

Abstract
In Escherichia coli, formation of new cells is mediated by the elongasome and divisome that govern cell elongation and septation, respectively. Proper transition between these events is essential to ensure viable progeny are produced; however, the components of each complex responsible for transmission of the cell signal to shift from elongation to septation are unclear. Recently, a region within the N-terminal domain of the essential divisome protein FtsK (FtsKN) was identified that points to a key role for FtsK as a checkpoint of cell envelope remodeling during division. Here, we used site-specific in vivo UV cross-linking to probe the periplasmic loops of FtsKN for protein interaction partners critical for FtsKN function. Mass spectrometry analysis of five unique FtsKN periplasmic cross-links revealed a network of potential FtsKN interactors, one of which included the septal peptidoglycan binding protein rare lipoprotein A (RlpA). This protein was further verified as a novel interaction partner of FtsKN by an in vitro pull-down assay. Deletion of rlpA from an FtsK temperature-sensitive E. coli strain partially restored cell growth and largely suppressed cellular filamentation compared to the wild-type strain. This suggests that interaction with RlpA may be critical in suppressing septation until proper assembly of the divisome.
Introduction
In Gram-negative, rod-shaped bacteria, the interplay between cell growth and division is highly coordinated. As with most Gram-negative bacteria, the cell envelope of Escherichia coli contains three layers; a thin peptidoglycan layer enclosed within a periplasmic space by two structurally distinct cell membranes1. Together, these layers form an essential, selectively permeable barrier to the external environment. Based on the indispensable nature of the cell envelope, the precise, simultaneous modification and rearrangement of all three layers during growth and division is necessary to ensure strict maintenance of its barrier function at all stages.
Through the combined action of two large macromolecular protein complexes, cells undergo two broad morphological changes during division. In E. coli, lateral insertion of peptidoglycan along the long axis of the cell is facilitated by the elongasome2,3. Following sufficient elongation, invagination of the cell envelope is driven by the divisome complex4,5. Collectively, the elongasome and divisome are made up of over 30 essential and non-essential proteins2,6. Each complex is composed of a membrane-anchored protein scaffold and associated peptidoglycan synthesis enzymes. During elongation, positioning of the elongasome peptidoglycan synthesis machinery is driven by MreB, a homolog of the eukaryotic protein actin7–10. Similarly, the septal peptidoglycan synthesis machinery is anchored at mid-cell by the Z-ring, which is mainly composed of the eukaryotic tubulin homolog FtsZ10–13.
Although many parallels exist between the functions and general organization of each complex2,3, it is still unclear how the elongasome and divisome interact to integrate essential information regarding the growth state of the cell. In particular, it is unknown how the switch from dispersed lateral peptidoglycan synthesis to concentrated synthesis at the new cell poles and invagination of the cell envelope during septation occurs. The prevailing theory is that transduction of a cell signal from the peptidoglycan synthesis machinery of the elongasome to the divisome in the periplasm is then transmitted by at least one transmembrane protein to the Z-ring in the cytoplasm to initiate constriction4,14–18. Co-localization and protein interaction analysis of several elongasome and divisome proteins indicates that these complexes are simultaneously present at mid-cell for approximately 40% of the cell division cycle, and that the peptidoglycan synthesis enzymes from each complex physically interact prior to visible cell constriction19. This organization would permit transduction of the signal between the peptidoglycan synthesis proteins in the periplasm (namely penicillin binding protein 2 [PBP2] of the elongasome and FtsI [PBP3] of the divisome), but would not account for constriction initiation by the Z-ring. In the absence of proper coordination, cells lose the ability to effectively replicate their cellular material, resulting in the inability to produce viable progeny. As such, elucidating the protein-protein interactions formed between these essential complexes is critical to understand how cells transmit information during division and ensure survival.
An intriguing candidate to mediate the transmission of this critical cell signal is the essential cell division protein FtsK. FtsK is a large, multi-spanning membrane protein that is composed of 1329 amino acids and has a molecular weight of approximately 147 kDa20,21. It belongs to the large, well-conserved SpoIIIE family of proteins that facilitates double-stranded DNA (dsDNA) translocation via its C-terminal domain during division and sporulation in E. coli and Bacillus subtilis, respectively22,23. In E. coli, the essential N-terminal, membrane anchor domain of FtsK (FtsKN) is involved in septation21,24,25, although the precise role of this domain is unclear.
Based on the bifunctional nature of FtsK, it has been suggested that it may serve as a critical checkpoint of cell division in E. coli26–30. However, until recently, this checkpoint function was thought to merely signal completion of chromosome segregation prior to septation. With recent evidence from our laboratory regarding a novel functional periplasmic loop of FtsKN, we proposed a revised role for FtsKN during division that involves the regulation of cell envelope remodeling events25. The uncoupling of cytoplasmic constriction from peptidoglycan and outer membrane invagination caused by mutation of periplasmic residues of FtsKN suggests that FtsKN might link the Z-ring to the periplasmic peptidoglycan synthesis machinery. While sufficient biochemical evidence suggests that FtsKN interacts with the Z-ring during division27,31–33, interaction between FtsK and proteins involved in peptidoglycan synthesis (e.g., FtsI) has not been unequivocally verified27,31,32.
In the following study, the periplasmic loops of FtsKN were probed by in vivo site-specific incorporation of an unnatural photoactivatable cross-linking residue to identify novel protein interaction partners of FtsK that may contribute to its proposed checkpoint function. Interestingly, an outer membrane lipoprotein of unknown function in E. coli, rare lipoprotein A (RlpA), was identified in all cross-linked samples within the functional periplasmic loop of FtsKN (i.e., residues D135, D136 and Y139). RlpA is one of four SPOR-domain containing proteins in E. coli (including FtsN, DedA and DamX) that bind peptidoglycan and are targeted to the septum during division34–36. Recently, RlpA was shown to function as a lytic transglycosylase in Pseudomonas aeruginosa37. Deletion of rlpA in P. aeruginosa caused slow growth and chaining of cells when grown in a low osmotic strength medium, suggesting a role for RlpA in cell-cell separation and rod shape maintenance37. Despite considerable effort by several groups to determine the function of RlpA in E. coli, null mutants of rlpA in this species have yielded no morphological defects, nor has purified E. coli RlpA shown any enzymatic activity towards peptidoglycan34,35,37. Here, we show that E. coli RlpA directly interacts with divisome protein FtsKN in vitro, and that deletion of rlpA partially bypasses the requirement for functional FtsK, as seen by growth and morphological analysis of an rlpA knockout strain.
Results
In vivo UV cross-linking approach
To identify FtsKN periplasmic interaction partners, a site-specific in vivo UV cross-linking approach was used. This technique has been successfully used in E. coli to probe various protein interaction surfaces, including mapping of the SecA dimer interface and its interaction with the Sec translocon38,39, transmembrane translocation by the Tat-pathway40, capsular polysaccharide export by Wza41 and formation of the essential divisome sub-complex FtsQ/B/L42. An exogenous photoactivatable amino acid, p-benzoyl-l-phenylalanine (pBpa), is incorporated into the amino acid sequence of FtsKN by a mutant aminoacyl-tRNA synthetase/tRNA pair derived from Methanococcus jannaschii43,44. This suppresses an amber codon (TAG) engineered within the sequence of ftsKN by incorporating the pBpa residue and generates a photo-modified protein variant (FtsKN*). When cells expressing the photo-modified variants are exposed to long wavelength UV light, a site-specific covalent link between FtsKN* and any interacting protein within 3.1 Å is formed45. Given that residues in the second periplasmic loop of FtsKN were previously found to be critical for function25, two functionally important residues (D135 and D136) were chosen for substitution with pBpa. Further, three additional residues (W51, Y139 and L158) were also chosen for substitution (Supplementary Fig. S1). Each position was selected to spread coverage over the periplasmic loops of FtsKN and verify the presence of additional protein binding sites at locations distinct from the functional region identified previously25. Large/aromatic amino acids were preferentially chosen for replacement to minimize disruption of protein contacts by incorporation of the bulky pBpa residue.
The goal of our cross-linking strategy was to capture endogenous interacting proteins. It was therefore critical to achieve near native expression levels of each FtsKN* variant. Using a highly tunable anhydrotetracycline (ATc)-inducible expression system41, expression of each FtsKN* variant in the presence and absence of pBpa was assessed by SDS-PAGE and Western blotting. This also verified production of full-length FtsKN. Full-length FtsKN could be detected in all samples only when pBpa was added to the culture medium (Fig. 1). In the absence of pBpa, truncated versions of FtsKN could be detected (with the exception of W51*), highlighting the specificity of the mutant tRNA/tRNA synthetase pair and proper incorporation of the amber stop codon into the sequence of ftsKN. All FtsKN constructs were only detectable in long exposures of the Western blot (5 min), indicating very low levels of expression were achieved.
Figure 1.

