

Abstract
The inhibitory checkpoint molecule programmed death (PD)‐1 plays a vital role in maintaining immune homeostasis upon binding to its ligands, PD‐L1 and PD‐L2. Several recent studies have demonstrated that soluble PD‐1 (sPD‐1) can block the interaction between membrane PD‐1 and PD‐L1 to enhance the antitumor capability of T cells. However, the affinity of natural sPD‐1 binding to PD‐L1 is too low to permit therapeutic applications. Here, a PD‐1 variant with approximately 3000‐fold and 70‐fold affinity increase to bind PD‐L1 and PD‐L2, respectively, was generated through directed molecular evolution and phage display technology. Structural analysis showed that mutations at amino acid positions 124 and 132 of PD‐1 played major roles in enhancing the affinity of PD‐1 binding to its ligands. The high‐affinity PD‐1 mutant could compete with the binding of antibodies specific to PD‐L1 or PD‐L2 on cancer cells or dendritic cells, and it could enhance the proliferation and IFN‐γ release of activated lymphocytes. These features potentially qualify the high‐affinity PD‐1 variant as a unique candidate for the development of a new class of PD‐1 immune‐checkpoint blockade therapeutics.
Keywords: inhibitory receptor, PD‐1, PD‐L1, PD‐L2, phage display
1. INTRODUCTION
Cancer immunotherapy, the exploitation of one's own immune system for fighting cancer, has become a major focus in medical research and industry. Several different approaches, such as immunotherapy using tumor‐infiltrating lymphocytes (TIL),1 checkpoint blockade,2, 3 engineered T cells with chimeric antigen receptors or T‐cell receptors,4, 5 and high‐affinity TCR‐based biologics,6 have been investigated and have demonstrated promising outcomes. Among these strategies, checkpoint blockade7, 8, 9 is pivotal for determining immune status.
As a CD28/B7 family member, programmed cell death protein 1 (PD‐1) is a monomeric type Ι transmembrane glycoprotein. The complete PD‐1 molecule comprises an extracellular immunoglobulin variable region (IgV)‐like domain, a transmembrane domain and a cytoplasmic tail. The PD‐1 inhibitory effects are achieved through the cytoplasmic tail, which contains 2 tyrosine‐based signal transmission motifs, 1 immune receptor tyrosine‐based inhibition motif (ITIM) and 1 immune receptor tyrosine‐based switch motif (ITSM).10, 11, 12 During the antigen engagement of TCR, PD‐1 regulates T‐cell responses by ligating its ligands, PD‐L1 (also known as B7‐H1 or CD274) or PD‐L2 (also known as B7‐DC or CD273). A cascade of signals leads to recruitment of Src homology 2‐based tyrosine phosphatases (SHP‐2) to the ITSM, which further induces dephosphorylation of proximal TCR signaling molecules, such as ZAP70, protein kinase C θ and CD3 ζ, to terminate the TCR signals.13, 14 The signal cascade exerts a strong negative effect on production of cytokines, such as IL‐2, IL‐4 and IFN‐γ, and on proliferation of T cells.13, 15 Yokosuka et al14 demonstrate that the PD‐1/PD‐L1 complex is functional in the immunological synapse formed at the contact site between T cells and antigen‐presenting cells. The colocalization brings the complex near the TCR‐peptide MHC complex to inhibit the TCR signaling pathway more efficiently during antigen recognition.
The expression patterns of PD‐L1 differ from those of PD‐L2. Unlike PD‐L2, which is only expressed on a few cell types such as macrophages and dendritic cells (DC), PD‐L1 is widely expressed on activated T and B cells, monocytes, DC and macrophages, and at low levels on organs such as the lungs, heart, kidneys and liver.9, 16 The PD‐L1 expression can also be upregulated by stimuli such as IFN‐γ, IL‐2, IL‐7, IL‐15 and IL‐21.16, 17 The expression pattern of PD‐L1 is thought to reflect the important physiological role of PD‐1/PD‐L1 signaling in preventing excessive T‐cell activation, protecting peripheral tissue damage during an immunity to infection, and limiting autoimmunity to maintain T‐cell tolerance.15, 18, 19, 20 However, as the tumor microenvironment harbors a mass of TIL with heightened levels of PD‐1, the PD‐1 can be exploited by tumor cells through the upsurge of PD‐L1 expression to induce cancer immune tolerance.21 Blockade of the PD‐1/PD‐L1 interaction will abate the negative signaling and release T cells from the inhibitory state, thus restoring their antitumor capability.22, 23 Several antibody drugs targeting PD‐1 or PD‐L1 have been approved by the US Food and Drug Administration for the treatment of malignant tumors, and many more are being tested in hundreds of clinical trials.21, 22
Five alternative forms of PD‐1, including the full‐length form and the soluble form, were described in 2005. They could be produced by peripheral blood mononuclear cells (PBMC) of healthy donors, and the levels of these transcripts were increased simultaneously during the activation of PBMC with CD3 and CD28 stimulation.24 Several studies have demonstrated that sPD‐1 could block the interaction between membrane PD‐1 and PD‐L1, or the engagement of PD‐L1/B7‐1 and PD‐L2/PD‐1, and lead to the promotion of T‐cell antitumor immunity.25, 26, 27 These features make sPD‐1 an attractive agent for blockading the PD‐1/PD‐L1 pathway.28
However, the binding of natural sPD‐1 to PD‐L1 is weak, with a dissociation constant (KD) of 8.2 μmol/L,29 making sPD‐1 a poor antagonist for blockading the PD‐1/PD‐L1 pathway. In previous studies, high‐affinity sPD‐1 molecules have been reported to blockade the PD‐1 axis.30, 31 However, we generated PD‐1‐based high‐affinity PD‐L1 binders with a unique mutation that played a dominant role in enhancing the binding strength. Our investigation demonstrated that the newly generated high‐affinity PD‐1 molecular could interact with PD‐L1 on tumor cell surfaces and compete with antibodies specific to PD‐L1 and PD‐L2. The high‐affinity PD‐1 variants provide us a new way to address the problem of immune inhibition in the tumor microenvironment.
2. MATERIALS AND METHODS
2.1. Construction of human PD‐1 libraries and screening for high‐affinity PD‐1 variants
The phage display libraries of human PD‐1 (hPD‐1) affinity maturation were constructed by PCR using primer mixes (Table 1). The mutated hPD‐1 fragments were cloned into pGZ196 with Escherichia coli TG1 as the host. Tenfold serial diluent of 10 μL in the transformant culture was spread onto TYE plates to for titer determining. The remaining culture was spread on a larger TYE plate to grow overnight.
Table 1.
