

Abstract
Cancer is currently viewed as a disease of evolving genomic instability and abnormal epigenomic modifications. Most solid cancers harbor oncogenic gene mutations driven by both extrinsic and intrinsic factors. Apolipoprotein B mRNA editing catalytic polypeptide‐like family (APOBEC) enzymes have an intrinsic deamination activity to convert cytosine to uracil during RNA editing and retrovirus or retrotransposon restriction. Beyond their natural defense in innate immunity, compelling evidence showed that a subclass of APOBEC3 can cause high mutation burden in various types of cancer genomes, and high expression subtypes of APOBEC3 may contribute to drug resistance and associate with clinical outcomes. The underlying molecular mechanisms of APOBEC‐mediated hypermutation phenotype are poorly understood. In this review, we discuss the linkage of activation‐induced deaminase (AID)/APOBEC3 enzymes to tumorigenesis, highlight the dysregulatory mechanisms of APOBEC3 activities during cancer development, and propose potential approaches to targeting APOBEC3‐mediated mutagenesis for cancer interventions.
Keywords: APOBEC, cancer genomics, mutagenesis, tumorigenesis
1. INTRODUCTION
Cancer is a disorder of genome alterations with lifetime accumulated single nucleotide changes, insertions, deletions and chromosomal structural aberrations.1 Genomic instability is one of the hallmarks of cancer and is known to cause both aberrant chromosomal architecture and mutational changes.2 Somatic mutations in a cancer genome are the aggregated outcome of 1 or more mutational processes including exogenous (environmental factors) and endogenous mutators during tumorigenesis. Two‐thirds of mutations occur randomly during the DNA replication process in self‐renewing tissues before tumor initiation3 suggesting mutational processing in mammalian genomes. Cancer genomic projects show that the majority of somatic mutations are passengers,4 and approximately 699 cancer‐related gene mutations have been identified from a catalogue of somatic mutations in a cancer project (COSMIC v83). Known exogenous mutational factors are UV light exposure, smoking, and aflatoxins; endogenous mutational factors are DNA mismatch repair defects, DNA damage repair defects and recent apolipoprotein B mRNA editing enzyme catalytic polypeptide‐like family genes (APOBEC) activation.5
APOBEC genes are a family of evolutionarily conserved cytidine deaminases.6 There are 11 human genes encoding members of the APOBEC family of enzymes, named as activation‐induced deaminase (AID), APOBEC1 gene on chromosome 12; APOBEC2 gene on chromosome 6; 7 APOBEC3 genes (APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, APOBEC3H) on chromosome 22, and APOBEC4 gene on chromosome 1.7 APOBEC enzymes contain 1 or 2 catalytic domains that recognize a specific DNA/RNA sequence8 (Figure 1). Crystal structure basis of APOBEC3B deaminase with strong 5′‐TC preference has recently been exploited: the U‐shaped conformation of ssDNA with the specificity‐conferring ‐1 thymidine (T) base flipped out, and targeted cytidine (C) resides in a zinc‐coordinated active size pocket, the T can fit into a groove between flexible loops and make direct hydrogen bonds with APOBEC3B enzyme,9 while APOBEC3A binds to ssDNA with TCG preference.9 APOBEC3B was previously detected as a major mutator deaminase in multiple types of cancer.10 Extended study in a yeast model and 6 different types of cancer showed that APOBEC3A‐like tumors have significantly greater APOBEC‐signature (YTCA, Y = pyrimidine) mutations than APOBEC3B‐like tumors (RTCA, R = purine),11 suggesting that APOBEC3A is the predominant mutagenic deaminase in certain types of cancer. The structure of APOBEC3F catalytic domain differs from other APOBEC deaminases in that the zinc is not required for the structural integrity of APOBEC3F.12
Figure 1.
