

Abstract
Runt (Runt domain)‐related transcription factor 1 (RUNX1) is a transcription factor belonging to the core‐binding factor (CBF) family. It is considered to be a master regulator of hematopoiesis and has been regarded as a tumor suppressor because it is essential for definitive hematopoiesis in vertebrates. It is one of the most frequent target genes of chromosomal translocation in leukemia, and germ line mutation of RUNX1 causes familial platelet disorder with associated myeloid malignancies. Somatic cell mutations and chromosomal abnormalities, including those of RUNX1, are observed in myelodysplastic syndrome, acute myeloid leukemia, acute lymphoblastic leukemia, and chronic myelomonocytic leukemia at a high frequency. In addition, recent studies reported by us and other groups suggested that WT RUNX1 is needed for survival and proliferation of certain types of leukemia. In this review, we describe the significance and paradoxical requirement of RUNX1 tumor suppressor in hematological malignancies based on recent findings such as “Genetic compensation of RUNX family transcription factors in leukemia,” “RUNX1 inhibition‐induced inhibitory effects on leukemia cells through p53 activation” and our novel promising theory “Cluster regulation of RUNX (CROX)” through the RUNX gene switch method using pyrrole‐imidazole polyamides as a new technique that could contribute to the next generation of leukemia treatment strategies.
Keywords: CBFβ, leukemia, p53, polyamides, RUNX cluster
1. INTRODUCTION
Runt (Runt domain)‐related transcription factor 1 (RUNX1), also known as acute myeloid leukemia 1 protein, is an important transcription factor involved in hematopoietic stem cell differentiation.1 It is one of the most frequent target genes of chromosomal translocation in leukemia, and germ line mutation of RUNX1 causes familial platelet disorder with associated myeloid malignancies. Somatic cell mutations and chromosomal abnormalities, including those of RUNX1, are observed in myelodysplastic syndrome, acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and chronic myelomonocytic leukemia at a high frequency.2, 3, 4, 5 RUNX1 is one of the RUNX family proteins (RUNX1, RUNX2, RUNX3), and each forms a heterodimer with core‐binding factor β (CBFβ:PEBP2B).6, 7 RUNX regulates transcription of target genes by recognizing and binding to the core consensus sequence, 5′‐TGTGGT‐3′, and in rare cases, to 5′‐TGCGGT‐3′, through the RUNT domain.6, 7 Core‐binding factor β is non‐DNA‐binding subunit that enhances the transcription activity of RUNX by potentiating the DNA‐binding ability and stability of the RUNX family (Fig. 1).
Figure 1.
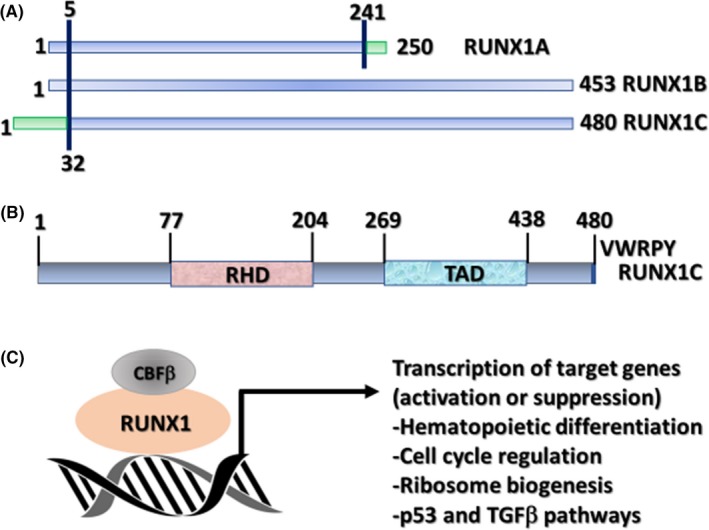
Runt (Runt domain)‐related transcription factor 1 (RUNX1) protein and its significant role as a transcription factor. (A) Schemas of the three major isoforms of RUNX1 (RUNX1A, B, and C). (B) Schematic of the protein encoded by the largest isoform (RUNX1C) with major functional domains marked: RUNX homology domain (RHD) and transactivation domain (TAD). Numbers above lines represent amino acid residues. (C) Schematic of RUNX1 heterodimerization with its non‐DNA‐binding partner, core‐binding factor β (CBFβ), and interaction with DNA at promoters of target genes that carry the specific binding site YGYGGTY, where Y is C or T TGFβ, transforming growth factor β. Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies, Blood. 2017;129:2070–20822, ©The American Society of Hematology
This image is not covered by the terms of the Creative Commons license of this publication. For permission to reuse, please contact the rights holder
Runt (Runt domain)‐related transcription factor 1 was previously found to be essential mainly for definitive hematopoiesis, based on the RUNX1‐knockout mouse phenotype.1 However, no consumption of hematopoietic stem cells was noted and the lifespan was not markedly influenced in the conditional knockout mouse, although slight change was noted in platelets, suggesting that RUNX1 is not essential for hematopoiesis in adults.8 It has been regarded as a tumor suppressor because mutation and chromosomal translocation of RUNX1 are observed in leukemia at a high rate, but this classical viewpoint has recently changed, based on our finding that WT RUNX1 is highly important for the developmental mechanisms of inv(16)(p13q22)/t(16;16)(p13;q22) leukemia (Inv16 leukemia) and mixed lineage leukemia (MLL).9, 10, 11 These studies described the carcinogenic properties of RUNX1 in the developmental mechanism of leukemia, but the neoplastic molecular basis of RUNX1, including that of other RUNX family members, for proliferation and maintenance of leukemia is unknown.
