliferation (Figure 2B). The apoptotic assay showed that cells transfected with miR-16 have significantly increased cell apoptosis compared with control and cells transfected with miR-Ctrl (Figure 2C and D). These data revealed that miR-16 could inhibit oral carcinoma cell proliferation and tumor growth via inducing cell apoptosis.
Figure 2.

miR-16 inhibited oral carcinoma cells growth via inducing apoptosis. (A) Observation of morphologic change of transfected with miR-16 (200× magnification). (B) The cells were transfected with miR-16, or inhibitor for 1, 2, 3, and 4 days. Cell viability was measured by MTT assay. (C and D) Flow cytometric analysis of apoptotic cell ratios after PI and Annex-V staining. Data presented as mean ± SD, n=3.**p<0.01, miR-16 group compared with control group.
miR-16 inhibits oral carcinoma cell growth in vivo
To evaluate the effect of miR-16 overexpression in vivo, the Cal-27 xenograft tumor model stably expressing miR-16 (lenti-miR-16) or control empty vector (lenti-vector) was used. The results showed that the tumor formed by lenti-miR16 cells had smaller volumes and lower weights than those formed by lenti-vector cells (Figure 3A–C). These data demonstrated that miR-16 could inhibit oral carcinoma cell proliferation and growth in vivo.
Figure 3.

miR-16 inhibits oral carcinoma tumor growth in vivo. BAL B/c null mice were subcutaneously injected with the oral squamous carcinoma cell with lenti-miR-16 or lenti-vector. (A) The tumor volumes of mice were measured every 5 days. (B and C) In the end of the study, the mice were sacrificed. Then, the tumors were excised from mice and weighted. Data presented as mean ± SD, n=8. *p<0.05, compared with lenti-vector group. **p<0.01.
miR-16 downregulated the overexpression of Wnt/β-catenin signaling in Cal-27
RT-PCR analysis was performed to further detect the mechanism of progression of oral carcinoma. As shown in the results, the gene expression of Wnt3a was increased in tumor tissues compared with the normal tissues. However, the Bcl-2, CCND1, CCNE1, Bmi-1, and YAP-1 gene expressions in tumor tissues had no obvious discrepancy compared with normal tissues (Figure 4A). In addition, the immunohistochemical and Western blotting analyses showed that the expression of β-catenin is decreased in the cells transfected with miR-16 compared with control or cells transfected with miR-16 inhibitor (Figure 4B and C). These results indicated that in Cal-27 cells, the Wnt/β-catenin signaling was activated, and we hypothesized that this signaling might be inhibited by miR-16.
Figure 4.

Wnt3a mRNA and proteins expression level in oral squamous carcinoma cell. (A) RT-PCR analysis of Wnt3a, Bcl-2, CCND1, CCNE1, Bmi-1, and YAP-1 mRNA expression in tumor tissues and adjacent normal tissues. (B) The expression levels of Wnt3a in tumor tissues and adjacent normal tissues were analyzed by immunohistochemistry, magnification ×200. (C) The expression of β-catenin in cells that are transferred with miR-16 or inhibitor. Data presented as mean ± SD, n=3. **p<0.01, compared with adjacent normal tissues.
miR-16 inhibited Cal-27 cell proliferation due to downregulation of Wnt/β-catenin signaling pathway
As shown in Figure 5A, miR-16 inhibited oral carcinoma cell growth in a time-dependent manner. Meanwhile, Wnt/β-catenin inhibitor XAV-939 also inhibited Cal-27 growth. These results indicated that inhibition of Wnt/β-catenin signaling pathway induced OSCC cells death. As we expected, the inhibitory effect of miR-16 on OSCC growth could be reversed by Wnt activator R-spondin 1 (Figure 5A). The results of Western blot indicated that the Wnt downstream proteins were overexpressed in the cells, which could be reversed by miR-16 transfection (Figure 5B and C). These data were consistent with the result of cell proliferation assay.
Figure 5.

miR-16 inhibited oral squamous carcinoma cell proliferation via downregulation of Wnt/β-catenin signaling pathway. (A) The Cal-27 cells were transferred with miR-16, inhibitor, or treated with Wnt signaling pathway inhibitor XAV-939, or treated with miR-16 plus R-spondin1 for 1, 2, 3, and 4 days. Cell viability was measured by MTT assay. (B) The protein levels of β-catenin, APC, and Axin-1 were measured by (C) semi-quantification of the results of Western blotting. Data presented as mean ± SD, n=3.