Growth of FtsKN amber mutants (FtsKN*) in the presence of pBpa restores expression of full-length FtsKN. Western blot analysis of FtsKN amber mutant expression. Temperature-sensitive E. coli strain LP11-1 (ftsK44) carrying wild-type (WT) FtsKN or each amber mutant was cultured at 42 °C for 2 h with (+) or without (−) 1 mM pBpa. Concentrated whole cell lysates were separated by SDS-PAGE, blotted onto a nitrocellulose membrane, and subsequently probed with mouse anti-His6 antibodies, followed by horseradish peroxidase-conjugated goat anti-mouse IgG. tRNA and pWQ743 represent control cells harbouring the mutant tRNA/tRNA synthetase and empty vector, respectively. Asterisks (*) indicate the presence of truncated FtsKN in cultures without pBpa. Samples detected on separate blots are delineated by the dotted lines, and all were developed using the parameters outlined in the ‘Methods’ section. The representative molecular weight ladder was captured by taking a reference image of the undeveloped blots.
Initially, we attempted to identify FtsKN periplasmic interaction partners by affinity-purifying FtsKN cross-linked products, separating the adducts by SDS-PAGE and identifying the resulting protein complex by mass spectrometry following in-gel digestion of the appropriate protein (as verified by Western blotting). However, given the low-level expression of the FtsKN* variants and limited abundance (50 to 300 copies per cell) of most endogenous cell division proteins4,46,47, repeated attempts using this process resulted in unreliable identification of adducts containing FtsKN. Therefore, we decided to use a more sensitive approach consisting of in-solution digestion of purified FtsKN cross-linked products followed by liquid chromatography tandem-mass spectrometry (LC-MS/MS) identification of the total FtsKN periplasmic interactome. This approach allowed us to identify a diverse interaction network of potential FtsKN interaction partners, as described below.
In vivo UV cross-linking reveals network of FtsKN periplasmic interaction partners
Whole cells expressing each of the five FtsKN* variants were irradiated with long wavelength UV light to capture endogenous protein interaction partners in vivo. The irradiated cells were lysed, and the protein complexes were purified from the membrane fraction under stringent conditions by immobilized metal affinity chromatography (IMAC). FtsKN and corresponding periplasmic interaction partners in each sample were then identified by LC-MS/MS. In total, 63 different proteins were identified, distributed across all five cross-linked samples (minimum 2 unique peptides; Fig. 2, Supplementary Table S1). All proteins were detected in UV-treated samples only and were absent from untreated and WT controls. Of the proteins identified, 92% were found in at least one cross-linked sample at or near the functional loop of FtsKN (residues D135, D136 or Y139), suggesting this region may be critical for the maintenance of multiple protein contacts during growth and division.
Figure 2.
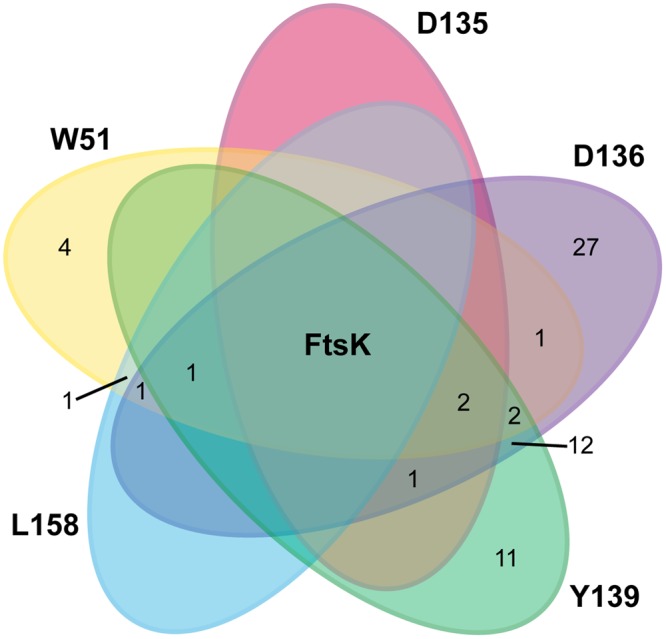
Distribution of protein groups detected by mass spectrometry of potential FtsKN interaction partners. FtsKN* variants W51*, D135*, D136*, Y139* and L158* were purified from cultures treated with (+) and without (−) UV light, and digested with chymotrypsin. The resulting peptides were analysed by LC-MS/MS on a Q-Exactive Orbitrap mass spectrometer (ThermoFisher) to detect cross-linked proteins. Numbers indicate proteins exclusively detected in UV-treated samples (absent in untreated and WT controls) and identified with a minimum of 2 unique peptides.
Our network of potential FtsKN interaction partners includes multiple protein groups of highly related function, including cell division proteins FtsN and DamX, and elongasome protein PBP1a (Fig. 3, Supplementary Table S1). Interestingly, a large proportion of the proteins detected are involved in cell wall organization and modification. As shown previously, disruption of the functional periplasmic loop of FtsKN leads to uncoupling of septation events, and improper invagination of the peptidoglycan and outer membrane layers of the cell envelope25. This suggests that the function of FtsK in division might be to couple cell envelope septation events and transition the cell from elongation to septation. Therefore, to carry out its role as this essential checkpoint, it stands to reason that the periplasmic loops of FtsKN would be in direct contact with the peptidoglycan synthesis machinery.
Figure 3.

STRING protein interaction network of potential FtsKN interaction partners identified by mass spectrometry. Proteins shown were identified exclusively in UV treated samples of FtsKN* variants W51*, D135*, D136*, Y139* and L158* (absent in untreated and WT controls). Interactions were determined using STRING 10.065. Lines indicate known or predicted protein-protein interactions, with thicker lines indicating higher levels of confidence (minimum interaction score, 0.4). Models of known protein structures are shown inside the corresponding protein spheres. Blue text highlights proteins involved in elongation and cell division, red text indicates proteins involved in general peptidoglycan modification. FtsK is circled and highlighted in yellow.
The use of mass spectrometry to identify purified protein complexes has notable limitations with respect to the specificity and sensitivity of the data provided. While affinity chromatography affords the ability to purify protein complexes that contain FtsKN, it cannot guarantee complete removal of other proteins or native complexes in tight association with the FtsKN cross-linked adduct. In conjunction with the attomolar sensitivity common with mass spectrometry analysis48, identification of even the smallest amount of contaminating proteins is unavoidable. Taken together with such a diverse network of proteins isolated, it is unlikely that all 63 proteins identified truly interact with FtsK in vivo. As such, further biochemical and in vivo verification of the potential FtsKN interaction partners is required.
To begin validation, we narrowed our list of candidate proteins by several criteria (Supplementary Table S1). First, proteins were ranked based on the average spectrum count and total number of unique peptides detected across all samples processed by LC-MS/MS to give a basic measure of protein abundance. While the LC-MS/MS method used was not quantitative with respect to absolute protein abundance, spectral counting, which counts and compares the number of fragment spectra identifying peptides of a given protein, can be used as a semi-quantitative, label free method for estimating protein abundance49. Ranking the protein list by the total number of unique peptides identified (i.e., the number of different amino acid sequences that are attributed to a single protein) also allowed us to roughly filter the putative FtsKN interaction partners by the confidence of identification. By this parameter, a greater number of unique peptides would denote increased confidence that the protein has been correctly identified in the sample. Second, proteins were also classified based on the total number of cross-linked samples in which they were detected. Finally, to identify partners critical for the checkpoint function of FtsKN, we focused on proteins identified in all cross-linked samples at or near the functional periplasmic region of FtsK (residues D135, D136 and Y139). Following these criteria, septal peptidoglycan binding protein RlpA was chosen for further analysis because it had the highest average spectrum count and most unique peptides among proteins detected in 4 of the 5 cross-linked samples and in all cross-linked samples at or near the functional periplasmic region of FtsK.
In vitro analysis of novel protein interaction between FtsKN and RlpA
Confirmation of direct interaction between FtsKN and RlpA was achieved by an in vitro pull-down assay using His10-FtsKN and a FLAG®-tagged soluble derivative of RlpA (FLAG-RlpA). Purified FLAG-RlpA was incubated with purified, IMAC resin-bound FtsKN and successively washed to remove unbound protein. Upon elution of His10-FtsKN, RlpA and FtsKN were detected in the elution fraction by Western blot analysis (Fig. 4A). To verify accuracy of the pull-down assay, RlpA was also incubated with IMAC resin in the absence of FtsKN as a negative control and was not detected in the elution fraction (Fig. 4B). Thus, this assay indicated direct interaction between FtsKN and RlpA. Additionally, purified FtsZ was used as a positive control of a known FtsKN interaction partner. FtsZ was successfully detected in the elution fraction at an increased proportion when FtsKN was bound to the IMAC resin (Fig. 4C, Elution), in comparison to the empty bead control (Fig. 4C, Bead), further verifying that the assay used can accurately detect the presence of a direct protein interaction with FtsKN and supports identification of a novel protein interaction between FtsKN and RlpA in vitro.
Figure 4.