Oligonucleotide primers used for generation of mutant libraries for affinity maturation
| Name | Primer 5′‐3′ |
|---|---|
| L1‐F | AATCAGACCGATAAACTGGCG |
| L1‐Rb | GCCAGTTTATCGGTCTGATTMNNCGGGCTMNNACGMNNCCAMNNCAGMNNAAACGATTCGCTC |
| L2‐Fb | GGTATCGTATGAGCCCGTCTNNKNNKNNKGATNNKCTGGCGNNKTTCCCGGAAGATCGCTCTCAGCC |
| L2‐R | GACGGGCTCATACGATACCAGTTCAG |
| L3‐F1b | CAGACCGATAAACTGGCGNNKTTCNNKGAAGATCGCTCTCAGNNKNNKCAAGACTGCCGTTTTCGC |
| L3‐R1 | CGCCAGTTTATCGGTCTGATTAG |
| L3‐F2b | CGCAATGACTCCGGCACCTACNNKTGTGGTGCAATTTCACTGGC |
| L3‐R2 | GTAGGTGCCGGAGTCATTGCG |
| L4‐Fb | CTCCGGCACCTACCTGTGTNNKNNKNNKTCANNKGCTNNKAAAGCCCAAATCAAAGAATCGC |
| L4‐R | ACACAGGTAGGTGCCGGAGTC |
| L5‐Fb | GGTGCAATTTCACTGGCTCCGNNKNNKNNKNNKAAANNKTCGCTGCGTGCGGAACTGCG |
| L5‐R | GAGCCAGTGAAATTGCACCACACAGG |
| Primer‐Fa | CTCATGCCATGGCAAATCCGCCGACG |
| Primer‐Ra | AAGGAAAAAAGCGGCCGCTTCGGCACGACGTTC |
NcoI/NotI are indicated by italic sequences.
In some of the primers, K was replaced with a mixture of G/T; M was replaced with a mixture of C/A; N was replaced with a mixture of A/T/C/G. The frequency of each base in each mixture was equal.
Phage display screening was performed as described in Liang et al32 except for the change in human PD‐L1 (hPD‐L1) concentrations, which were in order as 100, 50, 50, 10, 1, 1, 1 nmol/L, respectively, for each round of selection. After 7 rounds of selection, sublibraries obtained from each round of biopanning were used in polyclonal phage ELISA assays with biotinylated and immobilized hPD‐L1 (0.5 μg/mL) as antigen. For identification of high‐affinity PD‐1, 400 randomly picked clones were screened by monoclonal phage ELISA as above, and 99 ELISA‐positive PD‐1 variants were sequenced.
2.2. Phage western blotting
Approximately 2 × 1011 pfu of PD‐1 or helper phage particles were boiled for 10 min in sodium dodecyl sulfate (SDS) loading buffer and were clarified by centrifugation. After separation in a 12% SDS–PAGE, the gel was transferred to a PVDF membrane at 200 mA for 2 hours. The membrane was incubated with blocking buffer for 1 hour at room temperature, followed by incubation overnight with anti‐pIII Ab (NEB, Ipswich, MA, USA) at 1:1000 dilution in blocking buffer. After 3 washes, the membrane was incubated for 1 hour in blocking buffer containing HRP‐conjugated secondary antibody (Multi Sciences, Hangzhou, China) at 1:800 dilution. Color was developed with an ECL detection kit (Multi Sciences).
2.3. Proliferation and enzyme‐linked immunosorbent spot
For proliferation assays, PBMCs from healthy donors were obtained by Ficoll–Hypaque density gradient centrifugation and prestained with 1 μmol/L carboxyfluorescein diacetate succinimidyl ester (CFDA‐SE; Molecular Probes, Eugene, OR, USA) as described previously.33 The prestained PBMCs were cultured as 2 × 105 cells per well with RPMI‐1640 containing 10% FBS. The cells were stimulated with antibodies of 15 ng/mL anti‐CD3 (aCD3, clone: OKT3; BioLegend) and 7.5 ng/mL anti‐CD28 (aCD28, clone: CD28.2; R&D, Minneapolis, MN, USA) or 30 ng/mL aCD3 and 15 ng/mL aCD28. The culture was incubated for 4 days in the presence or absence of soluble hPD‐1, L5B7 or anti‐PD‐L1 Ab at the concentration of 5 μg/mL.
For IFN‐γ enzyme‐linked immunosorbent spot (ELISpot) assays, PBMCs in 2 × 104 per well were cultured and treated as above for 40 hours. IFN‐γ was detected according to the manufacturer's instructions (BD Pharmingen).
2.4. Extra materials and methods
Data S1 provide the information about cell lines, gene synthesis and vector construction, phage display of PD‐1 and phage ELISA, protein expression and affinity determination with surface plasmon resonance (SPR), dendritic cells preparation, flow cytometry analysis.
3. RESULTS
3.1. M13 phage displayed human PD‐1 bound human PD‐L1
The truncated hPD‐1 gene with a C93S mutation was fused to geneIII of M13 phage to encode a protein III N‐terminal fusion protein. The fusion protein displayed on phage was examined by western blotting, and it was clearly visible as an upper band above the thick pIII protein band in a well‐controlled setting (Figure 1A). We confirmed the hPD‐1 extracellular region displayed on phage retaining hPD‐L1 binding capability. As shown in Figure 1B, helper phage did not bind biotinylated hPD‐L1, but PD‐1‐displaying phage showed clear binding. These results demonstrated that the hPD‐1 displayed on M13 phage could be used for the molecular evolution of hPD‐1.
Figure 1.
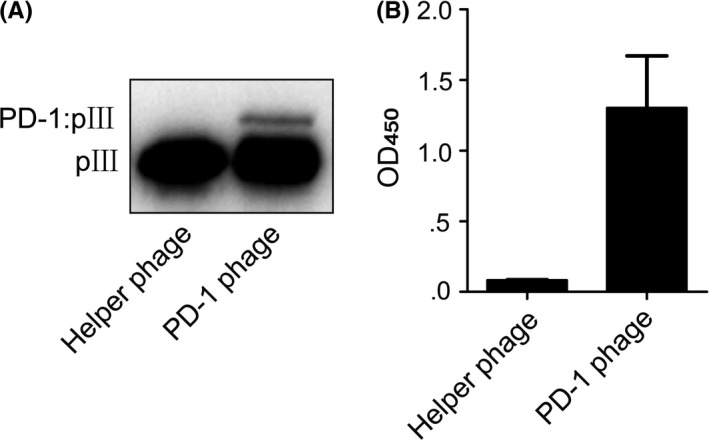
Display of hPD‐1 extracellular region on M13 phage. A, Western blot of PD‐1 phage. B, The binding of PD‐1 phage to hPD‐L1 detected by ELISA. Helper phage was run as a negative control. Error bars indicate SD (n = 3)
3.2. Library design and characterization
Mouse PD‐1 (mPD‐1) and hPD‐1 share 15 identical residues among a total of 18 interface residues in contact with hPD‐L1.29 Therefore, we used the mPD‐1/hPD‐L1 complex crystal structure (PDB number: 3BIK) to analyze the hPD‐1/hPD‐L1 interaction and selected the 18 residues for the library construction.
