

Abstract
Epigenetic modifications (e.g., DNA methylation or histone deacetylation) are commonly implicated in cancer chemoresistance. We previously showed that pretreating resistant MCF-7/ADR breast cancer cells with a demethylating agent (5-aza-2′-deoxycytidine; DAC) or with an inhibitor of histone deacetylase (suberoylanilide hydroxamic acid; SAHA) sensitized resistant cells to doxorubicin (DOX) treatment. However, even with increasing doses of DOX, a fraction of resistant cells remained nonresponsive to this pretreatment (~25% pretreated with DAC, ~45% with SAHA). We hypothesized that pretreating resistant cells with a combination of epigenetic drugs (DAC + SAHA) could more effectively overcome drug resistance. We postulated that delivery of epigenetic drugs encapsulated in biodegradable nanogels (NGs) would further enhance their efficacy. MCF-7/ADR cells were first treated with a single drug vs. a combination of epigenetic drugs, either as solutions or encapsulated in NGs, then subjected to DOX, either in solution or in NGs. Antiproliferative data showed that pretreatment with epigenetic drugs in NGs, then with DOX in NGs, was most effective in overcoming resistance; this treatment inhibited cell growth by >90%, even at low doses of DOX. Cell-cycle analysis showed that a major fraction of cells treated with a cocktail of epigenetic drugs + DOX, all in NG formulations, remained in the G2/M cell-cycle arrest phase for a prolonged period. The mechanism of better efficacy of epigenetic drugs in NGs could be attributed to their sustained effect. A similar strategy could be developed for other cancer cells in which drug resistance is due to epigenetic modifications.
Keywords: Cell-cycle analysis, Decitabine, Drug delivery, Nanoparticles, Polymers, Voronistat
Introduction
Aberrant epigenetic modifications, including DNA methylation, histone deacetylation, and nucleosome remodeling, are implicated in the development and progression of chemoresistance in most cancers [1]. These aberrations promote genomic instability and lead to silencing of tumor suppressor genes [2]. Further, epigenetic modifications cause reactivation of oncogenes and interference in growth regulatory and apoptotic pathways to maintain drug resistance [3, 4]. However, such epigenetic modifications are sometimes reversible. DNA methylation is promoted by DNA methyltransferase (DNMT) [5], and hence DNMT inhibitors (e.g., decitabine [DAC]) can reverse the process of DNA methylation [6]. Similarly, inhibitors of histone deacetylase (HDAC; e.g., suberoylanilide hydroxamic acid [SAHA]; generic name vorinostat) can reverse histone acetylation [3, 7].
In our previous study, we showed that pretreatment of cells with DAC or SAHA sensitized resistant breast cancer cells (MCF-7/ADR) to subsequent treatment with DOX [3]. Despite strong synergistic effects of this combination treatment, we found that some resistant cells remained nonresponsive to treatment. Of cells receiving DAC pretreatment, ~25% cells survived, whereas with SAHA pretreatment, ~45% cells survived, despite increasingly higher doses of DOX [3]. These results suggest a heterogeneous epigenetic profile of the resistant cell population. Pretreatment with DAC alone may not reactivate the expression of genes silenced by chromatin compaction caused by histone modifications; similarly, pretreatment with SAHA alone may not reactivate genes silenced due to DNA hypermethylation. In addition, most anticancer drugs act via one or more pathways that become inactivated because of epigenetic modifications [8, 9]. Based on the above rationale, we hypothesized that pretreatment with a combination of epigenetic drugs (DAC + SAHA) could be more synergistic than pretreatment with a single epigenetic drug (DAC or SAHA alone) in overcoming chemoresistance. Because epigenetic drugs are highly unstable in aqueous conditions, we tested their efficacy by first encapsulating them into sustained-release biodegradable nanogels (NGs). DOX was also encapsulated in NGs and evaluated for its efficacy, either alone and in combination with epigenetic drugs.
Materials and methods
Materials
DAC (5-aza-2′-deoxycytidine; generic name decitabine) and SAHA (generic name vorinostat), N-isopropylacrylamide (NIPAM), n-hexane, benzene, vinyl pyrrolidone (VP), sodium dodecyl sulfate (SDS), N,N’-cystamine bis-acrylamide (an S-S crosslinker), ammonium persulfate (APS), maleic anhydride, and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). Poly(ethylene glycol) (PEG; MW ~5000) was purchased from Polysciences, Inc. (Warrington, PA). Doxorubicin HCl (DOX) was purchased from Drug Source Co. LLC (subsidiary of Celgene Corp.; Westchester, IL). Cell-culture media, Dulbecco’s phosphate-buffered saline (DPBS), penicillin, and streptomycin were purchased from the Central Cell Services Media Laboratory at our institution. The CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) was purchased from Promega (Madison, WI). All organic solvents used were of high-performance liquid chromatography (HPLC) grade. HPLC columns and guard columns were purchased from Waters Corp. (Milford, MA).
Cell Culture
DOX resistant, MCF-7/ADR cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 15% fetal bovine serum (Gibco BRL, Grand Island, NY) and 100 μg/mL penicillin G and 100 μg/mL streptomycin and 100 ng/mL of DOX (to maintain resistance). Prior to any experiment, MCF-7/ADR cells were maintained in drug-free media for two passages. MCF-7 (DOX-sensitive) cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and antibiotics. Cells were cultured in an incubator maintained at 37 °C in a humidified and 5% CO2 atmosphere. Culture conditions for resistant and sensitive cells were optimized for proper growth.
NG Synthesis
NGs were synthesized as described in our previous study [10]. Briefly, NIPAM and VP were purified just before polymerization. NIPAM was purified by recrystallization from n-hexane:benzene (1:3, v/v). VP was distilled just before polymerization. All other chemicals such as N,N’-cystamine-bis-acrylamide (S-S cross linker), SDS, and APS were used without further purification. Pegylated maleic anhydride was prepared by reacting an equimolar ratio of poly(ethylene glycol) (MW ~5000) and maleic anhydride at an elevated temperature, as reported elsewhere [11].