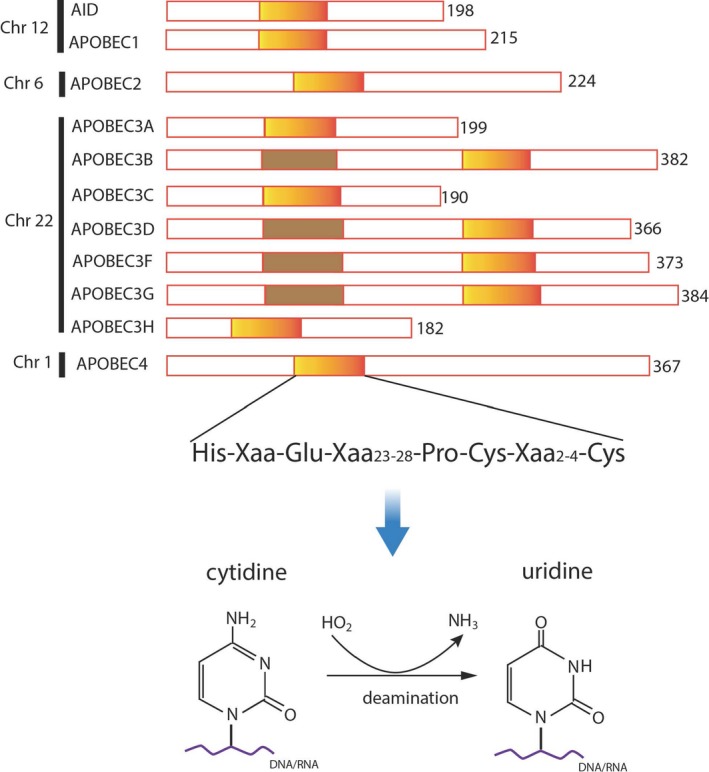
Schematic diagram displays apolipoprotein B mRNA editing enzyme catalytic polypeptide‐like (APOBEC) family proteins with cytidine deaminase domain. Human APOBEC family genes consist of 11 members in the genome: activation‐induced deaminase (AID) and APOBEC1 on chromosome 12, APOBEC2 on chromosome 6, 7 APOBEC3 (A‐H, except E) on chromosome 22 and APOBEC4 on chromosome 1. Encoded gene products contain 1 or 2 zinc‐dependent deaminase motifs (colored boxes). The cytidine deaminase domain in APOBEC is able to catalyze the biochemical reaction of conversion from cytidine to uridine on RNA or ssDNA substrates. Adapted from Ref. 8
APOBEC3 proteins are localized in the cytoplasm or nuclei depending on the features of individual proteins. AID and APOBEC1 localize mainly in the cytoplasm but act in the nuclei; APOBEC2, APOBEC3A, and APOBEC3C appear to reside in both cytoplasm and nuclei; APOBEC3B is predominantly located in the nucleus,13 possibly because APOBEC3B protein contains a nuclear localization sequence, and is able to actively enter nuclei.14 APOBEC3G is located in the cytoplasm against exogenous retroviruses. Thus, cells regulate APOBEC enzymatic activities through protein localizations.
2. FUNCTIONS OF APOBEC FAMILY GENES
Except for poorly characterized functions of APOBEC2 and APOBEC4, other members of APOBEC proteins generally function as: (i) innate immune response to viral infection (eg, HIV, hepatitis B virus [HBV], human papillomavirus [HPV]) such as AID, APOBEC3G; (ii) deamination of cytidine (C) to uridine (U) in RNA/ssDNA; (iii) generation of somatic hypermutations during cancer development; and (iv) selective deamination for methylated cytidines (mC) during epigenetic regulation such as APOBEC3A.15 We describe their functions in more detail below.
Activation‐induced deaminase is the oldest member of the APOBEC family and is essential for antigen‐driven B‐cell differentiation, antibody affinity maturation and diversification.16 Expression of AID in immune B cells causes hypermutations in the “variable region” of antibody genes, and produces antibody diversification with class‐switch recombination in response to infection.17 This biochemical process may collude with altered DNA repair pathways. AID also interacts with histone methyltransferases (eg, SUV4‐20H1.2) to increase methylation of cross‐switch recombination sites during Ig gene diversification.18 In addition to hypermutation of immunoglobulin genes, AID can create genome‐wide mutations and DNA strand breaks19 as seen in the translocation of Myc‐Ig genes in B‐cell lymphoma.
Activation‐induced deaminase expression is regulated by a number of proinflammatory cytokines, transforming growth factor (TGF)‐beta, tumor necrosis factor (TNF)‐alpha, and interleukin (IL)‐1 beta through the nuclear factor kappa B (NF‐κB) signaling pathway in human hepatocytes and reduced HBV infectivity in host cells,20 but increases tumorigenesis in a transgenic mouse model.21
APOBEC1 is the first member of APOBEC to be identified and characterized as the apolipoprotein B editing complex 1, a protein that is involved in editing C to U of the apolipoprotein B pre‐mRNA at nucleotide position 6666 and generates a stop translation codon that defines the carboxyl terminus of apolipoprotein B48.22, 23 In addition to editing apoB mRNA, a transcriptomic‐wide comparative RNA sequencing screen identified 32 new mRNA targets of APOBEC1 editing, all of them being located in AU‐rich segments of 3′ untranslated regions of transcripts.24 APOBEC1 was also reported to deaminate cytosine in DNA but with different preferred 5′ nucleotide content around cytosine (eg, C, T 5′ adjacent to cytosine).25 Hence, APOBEC1, like AID, can trigger DNA mutations through dC deamination and is a potential active dC/dG mutator.