Transcription factor p53 (TP53) is a tumor suppressor; its deficiency promotes cancerization and progression.12 Research into the role of TP53 in human cancer has been most intense. Cancers with defective p53 are characterized by dedifferentiation, genetic instability, and metastatic potential, and they show an enhanced malignant phenotype.13 The frequency of p53 mutation is <10% in de novo AML, but it increases depending on the cancer type.14, 15 The relationship between p53 and the RUNX family is often discussed based on radiosensitivity experiments using an overexpression system, but functional cross‐talk between RUNX1 and p53 in carcinogenicity, proliferation, and maintenance has not been elucidated at the physiological level.
Pyrrole‐imidazole (PI) polyamides (PI‐polyamides) enter the minor groove of the DNA double‐helix structure and recognize a specific nucleotide sequence.16, 17 Using an advanced technique to modify PI‐polyamides with a nitrogen mustard alkylating agent, chlorambucil (Chb), developed by our collaborator, Professor Hiroshi Sugiyama, at the Department of Chemistry, Kyoto University Graduate School of Science (Kyoto, Japan),18 we newly developed Chb‐M' and Chb‐50, which recognize and bind to the core consensus sequences (consensus RUNX‐binding sequences) common to the RUNX family: 5′‐TGTGGT‐3′ and 5′‐TGCGGT‐3′. Both Chb‐M' and Chb‐50 can comprehensively inhibit the target genes of the RUNX family by antagonistically inhibiting recruitment of the RUNX family to the RUNX‐binding consensus sequences.19
2. LATEST FINDINGS
2.1. Inhibition of RUNX1 induces cell cycle arrest and apoptosis in AML and ALL cells
When RUNX1 is specifically inhibited in MLL‐AF4 (+FLT3 internal tandem duplication [ITD]) AML and tyrosine kinase inhibitor‐resistant Philadelphia chromosome‐positive ALL cell lines, p53 protein is stabilized and the p53 target genes are activated, inducing cell cycle arrest at G0/G1 and apoptosis. Regarding stabilization of the p53 protein, we clarified that BCL11A and TRIM24, factors that degrade and inhibit p53,20, 21, 22 are directly transcriptionally regulated by the RUNX family, and expression of both factors is significantly correlated with RUNX1 expression in acute leukemia.19 Disappearance of both factors by specific inhibition of RUNX1 results in stabilization of the p53 protein.19 However, in the process of establishing a leukemia cell line with p53 mutation, the dominant‐negative effect of mutant p53 protein inhibited the function of WT p53 and reduced RUNX1 inhibition‐induced cell suppression, revealing that the RUNX1 inhibition‐induced inhibitory effects on leukemia cells are dependent on stabilization and activation of WT p53 protein.19 It was suggested that high expression of RUNX1 in leukemia induces high expression of p53‐degrading BCL11A and TRIM24, destabilizing p53 protein in leukemia cells. This indicates a previously unknown role of RUNX1 in leukemia.19
2.2. Marked cell suppression effects by comprehensively inhibiting the entire mutual compensatory system of the RUNX family cluster
Inhibition of RUNX1 alone effectively suppressed cells, but some leukemia cells retained proliferation activity. When expression of the other RUNX family members, RUNX2 and RUNX3, was analyzed after RUNX1 inhibition over time, their protein levels were found to have increased. Thus, based on the assumption of the presence of a complementary compensation mechanism among the RUNX family members, we closely investigated the transcriptional regulatory mechanism of each factor. In the RUNX family, the consensus RUNX‐binding sequences are present in the promotor region and control expression of each other through inhibitory transcriptional regulation, suggesting that regulation by mutual inhibition maintains the protein level of the RUNX family at a constant level.19 We named the mechanism “genetic compensation of RUNX family transcription factors.” Inhibition of RUNX1 alone increases RUNX2 and RUNX3 proteins, resulting in compensation for the total level of RUNX family expression. Comprehensive inhibition of RUNX1, RUNX2, and RUNX3 led to marked cell suppression compared with inhibition of RUNX1 alone, clarifying that the RUNX family members are important for proliferation and maintenance of leukemia cells, and comprehensive regulation of these “cluster regulation of RUNX” (CROX) is a very effective strategy to suppress leukemia cells (Fig. 2).19
Figure 2.