To prepare a batch of 1 g NGs, typically 700 mg NIPAM, 200 mg SDS, 200 mg VP, and 100 mg PEG-maleic anhydride were dissolved in 100 mL Milli-Q® water (Millipore Corp., Bedford, MA), and the polymerization was conducted in a three-necked flask under nitrogen flow at 70 °C for 6 h. The S-S crosslinker (60 mg in 2 mL methanol) followed by APS (80 mg in 5 mL Milli-Q water) was added to initiate the reaction. The reaction was continued, as noted above, for 6 h at 70 °C to form NGs. The reaction mixture was dialyzed against Milli-Q water (2 L) using Spectra/Por® dialysis tubing (MW cutoff, 12 kD; Spectrum® [Repligen], Laguna Hills, CA) for 2 weeks by changing water every day. The suspension of NGs collected from the dialysis tubing was lyophilized for 48 h at −55 °C, 3.5 Pa, using FreeZone 4.5 (Labconco Corp., Kansas City, MO) to obtain a dry powder.
Drug Loading into NGs
A suspension of NGs was prepared by adding 6 mL of Milli-Q water to 30 mg of lyophilized NGs in a 14.8-mL clean glass vial (Fisher Scientific, Pittsburgh, PA). The content was stirred slowly on a magnetic stir plate at room temperature until NGs formed a dispersion.
Methods for loading of each drug were optimized. For DAC loading, to an NG dispersion (30 mg/6 mL), DAC solution prepared in DMSO (300 μL, 8.1 mg/mL) was added dropwise, the vial cap was closed, and the dispersion was stirred for 3 h on a magnetic stirrer plate at 1200 rpm in a cold room maintained at 4 °C. This stirring in the cold room was carried out because of the instability of DAC at room temperature. For SAHA loading, to a NG dispersion (40 mg/8 mL) which was pre-chilled by keeping it in −20 °C for 5 min, a solution of SAHA (400 μL, 10 mg/mL in DMSO) was added; then pre-chilled methanol (1600 μL) was added dropwise over 30 min. The addition of methanol was shown to increase SAHA loading in NGs. The drug-NG suspension was stirred on a magnetic stirrer at 1200 rpm with the vial cap closed for 4 h in a cold room. The vial was uncapped and the drug-NG suspension was further stirred under a fume hood (air flow rate ~200 fpm) as above for an additional 2 h to evaporate the methanol. For DOX loading, to the NG dispersion (5 mg/ml, 8 mL Milli-Q water), 400 μL of DOX solution (10 mg/mL in DMSO) was added dropwise, and the DOX-NG suspension was stirred at 1200 rpm for 4 h in a glass vial with the cap closed. Following stirring, in each case, the NG dispersion was dialyzed in a cold room against Milli-Q water in dialysis tubing (MW cutoff, 12 kD, Spectrum®) for 1 h (4 h dialysis for SAHA) to remove any free drug. The content of the dialysis tubing was lyophilized as above and stored at −20 °C until used.
While loading, drug is dissolved in DMSO and added dropwise with stirring to NG suspension in water. In case of SAHA, DMSO and methanol are used as it is more soluble in the combination. DMSO or DMSO + methanol allows NGs to swell, allowing drug to diffuse into NGs and bind to polymer network via hydrophobic interaction. When DMSO is dialyzed out rapidly (methanol is evaporated in case of SAHA), bound drug is retained inside NGs.
Analysis of Drug Loading into NGs
To a borosilicate glass vial containing drug-loaded NGs, methanol (~1 mg NG/2 mL methanol) was added and stirred at 100 rpm on a magnetic stirrer in a cold room overnight (12–14 h). The samples were centrifuged at 14,000 rpm (Eppendorf 5417 R, Hauppauge, NY) for 10 min at 4 °C, and the supernatants were analyzed by HPLC (Shimadzu Scientific Instruments, Inc., Columbia, MD). The HPLC conditions were as follows:
For DAC analysis:
Column: C18 reversed-phase column, dimension: 4.6 × 250 mm; porosity 5 μm (Atlantis T3 Waters Corp., Milford, MA); Mobile phase: Sterile degassed methanol: water (60:40, v/v); Injection volume: 25 μL; Flow rate: 1.2 mL/min, isocratic elution mode for 6 min, wavelength −228 nm, by ultraviolet detector. Standard plot: 0–200 μg/mL.
For SAHA analysis:
Column: C18 reversed-phase column (dimension 2.1 × 50 mm; porosity 3.5 μm); Mobile phase: 25% acetonitrile; 75% water in the presence of 0.1% triethylamine, pH = 3. adjusted with orthophosphoric acid. Injection volume: 5 μL; Flow rate: 0.35 mL/min, isocratic elution mode for 6 min. The drug was detected at 260 nm using an ultraviolet detector. Standard plot: 0.97–500 μg/mL.
For DOX analysis:
Column: Nova-Pak C8 column (Waters; dimension: 2.1 × 150 mm, porosity 4 μm); Mobile phase: 25% acetonitrile; 75% water in the presence of 0.1% triethylamine pH-3 adjusted with orthophosphoric acid. Injection volume: 20 μL; Flow rate: 1.0 mL/min, isocratic elution mode for 6 min. Detector: fluorescence detector wavelength 480–560 nm (fluorescence gain 4). Standard plot: 0–160 ng/mL.
Physical Characterization of NGs
Briefly, each lyophilized NG sample (5 mg/mL) was dispersed in Milli-Q water and then diluted to 1:100 v/v in Milli-Q water prior to size determination. The hydrodynamic diameter of NGs pre- and post-loading with drug was measured in water by dynamic light scattering at a scattering angle of 90° at 25 °C using the NICOMP™ 380 ZLS (Particle Sizing Systems, Santa Barbara, CA). The undiluted NG suspension was used to measure the zeta potential in the phase-analysis mode and the current mode at a scattering angle of −14°.
DOX Release from NGs
DOX release from NGs in vitro was carried out in double diffusion chambers separated by a Millipore® hydrophilic membrane of 0.05 μm porosity. Each donor chamber was filled with 2.5 mL of NG suspension in a mannitol citrate buffer (recipe: Milli-Q water, 500 mL; D-mannitol,30.65 g; and sodium citrate tribasic dihydrate, 195 mg; pH 7.4) containing 0.1% (w/v) Tween-80 (Sigma); the receiver chamber contained the buffer without NGs. Diffusion cells were placed on an Environ orbital shaker (LabLine, Melrose Park, IL) rotating at 100 rpm at 37 °C. Diffusion cells were covered with an aluminium foil to prevent DOX degradation due to exposure to light. At several predetermined time intervals, the entire content of each receiver chamber was removed and replaced with fresh buffer. The contents of the receiver chambers at each time point were collected and lyophilized (as stated under the NG synthesis section above). Under the same condition, transport of DOX in solution was carried out to ensure that the memebrane placed between the donor and receiver chambers is not a transport barrier. After the transport experiment, total DOX recovered was measure to determine if there is any loss of DOX due to its binding to diffusion cells or membrane. It has to be noted that the release study was carried out in a buffer (no DMSO or methanol was used), hence the conditions used for drug loading and release study are different. DOX from the release study was extracted with methanol and quantified by HPLC using the protocol described above. A standard plot of DOX in solution (0–2 μg/mL; R2 = 0.993) was prepared under identical conditions, i.e., by dissolving the drug in Tween-80 solution, lyophilizing the samples, and extracting the drug as detailed above. The release of DAC or SAHA could not be carried out because of their instability in an aqueous medium at 37 °C.