Members of human APOBEC3 subfamily genes suppress virus infections and retro‐transposition of endogenous retro‐elements through mutagenic and non‐mutagenic mechanisms. These enzymes can cause mutations in the cellular genome at replication forks or within transcription bubbles depending on the physiological state of the cell and the phase of the cell cycle during which they are expressed (see review26). DNA viruses (eg, adeno‐associated virus [AAV], HBV, HPV, herpes simplex virus 1 [HSV‐1] and Epstein–Barr virus [EBV]) have been reported to be restricted by APOBEC3.27 For example, APOBEC3G was the first APOBEC3 protein demonstrated to have restriction activity of HIV infection through G to A mutations in the anti‐sense DNA strand; such changes by APOBEC3G enzyme create non‐infectious virions, possibly as a result of the degradation of viral DNA.28, 29
APOBEC/AID deaminase activities could produce “off‐target” mutations in the host genome associated with cancer development, progression, metastasis and drug resistance (see review30). A mammalian genome has DNA damage‐tolerant polymerase (PrimPol) to function as anti‐unwanted mutations induced by APOBEC3B,31 but this defense system might not efficiently block widespread dysregulated APOBEC3 effect. Accumulated evidence showed that aberrant APOBEC expressions and activities have been associated with cancer development. Understanding of regulation of APOBEC gene expressions at diverse cellular context is crucial to develop a novel avenue for cancer interventions.
3. APOBEC FAMILY GENES AND CANCER
Association between APOBEC and carcinogenesis is evidenced from multi‐dimensional observations. Mice with overexpression of APOBEC1 gene developed hepatocellular carcinoma;32 constitutive expression of AID not only causes T‐cell lymphoma with mutations of the Myc gene19 and Burkitt's lymphoma with IGH‐MYC translocation33 in a transgenic mouse model, but also causes non‐lymphoid tumors such as hepatocellular carcinoma34 and gastric cancer35 resulting from AID‐mediated dysregulation of class switch recombination and somatic hypermutations, suggesting AID plays clear roles in tumorigenesis. Aberrant expression of APOBEC led to enrichment for C >T mutations in tumor suppressor genes (eg, TP53 and APC)35, 36 Moreover, APOBEC activities are also responsible for the generation of helical domain hotspot mutations in the proto‐oncogene PIK3CA across multiple types of cancer.37 APOBEC‐mediated mutations not only occur in coding genes, but also take place in non‐coding regions resulting in oncogenic driver genes expression. For example, APOBEC‐like cytidine deaminase mutation at 4 kb upstream of LIM Domain Only 1 (LMO1) oncogene in T‐cell acute lymphoblastic leukemia generates a new MYB transcription factor binding site, which forms an aberrant transcriptional enhancer complex, leading to overexpression of the LMO1 gene.38
Increasing evidence show that a germline APOBEC3B deletion resulting in an APOBEC3A_APOBEC3B fusion variant increases the risk of breast cancer39, 40 and increases tumor mutational burden.41 Recent studies support that APOBEC3 activities are associated with tumorigenesis as a result of increased mutagenesis. In particular, APOBEC3B overexpressed in several human cancer types correlates with the presence of APOBEC3B mutational signature.10, 42 From whole exome sequence data analysis over 30 different types of cancer, a unique APOBEC3 mutational signature has been identified with cytosine mutation biases, particularly C to T transitions and C to G transversion, and predominantly in TCA or TCT trinucleotide contexts.10, 42, 43, 44 The APOBEC3B‐mediated mutational signature is often associated with breakpoint rearrangement in general, and HER2‐enriched subtype of breast cancer.42 Further analysis showed that APOBEC‐signature mutation load in cancer exons is statistically correlated with APOBEC3A and APOBEC3B transcript abundance.10, 42 Although APOBEC3B mRNA abundance tends to be greater than that of APOBEC3A in cancer samples, APOBEC3A presents a much greater potent inducer of DNA damage.11
APOBEC3 mutational signature may occur at different stages in different types of cancer. For example, the mutational signature of APOBEC3 is seen both in early superficial non‐invasive and subsequent invasive bladder tumors,45 whereas APOBEC3‐mediated mutagenesis contributes to later subclones in estrogen receptor‐negative (ER‐) breast cancer, lung adenocarcinoma, and head and neck squamous carcinoma as the tumors evolve.46, 47 High APOBEC3B expression in ER+ breast cancer showed short progression‐free time with tamoxifen treatment.48 All of this evidence supports that APOBEC, specifically APOBEC3A and APOBEC3B, act as drivers in cancer development and progression.
Notably, APOBEC family genes themselves are the victims of mutagenesis in cancer development. Missense mutations in APOBEC3B, APOBEC3F and APOBEC3G were detected in a subset (4/115 samples) of cervical cancers, which is associated with greater mutational burden.49 We further searched APOBEC gene alterations from cBioPortal (http://www.cbioportal.org/) (Figure 2). There are various APOBEC genetic alterations (mutation, deletion and amplification) across broad types of cancer, the frequency is from 0.5% to 9%. The genetic aberration of APOBEC may also contribute to tumorigenesis, but further investigation is needed.
Figure 2.