Suppression of Wnt/β-catenin signaling pathway inhibited Cal-27 xenograft tumor growth in vivo
To further determine the role of Wnt/β-catenin signaling pathway, we established Cal-27 xenograft tumor model using transfected lenti-vector cells. The mice were oral dosed with 10 mg/kg XAV939 daily for 25 days. The results showed that Cal-27 tumor growth was inhibited in vivo by using Wnt/β-catenin signaling pathway inhibitor XAV939 (Figure 6A–C). The body weights of xenografts had no obvious changes between vehicle and treatment groups (Figure 6D). All these data indicated that miR-16 plays a role of anti-tumor and exerts this effect by suppression of Wnt/β-catenin signaling pathway. These results were consistent with the result of cell proliferation assay.
Figure 6.

Suppressing of Wnt/β-catenin signaling inhibited oral carcinoma tumor growth in vivo. BAL B/c null mice were subcutaneously injected with Cal-27 cells, which transfer with lenti-vector. The mice were orally dosed with vehicle or 10 mg/kg XAV-939 for 25 days. (A) The tumor volumes of mice were measured every 5 days. (B) In the end of the study, the mice were sacrificed and tumors were weighted. (C) The tumors were isolated from xenografts. (D) The body weights of xenografts were monitored every 5 days. Data presented as mean ± SD, n=8. *p<0.05 compared with lenti-vector group. **p<0.01.
Discussion
The poor prognosis of oral carcinoma is the major problem over the past 20 years.26,31 This study elucidated that miR-16 can inhibit oral carcinoma proliferation and induce apoptosis via inhibition of Wnt signaling. Previous studies have demonstrated that dys-regulated expressions of miRNAs were involved in OSCC proliferation, migration, and invasion.32–35 In the present study, we identified that miR-16 expression level was significantly downregulated compared with normal tissue, demonstrating that miR-16 is involved in OSCC progression and tumorigenesis. miR-16 has been known as tumor suppressor, and the expression of miR-16 can affect cell proliferation, tumor apoptosis, tumor invasion and metastasis, and tumorigenicity. We further investigated its biological functions via transient or stable overexpression of miR-16 in OSCC cells. The results showed that forced expression of miR-16 suppressed OSCC tumor growth in vitro and in vivo via inducing OSCC apoptosis. Our finding is consistent with the report of Coutinho-Camillo et al, and we confirm that miR-16 is a pro-apoptotic miRNA in OSCC.16,36
In the recent days, numerous studies focused on the underlying mechanism of miR-16 and cancer cells. Huang et al reported that miR-16 target Rictor to induce autophagy and enhance chemo-sensitivity of camptothecin in HeLa cells.37 Jiang et al considered that miR-16 upregulation could reduce CDK4 expression by repressing CCND1 and sensitize nasopharyngeal carcinoma cells to chemotherapy.38 In addition, Wu et al reported that overexpression of miR-16 inhibited the proliferation, invasion, and metastasis of HepG2 cells via downregulation of PI3K/Akt signaling pathway.39 Moreover, Liang et al reported that miR-16 is targeting FEAT to promote the apoptosis of human cancer cells.40 In the current study, we also focused on the underlying mechanism of miR-16 in the process of OSCC tumor growth inhibition. It is well known that several oncogenes have been identified to be targets of miR-16, including BCL-2, CCND1, CCNE1, WNT3A, Bmi-1, and YAP1.39–44 In this study, we detected the expression of these oncogenes, while only Wnt3A was increased in the tumor tissues compared with normal tissues. WNT3A is a ligand of Wnt/β-catenin signaling, bind to Frizzled and LRP5/6 families of coreceptors.45 Hence, these results indicated that Wnt signaling pathway is involved in miR-16-induced OSCC tumor growth inhibition.
In canonical Wnt signaling, β-catenin is an essential downstream transcriptional effector and a subunit together with the scaffold proteins Axin and APC and the kinases GSK3 to compose the destruction complex.46 In this study, miR-16 transfection decreased the expression level of β-catenin, this means inhibition of Wnt/β-catenin signaling could also inhibit OSCC proliferation. Meanwhile, Wnt downstream proteins were overexpressed in OSCC, which could be reversed by miR-16 transfection. Collectively, these results indicated that miR-16 suppressed Wnt/β-catenin signaling to inhibit OSCC proliferation and induce apoptosis. However, the detailed interaction between miR-16 and Wnt/β-catenin signaling pathway is still unclear, and further investigations are needed.
Conclusion
In summary, this study provides evidence that overexpression of miR-16 inhibits OSCC cell growth by regulating Wnt/β-catenin signaling both in vitro and in vivo. Our findings suggest that overexpression of miR-16 could be a potential approach for gene therapy of OSCC in future.