In addition, using the YASARA program,34 we analyzed the potential contribution of the residues to the protein stability, especially their impacts on the hydrophobic core formation or structural integrity. As a result, six of such residues were selected, including Met70, Ala81, Pro83, Pro89, Ala125 and Gln133. Together with the 18 identified interface residues, a final list of 24 residues was generated in the hPD‐1 template to construct the phage display libraries (Figure 2A). The 24 target residues on hPD‐1 were assigned to 5 libraries (listed in Figure 2B) to keep each library theoretical variants <108. We demonstrated more than 92% in‐frame sequences by sequencing 20 randomly selected colonies from each library. All 5 individual libraries had the cell mixtures of more than 108 separated colonies, and were greater than the theoretical library size of approximately 3.3 × 107 encoding sequence diversity.
Figure 2.
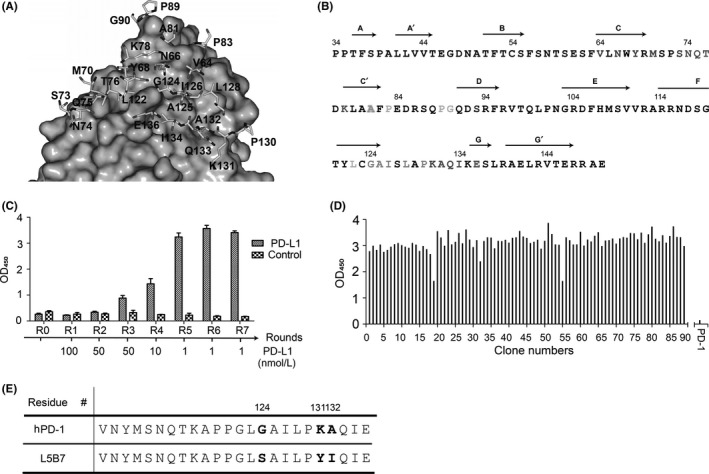
Design of hPD‐1 phage displays libraries and isolation of hPD‐L1 (hPD‐L1)‐binding phage by biopanning. A, The 24 selected residues for construction of phage display libraries. The structure of mPD‐1/hPD‐L1 (PDB number: 3BIK) was used as the template to build the hPD‐1/hPD‐L1 complex model. Residues of hPD‐1 are shown as stick models, whereas hPD‐L1 is represented by gray surface. B, Amino acid division of hPD‐1 for the library construction is shown in colors: Library 1, red; Library 2, green; Library 3, orange; Library 4, cyan; Library 5, pink. The Ala81 was assigned to Library 2 and Library 3. Residue Ser93 (blue) was a mutation from Cys. Secondary structural elements of hPD‐1 extracellular region were shown on top of the sequence. C, Polyclonal phage ELISA. The polyclonal phage particles from each round of biopannings were tested for their binding to 0.5 μg/mL immobilized hPD‐L1. An irrelevant protein (pHLA) was used as the negative control. The result of Library 5 was shown as a representative. D, Monoclonal phage ELISA. 400 individual colonies of each library from the 7th round of biopanning were tested for their binding to hPD‐L1 by monoclonal phage ELISA. The result of Library 5 is shown as a representative. E, Partial sequence alignment of hPD‐1 and L5B7. The 3 mutated amino acids are shown as G124S, K131Y and A132I
3.3. Isolation of high‐affinity PD‐1 variants
Seven rounds of biopanning were carried out for the selection of high‐affinity binders with the hPD‐L1 captured by streptavidin‐magnetic beads. Sublibraries generated from each round of biopanning were assessed for the hPD‐L1 binding with polyclonal phage ELISA (Figure 2C). We found that PD‐1 displayed on phage showed significant binding to immobilized hPD‐L1 after the screening, but no binding to a negative control antigen.
Over 400 randomly picked colonies from the 7th round of biopannings were evaluated for their binding to hPD‐L1, and nearly all the monoclonal phages tested were positive (Figure 2D). After sequence analysis, we found 19 unique sequences from the 5 libraries.
The soluble proteins of the 19 PD‐1 variants were expressed as inclusion bodies in E. coli with a pET28a vector. The functional proteins were purified with anion exchange and size exclusion chromatography after refolding. We obtained 1.4‐2.8 mg of proteins showing purity over 95% from 7‐mg inclusion bodies.
The affinity range of binding to hPD‐L1 for these variants varied greatly from micromolar to nanomolar determined by SPR (Table 2). The highest affinity PD‐1 variant was a clone from library 5, named L5B7, which containing 3 mutations of G124S, K131Y and A132I (Figure 2E). The KD of L5B7 binding to PD‐L1 was 0.69 nmol/L, showing an approximately 3000‐fold affinity increase. Its T1/2 was accordingly increased from <1 seconds to approximately 500 seconds (Table 2). These results indicated that the affinity‐based phage display biopanning was successful in isolating high‐affinity binders on a large background.
Table 2.
Kinetic and binding parameters for mutants of PD‐1 binding to hPD‐L1
| Protein | kon a (106 mol/L−1 s−1) | koff a (s−1) | KD b (μmol/L) | T1/2 (s) | |
|---|---|---|---|---|---|
| 0 | hPD‐1 | 0.431 | 1.1410 | 2.65 | <1 |
| 1 | L1G2 | 0.307 | 0.3081 | 1.00 | 3.25 |
| 2 | L1D3 | 0.553 | 0.4661 | 8.39 × 10−1 | 2.15 |
| 3 | L4H10 | 0.570 | 0.3375 | 5.92 × 10−1 | 2.96 |
| 4 | L4E1 | 0.438 | 0.0765 | 1.75 × 10−1 | 13.07 |
| 5 | L5D1 | 2.231 | 0.3500 | 1.57 × 10−1 | 2.86 |
| 6 | L5A4 | 1.380 | 0.2000 | 1.45 × 10−1 | 5.00 |
| 7 | L4B3 | 1.371 | 0.1802 | 1.31 × 10−1 | 5.55 |
| 8 | L4C6 | 0.883 | 0.8925 | 1.01 × 10−1 | 1.12 |
| 9 | L4C11 | 1.294 | 0.1212 | 9.37 × 10−2 | 8.25 |
| 10 | L4D7 | 1.476 | 0.1383 | 9.37 × 10−2 | 7.23 |
| 11 | L4F6 | 1.104 | 0.1017 | 9.21 × 10−2 | 9.83 |
| 12 | L4E8 | 0.723 | 0.0610 | 8.44 × 10−2 | 16.39 |
| 13 | L3E3 | 0.922 | 0.0599 | 6.49 × 10−2 | 16.69 |
| 14 | L3A7 | 1.853 | 0.0978 | 5.28 × 10−2 | 10.22 |
| 15 | L2B12 | 0.983 | 0.0386 | 3.93 × 10−2 | 25.91 |
| 16 | L1B2 | 0.818 | 0.0275 | 3.36 × 10−2 | 36.36 |
| 17 | L2F10 | 1.549 | 0.0085 | 5.49 × 10−3 | 117.65 |
| 18 | L2F8 | 2.586 | 0.0043 | 1.67 × 10−3 | 232.56 |
| 19 | L5B7 | 2.938 | 0.0020 | 6.87 × 10−4 | 500.00 |
Association (kon) and dissociation (koff) rate constants were measured with BIAcore T200.