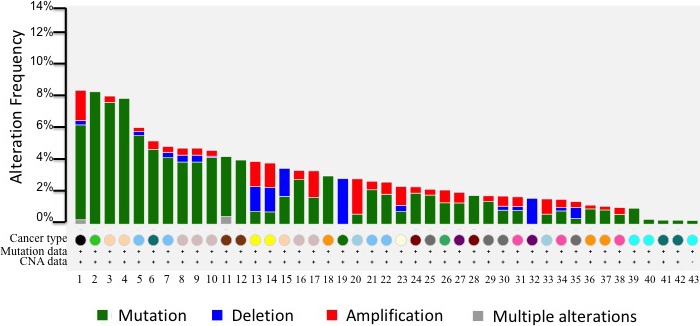
Apolipoprotein B mRNA editing enzyme catalytic polypeptide‐like (APOBEC) genetic alterations in cancer. cBioPortal website was used to search 11 APOBEC gene family members across broad types of cancer studied in The Cancer Genome Atlas (TCGA), and the frequency of APOBEC genetic alterations from high to low were plotted. Analyses include samples with both copy number alterations and mutations data. Color bars represent different types of alteration (green, APOBEC mutation; blue, APOBEC gene deletion; red, APOBCEC gene amplification; grey, APOBEC gene multiple alterations). Circles of different colors represent different types of cancer studied in TCGA. There are 43 different studies listed in this plot
4. REGULATION OF APOBEC3 GENE EXPRESSION
The downstream effects of APOBEC activities such as antivirus and the involvement of APOBEC3 in cancer have been documented. The upstream functional regulation of the APOBEC family of enzymes is less known. Several recent studies are beginning to elucidate the molecular basis of the regulation of APOBEC3 expression and their transcriptional activations or repressions. First, viral infection and immune responses in cells could induce overexpression of specific APOBEC genes. For instance, Helicobacter pylori infection in normal gastric epithelium induces aberrant AID gene expression through IKB kinase‐dependent NF‐κB signaling pathway activation. As a consequence of AID activation, tumor suppressor gene TP53 mutations and gastric carcinogenesis developed.35 Similarly, in hepatocyte cells, hepatitis C virus (HCV) infection or proinflammatory cytokines (eg, TNF‐α) activates the inhibitor of nuclear factor kappa‐B kinase subunit beta (NF‐κB/IKK‐β) signaling pathway, which can stimulate AID overexpression, resulting in genetic susceptibility to mutagenesis, and these pathological processes are responsible for the development of liver cancer.34 HPV16 infection is a major risk factor in cervical, head and neck cancers and oropharyngeal cancers. APOBEC3B mutation signature and expression of APOBEC3B were found to be enriched in HPV+ subtype of cancers,10, 37, 42 whereas APOBEC3A expression with uracil N‐glycosylase inhibition is responsible for HPV16 genome integration and hypermutations in oropharyngeal cancers.50 Mechanism of viral oncoprotein E6/E7‐mediated elevated APOBEC3B expression is due to removal of p53‐mediated repression of APOBEC3B expression.51 Interestingly, the promoter regions of APOBEC3 genes contain p53‐binding sites; p53 protein is a major transcriptional regulator of APOBEC3 genes in response to chromosomal stress, and p53 activation increases several APOBEC3 mRNAs and protein expression in a cellular context, except for APOBEC3B expression in a suppressive method.52 In contrast to wild‐type p53 activation, p53 hotspot mutants upregulate APOBEC3B expression in cancer cells.52 Therefore, inactivation of p53 by viral protein E6/E7 activation or loss of function of p53 mutations can activate APOBEC3B function, increase genome instability and promote tumor initiation.
Nuclear factor kappa B signaling pathway plays a crucial role in the transcriptional regulation of APOBEC genes such as AID expression.34, 35 A recent study showed that NF‐κB could bind to multiple promoter regions of the APOBEC3B gene and upregulate the expression of APOBEC3B mRNA under a variety of conditions (eg, PKC/IKK activation, oncogenic activation and IFN treatment).53 NF‐κB may cross‐talk with other oncogenic pathways to also upregulate APOBEC3B expression; for instance, DNA replication stress with a variety of stimulators can activate transcription of APOBEC3B through the ATR/Chk1‐dependent pathway.54 Several oncogenic signaling pathways such as PI3K, MAPK, AKT and mammalian target of rapamycin (mTOR) pathways are on the circuits of replication stress‐induced APOBEC3B activation.54 These signaling pathways are upstream molecules of the NF‐κB pathway; thus we speculate that NF‐κB may participate in replication stress‐mediated APOBEC3B upregulation.
In addition to transcriptional regulation of APOBEC gene expression by transcription factors, APOBEC are subject to post‐transcriptional regulation by microRNAs.55 Expression of the APOBEC3A_APOBEC3B variant is increased as a result of loss of 3′‐UTR of APOBEC3A, which is negatively targeted by microRNAs.30 Another example, microRNA 2909 binds to the 5′‐UTR region of APOBEC3G mRNA and decreases APOBEC3G translational expression.56 Conversely, APOBEC3G binds to 3′‐UTR of tumor suppressor gene KLF4 mRNA, resulting in decreased expression of KLF4, and increased expression of SP1 and other survival genes including c‐myc, Bmi‐1, BCL‐2 and MDM2.57 APOBEC3G overexpression produces truncated apoptosis antagonizing transcription factor (AATF, 23 kDa) through APOBEC3G binding AATF mRNA within its third exon.58 The microRNAs‐APOBEC‐mRNA network may contribute to oncogenic activations in some circumstances. Last, Hsp90 can stimulate APOBEC3B, APOBEC3C and APOBEC3G deamination activity59 as a result of stress responses. Taken together, APOBEC3 gene expression is transcriptionally regulated by multiple transcription factors and signaling pathways depending on cellular context (Figure 3).