Acknowledgments
This study was supported by China Postdoctoral Science fund (2015M572668XB).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45:309–316. doi: 10.1016/j.oraloncology.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Kumar SRP, Markusic DM, Biswas M, High KA, Herzog RW. Clinical development of gene therapy: results and lessons from recent successes. Mol Ther Methods Clin Dev. 2016;3:16034. doi: 10.1038/mtm.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Negre O, Eggimann AV, Beuzard Y, et al. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the β(A(T87Q))-globin gene. Hum Gene Ther. 2016;27:148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kay MA, Manno CS, Ragni MV, et al. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet. 2000;24:257–261. doi: 10.1038/73464. [DOI] [PubMed] [Google Scholar]
- 5.Larochelle A, Dunbar CE. Hematopoietic stem cell gene therapy. Assessing the relevance of pre-clinical models. Semin Hematol. 2013;50:101–130. doi: 10.1053/j.seminhematol.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu X, Li Z, Liu J. MiRNAs in primary cutaneous lymphomas. Cell Prolif. 2015;48:271–277. doi: 10.1111/cpr.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gargalionis AN, Basdra EK. Insights in microRNAs biology. Curr Top Med Chem. 2013;13:1493–1502. doi: 10.2174/15680266113139990098. [DOI] [PubMed] [Google Scholar]
- 10.Yu X, Li Z. The role of microRNAs expression in laryngeal cancer. Oncotarget. 2015;6:23297–23305. doi: 10.18632/oncotarget.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z, Yu X, Shen J, Wu WK, Chan MT. MicroRNA expression and its clinical implications in Ewing’s sarcoma. Cell Prolif. 2015;48:1–6. doi: 10.1111/cpr.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Z, Yu X, Shen J, Jiang Y. MicroRNA dysregulation in uveal melanoma: a new player enters the game. Oncotarget. 2015;6:4562–4568. doi: 10.18632/oncotarget.2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Tang DG. MicroRNA regulation of cancer stem cells. Cancer Res. 2011;71:5950–5954. doi: 10.1158/0008-5472.CAN-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okayama H, Schetter AJ, Harris CC. MicroRNAs and inflammation in the pathogenesis and progression of colon cancer. Dig Dis. 2012;30(Suppl 2):9–15. doi: 10.1159/000341882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Hernando C, Suarez Y, Rayner KJ, Moore KJ. MicroRNAs in lipid metabolism. Curr Opin Lipidol. 2011;22:86–92. doi: 10.1097/MOL.0b013e3283428d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coutinho-Camillo CM, Lourenco SV, de Araujo Lima L, Kowalski LP, Soares FA. Expression of apoptosis-regulating miRNAs and target mRNAs in oral squamous cell carcinoma. Cancer Genet. 2015;208:382–389. doi: 10.1016/j.cancergen.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 18.Yang E, Tacchelly-Benites O, Wang Z, et al. Wnt pathway activation by ADP-ribosylation. Nat Commun. 2016;7:11430. doi: 10.1038/ncomms11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 20.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 21.Yang K, Wang X, Zhang H, et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest. 2016;96:116–136. doi: 10.1038/labinvest.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Y, Han J, Xiao Z, et al. The miR-20-Rest-Wnt signaling axis regulates neural progenitor cell differentiation. Sci Rep. 2016;6:23300. doi: 10.1038/srep23300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Liu J, Cui J, et al. MiR218 modulates Wnt signaling in mouse cardiac stem cells by promoting proliferation and inhibiting differentiation through a positive feedback loop. Sci Rep. 2016;6:20968. doi: 10.1038/srep20968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan S, Yu Z, Liu Q, et al. GPC5, a novel epigenetically silenced tumor suppressor, inhibits tumor growth by suppressing Wnt/β-catenin signaling in lung adenocarcinoma. Oncogene. 2016;35:6120–6131. doi: 10.1038/onc.2016.149. [DOI] [PubMed] [Google Scholar]
- 25.Zhu L, Gao J, Huang K, et al. miR-34a screened by miRNA profiling negatively regulates Wnt/β-catenin signaling pathway in aflatoxin B1 induced hepatotoxicity. Sci Rep. 2015;5:16732. doi: 10.1038/srep16732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eckert AW, Lautner MH, Schutze A, et al. Co-expression of Hif1alpha and CAIX is associated with poor prognosis in oral squamous cell carcinoma patients. J Oral Pathol Med. 2010;39:313–317. doi: 10.1111/j.1600-0714.2009.00829.x. [DOI] [PubMed] [Google Scholar]