In vitro analysis of interaction between FtsKN and RlpA. (A) Representative Western blots of pull-down assay between FLAG-RlpA (prey, left panel) and His10-FtsKN (bait, right panel). Purified, immobilized His10-FtsKN was incubated with 320 µg of purified FLAG-RlpA, washed to remove unbound protein and subsequently eluted to assess its interaction with RlpA by Western blot. (B) Western blot of FLAG-RlpA incubated with empty IMAC resin (negative control). (C) Representative Western blot of pull-down assay between FtsZ (prey, left panel) and His10-FtsKN (bait, right panel). Purified, immobilized His10-FtsKN was incubated with 250 µg of purified FtsZ, washed to remove unbound protein and subsequently eluted to assess its interaction with FtsZ. ‘Bead’ lane represents the elution fraction of FtsZ incubated with empty IMAC resin (negative control). For all blots, the primary antibodies are indicated in the bottom right corner. In all panels: ‘FT’ – flow through; ‘W1, W2, W3’ – wash fractions 1, 2, and 3; ‘Elution’ – elution with 1 M imidazole. Samples detected on separate blots are delineated by the dotted lines, and all were developed using the parameters outlined in the ‘Methods’ section. The representative molecular weight ladders in the anti-FLAG® blots of panels A and B were captured by taking a reference image of the undeveloped blots.
Deletion of rlpA from LP11-1 (ftsK44) partially suppresses cellular filamentation
Although recent evidence has suggested that the septal peptidoglycan binding protein RlpA is a lytic transglycosylase involved in cell-cell separation in P. aeruginosa37, the exact function of RlpA in E. coli remains elusive. While several groups have attempted to uncover the role of RlpA in E. coli through the use of gene deletion mutants and enzymatic assays to probe the impact of purified RlpA on peptidoglycan degradation34,35,37, no study to date has been able to visualize any division-related phenotype for ∆rlpA mutants nor show enzymatic activity of the protein. In E. coli, rlpA is immediately upstream of the gene encoding penicillin binding protein 5 (dacA)50. Given deletion of dacA has been previously shown to suppress the loss of functional FtsK in temperature-sensitive ftsK44 strains of E. coli20, we reasoned that a unique division-related phenotype might also be observed if rlpA was deleted in conjunction with the loss of FtsK.
Therefore, we deleted rlpA in the temperature-sensitive E. coli strain LP11-1 (ftsK44) to generate LP11-1 ∆rlpA (Supplementary Fig. S2) and assessed the impact of this deletion on cell growth and morphology. We also created an rlpA knockout in the E. coli strain W3110 to compare the impact of rlpA deletion in an otherwise WT background. In both cases, the gene deletion was created within the boundaries of the rlpA open reading frame to minimize disruption of the surrounding genes or potential promoter sequences and minimize polar effects.
To probe the impact of rlpA deletion in both the presence and absence of functional FtsK, we completed growth and morphology analysis of the LP11-1 ∆rlpA and W3110 ∆rlpA strains at both permissive (30 °C) and nonpermissive (42 °C) temperatures, respectively (Figs 5 and 6). Although the WT E. coli strain W3110 grew more rapidly and to a higher absorbance than the temperature-sensitive strain LP11-1 (p < 0.05) at both temperatures, deletion of rlpA only had a significant impact on growth in the absence of functional FtsK (i.e., in the LP11-1 background during growth at 42 °C) (Fig. 5). In this case, rlpA deletion significantly alleviated the severe growth defect caused by the loss of FtsK, and partially suppressed the cellular filamentation typically associated with the temperature-sensitive ftsK44 mutation (Fig. 6). This partial restoration of growth and WT cell morphology could be negated by complementation of the LP11-1 ∆rlpA strain with a plasmid-encoded copy of rlpA (encoded on plasmid pSG002-1), suggesting that the combined loss of functional FtsK and RlpA was responsible for the observed phenotype.
Figure 5.

Deletion of rlpA partially alleviates growth inhibition of the temperature-sensitive E. coli strain LP11-1 (ftsK44). Growth curves of ΔrlpA knockout (W3110 ΔrlpA and LP11-1 ΔrlpA) and complemented (LP11-1 ΔrlpA pSG002-1) E. coli strains grown at permissive (30 °C) and nonpermissive (42 °C) temperatures (n = 3 per strain per temperature). The A600 of two technical replicates of each culture were sampled every hour using a SmartSpec™ Plus Spectrophotometer (Bio-Rad). In both panels, E. coli strains W3110 and LP11-1 were analyzed as WT controls. Pairwise comparison of growth differences was made using a one-way analysis of variance with Tukey-Kramer multiple comparison post-tests (*p < 0.05 versus LP11-1 at the same time point).
Figure 6.

Deletion of rlpA restores normal cell morphology of the temperature-sensitive E. coli strain LP11-1 (ftsK44). (A) Representative phase contrast micrographs, (B) mean cell length, and (C) cell length distribution of ΔrlpA knockout (W3110 ΔrlpA and LP11-1 ΔrlpA) and complemented (LP11-1 ΔrlpA pSG002-1) E. coli strains grown at 42 °C. In all panels, E. coli strains W3110 and LP11-1 were analyzed as WT controls. Micrographs depict typical cells of each strain after 0 or 8 h of growth at 42 °C. The genotype of each strain is indicated in the upper left corner of each micrograph (bar, 10 µm). Cells were measured for cell length using the ImageJ program and are reported as mean cell length (µm) ± S.E. (n = 150 cells per strain per time point). Pairwise comparisons of cell lengths were made using a one-way analysis of variance with Tukey-Kramer multiple comparison post-tests (*p < 0.001).
Discussion
Despite clear evidence that FtsK is essential during division, the limited amount of biochemical data currently available regarding the protein contacts it makes, or its biological function, gives only a crude picture of its role during division. With the cross-linking and deletion mutant data presented herein, we have uncovered an FtsKN interactome that again points to a role for FtsK in cell envelope remodeling during division.
The use of site-specific cross-linking by the photoactivatable amino acid pBpa in whole cells allows us to study the putative protein complexes FtsKN forms in its natural environment, with minimal alterations to the full-length proteins involved. While this technique allowed us to identify a large number of potential FtsKN interaction partners (Fig. 2, Supplementary Table S1), limitations regarding the specificity and high sensitivity of our LC-MS/MS approach makes further biochemical investigation of each protein as a direct FtsKN interactor necessary. Regardless, the list of proteins identified in our cross-linking analysis provides further insight into the role of FtsK in cell division, as the majority of proteins detected could be grouped into several small functional groups, with a high degree of known interaction among them (Fig. 3). In particular, this study is the first to report a confirmed, direct interaction between the OM lipoprotein RlpA and an essential member of the divisome (Fig. 4). The implication of this interaction is difficult to interpret in the absence of definitive evidence on the function of RlpA in E. coli. However, general localization data in E. coli34–36 and recent biochemical evidence on the enzymatic activity of RlpA in P. aeruginosa37 highlights the potential significance of this novel interaction.
Of all SPOR-domain containing proteins, RlpA is the most highly conserved across bacterial species37. This would suggest that RlpA plays an important role in bacteria. Based on its OM localization and ability to preferentially bind septal peptidoglycan34–36, one role for RlpA could be to physically link the OM to the peptidoglycan and IM during constriction. In this capacity, the interaction between RlpA and FtsK would function similarly to the Tol-Pal interaction. In E. coli, the trans-envelope Tol-Pal complex localizes to the site of division and helps to coordinate invagination of the OM with both the peptidoglycan and IM cell envelope layers51. Intriguingly, TolB was identified as a potential FtsK interaction partner by our mass spectrometry analysis (Fig. 3, Supplementary Table S1). Cells lacking an intact Tol-Pal system show delayed OM invagination during cell constriction51, which poses an attractive link to the inhibition of OM and peptidoglycan invagination we first observed upon the expression of non-functional FtsKN variants25. Interaction between the periplasmic loops of FtsKN and the Tol-Pal complex would create a link between the IM and OM that could facilitate coordinated invagination of the entire cell envelope during division. This connection might then account for the cell void phenotype seen upon the expression of non-functional periplasmic loop mutants of FtsKN25, as disruption of this interaction would directly uncouple IM division from OM invagination. This functional redundancy might also explain why null mutants of rlpA in WT E. coli strains have no morphological defects, as the loss of the OM-IM contact made between RlpA and FtsK upon deletion of RlpA could be accommodated by an intact interaction maintained by FtsK with the Tol-Pal complex. However, this scenario does not fully account for the restoration of growth seen in the absence of both RlpA and functional FtsKN (Fig. 5), indicating RlpA must play a larger role in division beyond simply bridging all three cell envelope layers.
A second role for RlpA may be in balancing lateral growth of the cell versus septal peptidoglycan synthesis. In addition to its septal localization, RlpA is also found at distinct foci along the cell cylinder34. Together with the fact that rlpA is encoded immediately downstream of two proteins involved in cell elongation, pbpA (encoding PBP2) and rodA50, it is proposed that RlpA might function as part of the elongation machinery, in addition to the divisome. Interaction between FtsK and RlpA would then represent another link between these two macromolecular complexes, and potentially facilitate the transition between elongation and septation in E. coli. The restoration of growth seen upon the loss of both RlpA and functional FtsK suggests that their interaction may be critical in suppressing septation until sufficient elongation and proper assembly of the divisome occurs.