Figure 3.

Summary of apolipoprotein B mRNA editing enzyme catalytic polypeptide‐like (APOBEC)3B transcriptional regulations in cancer cells. APOBEC3B is the main source of mutagenesis in multiple types of cancer. High expression of APOBEC3B and elevated activity have been reported in HER2+ breast cancer, human papillomavirus (HPV)+ cervical cancer, and head and neck squamous carcinoma. Nuclear factor kappa B (NF‐κB) pathway activation,53 DNA replication stress54 and HPV E6/7 oncoproteins and mutant p5352 are able to turn on APOBEC3B expression. In contrast, WT p53 activation suppresses APOBEC3B expression through p21‐mediated transcriptional suppressive complexes occupancy of promoter region of APOBEC3B.51, 52 AID, activation‐induced deaminase; IFN, interferon; TNF, tumor necrosis factor
5. APOBEC3 AS PROGNOSTIC MARKERS AND THERAPEUTIC TARGETS FOR CANCER TREATMENT
Accumulated evidence has shown that APOBEC3A and APOBEC3B are the main sources of somatic mutagenesis in human tumors, and APOBEC‐mediated mutagenesis is correlated with APOBEC mRNA levels.42 How the APOBEC mRNA expression is associated with clinical outcomes and overall survival is poorly documented. Chen et al60 reported that greater expression of APOBEC3A has better overall survival in Taiwanese oral squamous cell carcinoma (OSCC) patients, but not in The Cancer Genome Atlas (TCGA)‐OSCC patients. This unique clinical prognostic relevance of APOBEC3A expression in Taiwanese‐OSCC is due to greater (~50%) APOBEC3B deletion genotype. We explored the association of all APOBEC expression with overall survival in TCGA pan‐cancers (https://www.proteinatlas.org/), and found that greater expression of 5 APOBEC3 family genes (APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3G, APOBEC3H) are significantly correlated with better survival in TCGA‐cervical cancer, but with poor survival in TCGA‐renal cancer (Figure 4). This discrepancy of APOBEC expression‐mediated clinical benefit may, at least in part, depend on the level of APOBEC expression in the tumor. Now, the question is whether we can manage APOBEC activity for treatment. Development of chemical inhibitors to APOBEC3D/G/F/H enzymes for HIV‐1 treatment is in its early stage according to the interaction between APOBEC protein structure and virion infectivity factor, whereas screening chemical inhibitors for APOBEC3A and APOBEC3B is under consideration.61 Here we propose two strategies to inhibit AID, APOBEC3A and APOBEC3B enzymatic activities for cancer intervention. One is for direct inhibition using chemicals or biological approaches including shRNA or clustered regularly interspaced short palindromic repeats (CRISPR) technology. The mechanism of action of APOBEC enzymes is that these enzymes deaminate cytidine to uridine on RNA/ssDNA, and blocking the interaction between APOBEC and RNA/ssDNA substrates could alleviate unwanted mutations in the genome. Recent release of APOBEC3A‐ssDNA and APOBEC3B‐ssDNA co‐crystal structure will provide a foundation for structurally based design of APOBEC3 small molecule inhibitors.9, 62 Depletion of APOBEC3B mRNA with siRNA can reverse APOBEC3B‐mediated tamoxifen resistance in ER+ breast cancers.48 MicroRNAs have been reported to negatively regulate APOBEC3 genes,55 and these approaches are still in the experimental stage and not yet in any clinical trials. Similarly, CRISPR/Cas9‐derived transcriptional suppression approach might be used for AID, APOBEC3A, and APOBEC3B inactivation.
Figure 4.