KD was calculated as koff/kon.
3.4. L5B7 recognizes human PD‐L1 or human PD‐L2 on surfaces of tumor or dendritic cells
To assess the ability of binding to hPD‐L1 expressed on cancer cells, the purified hPD‐1 and L5B7 soluble proteins were used to stain MDA‐MB‐231 tumor cell lines, which showed over 90% PD‐L1+ detected by flow cytometry using an anti‐PD‐L1 Ab (29E.1A3) (data not shown). The half‐maximum concentrations (EC50) binding to hPD‐L1 on MDA‐MB‐231 cells were calculated as 4.86 μmol/L for hPD‐1 and 2.90 nmol/L for L5B7 (Figure 3A). We also found that L5B7 could compete with the binding of 29E.1A3 to PD‐L1 on the cells. With 29E.1A3 at the concentration of 3 × 10−8 mol/L used, we observed that the antibody signal reduction was clearly dependent on the L5B7 dose from 1.3 × 10−5 mol/L to 1.3 × 10−9 mol/L (Figure 3B).
Figure 3.
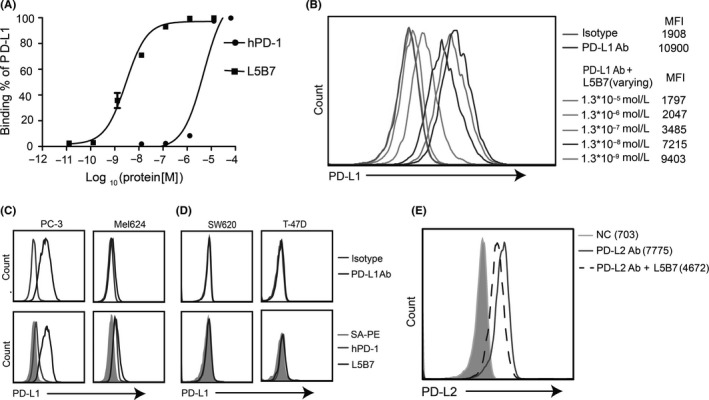
Interaction of hPD‐1 and L5B7 with hPD‐L1 or hPD‐L2 on the cell surface. A, Kinetic analysis of soluble hPD‐1 and L5B7 binding to hPD‐L1 on MDA‐MB‐231 cells. B, The soluble L5B7 competing with anti‐PD‐L1 Ab (29E.1A3) binding to hPD‐L1 on MDA‐MB‐231 cells. C,D, The soluble hPD‐1 and L5B7 binding to hPD‐L1 on various cells, (C) PD‐L1+ tumor cells PC‐3 and Mel624, (D) PD‐L1− tumor cells SW620 and T‐47D. Top panel, isotype control and anti‐PD‐L1 Ab. Bottom panel, SA‐PE, hPD‐1 and L5B7. E, The soluble L5B7 competing with anti‐PD‐L2 Ab (M1H18) binding to hPD‐L2 on mature dendritic cells
We further examined the binding of soluble L5B7 to various tumor cell lines. In PD‐L1+ cells, the strongest staining was detected on prostatic cancer cell line PC‐3, whereas the weakest staining was detected on melanoma cell line Mel624 (Figure 3C). These results coincided well with signal intensities of 29E.1A3 stained cells, indicating that the differential binding profiles of the cells were mainly due to the differential expression of PD‐L1. In contrast, both cells were stained weakly by hPD‐1. PD‐L1− cells SW620 and T‐47D showed negative binding by 29E.1A3, hPD‐1 and L5B7 (Figure 3D).
We also found that L5B7 retained hPD‐L2 binding capability, and the KD of L5B7 binding to hPD‐L2 was 13.7 nmol/L, showing an approximately70‐fold increase compared with hPD‐1 (Table 3). The supplementation of soluble L5B7 (1.0 × 10−5 mol/L) could reduce over 40% mean fluorescence intensity (MFI) generated by an anti‐PD‐L2 Ab (M1H18) binding to hPD‐L2 on mature DC (Figure 3E).
Table 3.
Kinetic and binding parameters for L5B7 binding to hPD‐L2
| Protein | kon a (106 mol/L−1 s−1) | koff a (s−1) | KD b (μmol/L) | T1/2 (s) |
|---|---|---|---|---|
| hPD‐1 | 0.356 | 0.3730 | 1.05 | 2.68 |
| L5B7 | 0.933 | 0.0128 | 1.37 × 10−2 | 78.13 |
Association (kon) and dissociation (koff) rate constants were measured with BIAcore T200.
KD was calculated as koff/kon.
3.5. The mutations at positions 124 and 132 play a key role in increasing affinity
The molecular basis of increased affinity for L5B7 was revealed by virtual mutations in silico with the help of the YASARA program and its plugin FoldX.34, 35 As indicated in Figure 4A, when glycine at position 124 was replaced with serine, the aromatic ring of Tyr123 of hPD‐L1 was rotated because of the enlarged side chain of serine compared with glycine. It caused the 2 phenol groups of Tyr68 (hPD‐1) and Tyr123 (hPD‐L1) to be oriented antiparallel in the complex. The π‐π stacking interaction strength was increased from 2.7 to 5.8, and an additional hydrogen bond was formed between Ser124 and Tyr123. Meanwhile, we also observed the variant L4C11, where a single‐site mutation from glycine to valine brought about a 28‐fold affinity increase compared to hPD‐1. The structural analysis showed that the site mutation increased the π‐π stacking interaction strength from 2.7 to 6.0.
Figure 4.