Perhaps the most intriguing aspect of the interaction between RlpA and FtsKN is its parallel with the functional interaction between FtsK and the carboxypeptidase DacA (PBP5). DacA catalyzes the removal of the terminal d-alanine from peptidoglycan side chains during cell wall synthesis2,52. Interestingly, DacA was also detected as a potential FtsKN interaction partner in our cross-linking analysis (Fig. 3, Supplementary Table S1). Deletion of dacA from E. coli carrying the temperature-sensitive ftsK44 allele has been shown to largely suppress filamentation of bacteria grown at a nonpermissive temperature (42 °C)20. This is in direct correlation with the phenotype we observe upon the deletion of rlpA from the same strain of E. coli (Fig. 6). In addition, and perhaps not surprisingly, rlpA also resides on the chromosome immediately upstream of dacA50. Given their proximity and evidence that RlpA is also involved in peptidoglycan remodeling37, it would not be unreasonable to estimate that the deletion of either of these genes would have a similar impact on the cell in the absence of functional FtsK. In the case of dacA, deletion of this gene is only able to compensate for the temperature-sensitive growth defect and not complete loss of FtsK53, suggesting that under these circumstances cells might be able to restore the function of the mutated FtsK44 protein rather than completely bypass its function. How this occurs is unclear, but we postulate that if DacA and FtsK form a direct protein interaction, deletion of DacA would free FtsK to form protein contacts with additional proteins that could stabilize its structure and restore its function. In our study, it would be pertinent to note that this may also be the case with the deletion of rlpA, and that further investigation into the ability of an rlpA deletion to permit the complete deletion of ftsK is necessary.
In addition to the verified interaction between FtsKN and RlpA described above, we were also able to detect several proteins involved in growth and division (e.g., divisome proteins FtsN and DamX, and elongasome protein PBP1a). These proteins represent novel potential FtsK interactors, as none of the proteins are previously reported in the literature as FtsK interaction partners. Both FtsN and DamX are SPOR-domain containing proteins that bind peptidoglycan and are targeted to the septum during division34–36. Specifically, FtsN is an essential component of the divisome that is responsible for activation of cell constriction during late stages of division54–56. In contrast, while DamX is an early recruit to the septum and interacts with several divisome proteins, it is not essential for cell division and currently has no known function34,35,57. During elongation, PBP1a acts as a bifunctional transglycosylase/transpeptidase that modifies and synthesizes the growing peptidoglycan layer58. Given limited knowledge about the functions of both FtsKN and DamX during division, the significance of an interaction between these two proteins is difficult to interpret. However, the interactions of FtsK with FtsN and PBP1a provide an intriguing link between the early and late stages of division, and the switch from cell elongation to septation in E. coli. The formation of a tripartite complex with FtsKN, FtsN, and PBP1a would physically link the elongasome with the divisome in the periplasm and allow for the direct transmission of a cell signal between these two essential complexes. Completion of cell elongation by the elongasome would be sensed through the interaction between PBP1a and FtsKN. In turn, this signal could be integrated with a signal for the completion of divisome formation and proper chromosome segregation sensed by FtsK, and then passed to FtsN through direct interaction of these two proteins to trigger constriction. While this report is the first to suggest direct interaction between FtsK and FtsN in E. coli, these proteins have been shown to interact in the Gram-negative bacterium Neisseria gonorrhoeae59. Together, these interactions shed light onto a potential mechanism of action for FtsKN in cell envelope remodeling during both growth and division.
Based on prior biochemical evidence, it is important to note that essential cell division proteins previously shown to interact with FtsK by bacterial two-hybrid analysis, including IM proteins FtsQ, FtsL, and FtsI27,31,32, were not detected in our cross-linking analysis. Given we have elected to only probe the periplasmic face of FtsKN, it is possible that these proteins interact with FtsK at locations distinct from our sites of inquiry, namely the transmembrane or cytoplasmic domains of the protein. Alternatively, it is possible that FtsK does not interact directly with these proteins, but rather they are bridged by a third interacting partner that would have confounded previous bacterial two-hybrid results. For example, FtsI has been shown to form a complex with the OM lytic transglycosylases MltA and MltB60,61; therefore, our detection of these two proteins by LC-MS/MS (Fig. 3, Supplementary Table S1) might explain formation of a tertiary complex between FtsK and FtsI bridged by these lytic transglycosylases.
As a whole, the novel periplasmic FtsKN interactome and verified interaction with RlpA described in this study provides further evidence for FtsK as a potential regulator of cell envelope remodeling during division. While the identity of the interactor responsible for the shift from cell elongation to septation remains unclear, we detected several proteins that could fulfill this role. In fact, it is most likely that a number of these proteins work in concert to accurately move the bacterium through its growth cycle. As with all of the potential FtsKN interaction partners identified, further biochemical verification of the specificity and validity of these interactions is needed to characterize each protein as a true interactor. Further investigation into the impact each interaction with FtsK has on both growth and division will undoubtedly enhance our understanding of both of these essential processes.
Methods
Bacterial strains, plasmids, and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 1. E. coli W3110, DH5α and Lemo21 cultures were grown at 37 °C in lysogeny broth (LB) (BD Biosciences) in a rotary shaker at 200 rpm. Media were supplemented with 150 µg/mL ampicillin, 30 µg/mL chloramphenicol or 50 µg/mL kanamycin where appropriate for plasmid-carrying strains. E. coli LP11-1 cultures were grown in Complementation Media (1% [w/v] tryptone, 0.5% [w/v] yeast extract, and 1% [w/v] NaCl [Fisher]) supplemented with 15 µg/mL tetracycline, as well as appropriate antibiotics for plasmid selection where applicable. Cultures were grown at 30 °C or 42 °C, as described previously25. Maintenance of the kanamycin resistance cassette in the ΔrlpA knockout strains was achieved by the addition of 25 µg/mL kanamycin to all overnight cultures. To assess the impact of deletion of rlpA on cell growth and morphology, overnight cultures of LP11-1 ΔrlpA and W3110 ΔrlpA were each diluted to an A600 of 0.1 (SmartSpec™ Plus Spectrophotometer; Bio-Rad) in two separate 25-mL aliquots of fresh Complementation Media and grown at 30 °C or 42 °C for 8 h (n = 3 per strain per temperature). The A600 of two technical replicates of each culture were sampled every hour and cells were imaged by phase contrast microscopy (Leica DM2000 LED, ProgRes CT3 camera; Jenoptik AG). Cell length was assessed for 150 random cells from each culture using the ImageJ program (version 1.46r, National Institutes of Health) and is reported as mean length ± S.E. Statistical analysis for cell growth (A600) and mean length were completed using one-way analysis of variance with Tukey-Kramer multiple comparison post-tests by Prism 5 software (GraphPad Software, Inc.) using a level of significance of α = 0.05 for all tests. E. coli K12 W3110 and LP11-1 were also grown at 30 °C and 42 °C as described above for wild-type (WT) controls. Deletion of rlpA was complemented in trans by expression of FLAG-rlpA from plasmid pSG002-1 in strain LP11-1 ΔrlpA using 80 ng/mL anhydrotetracycline (ATc).
Table 1.
Bacterial strains and plasmids.
| Strain or plasmid | Description | Source or Reference |
|---|---|---|
| E. coli strain | ||
| W3110 | rph-1IN (rrnD-rrnE) | Coli Genetic Stock Center |
| Lemo21 (DE3) | fhuA2 [lon] ompT gal (λDE3) [dcm] ΔhsdS/ pLemo(CamR) λ DE3 = λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5 pLemo = pACYC184-PrhaBAD-lysY | New England Biolabs |
| DH5α | F−Φ80lacZΔM15Δ (lacZYA-argF) U169 recA1 endA1 hsdR17 | Invitrogen |
| (rK−, mK+) phoA supE44 λ-thi-1 gyrA96 relA1 | ||
| LP11-1 | W3110 ftsK44 aroA::Tn10 | K. Young69 |
| LP11-1 ΔrlpA | W3110 ftsK44 aroA::Tn10 ΔrlpA::kan | This study |
| W3110 ΔrlpA | W3110 ΔrlpA::kan | This study |
| Plasmids | ||
| pEVOL-pBpF | Expression vector encoding tRNA/aminoacyl-tRNA synthetase pair derived from Methanococcus jannaschii for in vivo incorporation of the photo-crosslinker p-benzoyl-l-phenylalanine (pBpa) into proteins in E. coli; CamR | Addgene (no. 31190)43 |
| pKD4 | Template plasmid for λ red mediated gene deletion; KanR | 68 |
| pKD46 | Red recombinase expression plasmid under the control of Para; AmpR | 68 |
| pBAD24 | Protein expression vector under the control of Para; AmpR | Addgene |
| pWQ743 | Protein expression vector under the control of Ptet; AmpR | C. Whitfield41 |
| pET28a | Protein expression vector under the control of Plac; KanR | EMD Millipore |
| pWQ572 | Protein expression vector under the control of Ptet; CamR | C. Whitfield41 |
| pAB006-2 | pBAD24 derivative encoding amino acids 1-220 of FtsK (His10-FtsKN(220)) from E. coli | 25 |
| pAB008-1 | pWQ743 derivative encoding amino acids 1-220 of FtsK (His10-FtsKN(220)) from E. coli | This study |
| pSG001-1 | pET28a derivative encoding amino acids 18-362 of RlpA with an N-terminal FLAG-tag (FLAG-RlpA) from E. coli | This study |
| pSG002-1 | pWQ572 derivative encoding amino acids 18-362 of RlpA with an N-terminal FLAG-tag (FLAG-RlpA) from E. coli | This study |
The abbreviations used are as follows: Amp, ampicillin; Cam, chloramphenicol; Kan, kanamycin.