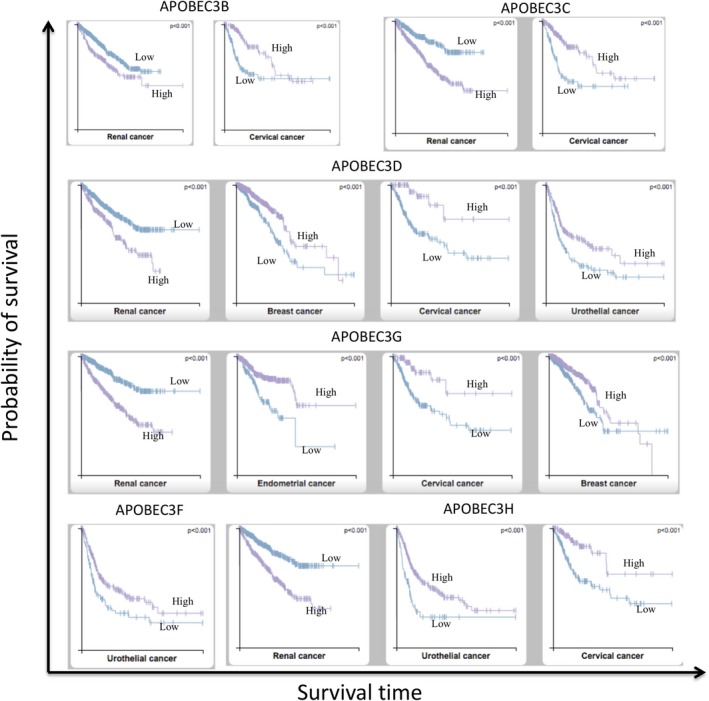
Apolipoprotein B mRNA editing enzyme catalytic polypeptide‐like (APOBEC) family gene expression‐associated clinical outcomes in TCGA pan‐cancers. APOBEC family gene expression in database (http://www.proteinatlas.org/pathology)67 with 17 major cancer types with respect to clinical outcome was analyzed for Kaplan‐Meier survival plot. No APOBEC3A expression is associated with clinical outcome; however, higher expression of APOBEC3B, C, D, G, H genes show poor survival time in renal cancer. In contrast, better survival time is seen in cervical cancer. Breast cancer benefits from higher expression of APOBEC3D and APOBEC3G; urothelial cancer benefits from higher expression of APOBEC3D, F, H; and endometrial cancer show better survival time with higher expression of APOBEC3G. Purple lines represent higher expression of APOBEC3; blue lines represent lower expression of APOBEC3
An alternative strategy is to inhibit known AID and APOBEC3B regulatory pathways with known small molecule inhibitors. As we summarized in Figure 3, NF‐κB inhibitors, SN50 and MG132, reduce the expression of AID in liver cancer34 or PKC/IKK inhibitors suppress NF‐κB activation; downregulated APOBEC3B expression might control cancer progression and metastasis mediated by APOBEC3B‐induced mutagenesis.53 APOBEC‐driven replication stress in cancer cells may provide a potential opportunity for ATR‐targeted therapy.63 Targeting replication stress‐directed APOBEC3B activation has broad implications for many inhibitors including afatinib and lapatinib for ERBB2 amplification, LY294002 and rapamycin for PI3K/mTOR signaling, U0126 for MAPK, CHK1 inhibitor, and ATM/ATR inhibitor.54 P53 defective cells with high expression of APOBEC3B are more sensitive to DNA damage response inhibitors (eg, PARP inhibitor), which have been used in clinical trials.64 In addition, APOBEC3 highly expressed tumors are correlated with high mutation burden, and overexpression of APOBEC3 paralogs appear to play pivotal roles in the regulation of programmed death‐ligand 1 (PD‐L1) expression,65, 66 which is the predictive biomarker for subset cancer immunotherapy. It is reasoned that these subtypes of patients may respond to immune checkpoint blockage therapies such as programmed cell death protein 1 (PD‐1)/PD‐L1 and cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) inhibitors. Taken together, targeting APOBEC with various approaches for subsets of patients may improve clinical outcomes. Meanwhile, potential adverse effects derived from the inhibition of APOBEC may arise, such as decreasing host immune defense against viral infection.
6. SUMMARY AND FUTURE PERSPECTIVES
Cancer is currently viewed as a disease of evolving genomic instability and abnormal epigenomic modifications. Except for known exogenous mutagens causing cancer, endogenous mutators, such as APOBEC family genes, are found to associate with cancer development, and their high expressions correlate to mutational burden in diverse types of cancer and poor survival time in subtypes of cancer. Several studies indicate that NF‐κB pathway activation, p53 inactivation by HPV oncoprotein E6/E7 activation or loss‐of‐function mutations in the TP53 gene and replication stress activation are responsible for transcriptional activation of APOBEC, in particular, APOBEC3B. We proposed different strategies to directly or indirectly target APOBEC3B as a prototype regimen for inactivation of aberrant expression of APOBEC for potential interventions. We are only beginning to appreciate the effects of APOBEC‐mediated mutagenesis in cancer; many questions remain to be answered. For example, whether elevated expressions of APOBEC3 are causative factors for cancer initiation or just a bystander consequence during cancer development; how can we effectively prevent the off‐target harmful mutations by APOBEC at the precancerous stage; why elevated expression of APOBEC has a paradoxically clinical benefit on cancers; and how can we quickly apply hypothetical approaches to manage subtypes of cancer patients with aberrant APOBEC expression in the clinical setting. With more research focused on APOBEC biology, we hope these answers will be addressed in the near future.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
The National Natural Science Foundation of China (no. 81171992), Henan Natural Science Foundation (no. 162300410279) supported this work. We thank all scientists who contribute their work in this field and we apologize to our colleagues whose outstanding contributions to the growing APOBEC field were not cited as primary references because of space constraints.
Gao J, Choudhry H, Cao W. Apolipoprotein B mRNA editing enzyme catalytic polypeptide‐like family genes activation and regulation during tumorigenesis. Cancer Sci. 2018;109:2375–2382. 10.1111/cas.13658
Funding information The National Natural Science Foundation of China (Grant/Award Number: ‘81171992’), Henan Natural Science Foundation (Grant/Award Number: ‘162300410279’).