Antiproliferative Effect of Treatments
The synergistic effects of the combination of epigenetic drugs (SAHA + DAC) or the combination of epigenetic drugs with DOX were assessed using an in vitro cytotoxicity (MTS) assay of both drug-resistant and drug-sensitive breast cancer cells. Drug solutions and dispersions of drug-loaded NGs were prepared as follows: DAC solution (8.1 mg/mL in DMSO); SAHA solution (10 mg/mL in DMSO) or DOX solution (3.8 mg/mL in ethanol: Milli-Q water; 2:1, v/v). The dispersions of drug-loaded NGs were prepared at 5 mg/mL in cell-culture medium. Prior to treatment, the stock drug solutions or NG dispersions were mixed in cell-culture medium to achieve the desired doses. Controls were either medium without drug or medium containing empty NGs.
Prior to evaluating the effects of different treatments, half maximal inhibitory concentration (IC50) values of each epigenetic drug and DOX either in solutions or in the respective NG formulations were assessed at 5 or 10 days post treatment. Briefly, resistant cells in logarithmic growth phase were seeded at a density of 3000 cells/0.1 mL/well in 96-well flat-bottom plates (Microtest, Becton Dickinson Labware, Franklin Lakes, NJ) and were allowed to adhere overnight prior to treatments. Cell-culture media from wells was replaced with the media containing either drug(s) in solution or in NGs and allowed to culture for 72 h in a cell-culture incubator maintained at 37 °C in a humidified 5% CO2 atmosphere. The cells were then washed with 1×DPBS, and the medium in the plates was replaced with drug-free medium every 48 h until the endpoint (5 or 10 days). Cell viability was assessed using a standard MTS assay according to manufacturer’s instructions. Prior to adding CellTiter 96® AQueous One Solution Reagent (MTS reagent), cell-culture media in the plate was replaced with fresh media (0.1 mL) and incubated for 1 h to equilibrate; then 20 μl/well of MTS reagent was added, the plates were incubated for 2 h at 37 °C, and the color intensity was measured at 490 nm using a plate reader (BioTek Instruments, Inc., Winooski, VT). Cell viability was calculated as the percentage of treated cells’ growth vs. growth of the respective controls. For cell viability analyzed at 10 days post treatment, cells were seeded at a lower cell density (1500 cells/0.1 mL/well) than for a 5-day treatment protocol to avoid over-confluency in untreated groups. Apart from this adjustment, all other procedures followed were the same for the cell-viability analysis at both time points.
The antiproliferative effects of the treatments were calculated as the percentage of treated cells’ growth vs. growth of control cells that had received no drug treatment, using the following
where x = drug concentration, y = % cell growth as determined by MTS assay, A1 = % growth on the top plateau region of the growth curve, A2 = % growth at the bottom plateau region of the curve, x0 = inflection point of the curve, and p = slope. The data points were fit to this equation using OriginPro 8 (OriginLab Corp., Northampton, MA). IC50 was determined by using y = 50 in the above equation and calculating x using the parameters obtained after curve fitting. Similarly, IC75 and IC90 were calculated using y = 75 or 90.
Sequential or simultaneous treatment
Half of the IC50 dose as determined above for DAC or SAHA was considered a nontoxic dose because at this dose, there is insignificant cell death. For sequential treatment with the combination SAHA + DAC either in solution or in NGs, cells were exposed to a fixed nontoxic dose of SAHA (100 ng/mL; half the IC50 dose) for 24 h prior to exposing them to varying doses of DAC for 48 h. For simultaneous treatment, a fixed nontoxic dose of SAHA (100 ng/mL) and varying doses of DAC were mixed in 0.1 mL cell-culture medium and added to the wells at the same time. For all other combinations containing DOX, a fixed nontoxic dose of SAHA (100 ng/mL) and/or DAC (50 ng/mL; half the IC50 dose) was added 24 h before adding varying doses of DOX. Cell-seeding density and the protocol followed were identical to those mentioned above. Synergism/antagonism of the drug combinations were analyzed by CalcuSyn 3.0 software (BIOSOFT, Cambridge, UK), based on the analytical method of Chou and Talalay [12].
Cell-Cycle Analysis
To understand the antiproliferative efficacy of the combination treatments, cells were analyzed for cell-cycle analysis at 1, 3, 5, or 8 days post treatment, as described previously [10]. Briefly, 1 × 106 cells were seeded in a 100-mm-diameter cell-culture dish (Becton Dickinson, San Jose, CA). For 8-day treatment, the initial cell seeding was 0.5 × 106 cells. Cells were allowed to attach for 24 h prior to treatment. At different time points post treatment, cells were harvested by trypsinization and centrifuged at 1300 rpm for 3 min at 4 °C (Sorvall Legend RT centrifuge, Thermo Electron Corp., Waltham, MA). The supernatant was decanted, the cell pellet was washed twice with ice-cold 1×DPBS (pH 7.4), and then the pellet was resuspended in a solution (12.5 mg propidium iodide [Promega, Madison, WI], 250 mg sodium-citrate, and 250 μL Triton™ X-100 in 250 mL of water). The cells in the propidium iodide solution were then incubated for 2 h in the dark in a cold room and then analyzed by flow cytometry (FACScan flow cytometer, BD Biosciences, San Jose, CA). ModFit LT software (Verity Software House, Inc., Topsham, ME) was used for data analysis.
Statistical Analysis
Data are expressed as mean ± standard error of the mean (s.e.m.). Statistical analyses were performed using Student’s t test. Differences were considered significant for p < 0.05.
Results
Characterization of NGs
Hydrodynamic diameters of different formulations of NGs ranged from 228 to 293 nm, with loaded drug marginally influencing their physical characteristics. The polydispersity index of NGs increased following drug loading but remained within an acceptable range (0.1–0.2) to be considered uniformly dispersed. All NG formulations demonstrated negative zeta potentials; however, drug-encapsulated NGs showed slightly reduced negative zeta potentials compared with empty NGs. Depending on the drug, the encapsulation efficiency and hence the drug loading in NGs also varied slightly (Table 1).