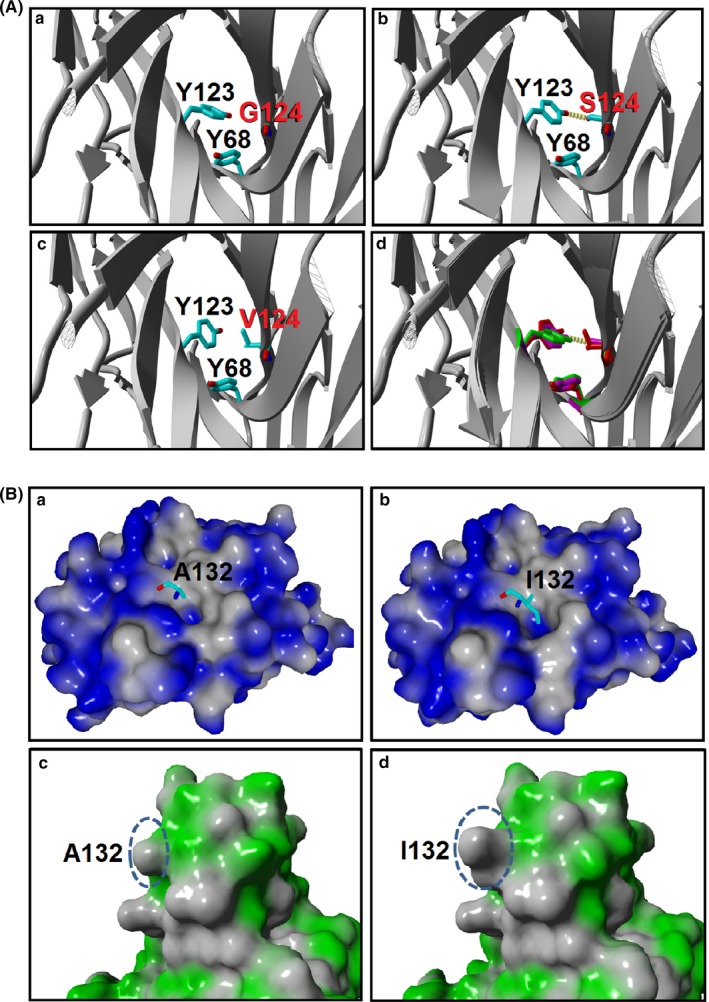
Close‐up views of π‐π interaction in Gly124 and hydrophobic interaction in Ala132. A, The enhancement of π‐π interaction in G124S and G124V PD‐1 variants. The hPD‐1/hPD‐L1 (PDB: 4ZQK) served as the template to build G124S and G124V mutation models. The complex was shown in ribbon representation, and the Tyr123 of hPD‐L1, Tyr68, Gly/Ser/Val124 of hPD‐1 are shown in a stick model. Hydrogen bonds are depicted as yellow dashed lines: (a) Gly124; (b) Ser124; (c) Val124; and (d) superposition of Gly124 (green), Ser124 (magenta) and Val124 (red) in hPD‐1 variants. B, Hydrophobic interaction was enhanced in the hPD‐1 variants. Molecular surfaces of hPD‐L1 (a,b) and hPD‐1 (c,d) are represented with blue or green color, respectively, whereas the hydrophobic part is colored gray. Residues Ala/Ile132 of hPD‐1/variant are shown as stick models (a,b), and the corresponding molecular surfaces are indicated as dashed circles (c,d)
In the case of residue Ala132 of hPD‐1, the A132I mutation caused the hPD‐L1 interface hydrophobic local surface to enlarge from 628 Å2 to 682 Å2, because of the larger side chain (Figure 4B). The hydrophobic pocket (formed by Ile54, Val68, Tyr56 and Met115) of hPD‐L1 became more compact when accommodating Ile132 compared to Ala132. The hydrophobic interaction strength increased from 66.7 to 81.3, further implying the A132I mutation's possible contribution to the increased affinity. To verify this speculation, we introduced point mutations on the hPD‐1, A132I and a combined mutation of G124V and A132I that showed KD values of 9.9 and 0.97 nmol/L, respectively. It indicated that a single A132I mutation could cause the affinity to increase over 200‐fold, and addition of G124V mutation delivered a further 10‐fold increase. This result showed the importance of the mutations at these 2 positions for improving the affinity of hPD‐1/hPD‐L1 interaction.
To analyze the structural basis of L5B7 binding to hPD‐L2, we built a homology model of hPD‐1/hPD‐L2 using an available crystal structure of mouse PD‐1/PD‐L2 (PDB number: 3BP5) (Figure 5A), which showed high homology scores with the human counterparts. We applied a structure‐based sequence alignment of apo‐hPD‐1 (PDB number: 3RRQ), hPD‐1/hPD‐L1 (PDB number: 4ZQK), and mPD‐1/mPD‐L2. We found that the FG loop orientation of PD‐1 in the PD‐1/PD‐L1 complex was different from that of the PD‐1/PD‐L2 complex (Figure 5B), which might result from a missing sequence in PD‐L2 between positions 70 and 71 in comparison with PD‐L1 (Figure 5B,C). When glycine at position 124 was substituted by serine, an additional hydrogen bond could be formed between Ser124 of hPD‐1 and Tyr112 of hPD‐L2 to secure the phenol group of Tyr112. This interaction and the residue Trp110 of hPD‐L2 composed the hydrophobic core at the hPD‐1/hPD‐L2 interface to stabilize the complex of hPD‐1 and hPD‐L2 (Figure 5D). For A132I mutation, the hydrophobic interaction of Ile132 with Ile105 of hPD‐L2 could be produced, and the mutation increased the stability of the side chain of Leu128 of hPD‐1 (Figure 5D). Compared to the effect on the affinity binding to hPD‐L1, the role of the mutations in enhancing the affinity of hPD‐1 binding to hPD‐L2 was very weak. This weakness might be due to the orientation discrepancy of the FG loop caused by the missing sequence in hPD‐L2.
Figure 5.