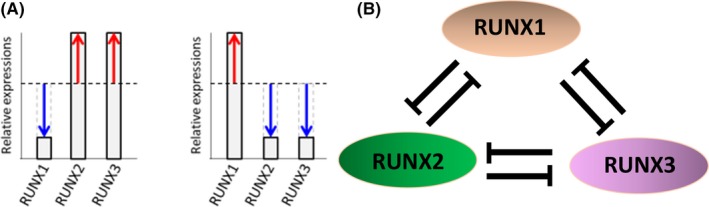
Schematic abstracts showing the mechanism “genetic compensation of Runt (Runt domain)‐related transcription factor 1 (RUNX) family transcription factors”
2.3. Development and proposal of a RUNX family cluster regulation method using PI‐polyamides
Pyrrole‐imidazole polyamide is a superior medium‐sized compound with selective binding ability to the specific nucleotide sequence in the DNA double‐helix structure. We designed original molecules, PI‐polyamide Chb‐M' and Chb‐50s, which specifically bind to the RUNX‐binding consensus sequences, and evaluated the gene expression pattern of the comprehensively regulated RUNX family. The RUNX family pathway was negatively controlled and strong cell suppression effects were observed at the nanomolar concentration (nmol/L) level in leukemia cell lines.19 In addition, the survival rate of a xenograft treatment model using immunodeficient mice was improved and no toxicity was noted in several preliminary toxicity tests. As all RUNX family members share the RUNX‐binding consensus sequences 5′‐TGTGGT‐3′ or 5′‐TGCGGT‐3′, CROX was possible. This was not possible with the RUNX1 inhibitor alone, and the compensatory mechanism of the RUNX family was successfully overcome (Fig. 3).19
Figure 3.
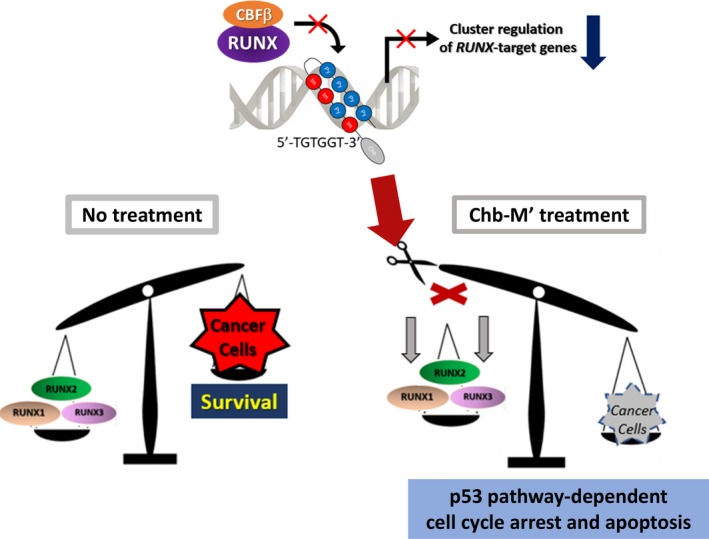
Modified chlorambucil (Chb‐M') as a safe lead compound of novel anticancer drug therapy. Chb‐M' targets the consensus Runt (Runt domain)‐related transcription factor (RUNX)‐binding sequences on the genome DNA and achieves “cluster regulation of RUNX,” which offers efficient antitumor activity through potentiating the pro‐apoptotic p53‐mediated cell death pathway. CBFβ, core‐binding factor β. Modified from Morita K, Suzuki K, Maeda S, et al. Genetic regulation of the RUNX transcription factor family has antitumor effects. J Clin Invest. 2017;127:2815–282819, by permission of American Society for Clinical Investigation.