Plasmid construction
An oligonucleotide encoding the N-terminal 220 amino acids of FtsK (FtsKN(220)) with an N-terminal decahistidine tag was cloned into the ATc-inducible expression vector pWQ743 to produce pAB008-1, as described previously25. For over-expression and purification of RlpA, an oligonucleotide encoding a soluble derivative of RlpA containing an N-terminal FLAG®-tag in place of its type II signal sequence (FLAG-RlpA) was cloned into expression vector pET28a as described previously25, using custom primers AMB006Fa (5′-TGGACCATGGATTATAAAGATGATGATGATAAATCCAGTACAAGCGATGATGGTCAGC-3′) and AMB006R (5′-AAGGTCAAGCTTACTTTACTGCGCGGTAGTAATAAATGACTGTAATTGGGCTTC-3′) to produce pSG001-1. Similarly, for complementation of the ΔrlpA knockout strains, the oligonucleotide encoding FLAG-RlpA was also cloned into the ATc-inducible expression vector pWQ572 using the above primers, producing pSG002-1. All constructs were verified by DNA sequencing (Genomics Facility, Advanced Analysis Center, University of Guelph).
Site-directed mutagenesis
To capture FtsKN protein interaction partners, five site-specific amber stop codon variants of FtsKN (FtsKN*) were generated by site-directed mutagenesis. A single amber codon (TAG) was introduced into the sequence of ftsKN corresponding to the periplasmic amino acid positions Trp-51, Asp-135, Asp-136, Tyr-139 or Leu-158 using a QuikChange Lightning site-directed mutagenesis kit (Stratagene). All primers used for mutagenesis and resulting plasmids are shown in Table 2. All single amber codon variants were confirmed by DNA sequencing (Genomics Facility, Advanced Analysis Center, University of Guelph).
Table 2.
Oligonucleotide pairs used for site-directed mutagenesis.
| Mutation | Sequence of mutagenic oligonucleotide (5′ to 3′)a | Plasmid |
|---|---|---|
| W51* | CGGACCCCAGCTAGTCGCAAACGGC | pAB008-W51* |
| GCCGTTTGCGACTAGCTGGGGTCCG | ||
| D135* | CTGGCGGCAATCAACGCTTAGGATATCTGGTATTTTCGG | pAB008-D135* |
| GGCAAAATACCAGATATCCTAAGCGTTGATTGCCGCCAG | ||
| D136* | GCGGCAATCAACGCTGACTAGATCTGGTATTTTGCCTCC | pAB008-D136* |
| GGAGGCAAAATACCAGATCTAGTCAGCGTTGATTGCCGC | ||
| Y139* | CTGACGATATCTGGTAGTTTGCCTCCGGTGGCG | pAB008-Y139* |
| CGCCACCGGAGGCAAACTACCAGATATCGTCAG | ||
| L158* | CACTACGCTACAACCACTGTAGCACAGTAGCGGGGGAACTA | pAB008-L158* |
| TAGTTCCCCCGCTACTGTGCTACAGTGGTTGTAGCGTAGTG |
aBase changes are underlined and in boldface.
In vivo UV cross-linking
For in vivo cross-linking of FtsKN interacting proteins, the photoactivatable amino acid p-benzoyl-l-phenylalanine (pBpa) was incorporated into FtsKN by a mutant tRNA/tRNA synthetase pair from Methanococcus jannaschii that has been engineered to recognize the amber stop codon (TAG)43. Cross-linking of whole bacterial cells expressing site-specific amber stop codon variants of FtsKN (FtsKN*) in the presence of pBpa and the mutant tRNA/tRNA synthetase (encoded on plasmid pEVOL-pBpF) was performed as described by Nickerson et al.41, with minor modifications. Briefly, overnight cultures of E. coli LP11-1 strains carrying FtsKN* variants were diluted to an A600 of 0.1 (SmartSpec™ Plus Spectrophotometer; Bio-Rad) in 400 mL Complementation Media supplemented with ampicillin and chloramphenicol (75 μg/mL and 15 μg/mL, respectively), 1 mM pBpa (Bachem), as well as 0.02% (w/v) l-arabinose to induce synthesis of the tRNA/tRNA synthetase pair from pEVOL-pBpF. FtsKN* variants were induced with 40 ng/mL ATc. Cultures were incubated under reduced lighting at 42 °C for 2 h in a rotary shaker at 200 rpm. Identical cultures grown in the absence of 1 mM pBpa were prepared to assess the efficiency of pBpa incorporation into full-length FtsKN. One-millilitre aliquots of cultures grown in the presence and absence of pBpa were concentrated 1:100, mixed with 20 µL of 5 × Loading Buffer (250 mM Tris-HCl, pH 6.8, 10% [w/v] SDS, 30% [v/v] glycerol, 5% [v/v] β-mercaptoethanol, 0.1% [w/v] bromophenol blue), and boiled for 10 min in a covered beaker. Samples were analyzed by SDS-PAGE and Western blotting as described below.
To induce in vivo cross-linking of FtsKN* variants, cultures grown in the presence of pBpa were harvested by centrifugation and washed twice in 10 mL sterile PBS. The resulting cell pellets were suspended into 10 mL phosphate-buffered saline (PBS) for every A600 0.1 of the original culture (e.g., a culture with an A600 of 0.5 was suspended in 50 mL PBS) before splitting into two equal aliquots. Aliquot #1 was placed into a 6-well tissue culture plate on ice (5 mL suspension per well) and the whole cells were irradiated at 365 nm with a handheld UV lamp (Spectroline Model EN-180L; 2.5 cm from surface of the cell suspension) for 15 min. Aliquot #2 was covered in tinfoil and incubated on ice for 15 min as a non-crosslinked control. Both aliquots were harvested by centrifugation and the pellets were stored at −20 °C. All samples were protected from additional light exposure, where possible, during storage and purification of FtsKN. All samples were prepared in duplicate from biological replicate cultures. An additional culture expressing WT FtsKN(220) was harvested and processed in parallel with cross-linked and non-crosslinked control cells as described below to account for contaminant proteins inherent to the purification of FtsKN.
Cross-linked (aliquot #1) and non-crosslinked control cells (aliquot #2) were thawed in 3 mL SDS Lysis Buffer (20 mM Tris-HCl, pH 7.0, 100 mM NaCl, 1% [w/v] SDS) and sonicated on ice for 1 min (10 s on, 10 s off; Misonix Ultrasonic Processor XL2020, Misonix, Inc.) to complete cell lysis. Samples were then diluted with 27 mL Purification Buffer (20 mM Tris-HCl, pH 7.4, 300 mM NaCl, 25 mM imidazole) to reach a final SDS concentration of 0.1% (w/v). Profinity Ni2+-charged immobilized metal affinity chromatography (IMAC) resin (Bio-Rad) was added to each sample at a bed volume of 100 µL and then incubated at 4 °C for 1.5 h with rocking. Cell lysate/resin mixtures were collected by centrifugation at 4,500 × g for 5 min at 4 °C and washed five times with 1 mL of Purification Buffer containing 0.1% (w/v) SDS before performing on-bead digestion for protein analysis.
On-bead digestion and liquid chromatography tandem-mass spectrometry (LC-MS/MS)
WT FtsKN and FtsKN* from cross-linked and non-crosslinked controls were subjected to on-bead proteolytic digestion with chymotrypsin as described by Park et al.62. Briefly, resin mixtures containing FtsKN complexes were washed five times in 1 mL freshly prepared 50 mM ammonium bicarbonate (ABC) buffer (pH 8.0). Beads were then suspended in Denaturation Buffer (6 M urea/2 M thiourea in 10 mM HEPES, pH 8.0) and proteins were subsequently reduced by incubation at 50 °C for 30 min in Reduction Buffer (10 mM dithiothreitol in ABC buffer). Samples were then incubated in Alkylation Buffer (55 mM iodoacetamide in ABC buffer) for 30 min at room temperature in the dark. Reduced and alkylated proteins were diluted with ABC buffer and digested with 1 µg chymotrypsin (Princeton Separations) in the presence of 0.01% (w/v) ProteaseMAX surfactant (Promega) at 30 °C overnight. The IMAC resin was removed by centrifugation and digested peptides in the supernatant were lyophilized using a speed-vacuum concentrator. Samples were reconstituted in 1% trifluoroacetic acid in water and passed through a C18 stage tip to remove any trace detergent and salt from the purification process, and lyophilized.