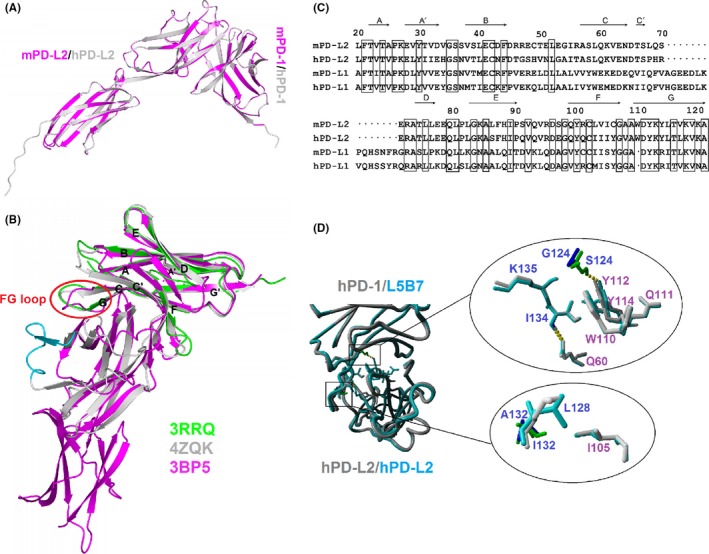
Structural analysis of interaction between L5B7 and hPD‐L2. A, Structure of hPD‐1/hPD‐L2 superimposed on mPD‐1/mPD‐L2 (PDB number: 3BP5) represented as ribbon models. mPD‐1/mPD‐L2 are colored magenta. B, Structure‐based sequence alignment of apo‐hPD‐1 (green, PDB number: 3RRQ), hPD‐1/hPD‐L1 (gray, PDB number: 4ZQK), mPD‐1/mPD‐L2 (magenta, PDB number: 3BP5). The black letters label the secondary structural elements of hPD‐1. The red circle represents the orientation discrepancy of the FG loop of PD‐1. The cyan represented redundant hPD‐L1 sequence on structure compared to mPD‐L2. C, The sequence alignment of extracellular IgV domains of mouse/human PD‐L2 and PD‐L1. The black dot shows the missing residues in PD‐L2 compared to PD‐L1. The black box represents a conservative sequence in PD‐L1 and PD‐L2. The secondary structural elements of the extracellular IgV region of mPD‐L2 are shown on top of the sequence. D, The role of G124S and A132I in enhancing L5B7 binding to hPD‐L2. The mutations are located at the key position related to interaction of hPD‐1 and hPD‐L2 (left). The physical impact of G124S and A132I in enhancing L5B7 binding to hPD‐L2 (right). Top, G124S (blue/green); bottom, A132I (blue/green)
3.6. L5B7 enhanced the proliferation and IFN‐γ release of activated peripheral blood mononuclear cells
We used aCD3 and aCD28 stimulated PBMC as a T‐cell activation model to examine the immune‐checkpoint blocking activity of soluble hPD‐1, L5B7 and anti‐PD‐L1 Ab respectively at the concentration of 5‐μg/mL. L5B7 promoted a significantly improved PBMC proliferation from 27.1% to 55.4%, in comparison with that of hPD‐1 (to 30.8%) and anti‐PD‐L1 Ab (to 45.3%) when PBMC were stimulated with low‐dose antibodies of 15 ng/mL aCD3 and 7.5 ng/mL aCD28. Such significance could not be observed when the PBMC were stimulated with high‐dose antibodies of 30 ng/mL aCD3 and 15 ng/mL aCD28. In this condition, L5B7 promoted the cell proliferation from 63.1% to 76.4%, which was similar to that of anti‐PD‐L1 Ab (to 74.9%) but was higher than that of hPD‐1 (to 63.7%) (Figure 6A,B). The result was consistent with the previous result that the strength of TCR signals would determine the PD‐1‐mediated inhibitory effects on T‐cell function.15
Figure 6.
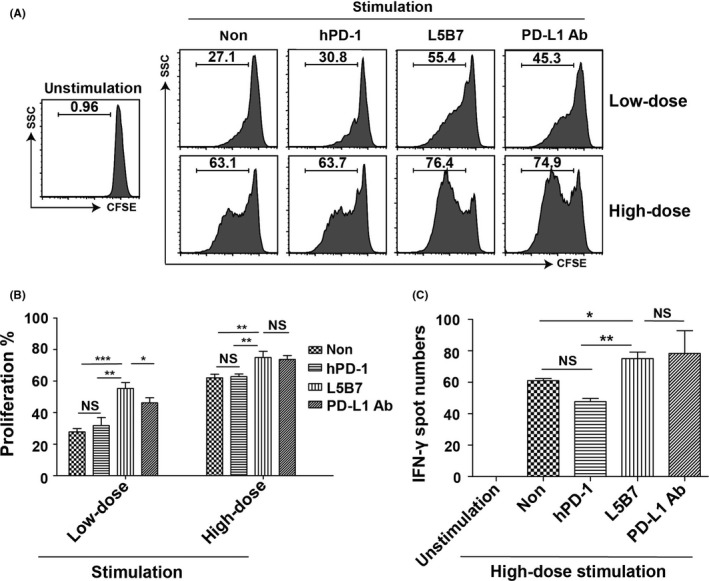
The enhancement of proliferation and IFN‐γ release of activated PBMC. A, The flow cytometry data of proliferation of activated PBMC with anti‐CD3 Ab (aCD3) and anti‐CD28 Ab (aCD28) in the presence or absence of soluble hPD‐1, L5B7 and anti‐PD‐L1 Ab (5 μg/mL). PBMCwere stimulated with low‐dose antibodies of 15 ng/mL aCD3 and 7.5 ng/mL aCD28 or high‐dose antibodies of 30 ng/mL aCD3 and 15 ng/mL aCD28. B, The statistical data of proliferating cells in (A). Error bars indicated SD (n = 3). C, ELISpot assays showing IFN‐γ release of PBMC activated with 30 ng/mL aCD3 and 15 ng/mL aCD28 in the presence or absence of soluble hPD‐1, L5B7 and anti‐PD‐L1 Ab (5 μg/mL). Error bars indicated SD (n = 3). Unpaired Student's t test, NS, P > .05; *, P < .05; **, P < .01; ***, P < .001
Enhanced cell activation was also revealed with increasing IFN‐γ release of activated PBMC followed by the treatment of soluble L5B7. As shown in Figure 6C, when activated PBMC were treated with L5B7 and anti‐PD‐L1 Ab at the concentration of 5 μg/mL, L5B7 increased the IFN‐γ release significantly as well as that of anti‐PD‐L1 Ab in comparison with the cells stimulated only with high‐dose antibodies of 30 ng/mL aCD3 and 15 ng/mL aCD28. In contrast, hPD‐1 significantly reduced IFN‐γ release. The results indicated that L5B7 could effectively promote IFN‐γ release of activated PBMC.
4. DISCUSSION
The PD‐1/PD‐L1 signaling pathway is crucial to the suppression of T‐cell responses, and it has emerged as an important target for cancer immunotherapy. To generate a unique PD‐1 axis blockade reagent, we produced soluble high‐affinity PD‐1 molecules for hPD‐L1 binding.