This image is not covered by the terms of the Creative Commons license of this publication. For permission to reuse, please contact the rights holder
2.4. Core‐binding factor β as a pan‐cancer marker
To establish the RUNX family cluster regulation method, we analyzed the protein and mRNA expression levels of RUNX1, RUNX2, RUNX3, and CBFβ in leukemia and different cancer cell lines. Compared with that in normal‐type cells, the CBFβ expression level in tumor cells was significantly higher in both leukemia and different cancers.19 In addition, the total protein level of the RUNX family (RUNX1 + RUNX2 + RUNX3) was positively correlated with the CBFβ protein level. Moreover, the outcome was significantly poorer in the high CBFβ expression group for all cancer types. These findings suggested that the total RUNX family expression level is correlated with CBFβ expression, and the RUNX family cluster is a common antitumor target among cancers.19
2.5. Enhancement of the leukemia proliferation mechanism by moderate inhibition of RUNX1
Inv16 leukemia (FAB M4 Eo) induced by inv(16)(p13q22) is caused by the CBFβ– smooth muscle myosin heavy chain (SMMHC) fusion protein.23, 24, 25 The RUNX1 high‐affinity‐binding domain (HABD) is present in the fusion protein and it markedly enhances the binding ability between the fusion protein and RUNX1 compared with that between CBFβ (WT) and RUNX1. The fusion protein captures RUNX1 and transfers it to the cytoplasm from the nucleus, and RUNX1, previously considered a tumor suppressor, is dominantly inhibited by the fusion protein. These have been considered as the main causes of disease development.25 We extracted the most important RUNX1 inhibition domain in Inv16 leukemia by establishing five types of knockout mice in which the core RUNX1 inhibition domain present in the long tail of the fusion protein was deleted.9, 26 In D43 mice with the HABD deletion, the RUNX1‐transferring effect was inhibited and RUNX1 remained in the nucleus, which moderately improved the dominant RUNX1 inhibition. However, unexpectedly, D43 chimeric mice developed Inv16 leukemia at approximately 1 week after birth, even though no additional gene abnormality was present, suggesting that dominant RUNX1 inhibition is not essential for Inv16 leukemia and RUNX1 could be targeted by treatment.9
To investigate the correlation between the RUNX1 expression level and outcome of AML, the outcomes of three groups, RUNX1 high, RUNX1 intermediate, and RUNX1 low, were analyzed using the de novo AML cohort of The Cancer Genome Atlas clinical dataset. The outcome was the poorest in the RUNX1 intermediate group and most favorable in the RUNX1 low group, different from the previous one‐dimensional consideration of RUNX1 as a tumor suppressor; the presence of a mechanism enhancing leukemia proliferation due to moderately attenuated RUNX1 expression (moderate inhibition) was suggested. To elucidate this mechanism, we established cell lines with moderate and marked RUNX1 protein knockdown in two MLL leukemia + FLT3‐ITD cell lines and two non‐MLL leukemia cell lines. When the influence of the RUNX1 protein level on cell proliferation was evaluated, cell cycle arrest at G0/G1 and apoptosis induction were enhanced, and cell proliferation was inhibited in the marked knockdown cell lines, but the cell cycle was promoted and apoptosis induction was reduced in the moderate knockdown cell lines.27 We newly extracted GSTA2 (intracellular ROS scavenger and cell cycle promotion factor), belonging to the GST family, as a causative gene. It was clarified that under the condition of moderate knockdown of RUNX1 protein, GSTA2 markedly increased and its ROS scavenger action was promoted, decreasing ROS and intracellular free radicals (H2O2); this condition promoted the cell cycle.27 In addition, the RUNX‐binding consensus sequence is present in the GSTA2 promotor region and GSTA2 was found to be directly transcriptionally regulated by the RUNX family through efficient binding by RUNX1, RUNX2, and RUNX3. Moderate inhibition of RUNX1 most markedly increased the total RUNX family level through “genetic compensation of RUNX family transcription factors” and maximized the GSTA2 protein level, thereby reducing cell cycle promotion and induction of apoptosis. Indeed, in a mouse treatment model using a GST inhibitor, ethacrynic acid, the survival time was markedly extended (Fig. 4).27
Figure 4.