Contributor Information
Jianlong Gao, Email: 15298570305@163.com.
Wei Cao, Email: caoweiyu@hotmail.com.
REFERENCES
- 1. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 3. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuong KJ, Loeb LA. APOBEC3B mutagenesis in cancer. Nat Genet. 2013;45:964‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Refsland EW, Harris RS. The APOBEC3 family of retroelement restriction factors. Curr Top Microbiol Immunol. 2013;371:1‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868‐877. [DOI] [PubMed] [Google Scholar]
- 8. Salter JD, Bennett RP, Smith HC. The APOBEC protein family: united by structure, divergent in function. Trends Biochem Sci. 2016;41:578‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi K, Carpenter MA, Banerjee S, et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat Struct Mol Biol. 2017;24:131‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan K, Roberts SA, Klimczak LJ, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47:1067‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shaban NM, Shi K, Li M, Aihara H, Harris RS. 1.92 angstrom zinc‐free APOBEC3F catalytic domain crystal structure. J Mol Biol. 2016;428:2307‐2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008;9:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kinomoto M, Kanno T, Shimura M, et al. All APOBEC3 family proteins differentially inhibit LINE‐1 retrotransposition. Nucleic Acids Res. 2007;35:2955‐2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ito F, Fu Y, Kao SA, Yang H, Chen XS. Family‐wide comparative analysis of cytidine and methylcytidine deamination by eleven human APOBEC proteins. J Mol Biol. 2017;429:1787‐1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muramatsu M, Sankaranand VS, Anant S, et al. Specific expression of activation‐induced cytidine deaminase (AID), a novel member of the RNA‐editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470‐18476. [DOI] [PubMed] [Google Scholar]
- 17. Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation‐induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295:1301‐1306. [DOI] [PubMed] [Google Scholar]
- 18. Rodriguez‐Cortez VC, Martinez‐Redondo P, Catala‐Moll F, et al. Activation‐induced cytidine deaminase targets SUV4‐20‐mediated histone H4K20 trimethylation to class‐switch recombination sites. Sci Rep. 2017;7:7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Okazaki IM, Hiai H, Kakazu N, et al. Constitutive expression of AID leads to tumorigenesis. J Exp Med. 2003;197:1173‐1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Watashi K, Liang G, Iwamoto M, et al. Interleukin‐1 and tumor necrosis factor‐alpha trigger restriction of hepatitis B virus infection via a cytidine deaminase activation‐induced cytidine deaminase (AID). J Biol Chem. 2013;288:31715‐31727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kou T, Marusawa H, Kinoshita K, et al. Expression of activation‐induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int J Cancer. 2007;120:469‐476. [DOI] [PubMed] [Google Scholar]
- 22. Navaratnam N, Morrison JR, Bhattacharya S, et al. The p27 catalytic subunit of the apolipoprotein B mRNA editing enzyme is a cytidine deaminase. J Biol Chem. 1993;268:20709‐20712. [PubMed] [Google Scholar]
- 23. Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816‐1819. [DOI] [PubMed] [Google Scholar]
- 24. Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. Transcriptome‐wide sequencing reveals numerous APOBEC1 mRNA‐editing targets in transcript 3’ UTRs. Nat Struct Mol Biol. 2011;18:230‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harris RS, Petersen‐Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10:1247‐1253. [DOI] [PubMed] [Google Scholar]
- 26. Siriwardena SU, Chen K, Bhagwat AS. Functions and malfunctions of mammalian DNA‐Cytosine deaminases. Chem Rev. 2016;116:12688‐12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vieira VC, Soares MA. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed Res Int. 2013;2013:683095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV‐1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646‐650. [DOI] [PubMed] [Google Scholar]
- 29. Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99‐103. [DOI] [PubMed] [Google Scholar]
- 30. Henderson S, Fenton T. APOBEC3 genes: retroviral restriction factors to cancer drivers. Trends Mol Med. 2015;21:274‐284. [DOI] [PubMed] [Google Scholar]
- 31. Pilzecker B, Buoninfante OA, Pritchard C, et al. PrimPol prevents APOBEC/AID family mediated DNA mutagenesis. Nucleic Acids Res. 2016;44:4734‐4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamanaka S, Balestra ME, Ferrell LD, et al. Apolipoprotein B mRNA‐editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc Natl Acad Sci USA. 1995;92:8483‐8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dorsett Y, McBride KM, Jankovic M, et al. MicroRNA‐155 suppresses activation‐induced cytidine deaminase‐mediated Myc‐Igh translocation. Immunity. 2008;28:630‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Endo Y, Marusawa H, Kinoshita K, et al. Expression of activation‐induced cytidine deaminase in human hepatocytes via NF‐kappaB signaling. Oncogene. 2007;26:5587‐5595. [DOI] [PubMed] [Google Scholar]