To identify cross-linked proteins, samples were analyzed by liquid chromatography tandem-mass spectrometry (LC-MS/MS) on an Orbitrap analyzer (Q-Exactive, ThermoFisher) outfitted with a nanospray source and EASY-nLC nano-LC system (ThermoFisher). Lyophilized peptide mixtures were dissolved in 0.1% formic acid and loaded onto a 75 µm × 50 cm PepMax RSLC EASY-Spray column filled with 2 µm C18 beads (ThermoFisher) at a pressure of 800 Bar heated to 60 °C. Peptides were eluted over 90 min at a rate of 250 nL/min using a 0 to 42% Buffer A to Buffer B gradient (Buffer A: 0.1% formic acid; Buffer B: 80% acetonitrile, 0.1% formic acid).
Peptides were introduced by nano-electrospray into the Q-Exactive mass spectrometer (ThermoFisher). The instrument method consisted of one MS full scan (400–1500 m/z) in the Orbitrap mass analyzer with an automatic gain control (AGC) target of 1 × 106, maximum ion injection time of 120 ms and a resolution of 70,000, followed by 10 data-dependent MS/MS scans with a resolution of 17,500, an AGC target of 1 × 106, maximum ion time of 120 ms, and one microscan. The intensity threshold to trigger an MS/MS scan was set to 6.7 × 104. Fragmentation occurred in the higher-energy collisional dissociation (HCD) trap with normalized collision energy set to 27. The dynamic exclusion was applied using a setting of 10 seconds.
Raw data files were loaded into PEAKS 7 software (Bioinformatics Solutions, Inc.) for peptide and protein analysis using the UniProtKB E. coli K12 database. The generated protein and peptide identifications were then visualized using Scaffold (version 4.4.8, Proteome Software Inc., Portland, OR). Proteins identified with >95% probability using the Peptide63 and Protein64 Prophet algorithms, and a minimum of 2 unique peptides in both biological replicates, were accepted as positive identifications. Proteins detected in the WT preparation of FtsKN and non-crosslinked negative controls were used as an exclusion list for proteins found in the cross-linked samples. Proteins conclusively reported to have a cytoplasmic cellular localization were also excluded. Protein-protein interaction networks of all cross-linked proteins were built using STRING version 10.065 using all available prediction methods and a minimum confidence level of 0.4.
Expression and purification of His10-FtsKN(220)
Overnight cultures of E. coli Lemo21 cells carrying WT His10-FtsKN(220) encoded by a pBAD24 derivative (plasmid pAB006-2) were diluted 1:50 into 250 mL LB media and grown at 37 °C for 1.5 h. l-arabinose was added to a final concentration of 0.2% (w/v) and incubated for another hour. Following induction, the entire culture was harvested by centrifugation (8,000 × g, 10 min, 4 °C) and cells were resuspended in 5 mL of Lysis Buffer (20 mM Tris-HCl pH 7.0, 100 mM NaCl, 4 mM EDTA, 40 µg/mL DNaseI, 40 µg/mL RNase A, and 300 µg/mL lysozyme). Samples were incubated at room temperature for 10 min with rocking, then sonicated for 1 min (10 s on, 10 s off) to complete cell lysis. The total membrane fraction was collected by ultracentrifugation at 120,000 × g for 1 h at 4 °C (Beckman L8-55 M ultracentrifuge, Ti70 rotor). Membranes were suspended in 3 mL Purification Buffer (20 mM Tris-HCl pH 7.4, 300 mM NaCl, 25 mM imidazole, 10% [v/v] glycerol) containing 2% lauryldimethylamine-oxide (LDAO) and 100 µL of Profinity Ni2+-charged IMAC resin (Bio-Rad) and were incubated at 4 °C for 1.5 h with rocking. Solubilized membrane/resin mixtures were collected by centrifugation at 14,000 × g for 1 min at 4 °C in microcentrifuge tubes and washed ten times with 1 mL of Purification Buffer containing 0.05% LDAO. Purified, resin-bound FtsKN was then used during in vitro characterization of protein interaction between FtsK and RlpA by pull-down assay.
Expression and purification of FLAG-RlpA
Overnight cultures of E. coli Lemo21 cells carrying soluble FLAG-RlpA encoded by a pET28a derivative (plasmid pSG001-1) were diluted 1:100 into 1 L LB media and grown at 37 °C for 1 h before transferring to 30 °C for an additional hour of growth. Isopropyl-β-d-thiogalactoside (IPTG) was added to 1 mM and the culture was incubated for an additional 3 h at 30 °C. Following induction, the entire culture was harvested by centrifugation (8,000 × g, 10 min, 4 °C) and resuspended in 25 mL of Lysis Buffer before lysing by three passes through a French pressure cell (operating at 1000 psi). To remove cellular debris, samples were centrifuged at 8,000 × g for 10 min at 4 °C. The resulting supernatant was collected and ultracentrifuged at 120,000 × g for 1 h at 4 °C (Beckman L8-55 M ultracentrifuge, Ti70 rotor) to remove the membrane fraction. The resulting supernatant containing soluble FLAG-RlpA was added to 300 µL of anti-FLAG® M2 affinity gel (Sigma-Aldrich) and incubated overnight at 4 °C with rocking. FLAG-RlpA/resin mixtures were collected by centrifugation at 100 × g for 2 min at 4 °C and washed five times with 1 mL of Wash Buffer (20 mM Tris-HCl pH 7.0, 100 mM NaCl). FLAG-RlpA was successively eluted in four washes of 250 µL Wash Buffer containing 100 µg/mL FLAG® peptide (Sigma-Aldrich). Eluted FLAG-RlpA was pooled and the total protein concentration was determined by a bicinchoninic acid protein assay as per manufacturer’s instructions (Thermo Scientific) using bovine serum albumin (BSA) as a standard.
Pull-down assays
To assess the interaction between FtsK and RlpA, 320 µg of purified FLAG-RlpA was incubated with 100 µL of IMAC resin-bound His10-FtsKN at 4 °C overnight with rocking. The beads were collected by centrifugation and washed three times in 1 mL of Wash Buffer containing 25 mM imidazole and 0.05% LDAO. Bound proteins were eluted in 240 µL of Wash Buffer containing 1 M imidazole and 0.05% LDAO, mixed with 60 µL of 5 × Loading Buffer (250 mM Tris-HCl, pH 6.8, 10% [w/v] SDS, 30% [v/v] glycerol, 5% [v/v] β-mercaptoethanol, 0.1% [w/v] bromophenol blue) and boiled for 10 min. The presence of both proteins was verified by loading 15-µL aliquots onto duplicate 13% SDS-polyacrylamide gels followed by Western blotting. Unbound FLAG-RlpA in the flow through was diluted 1:20 before loading. Purified FLAG-RlpA was also incubated with 100 µL of unbound IMAC resin and washed as above to verify the absence of non-specific binding of FLAG-RlpA to the resin itself. Pull-down assays were also performed with IMAC resin-bound His10-FtsKN and 250 µg of purified FtsZ (purified as in ref.66) as a positive interaction control. All experiments were conducted in duplicate.
Western blotting
Expression of each FtsKN* construct was verified by preparing whole cell lysates for SDS-PAGE and Western blotting. FtsKN* variants expressed in the presence and absence of pBpa were diluted to an A600 of 0.25, equal volumes separated using a 13% SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane (Bio-Rad) using a Trans-Blot® Turbo transfer system (Bio-Rad) on the Turbo Transfer setting. The blot was developed using a SNAP i.d.® 2.0 Protein Detection System (EMD Millipore) as per the manufacturer’s instructions. Primary and secondary antibodies used were mouse anti-His6 (Clontech) and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (H + L) (Bio-Rad), respectively. Chemiluminescence was detected using 1 mL of HRP Detection Buffer (100 mM Tris-HCl pH 8.8, 1.25 mM luminol, 2 mM 4-iodophenylboronic acid, 5.3 mM hydrogen peroxide)67 with long exposure (5 min) in a Bio-Rad Gel Doc™ XR imaging system. Full-length western blots are shown in Supplementary Fig. S3.
To verify the presence of FtsKN, RlpA, and FtsZ in the pull-down assays, samples were separated using 13% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes that were then blocked in 5% (w/v) skim milk in Tris-buffered saline (TBS) at 4 °C overnight. Mouse anti-His6 (Clontech) and rabbit anti-FtsZ (Cedarlane) primary antibodies were used for the detection of FtsKN and FtsZ, respectively, in conjunction with the secondary antibodies alkaline phosphatase-conjugated goat anti-mouse IgG (H + L) (Sigma) and alkaline phosphatase-conjugated goat anti-rabbit IgG (H + L) (Sigma). FtsKN and FtsZ blots were developed using a solution of 3.3 mg of nitro blue tetrazolium and 1.7 mg of 5-bromo-4-chloro-3-indolyl phosphate in 10 mL of alkaline phosphatase substrate buffer (100 mM Tris-HCl pH 9.5, 100 mM NaCl, 5 mM MgCl2). The full-length FtsZ western blot is shown in Supplementary Fig. S4. RlpA was probed directly using an HRP-conjugated mouse anti-FLAG® (Sigma-Aldrich) antibody and chemiluminescence was detected using 1 mL of HRP Detection Buffer in a Bio-Rad Gel Doc™ XR imaging system.