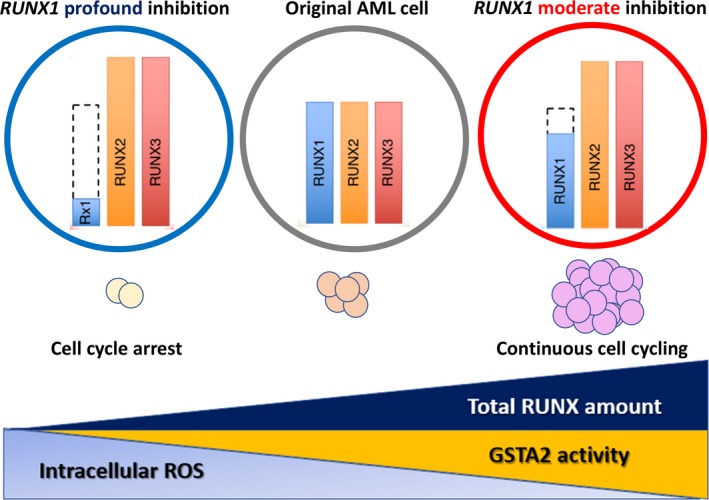
Schematic abstract showing that moderately attenuated Runt (Runt domain)‐related transcription factor 1 (RUNX1) expressions paradoxically enhance leukemogenesis in acute myeloid leukemia (AML) cells through intracellular environmental change by GSTA2, which depends on total RUNX amount, could be a novel therapeutic target in antileukemia strategies. ROS, reactive oxygen species. Morita K, Maeda S, Suzuki K, et al. Paradoxical enhancement of leukemogenesis in acute myeloid leukemia with moderately attenuated RUNX1 expressions. Blood Adv. 2017;1:1440–14527, ©The American Society of Hematology
This image is not covered by the terms of the Creative Commons license of this publication. For permission to reuse, please contact the rights holder
3. DISCUSSION AND CONCLUSION
Dominant inhibition of the tumor suppressor RUNX1 was previously considered to be the cause of the disease‐developing proliferation maintenance mechanism, mainly in CBF leukemia. However, our study clarified that WT RUNX1 is needed for survival and proliferation of leukemia cells, suggesting that a treatment strategy targeting RUNX1 is more effective. As the RUNX family has a mechanism to compensate for loss among the family members, moderate inhibition of RUNX1 increases the total RUNX family protein level (genetic compensation of RUNX family transcription factors), and the RUNX family transcriptionally regulates the p53 inhibitor. Therefore, several groups have tried to regulate RUNX1 alone, but suppression of leukemia cells was incomplete. To overcome this, we propose comprehensive RUNX family cluster regulation (CROX) as a leukemia treatment strategy because it is difficult to individually inhibit RUNX family proteins. As PI‐polyamide Chb‐M' and Chb‐50, currently in development, target the common consensus sequences of the RUNX family, individual inhibition of the members is not necessary. Although the comprehensive RUNX family cluster regulation has yet to be applied, we suggest the RUNX gene switch method using PI‐polyamides as a new technique that could contribute to the next generation of leukemia treatment strategies.
DISCLOSURE STATEMENT
The authors have no conflict of interest.
ACKNOWLEDGMENTS
This review depends on the results of research supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)), and Basic Science and Platform Technology Program for Innovative Biological Medicine from Japan Agency for Medical Research and Development (Grant No. 15am0301005h0002), Grant‐in‐Aid for Scientific Research (KAKENHI Grant Nos. 17H03597 and 16K14632) from the Japan Society for the Promotion of Science.