- 35. Matsumoto Y, Marusawa H, Kinoshita K, et al. Helicobacter pylori infection triggers aberrant expression of activation‐induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470‐476. [DOI] [PubMed] [Google Scholar]
- 36. Beale RC, Petersen‐Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context‐dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2004;337:585‐596. [DOI] [PubMed] [Google Scholar]
- 37. Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC‐mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus‐driven tumor development. Cell Rep. 2014;7:1833‐1841. [DOI] [PubMed] [Google Scholar]
- 38. Li Z, Abraham BJ, Berezovskaya A, et al. APOBEC signature mutation generates an oncogenic enhancer that drives LMO1 expression in T‐ALL. Leukemia. 2017;31:2057‐2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Long J, Delahanty RJ, Li G, et al. A common deletion in the APOBEC3 genes and breast cancer risk. J Natl Cancer Inst. 2013;105:573‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xuan D, Li G, Cai Q, et al. APOBEC3 deletion polymorphism is associated with breast cancer risk among women of European ancestry. Carcinogenesis. 2013;34:2240‐2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nik‐Zainal S, Wedge DC, Alexandrov LB, et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC‐dependent mutations in breast cancer. Nat Genet. 2014;46:487‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roberts SA, Lawrence MS, Klimczak LJ, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nik‐Zainal S, Alexandrov LB, Wedge DC, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nordentoft I, Lamy P, Birkenkamp‐Demtroder K, et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 2014;7:1649‐1663. [DOI] [PubMed] [Google Scholar]
- 46. de Bruin EC, McGranahan N, Mitter R, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Law EK, Sieuwerts AM, LaPara K, et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER‐positive breast cancer. Sci Adv. 2016;2:e1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ojesina AI, Lichtenstein L, Freeman SS, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kondo S, Wakae K, Wakisaka N, et al. APOBEC3A associates with human papillomavirus genome integration in oropharyngeal cancers. Oncogene. 2017;36:1687‐1697. [DOI] [PubMed] [Google Scholar]
- 51. Periyasamy M, Singh AK, Gemma C, et al. p53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res. 2017;45:11056‐11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Menendez D, Nguyen TA, Snipe J, Resnick MA. The Cytidine Deaminase APOBEC3 Family Is Subject to Transcriptional Regulation by p53. Mol Can Res. 2017;15:735‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maruyama W, Shirakawa K, Matsui H, et al. Classical NF‐kappaB pathway is responsible for APOBEC3B expression in cancer cells. Biochem Biophys Res Comm. 2016;478:1466‐1471. [DOI] [PubMed] [Google Scholar]
- 54. Kanu N, Cerone MA, Goh G, et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol. 2016;17:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cao W, Wu W. MicroRNAs regulate APOBEC gene expression. Histol Histopathol. 2018;33:117‐120. [DOI] [PubMed] [Google Scholar]
- 56. Kaul D, Arora M, Garg A, Sharma S. MALT1 induced immune response is governed by miR‐2909 RNomics. Mol Immunol. 2015;64:210‐217. [DOI] [PubMed] [Google Scholar]
- 57. Garg A, Kaul D, Chauhan N. APOBEC3G governs to ensure cellular oncogenic transformation. Blood Cells Mol Dis. 2015;55:248‐254. [DOI] [PubMed] [Google Scholar]
- 58. Sharma S, Garg A, Dhanda RS, Kaul D. APOBEC3G governs the generation of truncated AATF protein to ensure oncogenic transformation. Cell Biol Int. 2016;40:1366‐1371. [DOI] [PubMed] [Google Scholar]
- 59. Chen Z, Eggerman TL, Bocharov AV, et al. Heat shock proteins stimulate APOBEC‐3‐mediated cytidine deamination in the hepatitis B virus. J Biol Chem. 2017;292:13459‐13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen TW, Lee CC, Liu H, et al. APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat Commun. 2017;8:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Olson ME, Harris RS, Harki DA. APOBEC enzymes as targets for virus and cancer therapy. Cell Chem Biol. 2018;25:36‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kouno T, Silvas TV, Hilbert BJ, et al. Crystal structure of APOBEC3A bound to single‐stranded DNA reveals structural basis for cytidine deamination and specificity. Nat Commun. 2017;8:15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Buisson R, Lawrence MS, Benes CH, Zou L. APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Can Res. 2017;77:4567‐4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nikkila J, Kumar R, Campbell J, et al. Elevated APOBEC3B expression drives a kataegic‐like mutation signature and replication stress‐related therapeutic vulnerabilities in p53‐defective cells. Br J Cancer. 2017;117:113‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mullane SA, Werner L, Rosenberg J, et al. Correlation of Apobec Mrna expression with overall survival and pd‐l1 expression in urothelial carcinoma. Sci Rep. 2016;6:27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Boichard A, Tsigelny IF, Kurzrock R. High expression of PD‐1 ligands is associated with kataegis mutational signature and APOBEC3 alterations. Oncoimmunology. 2017;6:e1284719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Uhlen M, Zhang C, Lee S, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]