Table 1.
Characterization of nanogel formulations
| Nanogel formulations | Loading efficiency(%) | Loading content(%) | DLS diameter(nm) | PI | Zeta potential (mV) |
|---|---|---|---|---|---|
| Without drug | – | – | 233 | 0.06 | −25 ± 4.0 |
| With SAHA | 45.2 ± 1.8 | 4.5 ± 0.2 | 228 | 0.2 | −10 ± 1.0 |
| With DOX | 68.5 ± 8.5 | 6.8 ± 0.9 | 293 | 0.139 | −18 ± 0.3 |
| With DAC | 79.9 ± 5.3 | 6.4 ± 0.4 | 244 | 0.11 | −19 ± 1.0 |
Data as mean ± s.e.m; n = 2 for DOX and SAHA; n = 4 for DAC. DLS, dynamic light scattering, PI, polydispersity index.
Synergistic Effects of SAHA and DAC
The dose-response study at 5 days post treatment showed that DAC in solution exerted greater antiproliferative effects than did SAHA in solution (Fig. 1a; Table 2). The sequential and simultaneous treatment of both epigenetic drugs in combination had identical effects either when the cells were treated with solution or with NGs (Fig. 1). Based on IC50 values, the epigenetic drug combination showed a stronger antiproliferative effect in NGs than in solution at 5 days post treatment (11 ng/mL in NG vs. 32 ng/mL in solution; Table 2). The 10-day study showed that the DAC in NGs had a significantly greater antiproliferative effect than DAC in solution, as evident from their respective IC50 values (3 ng/mL vs. 887 ng/mL; p < 0.05, Table 2). Similar improved efficacy was seen when SAHA alone was delivered in NGs (Table 2). Further, the effects of epigenetic drugs in NGs were sustained much longer than drugs in solution (Fig. 2b). Although the effects of the combination of DAC + SAHA were synergistic in solution, such synergistic effects were seen at lower doses when used in NGs (Fig. 2c; Table 2).
Fig. 1. Synergistic effects of combination treatment with epigenetic drugs.
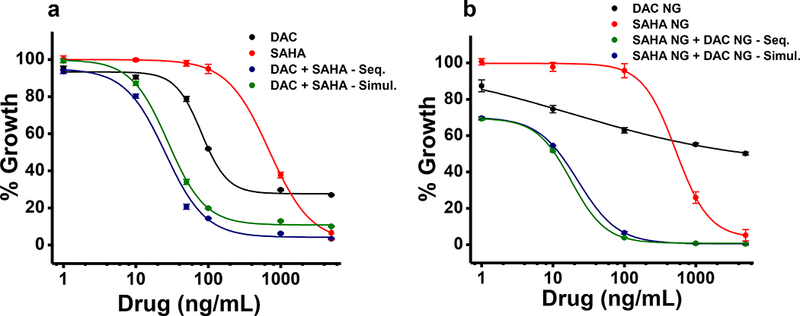
Synergistic effects on MCF-7/ADR cells of the DNA methyltransferase inhibitor DAC and the histone deacetylase inhibitor SAHA, both in solution (left) vs. loaded in NGs (right). The antiproliferative effects of SAHA and DAC were assessed as a) solution or b) loaded in NGs on cells treated sequentially or simultaneously, here after 5 days. Sequential or simultaneous treatment showed no significant differences in efficacy. Combination treatment showed greater efficacy than treatment with either drug alone. Data are expressed as mean ± s.e.m., n = 6.
Table 2.
Comparison of IC50 and combination index values of drugs post treatment in drug-resistant breast cancer cells
| Drug (alone or in combination) | Mode of delivery | IC50 (ng/mL) | Combination index values | ||
|---|---|---|---|---|---|
| 5 days | 10 days | 5 days | 10 days | ||
| DAC alone | Soln. | 105 ± 4* | 923 ± 114 | – | – |
| NG | 4312 ± 606 | 10 ± 0.8* | – | – | |
| SAHA alone | Soln. | 738 ± 45 | 4038 ± 67 | – | – |
| NG | 543 ± 76 | 379 ± 4.9* | – | – | |
| SAHA + DAC | Soln. | 32 ± 1.4 | 887 ± 135 | 0.24 ± 0.02 | 0.77 ± 0.09 |
| NG | 11 ± 0.3* | 3 ± 0.3* | 0.19 ± 0.004 | 0.21 ± 0.03* | |
Data expressed as mean ± s.e.m; n = 6.
p < 0.05 for drug solution vs. drug in nanogels.
Fig. 2. Comparison of sustained cytotoxicity of co-treatment of epigenetic drugs in solution vs. in NGs.
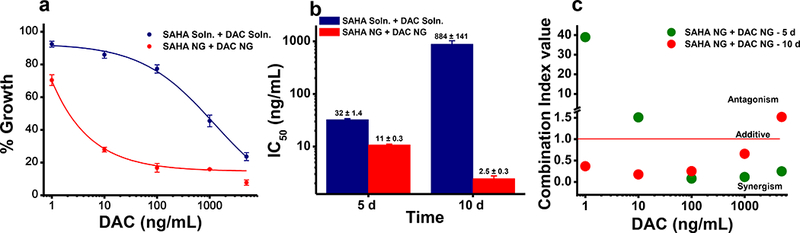
a) Co-treatment of SAHA + DAC in NGs showed greater cytotoxicity than a combination of drugs in solution at 10 days post treatment. b) Half maximal inhibitory concentration (IC50) values of the SAHA + DAC combination at different time points. Combination of drugs in NGs showed, respectively, an ~3-fold and 350-fold decrease in drug concentration required to achieve IC50 at 5 days and 10 days post treatment in comparison with a drug combination in solution. Data are expressed as mean ± s.e.m., n = 6. c) Combination index values (measure of synergism) of SAHA + DAC-loaded NGs at 5 vs. 10 days post treatment. Synergism between the drugs was calculated using CalcuSyn 3.0 software (BIOSOFT, Cambridge, UK).