Lambda red deletion of rlpA
Deletion of rlpA from E. coli LP11-1 and W3110 was achieved using the Lambda red recombinase method described by Datsenko and Wanner68. Briefly, a PCR fragment containing a kanamycin resistance cassette flanked by 50 bp homology extensions of the upstream and downstream regions of rlpA was generated from template pKD4 using primers rlpA_P1 (5′-AATCCACACCCACAGGAAAATGTTGTCGAAAAGCGTGTAAGAGGTGCGCAGTGTAGGCTGGAGCTGCTTC-3′) and rlpA_P2 (5′-TTTGTTAACGTCATTTACAGAAATTGACACATCAGATGCCTGCTTTACGATGAATATCCTCCTTAG-3′). An overnight culture of E. coli LP11-1 or W3110 carrying the Lambda red helper plasmid pKD46 was diluted 1:100 into 20 mL Complementation Media supplemented with 150 µg/mL ampicillin and 10mM l-arabinose and grown for 2 h at 30 °C. Cells were made electrocompetent by washing three times in ice-cold filtered Milli-Q H2O and suspended in 200 µL ice-cold filtered Milli-Q H2O (100-fold concentration). Fifty-microlitres of competent cells and 100 ng of PCR product were added to a 1 mm gap electroporation cuvette and incubated on ice for 5 min. Electroporation was done using a Gene Pulser Xcell™ electroporation system (Bio-Rad) using the following parameters: 1800 V, 25 µF, 200 Ω. Shocked cells were recovered for 1 h at 30 °C in 1 mL Super Optimal Broth with catabolite repression (SOC) medium (2% [w/v] tryptone, 0.5% [w/v] yeast extract, 8.56 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 20 mM glucose [Fisher]), then allowed to stand overnight at room temperature before spreading onto LB agar containing 25 µg/mL kanamycin to select for kanamycin resistant transformants. Deletion of rlpA and insertion of the kanamycin resistance cassette in each strain was verified by colony PCR using primers homologous to a region 500 bp upstream of rlpA (ΔrlpAFor; 5′-CCAGCGCGTAATGATGCTCCTGGAC-3′) and within the kanamycin resistance cassette (k1; 5′-CAGTCATAGCCGAATAGCCT-3′)68, as well as primers homologous to regions 50 bp upstream (ΔrlpAUp; 5′-CAATCCACACCCACAGGAAAATGTTCGTC-3′) and 50 bp downstream of rlpA (ΔrlpADown; 5′-ACTTTGTTAACGTCATTTACAGAAATTGACACA-3′). PCR verification of rlpA deletion in each strain was completed in duplicate along with control colonies of WT E. coli W3110 and LP11-1.
Electronic supplementary material
Acknowledgements
We acknowledge the kind gift of bacterial strain LP11-1 from Dr. Kevin Young. We thank Dr. Dyanne Brewer of the Mass Spectrometry Facility at the University of Guelph Advanced Analysis Centre for assistance with LC-MS/MS data analysis. We are also grateful to Dr. Chris Whitfield for plasmids pWQ743 and pWQ572, as well as for critical reading of the manuscript. This work was supported by Grant 371639 (to CMK), Canada Graduate Scholarship-Doctoral (to AMB), and Undergraduate Student Research Awards (to SG and MCG) from the Natural Sciences and Engineering Research Council of Canada.
Author Contributions
Conceived and designed experiments – A.M.B., J.R.K., and C.M.K. Performed the experiments – A.M.B., S.G., E.J.R., M.C.G., J.R.K., and C.M.K. Analyzed the data – A.M.B., J.R.K., and C.M.K. Wrote the manuscript – A.M.B., J.R.K., and C.M.K. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-30979-5.
References
- 1.Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010;2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 2012;10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szwedziak P, Löwe J. Do the divisome and elongasome share a common evolutionary past? Curr. Opin. Microbiol. 2013;16:745–751. doi: 10.1016/j.mib.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Goehring NW, Beckwith J. Diverse paths to midcell: Assembly of the bacterial cell division machinery. Curr. Biol. 2005;15:R514–R526. doi: 10.1016/j.cub.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 5.Haeusser DP, Margolin W. Splitsville: Structural and functional insights into the dynamic bacterial Z ring. Nat. Rev. Microbiol. 2016;14:305–319. doi: 10.1038/nrmicro.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randich AM, Brun YV. Molecular mechanisms for the evolution of bacterial morphologies and growth modes. Front. Microbiol. 2015;6:1–13. doi: 10.3389/fmicb.2015.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Errington J. Bacterial morphogenesis and the enigmatic MreB helix. Nat. Rev. Microbiol. 2015;13:241–248. doi: 10.1038/nrmicro3398. [DOI] [PubMed] [Google Scholar]
- 8.van den Ent F, Amos LA, Löwe J. Prokaryotic origin of the actin cytoskeleton. Nature. 2001;413:39–44. doi: 10.1038/35092500. [DOI] [PubMed] [Google Scholar]
- 9.Young KD. Bacterial shape: Two-dimensional questions and possibilities. Annu. Rev. Microbiol. 2010;64:223–240. doi: 10.1146/annurev.micro.112408.134102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michie KA, Löwe J. Dynamic filaments of the bacterial cytoskeleton. Annu. Rev. Biochem. 2006;75:467–492. doi: 10.1146/annurev.biochem.75.103004.142452. [DOI] [PubMed] [Google Scholar]
- 11.Bisson-Filho AW, et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science. 2017;355:739–743. doi: 10.1126/science.aak9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukherjee A, Dai K, Lutkenhaus J. Escherichia coli cell division protein FtsZ is a guanine nucleotide binding protein. Proc. Natl. Acad. Sci. USA. 1993;90:1053–1057. doi: 10.1073/pnas.90.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bi E, Lutkenhaus J. FtsZ ring structure associated with division in Escherichia coli. Nature. 1991;354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 14.Rothfield L. New insights into the developmental history of the bacterial cell division site. J. Bacteriol. 2003;185:1125–1127. doi: 10.1128/JB.185.4.1125-1127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strauss MP, et al. 3D-SIM super resolution microscopy reveals a bead-like arrangement for FtsZ and the division machinery: Implications for triggering cytokinesis. PLoS Biol. 2012;10:e1001389. doi: 10.1371/journal.pbio.1001389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wientjes FB, Nanninga N. Rate and topography of peptidoglycan synthesis during cell division in Escherichia coli: Concept of a leading edge. J. Bacteriol. 1989;171:3412–3419. doi: 10.1128/jb.171.6.3412-3419.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pogliano J, Pogliano K, Weiss DS, Losick R, Beckwith J. Inactivation of FtsI inhibits constriction of the FtsZ cytokinetic ring and delays the assembly of FtsZ rings at potential division sites. Proc. Natl. Acad. Sci. USA. 1997;94:559–564. doi: 10.1073/pnas.94.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nanninga N. Cell division and peptidoglycan assembly in Escherichia coli. Mol. Microbiol. 1991;5:791–795. doi: 10.1111/j.1365-2958.1991.tb00751.x. [DOI] [PubMed] [Google Scholar]
- 19.van der Ploeg R, et al. Colocalization and interaction between elongasome and divisome during a preparative cell division phase in Escherichia coli. Mol. Microbiol. 2013;87:1074–1087. doi: 10.1111/mmi.12150. [DOI] [PubMed] [Google Scholar]
- 20.Begg KJ, Dewar SJ, Donachie WD. A new Escherichia coli cell division gene, ftsK. J. Bacteriol. 1995;177:6211–6222. doi: 10.1128/jb.177.21.6211-6222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu G, Draper GC, Donachie WD. FtsK is a bifunctional protein involved in cell division and chromosome localization in Escherichia coli. Mol. Microbiol. 1998;29:893–903. doi: 10.1046/j.1365-2958.1998.00986.x. [DOI] [PubMed] [Google Scholar]
- 22.Barre F-X. FtsK and SpoIIIE: The tale of the conserved tails. Mol. Microbiol. 2007;66:1051–1055. doi: 10.1111/j.1365-2958.2007.05981.x. [DOI] [PubMed] [Google Scholar]
- 23.Demarre, G., Galli, E. & Barre, F.-X. In DNA Helicases and DNA Motor Proteins (ed. Spies, M.) 973, 245–262 (Springer New York, 2013).