- 27.Yu T, Wang XY, Gong RG, et al. The expression profile of microRNAs in a model of 7,12-dimethyl-benz[a]anthrance-induced oral carcinogenesis in Syrian hamster. J Exp Clin Cancer Res. 2009;28:64. doi: 10.1186/1756-9966-28-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manikandan M, Deva Magendhra Rao AK, Arunkumar G, et al. Oral squamous cell carcinoma: microRNA expression profiling and integrative analyses for elucidation of tumourigenesis mechanism. Mol Cancer. 2016;15:28. doi: 10.1186/s12943-016-0512-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawasaki H, Mizuseki K, Nishikawa S, et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 2000;28:31–40. doi: 10.1016/s0896-6273(00)00083-0. [DOI] [PubMed] [Google Scholar]
- 30.Toda T, Homma D, Tokuoka H, et al. Birth regulates the initiation of sensory map formation through serotonin signaling. Dev Cell. 2013;27:32–46. doi: 10.1016/j.devcel.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Schrijvers D, Johnson J, Jiminez U, et al. Phase III trial of modulation of cisplatin/fluorouracil chemotherapy by interferon alfa-2b in patients with recurrent or metastatic head and neck cancer. Head and Neck Interferon Cooperative Study Group. J Clin Oncol. 1998;16:1054–1059. doi: 10.1200/JCO.1998.16.3.1054. [DOI] [PubMed] [Google Scholar]
- 32.Cheng CM, Shiah SG, Huang CC, Hsiao JR, Chang JY. Up-regulation of miR-455-5p by the TGF-beta-SMAD signalling axis promotes the proliferation of oral squamous cancer cells by targeting UBE2B. J Pathol. 2016;240:38–49. doi: 10.1002/path.4752. [DOI] [PubMed] [Google Scholar]
- 33.Irani S. miRNAs signature in head and neck squamous cell carcinoma metastasis: a literature review. J Dent (Shiraz) 2016;17:71–83. [PMC free article] [PubMed] [Google Scholar]
- 34.Hou YY, You JJ, Yang CM, et al. Aberrant DNA hypomethylation of miR-196b contributes to migration and invasion of oral cancer. Oncol Lett. 2016;11:4013–4021. doi: 10.3892/ol.2016.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu XY, Liu ZJ, He H, et al. MicroRNA-101-3p suppresses cell proliferation, invasion and enhances chemotherapeutic sensitivity in salivary gland adenoid cystic carcinoma by targeting Pim-1. Am J Cancer Res. 2015;5:3015–3029. [PMC free article] [PubMed] [Google Scholar]
- 36.Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 2010;17:215–220. doi: 10.1038/cdd.2009.69. [DOI] [PubMed] [Google Scholar]
- 37.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonci D, Coppola V, Musumeci M, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–1277. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- 39.Kang W, Tong JH, Lung RW, et al. Targeting of YAP1 by microRNA-15a and microRNA-16-1 exerts tumor suppressor function in gastric adenocarcinoma. Mol Cancer. 2015;14:52. doi: 10.1186/s12943-015-0323-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogalinska M, Kilianska ZM. Targeting Bcl-2 in CLL. Curr Med Chem. 2012;19:5109–5115. doi: 10.2174/092986712803530566. [DOI] [PubMed] [Google Scholar]
- 41.Jiao LR, Frampton AE, Jacob J, et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PLoS One. 2012;7:e32068. doi: 10.1371/journal.pone.0032068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu F, Ahmed AS, Kang X, et al. MicroRNA-15b/16 attenuates vascular neointima formation by promoting the contractile phenotype of vascular smooth muscle through targeting YAP. Arterioscler Thromb Vasc Biol. 2015;35:2145–2152. doi: 10.1161/ATVBAHA.115.305748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang S, Zou X, Zhu JN, et al. Attenuation of microRNA-16 derepresses the cyclins D1, D2 and E1 to provoke cardiomyocyte hypertrophy. J Cell Mol Med. 2015;19:608–619. doi: 10.1111/jcmm.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen F, Chen L, He H, et al. Up-regulation of microRNA-16 in glioblastoma inhibits the function of endothelial cells and tumor angiogenesis by targeting Bmi-1. Anticancer Agents Med Chem. 2016;16:609–620. doi: 10.2174/1871520615666150916092251. [DOI] [PubMed] [Google Scholar]
- 45.Zimmerman ZF, Kulikauskas RM, Bomsztyk K, Moon RT, Chien AJ. Activation of Wnt/beta-catenin signaling increases apoptosis in melanoma cells treated with trail. PLoS One. 2013;8:e69593. doi: 10.1371/journal.pone.0069593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saito-Diaz K, Chen TW, Wang X, et al. The way Wnt works: components and mechanism. Growth Factors. 2013;31:1–31. doi: 10.3109/08977194.2012.752737. [DOI] [PMC free article] [PubMed] [Google Scholar]