Cytotoxicity of DOX-NGs
DOX release from NGs was sustained for a prolonged time, with ~ 60% of the encapsulated cumulative drug release occurring in the first 9 days and 80% cumulative release in 12 days (Fig. 3a). Complete transport of DOX from donor to receiver chamber occurred within 24 hrs. Since the DOX release study from NGs was carried over few weeks and shows sustained drug release, we consider that the membrane is not a limiting factor in the transport process (Fig. 3a). Further, while calculating the cumulative release, we considered the amount of the drug left in the donor chamber for each time. The release never reached 100%, possibly due to partial degradation of DOX in an aqueous condition at 37 °C. We also determined that ~4% loss occurs due to DOX binding to diffusion cells and membrane which may also be contributing to the incomplete release value noted above. In a dose-response study, DOX in NGs was seen to be more effective in causing cytotoxicity than DOX in solution (Fig. 3b), and the drug effects in NGs were more sustained than drug in solution (Fig. 3c). Although DOX alone in NGs was effective in achieving IC50, it was not effective in achieving IC75 or IC90 in a 10-day treatment study, even at the highest drug dose (10,000 ng/mL) used in this study (Table 3).
Fig. 3. Sustained antiproliferative effect of DOX-loaded NGs in resistant cells.
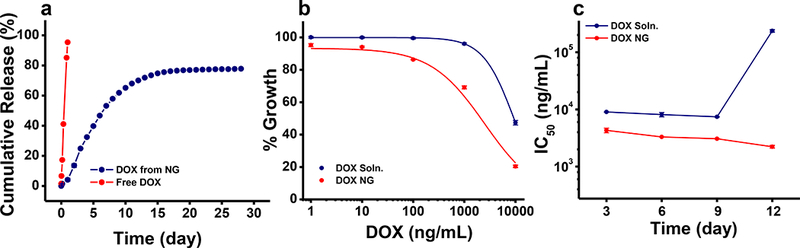
a) Release of DOX in vitro from NGs under sink conditions. Data are shown as mean ± s.e.m., n = 3. DOX transport when used in solution (not encapsulated in NGs) from donor to receiver chamber (n=1) b) Cytotoxic effects of DOX NGs vs. DOX in solution in MCF-7/ADR cells at 5 days post treatment. DOX NGs showed greater antitumor effects than DOX solution. c) Antiproliferative effects of DOX NG vs. DOX solution in MCF-7/ADR cells at different time points. DOX NG showed increased cytotoxicity and sustained effects compared with DOX solution. Data are expressed as mean ± s.e.m., n = 6.
Table 3.
Cytotoxicity of DOX in drug-resistant breast cancer cells pretreated with DAC and/or SAHA
| Drug (alone/combination) | Mode of delivery | IC50 (ng/mL) | IC75 (ng/mL) | IC90(ng/mL) | |||
|---|---|---|---|---|---|---|---|
| 5 days | 10 days | 5 days | 10 days | 5 days | 10 days | ||
| DOX alone | Soln. | 9415 ± 415 | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| NG | 2399 ± 98* | 3586 ± 116* | 7940 ± 350* | >10,000 | >10,000 | >10,000 | |
| DAC + DOX | Soln. | <1 | <1 | 4115 ± 777 | >10,000 | >10,000 | >10,000 |
| NG | <1 | 1.1 ± 0.1 | 0.4 ± 0.2* | 12 ± 0.6* | >10,000 | 186 ± 13* | |
| SAHA + DOX | Soln. | 7220 ± 89 | 2499 ± 714 | >10,000 | >10,000 | >10,000 | >10,000 |
| NG | 1504 ± 298* | 129 ± 22* | 6.3 ± 1.5* | 751 ± 59* | >10,000 | >10,000 | |
| SAHA + DAC + DOX | Soln. | <1 | 91 ±7.2 | 4546 ± 429 | >10,000 | >10,000 | >10,000 |
| NG | <1 | <1* | <1* | 7 ± 1.6* | 1722 ± 506* | 57 ± 8* | |
Data expressed as mean ± s.e.m; n = 6.
p < 0.05 for drug(s) in solution vs. drug(s) in NGs.
Efficacy of a Cocktail of Drugs
Pretreatment with a combination of epigenetic drugs (DAC [50 ng/mL] + SAHA [100 ng/mL]) in solution reduced the IC50 of DOX when used as solutions in 5- and 10-day treatment studies (Table 3); however, the treatment required high doses of DOX to achieve IC75 and was not able to achieve IC90. Furthermore, the effects of DOX in solution in combination treatment were not sustained (Table 3). In contrast, pretreatment with both epigenetic drugs in NGs followed by treatment with DOX in NGs was not only effective in achieving IC90 but this antiproliferative effect was sustained; the treatment continued to show lower IC90 values for 5- and 10-day treatment compared with all other treatments (Table 3).
Cell-Cycle Analysis
At 3 days post treatment, cells treated with DAC alone showed a greater percentage of cells in the G2/M arrest phase than control cells. Cells treated with SAHA alone showed no effects on the cell cycle; the results were the same as in untreated control cells. However, cells treated with the combination of DAC + SAHA showed a greater percentage of cells in the G2/M arrest phase than in cells treated with DAC alone. The combination of all three drugs (DAC + SAHA + DOX) in solution was seen to further increase the percentage of cells in the G2/M arrest phase (Fig. 4a). The effects of DAC, including the combination of drugs, was sustained when the agents were delivered in NGs because a greater fraction of cells remained in the G2/M arrest phase than when treated with the respective drug or drug combination in solution (Fig. 4 a vs. b). The effects of DAC alone in NGs declined over time, but the effects of the combination of DAC + SAHA or the cocktail of all three drugs in NGs was sustained (Fig. 4b). The difference between the cells treated with drugs in solution and in NG formulations is clearly evident from the 8-day cell-cycle analysis data (Fig. 4c).
Fig. 4. Efficacy of the combination treatment in inducing G2/M arrest in MCF-7/ADR cells at different time points post treatment.