- 24.Draper GC, McLennan N, Begg K, Masters M, Donachie WD. Only the N-terminal domain of FtsK functions in cell division. J. Bacteriol. 1998;180:4621–4627. doi: 10.1128/jb.180.17.4621-4627.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berezuk AM, Goodyear M, Khursigara CM. Site-directed fluorescence labeling reveals a revised N-terminal membrane topology and functional periplasmic residues in the Escherichia coli cell division protein FtsK. J. Biol. Chem. 2014;289:23287–23301. doi: 10.1074/jbc.M114.569624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bigot S, Sivanathan V, Possoz C, Barre F-X, Cornet F. FtsK, a literate chromosome segregation machine. Mol. Microbiol. 2007;64:1434–1441. doi: 10.1111/j.1365-2958.2007.05755.x. [DOI] [PubMed] [Google Scholar]
- 27.Dubarry N, Possoz C, Barre F-X. Multiple regions along the Escherichia coli FtsK protein are implicated in cell division. Mol. Microbiol. 2010;78:1088–1100. doi: 10.1111/j.1365-2958.2010.07412.x. [DOI] [PubMed] [Google Scholar]
- 28.Dubarry N, Barre F-X. Fully efficient chromosome dimer resolution in Escherichia coli cells lacking the integral membrane domain of FtsK. EMBO J. 2010;29:597–605. doi: 10.1038/emboj.2009.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grainge I. FtsK - a bacterial cell division checkpoint? Mol. Microbiol. 2010;78:1055–1057. doi: 10.1111/j.1365-2958.2010.07411.x. [DOI] [PubMed] [Google Scholar]
- 30.Lesterlin C, Pages C, Dubarry N, Dasgupta S, Cornet F. Asymmetry of chromosome replichores renders the DNA translocase activity of FtsK essential for cell division and cell shape maintenance in Escherichia coli. PLoS Genet. 2008;4:e1000288. doi: 10.1371/journal.pgen.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grenga L, Luzi G, Paolozzi L, Ghelardini P. The Escherichia coli FtsK functional domains involved in its interaction with its divisome protein partners. FEMS Microbiol. Lett. 2008;287:163–167. doi: 10.1111/j.1574-6968.2008.01317.x. [DOI] [PubMed] [Google Scholar]
- 32.Di Lallo G, Fagioli M, Barionovi D, Ghelardini P, Paolozzi L. Use of a two-hybrid assay to study the assembly of a complex multicomponent protein machinery: bacterial septosome differentiation. Microbiology. 2003;149:3353–3359. doi: 10.1099/mic.0.26580-0. [DOI] [PubMed] [Google Scholar]
- 33.Butland G, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 34.Gerding MA, et al. Self-enhanced accumulation of FtsN at division sites and roles for other proteins with a SPOR domain (DamX, DedD, and RlpA) in Escherichia coli cell constriction. J. Bacteriol. 2009;191:7383–7401. doi: 10.1128/JB.00811-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arends SJR, et al. Discovery and characterization of three new Escherichia coli septal ring proteins that contain a SPOR domain: DamX, DedD, and RlpA. J. Bacteriol. 2010;192:242–255. doi: 10.1128/JB.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yahashiri A, Jorgenson MA, Weiss DS. Bacterial SPOR domains are recruited to septal peptidoglycan by binding to glycan strands that lack stem peptides. Proc. Natl. Acad. Sci. USA. 2015;112:11347–11352. doi: 10.1073/pnas.1508536112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jorgenson MA, Chen Y, Yahashiri A, Popham DL, Weiss DS. The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in Pseudomonas aeruginosa. Mol. Microbiol. 2014;93:113–128. doi: 10.1111/mmi.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mori H, Ito K. Different modes of SecY-SecA interactions revealed by site-directed in vivo photo-cross-linking. Proc. Natl. Acad. Sci. USA. 2006;103:16159–16164. doi: 10.1073/pnas.0606390103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu D, Wowor AJ, Cole JL, Kendall DA. Defining the Escherichia coli SecA dimer interface residues through in vivo site-specific photo-cross-linking. J. Bacteriol. 2013;195:2817–2825. doi: 10.1128/JB.02269-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panahandeh, S. & Müller, M. In Protein Secretion (ed. Economou, A.) 619, 217–240 (Humana Press, 2010).
- 41.Nickerson NN, et al. Trapped translocation intermediates establish the route for export of capsular polysaccharides across Escherichia coli outer membranes. Proc. Natl. Acad. Sci. USA. 2014;111:8203–8208. doi: 10.1073/pnas.1400341111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van den Berg van Saparoea HB, et al. Fine-mapping the contact sites of the Escherichia coli cell division proteins FtsB and FtsL on the FtsQ protein. J. Biol. Chem. 2013;288:24340–24350. doi: 10.1074/jbc.M113.485888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chin JW, Martin AB, King DS, Wang L, Schultz PG. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. USA. 2002;99:11020–11024. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chin JW, Schultz PG. In vivo photocrosslinking with unnatural amino acid mutagenesis. ChemBioChem. 2002;3:1135–1137. doi: 10.1002/1439-7633(20021104)3:11<1135::AID-CBIC1135>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 45.Dorman G, Prestwich G. Benzophenone photophores in biochemistry. Biochemistry. 1994;33:5661–5673. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- 46.Karimova G, Dautin N, Ladant D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 2005;187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li GW, Burkhardt D, Gross C, Weissman JS. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urban PL. Clarifying misconceptions about mass and concentration sensitivity. J. Chem. Educ. 2016;93:984–987. doi: 10.1021/acs.jchemed.5b00986. [DOI] [Google Scholar]
- 49.Lundgren DH, Hwang S, II, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert Rev. Proteomics. 2010;7:39–53. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- 50.Takase I, et al. Genes encoding two lipoproteins in the leuS-dacA region of the Escherichia coli chromosome. J Bacteriol. 1987;169:5692–5699. doi: 10.1128/jb.169.12.5692-5699.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PAJ. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol. Microbiol. 2007;63:1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Potluri L, et al. Septal and lateral wall localization of PBP5, the major D, D-carboxypeptidase of Escherichia coli, requires substrate recognition and membrane attachment. Mol. Microbiol. 2010;77:300–323. doi: 10.1111/j.1365-2958.2010.07205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, Lutkenhaus J. FtsK is an essential cell division protein that is localized to the septum and induced as part of the SOS response. Mol. Microbiol. 1998;29:731–740. doi: 10.1046/j.1365-2958.1998.00958.x. [DOI] [PubMed] [Google Scholar]
- 54.Pichoff S, Du S, Lutkenhaus J. The bypass of ZipA by overexpression of FtsN requires a previously unknown conserved FtsN motif essential for FtsA-FtsN interaction supporting a model in which FtsA monomers recruit late cell division proteins to the Z ring. Mol. Microbiol. 2015;95:971–987. doi: 10.1111/mmi.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weiss DS. Last but not least: New insights into how FtsN triggers constriction during Escherichia coli cell division. Mol. Microbiol. 2015;95:903–909. doi: 10.1111/mmi.12925. [DOI] [PubMed] [Google Scholar]
- 56.Liu B, Persons L, Lee L, de Boer PAJ. Roles for both FtsA and the FtsBLQ subcomplex in FtsN-stimulated cell constriction in Escherichia coli. Mol. Microbiol. 2015;95:945–970. doi: 10.1111/mmi.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lopez-Garrido J, Casadesus J. The DamX protein of Escherichia coli and Salmonella enterica. Gut Microbes. 2010;1:285–288. doi: 10.4161/gmic.1.4.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Born P, Breukink E, Vollmer W. In vitro synthesis of cross-linked murein and its attachment to sacculi by PBP1A from Escherichia coli. J. Biol. Chem. 2006;281:26985–26993. doi: 10.1074/jbc.M604083200. [DOI] [PubMed] [Google Scholar]
- 59.Zou Y, Li Y, Dillon J-AR. The distinctive cell division interactome of Neisseria gonorrhoeae. BMC Microbiol. 2017;17:232. doi: 10.1186/s12866-017-1140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.von Rechenberg M, Ursinus A, Höltje J-V. Affinity chromatography as a means to study multienzyme complexes involved in murein synthesis. Microb. Drug Resist. 1996;2:155–157. doi: 10.1089/mdr.1996.2.155. [DOI] [PubMed] [Google Scholar]
- 61.Vollmer W, von Rechenberg M, Höltje JV. Demonstration of molecular interactions between the murein polymerase PBP1B, the lytic transglycosylase MltA, and the scaffolding protein MipA of Escherichia coli. J. Biol. Chem. 1999;274:6726–6734. doi: 10.1074/jbc.274.10.6726. [DOI] [PubMed] [Google Scholar]
- 62.Park AJ, et al. A temporal examination of the planktonic and biofilm proteome of whole cell Pseudomonas aeruginosa PAO1 using quantitative mass spectrometry. Mol. Cell. Proteomics. 2014;13:1095–1105. doi: 10.1074/mcp.M113.033985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 64.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 65.Szklarczyk D, et al. STRING v10: protein – protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roach EJ, Kimber MS, Khursigara CM. Crystal structure and site-directed mutational analysis reveals key residues involved in Escherichia coli ZapA function. J. Biol. Chem. 2014;289:23276–23286. doi: 10.1074/jbc.M114.561928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haan C, Behrmann I. A cost effective non-commercial ECL-solution for Western blot detections yielding strong signals and low background. J. Immunol. Methods. 2007;318:11–19. doi: 10.1016/j.jim.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 68.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Potluri L-P, Kannan S, Young KD. ZipA is required for FtsZ-dependent preseptal peptidoglycan synthesis prior to invagination during cell division. J. Bacteriol. 2012;194:5334–5342. doi: 10.1128/JB.00859-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.