Kamikubo Y. Genetic compensation of RUNX family transcription factors in leukemia. Cancer Sci. 2018;109:2358–2363. 10.1111/cas.13664
REFERENCES
- 1. Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321‐330. [DOI] [PubMed] [Google Scholar]
- 2. Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017;129:2070‐2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu P, Tarl′e SA, Hajra A, et al. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041‐1044. [DOI] [PubMed] [Google Scholar]
- 4. Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci USA. 1991;88:10431‐10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer. 2015;15:81‐95. [DOI] [PubMed] [Google Scholar]
- 6. Warren AJ, Bravo J, Williams RL, Rabbitts TH. Structural basis for the heterodimeric interaction between the acute leukaemia‐associated transcription factors AML1 and CBFbeta. EMBO J. 2000;19:3004‐3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan J, Liu Y, Lukasik SM, Speck NA, Bushweller JH. CBFbeta allosterically regulates the Runx1 Runt domain via a dynamic conformational equilibrium. Nat Struct Mol Biol. 2004;11:901‐906. [DOI] [PubMed] [Google Scholar]
- 8. Ichikawa M, Asai T, Saito T, et al. AML‐1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med. 2004;10:299‐304. [DOI] [PubMed] [Google Scholar]
- 9. Kamikubo Y, Zhao L, Wunderlich M, et al. Accelerated leukemogenesis by truncated CBF beta‐SMMHC defective in high‐affinity binding with RUNX1. Cancer Cell. 2010;17:455‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goyama S, Schibler J, Cunningham L, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123:3876‐3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hyde RK, Zhao L, Alemu L, Liu PP. Runx1 is required for hematopoietic defects and leukemogenesis in Cbfb‐MYH11 knock‐in mice. Leukemia. 2015;29:1771‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berardi MJ, Sun C, Zehr M, et al. The Ig fold of the core binding factor alpha Runt domain is a member of a family of structurally and functionally related Ig‐fold DNA binding domains. Structure. 1999;7:1247‐1256. [DOI] [PubMed] [Google Scholar]
- 13. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53‐mediated tumour suppression. Nat Rev Cancer. 2014;14:359‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Renneville A, Roumier C, Biggio V, et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008;22:915‐931. [DOI] [PubMed] [Google Scholar]
- 16. Trauger JW, Baird EE, Dervan PB. Recognition of DNA by designed ligands at subnanomolar concentrations. Nature. 1996;382:559‐561. [DOI] [PubMed] [Google Scholar]
- 17. Best TP, Edelson BS, Nickols NG, Dervan PB. Nuclear localization of pyrrole‐imidazole polyamide‐fluorescein conjugates in cell culture. Proc Natl Acad Sci USA. 2003;100:12063‐12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bando T, Sugiyama H. Synthesis and biological properties of sequence‐specific DNA‐alkylating pyrrole‐imidazole polyamides. Acc Chem Res. 2006;39:935‐944. [DOI] [PubMed] [Google Scholar]
- 19. Morita K, Suzuki K, Maeda S, et al. Genetic regulation of the RUNX transcription factor family has antitumor effects. J Clin Invest. 2017;127:2815‐2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allton K, Jain AK, Herz HM, et al. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci USA. 2009;106:11612‐11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jain AK, Allton K, Duncan AD, Barton MC. TRIM24 is a p53‐induced E3‐ubiquitin ligase that undergoes ATM‐mediated phosphorylation and autodegradation during DNA damage. Mol Cell Biol. 2014;34:2695‐2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jain AK, Barton MC. Regulation of p53: TRIM24 enters the RING. Cell Cycle. 2009;8:3668‐3674. [DOI] [PubMed] [Google Scholar]
- 23. Liu P, Tarle SA, Hajra A, et al. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041‐1044. [DOI] [PubMed] [Google Scholar]
- 24. Liu PP, Hajra A, Wijmenga C, Collins FS. Molecular pathogenesis of the chromosome 16 inversion in the M4Eo subtype of acute myeloid leukemia. Blood. 1995;85:2289‐2302. [PubMed] [Google Scholar]
- 25. Lukasik SM, Zhang L, Corpora T, et al. Altered affinity of CBF beta‐SMMHC for Runx1 explains its role in leukemogenesis. Nat Struct Biol. 2002;9:674‐679. [DOI] [PubMed] [Google Scholar]
- 26. Kamikubo Y, Hyde RK, Zhao L, et al. The C‐terminus of CBFβ‐SMMHC is required to induce embryonic hematopoietic defects and leukemogenesis. Blood. 2013;121:638‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morita K, Maeda S, Suzuki K, et al. Paradoxical enhancement of leukemogenesis in acute myeloid leukemia with moderately‐attenuated RUNX1 expressions. Blood Adv.2017;1:1440‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]