The structure of the hPD‐1/hPD‐L1 complex (PDB number: 4ZQK) was published36 after this work had been started, which provided a judgment for our rational phage display design. In light of the new data, we noted that all residues on hPD‐1 with major contributions to hPD‐1/hPD‐L1 binding were selected (Asn66, Tyr68, Gly124, Ile126, Leu128, Ile134 and Glu136). Moreover, approximately 60% of residues relating to hPD‐1 plasticity (from Met70 to Asp77) in the hPD‐1/hPD‐L1 complex were also selected.
During the current study, Maute et al30 developed a series of high‐affinity PD‐1 mutants through yeast surface display. The highest affinity mutant was HAC PD‐1, with 10 residues mutated for the KD to reach approximately 0.1 nmol/L, but it showed no binding to hPD‐L2. The binding is enthalpy driven, and Y68H, M70E and K78T contributed greatly to the stability improvement of the interface for HAC PD‐1 binding to hPD‐L1 through hydrogen bonding and salt bridge formation. However, when the 3 residues were alone or grouped together, no significant stability or affinity increase was traceable except for M70E. It was argued that the 3 substitutions in HAC PD‐1 worked with the other mutations synergistically to increase the stability and affinity to bind hPD‐L1.37
We found that the affinity of L5B7 with 3 residue mutations could attain a KD with 0.69 nmol/L for binding hPD‐L1. After analyzing the complex structure, we found that the key forces in the affinity improvement were the increased strength of the π‐π stacking and hydrophobic interactions in L5B7 binding to hPD‐L1. It is advantageous that the affinity increase was produced from a minimum number of mutations, as introducing too many mutations can potentially make the original human protein more immunogenic.
We demonstrated the binding capacity of soluble L5B7 to PD‐L1 on the cell surface using direct staining and anti‐PD‐L1 Ab competitive assays. Interestingly, soluble L5B7 can bind to hPD‐L2 with an approximately 70‐fold affinity increase in comparison with hPD‐1. As PD‐L2 expression is limited to only a few cell types, it attracts less concern than PD‐L1. In fact, the affinity of PD‐1/PD‐L2 is 3‐fold to 4‐fold higher than that of PD‐1/PD‐L1, suggesting that the binding of PD‐L2 to PD‐1 is better than that of PD‐L1. Normally PD‐L2 is mainly expressed on APC, indicating its vital role during T‐cell priming,19 whereas the signaling pathway may be exploited by tumor cells to enhance the immunosuppressive mechanism in the tumor environment.38, 39 Increasing evidence also demonstrates that PD‐L2 can be expressed on many tumor cell types,40 and the expression is related negatively to tumor‐infiltrating lymphocytes,41 prognosis42 and survival.43 Targeting both PD‐L1 and PD‐L2 with 1 molecule may, therefore, prove beneficial in clinical practice. In head and neck squamous cell carcinoma patients treated with immune‐checkpoint blockade, PD‐L1/2 double‐positive patients had better responses and longer median progression‐free survival in comparison with those PD‐L1 single‐positive patients.40 In our study, with the feature of high affinity to both PD‐L1 and PD‐L2, soluble L5B7 might have an enhanced role in the suppression of PD‐1/PD‐ligand pathways. It should benefit from blockading not only PD‐1/PD‐L1 and PD‐L1/CD80 but also PD‐1/PD‐L2 interactions, and enable better antitumor effects.
The therapeutic essence of anti‐PD Abs as the immune‐checkpoint blockade is dependent on 2 features of the binding affinity and the location of the epitope for preventing the PD‐1/PD‐L1 interaction.44, 45, 46 For soluble PD‐1 based immune‐checkpoint blockade reagent, the “epitope” location is perfect for preventing the membrane PD‐1 and PD‐L1 interaction; the key is the binding affinity for effective therapeutic molecules. It is not difficult to generate antibodies specific to PD‐1 or PD‐L1, but not all the antibodies are suitable as the PD‐1 axis blockade for the clinical applications. One of the reasons is that the epitope for the antibodies should overlap the interface of PD‐1/PD‐L1 interaction. Thus, compared with antibody‐based therapies, our approach may provide an effective way to develop PD‐1 axis blockades.
In the clinic, the immunity enhancement by the blockade of PD‐1 signaling pathway with high‐affinity anti‐PD‐L1 Abs may generate a unique inflammatory toxicity, known as immune‐related adverse events (irAE). It has been reported that the KDs of durvalumab, atezolizumab, avelumab and BMS‐936559 binding to PD‐L1 are 0.667, 1.75, 0.0467 and 0.83 nmol/L, respectively.47 There is no report that can indicate the relation between the affinity and irAE. Although L5B7 has a KD of 0.69 nmol/L for binding to PD‐L1, which is in the range of these antibodies, we have no idea how L5B7 will behave in terms of irAE. We should pay more attention to this issue in future research.
In summary, using directed molecular evolution and high‐throughput phage screening, we identified a PD‐1 mutant L5B7 from a series of high‐affinity PD‐1 mutants that coData Data uld be used to block the PD‐1/PD‐L1 and PD‐1/PD‐L2 interactions. The high‐affinity PD‐1 with further modification may be developed as biologics for clinic applications. Our study also provides additional strategies utilizing the checkpoint proteins, such as CTLA‐4 and Tim‐3, or their ligands, for developing new types of immune‐checkpoint protein blockades.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Supporting information
ACKNOWLEDGMENTS
We thank Peipei Zhou, Synat Kang, Yajing Zhang, Yongjie Sun and Siqi Peng for experiment preparation, and Dr Jianbin Zhang for initial editing of the manuscript.