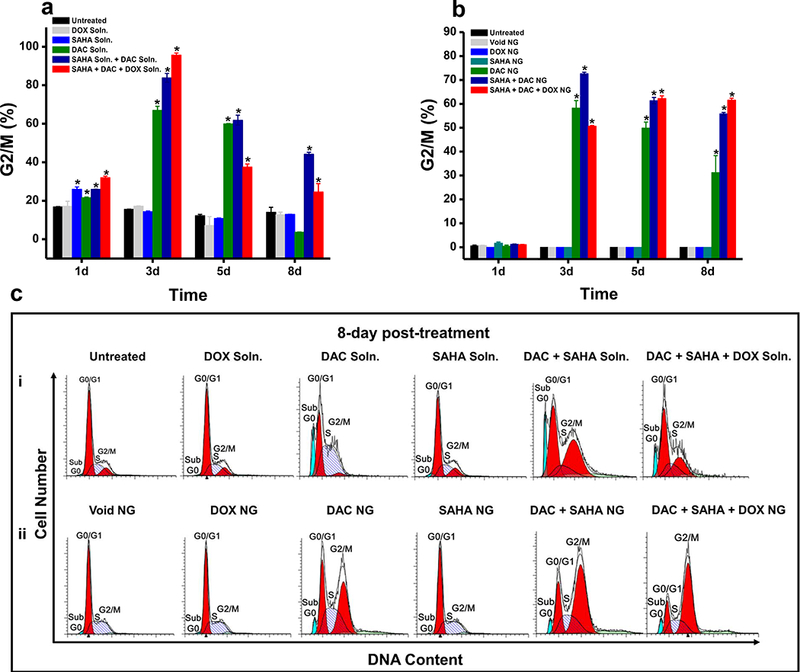
Cell-cycle analysis of MCF-7/ADR cells treated with a combination of SAHA + DAC and DOX-loaded NGs at different time points showed the following results. a) Quantification of percentage of cells in G2/M arrest phase treated with a combination of drugs in solution over time. b) Quantification of percentage of cells in G2/M arrest phase treated with a combination of drugs loaded in NGs and analyzed at different time periods. c) Representative histogram from two independent experiments showing increased G2/M arrest in cells at 8 days post treatment with all three drugs loaded in NGs (bottom) than in solution (top) or with their respective controls. *p values < 0.05 were considered statistically significant.
Effects in Sensitive Breast Cancer Cells
Although sensitive cells respond better to the drug cocktail delivered in NGs than DOX alone in NGs, the improvement was marginal compared to that seen in resistant cells (Fig. 5).
Fig. 5. Effect of sequential treatment of epigenetic drugs (SAHA + DAC NGs) and DOX NGs in DOX-sensitive breast cancer (MCF-7) cells.
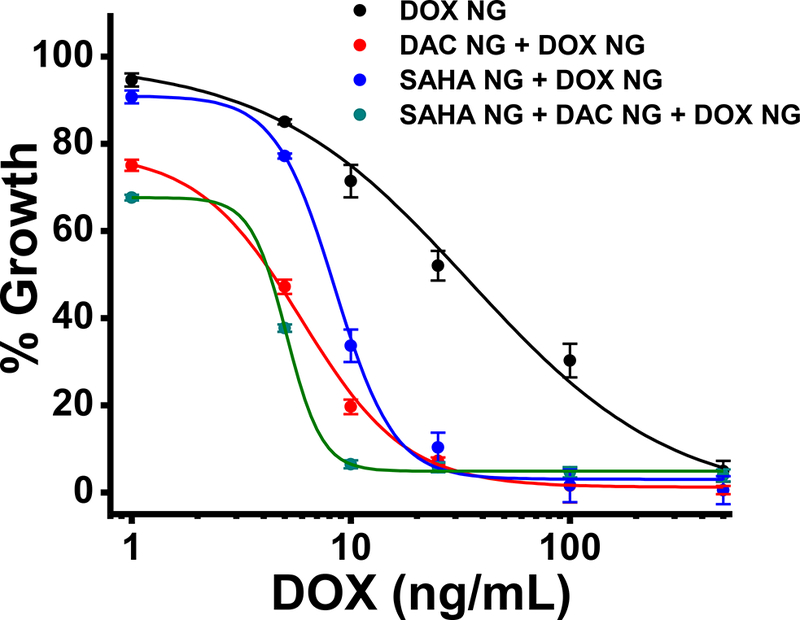
Cytotoxicity of DOX NG in cells pretreated with a combination of epigenetic drugs (SAHA + DAC) in NGs. The cocktail of drugs I n NGs showed greater antiproliferative effects than DOX alone in NGs. Data are expressed as mean ± s.e.m., n = 6.
Discussion
It is now well recognized that tumor cell populations are quite heterogeneous in nature [13], and hence monotherapy may not be effective in completely regressing tumor growth or in preventing tumors from developing drug resistance and relapsing [14]. Genetic and epigenetic variations in the cell population are implicated in the differential effects of anticancer therapy; this variability remains a major challenge in developing effective chemotherapy for cancer [15].
Aberrant epigenetic modifications in cancer cell populations could be inherent or acquired following chemotherapy [16]. It is known that epigenetic changes could influence interactions of cancer chemotherapeutic agents with different cellular targets, drug transport and efflux, drug metabolism, apoptotic pathways, etc. [17]. Thus these epigenetic changes are considered one of the key mechanisms of cancer drug resistance [17, 18]. Epigenetic changes can be of different types (e.g., DNA methylation and chromatin modifications [19]), and hence a single epigenetic drug, acting via a single mechanism, may not be effective in completely reversing epigenetic modifications responsible for drug resistance. The data from the present study clearly show that the combination of epigenetic drugs was more effective in sensitizing resistant cells to the anticancer effects of DOX than a single epigenetic drug could be (Table 2).
Several major hurdles are encountered when exploring epigenetic drugs as potential anticancer agents: the drugs’ own stability, their specificity in relation to the cell cycle, and the effect of epigenetic drugs on cell membrane lipids. Epigenetic drugs, particularly DAC and SAHA, are highly unstable in aqueous conditions (half-life of DAC in culture medium, 17.5 h [20]; of SAHA in buffers, <24 h); thus their long-term efficacy is limited [21, 22]. Prior studies have also shown the transient re-expression of methylation-silenced genes (such as p16, melanoma-associated antigen 1 [MAGE-A1], and MutL homolog 1, colon cancer, nonpolyposis type 2 [E. coli][MLH1]) post treatment with DAC in solution, even in the presence of histone deacetylase inhibitors [23–25]. Previously, we have demonstrated that encapsulating epigenetic drugs in NGs is more effective than delivering drugs in solution, particularly in the context of a long-term antiproliferative study [10]. In this study, compared to 5-day antiproliferative effects, the 10-day data with the combination of the epigenetic drugs DAC + SAHA in NGs showed greater antiproliferative effects than the same drug combination in solution (Table 2). The prolonged effects of the drug combination are also evident from cell-cycle analysis data, which showed a greater percent of cells remaining in the cell-cycle arrest phase when treated with drugs in NG than drugs in solution (Fig. 4). Even the epigenetic drugs alone loaded in NGs (without DOX treatment) were found to be effective in sustaining the cell-cycle arrest phase. Previously, we showed that DAC delivered in NGs to MCF-7/ADR cells was effective in depleting DNMT1, an enzyme responsible for DNA methylation, for a sustained period, whereas DAC in solution produced only transient effects [10]. Sustained delivery of DAC in NGs also yielded greater antiproliferative effects not only in MCF-7/ADR cells but also in DAC-resistant melanoma and leukemia cells [10].
A second issue is that epigenetic drugs are mostly S-phase specific, so their transient effects in solution may occur because not all cells are at S-phase at the time of treatment, and over time there may not be enough drug left in the culture medium (due to their rapid degradation) to cause antiproliferative effects [10]. Therefore, not all treated cells or slowly dividing cells are affected by epigenetic drug treatment. Further, it appears that the use of NGs improves drug delivery to cells. For example, DOX delivered in NGs produced greater antiproliferative effects than DOX in solution (Fig. 3b and c). The combination treatment in NGs not only prolonged the antiproliferative effects but achieved IC90 at a comparatively low dose of DOX (Table 3). Giving lower doses of DOX in the combination treatment can reduce the drug’s toxic effects. Thus, there could be several mechanisms via which the cocktail of epigenetic drugs and DOX in NGs might achieve the goal of overcoming drug resistance.
Interestingly, we have recently shown a link between epigenetic changes and membrane lipid composition [26]. MCF-7/ADR cells have been shown to have an unexpected membrane lipid profile, making the membrane more rigid than the lipid membranes of sensitive MCF-7 cells, thus impeding drug transport and endocytic function [26]. The treatment of resistant cells with DAC partially reversed this lipid profile, but the effects of DAC in NGs were more pronounced and sustained [27]. Thus, it is quite possible that the sustained NG-mediated delivery of epigenetic drugs or combinations of drugs regained endocytotic function, thus resulting in more effective delivery to cells of DOX in NGs and hence the greater cytotoxicity with pretreatment than without pretreatment with epigenetic drugs. More importantly, the cocktail/combination treatment delivered in NGs can achieve IC90, which a single epigenetic drug pretreatment cannot achieve. Furthermore, NG-mediated delivery of epigenetic drugs could potentially show better efficacy in an in vivo study because of the unstable nature of DAC and SAHA in serum due to the activity of cytidine deaminase and uridine 5′-diphosphate-glucuronosyltransferases in the liver [28, 29]. NGs can potentially stabilize the epigenetic drugs and also reduce the likelihood of their too-early clearance. NGs have the advantage as all the three drugs used in this study could be encapsulated in preformed NGs under optimized condition (Table 1), thus not requiring to develop different nanocarrier formulations for each drug.
Epigenetic drugs can reactivate silenced tumor suppressor genes. Previously, we have shown that DAC treatment activated the expression of p21, which is responsible for cell-cycle arrest [3]. In this study, we observed that epigenetic drugs, particularly in NGs, kept the cells in the G2/M arrest phase for a sustained period of time. Although SAHA alone seemed to have no effect on cell-cycle arrest, SAHA + DAC increased the percentage of cells in the G2/M arrest phase (Fig. 4). Earlier studies have shown that the addition of SAHA further increases the demethylating activity of DAC, and hence its effects are not due to gene methylation but due to increased access of the transcription factors to the demethylated genes as a result of increased levels of histone acetylation and the consequent chromatin remodeling [23]. As a result, DOX could have better access to DNA for intercalation when the cells are pretreated with epigenetic drugs than with DOX alone. Although not determined in this study, there could be other two potential mechanisms for greater efficacy with the cocktail drugs in NGs: a) cells remaining in G2/M cell-cycle phase for a prolonged period are prone to apoptosis [30] and b) there could greater accumulation of topoisomerase IIα inside cells during G2/M cell-cycle phase [31] that DOX utilizes to induce DNA damage [32, 33]. Thus there is could be multiple but complementary mechanisms via which cocktail of epigenetic drugs and DOX in sustained release NGs can overcome drug resistance.
Concluding Remarks
The results of the present study show that the combination of epigenetic drugs acts synergistically to overcome DOX drug resistance in breast cancer cells. The effects of the combination are more pronounced when the drugs are encapsulated in NGs. The improved efficacy with NG-mediated delivery could be due to a combination of factors, including greater stability of drugs, better cellular uptake, and the greater sustained effects of drugs in NGs than drugs in solution. The results of the study signify that a cocktail of drugs acting via different pathways may be required in a sustained-release formulation to overcome drug resistance. Further studies evaluating the efficacy of the combination of epigenetic drugs and chemotherapy in different drug-resistant cell lines are needed to generalize the concept. However, our study outlines a strategy for how to evaluate and determine optimal drug combinations for treatment.
Acknowledgments
This study was funded by grant R01 CA149359–01 (to VL) from the National Cancer Institute of the U.S. National Institutes of Health.
Financial support: This study was funded by grant ROI CA149359–01 (to VL) from the National Cancer Institute of the U.S. National Institutes of Health.
ABBREVIATIONS
- APS
Ammonium persulfate
- DAC
Decitabine (trade name Dacogen)
- DNMT
DNA Methyltransferase
- DOX
Doxorubicin hydrochloride
- DMSO
Dimethyl sulfoxide
- DPBS
Dulbecco’s phosphate-buffered saline
- HPLC
High-performance liquid chromatography
- IC50/75/90
Inhibitory concentration required for 50%/75%/90% cell death
- NIPAM
N-Isopropylacrylamide
- NG
Nanogel
- SAHA
Suberoylanilide hydroxamic acid (international proprietary name vorinostat)
- SDS
Sodium dodecyl sulfate
- VP
Vinyl pyrrolidone
Footnotes
Ethical consideration: This study does not involve animal use.
Conflict of interest Both authors are coinventors on pending U.S. patent applications that involve delivery of epigenetic drugs using nanogels. Potential conflict of interest matters are managed according to guidelines of the Conflict of Interest committee of Cleveland Clinic.
References
- 1.Hu Q, Baeg GH. Role of epigenome in tumorigenesis and drug resistance. Food Chem Toxicol. 2017;109(Pt 1):663–8. doi: 10.1016/j.fct.2017.07.022. [DOI] [PubMed] [Google Scholar]
- 2.Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim Biophys Acta. 2016;1865(2):275–88. doi: 10.1016/j.bbcan.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Vijayaraghavalu S, Dermawan JK, Cheriyath V, Labhasetwar V. Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm. 2013;10(1):337–52. doi: 10.1021/mp3004622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young CS, Clarke KM, Kettyle LM, Thompson A, Mills KI. Decitabine-Vorinostat combination treatment in acute myeloid leukemia activates pathways with potential for novel triple therapy. Oncotarget. 2017;8(31):51429–46. doi: 10.18632/oncotarget.18009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang W, Xu J. DNA methyltransferases and their roles in tumorigenesis. Biomark Res. 2017;5:1. doi: 10.1186/s40364-017-0081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oki Y, Aoki E, Issa JP. Decitabine--bedside to bench. Crit Rev Oncol Hematol. 2007;61(2):140–52. doi: 10.1016/j.critrevonc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Fedele P, Orlando L, Cinieri S. Targeting triple negative breast cancer with histone deacetylase inhibitors. Expert Opin Investig Drugs. 2017;26(11):1199–206. doi: 10.1080/13543784.2017.1386172. [DOI] [PubMed] [Google Scholar]
- 8.Calcagno AM, Ambudkar SV. Molecular mechanisms of drug resistance in single-step and multi-step drug-selected cancer cells. Methods Mol Biol. 2010;596:77–93. doi:10.1007/978–1-60761–416-6_ 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boettcher M, Kischkel F, Hoheisel JD. High-definition DNA méthylation profiles from breast and ovarian carcinoma cell lines with differing doxorubicin resistance. PLoS One. 2010;5(6):e11002. doi: 10.1371/journal.pone.0011002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vijayaraghavalu S, Labhasetwar V. Efficacy of decitabine-loaded nanogels in overcoming cancer drug resistance is mediated via sustained DNA methyltransferase 1 (DNMT1) depletion. Cancer Lett. 2013;331(1):122–9. doi: 10.1016/j.canlet.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan CK, Chu IM. In vitro degradation of poly(sebacic anhydride-co-ethylene glycol). Mater Chem Phys. 2004;88(1):59–66. [Google Scholar]
- 12.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 13.Sun XX, Yu Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. 2015;36(10):1219–27. doi: 10.1038/aps.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavi O, Greene JM, Levy D, Gottesman MM. The role of cell density and intratumoral heterogeneity in multidrug resistance. Cancer Res. 2013;73(24):7168–75. doi: 10.1158/0008-5472.CAN-13–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones HG, Jenkins G, Williams N, Griffiths P, Chambers P, Beynon J et al. Genetic and Epigenetic Intra-tumour Heterogeneity in Colorectal Cancer. World J Surg. 2017;41(5):1375– 83. doi: 10.1007/s00268-016-3860-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calcagno AM, Fostel JM, To KK, Salcido CD, Martin SE, Chewning KJ et al. Single-step doxorubicin-selected cancer cells overexpress the ABCG2 drug transporter through epigenetic changes. Br J Cancer. 2008;98(9):1515–24. doi: 10.1038/sj.bjc.6604334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer. 2014;14(11):747–53. doi: 10.1038/nrc3819. [DOI] [PubMed] [Google Scholar]
- 18.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54(5):716–27. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biswas S, Rao CM. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol Ther. 2017;173:118–34. doi: 10.1016/j.pharmthera.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Covey JM, Zaharko DS. Effects of dose and duration of exposure on 5-aza-2’-deoxycytidine cytotoxicity for L1210 leukemia in vitro. Cancer Treat Rep. 1984;68(12):1475–81. [PubMed] [Google Scholar]
- 21.Rogstad DK, Herring JL, Theruvathu JA, Burdzy A, Perry CC, Neidigh JW et al. Chemical decomposition of 5-aza-2’-deoxycytidine (Decitabine): kinetic analyses and identification of products by NMR, HPLC, and mass spectrometry. Chem Res Toxicol. 2009;22(6):1194–204. doi: 10.1021/tx900131u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, Shaw PG, Davidson NE. Inhibition of histone deacetylases. Methods Mol Biol. 2011;791:297–311. doi: 10.1007/978-1-61779-316-5_22. [DOI] [PubMed] [Google Scholar]
- 23.Steele N, Finn P, Brown R, Plumb JA. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br J Cancer. 2009;100(5):758–63. doi: 10.1038/sj.bjc.6604932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egger G, Aparicio AM, Escobar SG, Jones PA. Inhibition of histone deacetylation does not block resilencing of p16 after 5-aza-2’-deoxycytidine treatment. Cancer Res. 2007;67(1):346–53. doi: 10.1158/0008-5472.CAN-06-2845. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31(2):141–9. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- 26.Vijayaraghavalu S, Peetla C, Lu S, Labhasetwar V. Epigenetic modulation of the biophysical properties of drug-resistant cell lipids to restore drug transport and endocytic functions. Mol Pharm. 2012;9(9):2730–42. doi: 10.1021/mp300281t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raghavan V, Vijayaraghavalu S, Peetla C, Yamada M, Morisada M, Labhasetwar V. Sustained Epigenetic Drug Delivery Depletes Cholesterol-Sphingomyelin Rafts from Resistant Breast Cancer Cells, Influencing Biophysical Characteristics of Membrane Lipids. Langmuir. 2015;31(42):11564–73. doi: 10.1021/acs.langmuir.5b02601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer. 2008;112(11):2341– 51. doi: 10.1002/cncr.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang SP, Ramirez J, House L, Zhang W, Mirkov S, Liu W et al. A pharmacogenetic study of vorinostat glucuronidation. Pharmacogenet Genomics. 2010;20(10):638–41. doi: 10.1097/FPC.0b013e32833e1b37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Y, Wang G, Chen Q, Lin T, Zeng Z, Luo Q et al. Intrinsic apoptotic pathway and G2/M cell cycle arrest involved in tubeimoside I-induced EC109 cell death. Chin J Cancer Res. 2013;25(3):312–21. doi: 10.3978/j.issn.1000-9604.2013.06.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melixetian MB, Beryozkina EV, Pavlenko MA, Grinchuk TM. Altered expression of DNA-topoisomerase IIalpha is associated with increased rate of spontaneous polyploidization in etoposide resistant K562 cells. Leuk Res. 2000;24(10):831–7. [DOI] [PubMed] [Google Scholar]
- 32.Walker JV, Nitiss JL. DNA topoisomerase II as a target for cancer chemotherapy. Cancer Invest. 2002;20(4):570–89. [DOI] [PubMed] [Google Scholar]
- 33.Quan ZW, Yue JN, Li JY, Qin YY, Guo RS, Li SG. Somatostatin elevates topoisomerase II alpha and enhances the cytotoxic effect of doxorubicin on gallbladder cancer cells. Chemotherapy. 2008;54(6):431–7. doi: 10.1159/000158662. [DOI] [PubMed] [Google Scholar]