Li Y, Liang Z, Tian Y, et al. High‐affinity PD‐1 molecules deliver improved interaction with PD‐L1 and PD‐L2. Cancer Sci. 2018;109:2435–2445. 10.1111/cas.13666
REFERENCES
- 1. Lee S, Margolin K. Tumor‐infiltrating lymphocytes in melanoma. Curr Oncol Rep. 2012;14:468‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185‐202. [DOI] [PubMed] [Google Scholar]
- 3. Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. A systematic review of immunotherapy in urologic cancer: evolving roles for targeting of CTLA‐4, PD‐1/PD‐L1, and HLA‐G. Eur Urol. 2015;68:267‐279. [DOI] [PubMed] [Google Scholar]
- 4. Brentjens RJ, Davila ML, Riviere I, et al. CD19‐targeted T cells rapidly induce molecular remissions in adults with chemotherapy‐refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robbins PF, Kassim SH, Tran TL, et al. A pilot trial using lymphocytes genetically engineered with an NY‐ESO‐1‐reactive T‐cell receptor: long‐term follow‐up and correlates with response. Clin Cancer Res. 2015;21:1019‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oates J, Hassan NJ, Jakobsen BK. ImmTACs for targeted cancer therapy: why, what, how, and which. Mol Immunol. 2015;67:67‐74. [DOI] [PubMed] [Google Scholar]
- 7. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sledzinska A, Menger L, Bergerhoff K, Peggs KS, Quezada SA. Negative immune checkpoints on T lymphocytes and their relevance to cancer immunotherapy. Mol Oncol. 2015;9:1936‐1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD‐1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887‐3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang X, Schwartz JC, Guo X, et al. Structural and functional analysis of the costimulatory receptor programmed death‐1. Immunity. 2004;20:337‐347. [DOI] [PubMed] [Google Scholar]
- 12. Chen L. Co‐inhibitory molecules of the B7‐CD28 family in the control of T‐cell immunity. Nat Rev Immunol. 2004;4:336‐347. [DOI] [PubMed] [Google Scholar]
- 13. Latchman Y, Wood CR, Chernova T, et al. PD‐L2 is a second ligand for PD‐1 and inhibits T cell activation. Nat Immunol. 2001;2:261‐268. [DOI] [PubMed] [Google Scholar]
- 14. Yokosuka T, Takamatsu M, Kobayashi‐Imanishi W, Hashimoto‐Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dai S, Jia R, Zhang X, Fang Q, Huang L. The PD‐1/PD‐Ls pathway and autoimmune diseases. Cell Immunol. 2014;290:72‐79. [DOI] [PubMed] [Google Scholar]
- 17. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015;15:45‐56. [DOI] [PubMed] [Google Scholar]
- 18. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD‐L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Latchman YE, Liang SC, Wu Y, et al. PD‐L1‐deficient mice show that PD‐L1 on T cells, antigen‐presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA. 2004;101:10691‐10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen L, Han X. Anti‐PD‐1/PD‐L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384‐3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti‐PD‐L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558‐562. [DOI] [PubMed] [Google Scholar]
- 23. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167‐3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nielsen C, Ohm‐Laursen L, Barington T, Husby S, Lillevang ST. Alternative splice variants of the human PD‐1 gene. Cell Immunol. 2005;235:109‐116. [DOI] [PubMed] [Google Scholar]
- 25. Liu C, Jiang J, Gao L, et al. Soluble PD‐1 aggravates progression of collagen‐induced arthritis through Th1 and Th17 pathways. Arthritis Res Ther. 2015;17:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shin SP, Seo HH, Shin JH, et al. Adenovirus expressing both thymidine kinase and soluble PD1 enhances antitumor immunity by strengthening CD8 T‐cell response. Mol Ther. 2013;21:688‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He YF, Zhang GM, Wang XH, et al. Blocking programmed death‐1 ligand‐PD‐1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. J Immunol. 2004;173:4919‐4928. [DOI] [PubMed] [Google Scholar]
- 28. Song MY, Park SH, Nam HJ, Choi DH, Sung YC. Enhancement of vaccine‐induced primary and memory CD8(+) T‐cell responses by soluble PD‐1. J Immunother. 2011;34:297‐306. [DOI] [PubMed] [Google Scholar]
- 29. Lin DY, Tanaka Y, Iwasaki M, et al. The PD‐1/PD‐L1 complex resembles the antigen‐binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci USA. 2008;105:3011‐3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maute RL, Gordon SR, Mayer AT, et al. Engineering high‐affinity PD‐1 variants for optimized immunotherapy and immuno‐PET imaging. Proc Natl Acad Sci USA. 2015;112:E6506‐E6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lazar‐Molnar E, Scandiuzzi L, Basu I, et al. Structure‐guided development of a high‐affinity human Programmed Cell Death‐1: implications for tumor immunotherapy. EBioMedicine. 2017;17:30‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang Z, Tian Y, Cai W, et al. High‐affinity human PD‐L1 variants attenuate the suppression of T cell activation. Oncotarget. 2017;8:88360‐88375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Younes SA, Yassine‐Diab B, Dumont AR, et al. HIV‐1 viremia prevents the establishment of interleukin 2‐producing HIV‐specific memory CD4+ T cells endowed with proliferative capacity. J Exp Med. 2003;198:1909‐1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA–a self‐parameterizing force field. Proteins. 2002;47:393‐402. [DOI] [PubMed] [Google Scholar]
- 35. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382‐W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zak KM, Kitel R, Przetocka S, et al. Structure of the complex of human programmed death 1, PD‐1, and Its ligand PD‐L1. Structure. 2015;23:2341‐2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pascolutti R, Sun X, Kao J, et al. Structure and dynamics of PD‐L1 and an ultra‐high‐affinity PD‐1 receptor mutant. Structure. 2016;24:1719‐1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kenkel JA, Tseng WW, Davidson MG, et al. An immunosuppressive dendritic cell subset accumulates at secondary sites and promotes metastasis in pancreatic cancer. Cancer Res. 2017;77:4158‐4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Horlad H, Ma C, Yano H, et al. An IL‐27/Stat3 axis induces expression of programmed cell death 1 ligands (PD‐L1/2) on infiltrating macrophages in lymphoma. Cancer Sci. 2016;107:1696‐1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yearley JH, Gibson C, Yu N, et al. PD‐L2 expression in human tumors: relevance to anti‐PD‐1 therapy in cancer. Clin Cancer Res. 2017;23:3158‐3167. [DOI] [PubMed] [Google Scholar]
- 41. Sridharan V, Gjini E, Liao X, et al. Immune profiling of adenoid cystic carcinoma: PD‐L2 expression and associations with tumor‐infiltrating lymphocytes. Cancer Immunol Res. 2016;4:679‐687. [DOI] [PubMed] [Google Scholar]
- 42. Wang H, Yao H, Li C, et al. PD‐L2 expression in colorectal cancer: independent prognostic effect and targetability by deglycosylation. Oncoimmunology. 2017;6:e1327494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang H, Kim JS, Choi YJ, et al. Overexpression of PD‐L2 is associated with shorter relapse‐free survival in patients with malignant salivary gland tumors. Onco Targets Ther. 2017;10:2983‐2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tan S, Zhang H, Chai Y, et al. An unexpected N‐terminal loop in PD‐1 dominates binding by nivolumab. Nat Commun. 2017;8:14369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Na Z, Yeo SP, Bharath SR, et al. Structural basis for blocking PD‐1‐mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res. 2017;27:147‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu K, Tan S, Chai Y, et al. Structural basis of anti‐PD‐L1 monoclonal antibody avelumab for tumor therapy. Cell Res. 2017;27:151‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tan S, Liu K, Chai Y, et al. Distinct PD‐L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein Cell. 2018;9:135‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials