

Abstract
Cancer‐associated fibroblasts (CAF), derived from stroma of cancer tissues, interact with cancer cells and play an important role in cancer initiation, growth, and metastasis. Nab‐paclitaxel (nab‐PTX) is a 130 nm albumin‐binding paclitaxel and recommended for many types of cancer chemotherapy. The nab‐PTX stromal‐disrupting effect during pancreatic cancer treatment has been reported. The aim of the present study was to determine the role of nab‐PTX in cancer cells and CAF interaction. Cancer cells (MIA PaCa‐2 and Panc‐1) were cocultured with CAF or treated with CAF conditioned medium, after which their migration and invasion ability, epithelial‐mesenchymal transition (EMT)‐related marker expression and C‐X‐C motif chemokine 10 (CXCL10) expression and secretion were detected. Nab‐PTX treatment was carried out during the coculture system or during preparation of CAF conditioned medium. Then cancer cell migration and invasion ability, EMT‐related marker expression, CXCL10 expression and secretion, and interleukin‐6 (IL‐6) expression and secretion by CAF were checked After coculture with CAF, migration and invasion ability of cancer cells increased. CAF also downregulated E‐cadherin and upregulated N‐cadherin and vimentin expression in cancer cells. During coculture or stimulation with cancer cell‐cultured medium, CAF significantly increased IL‐6 expression and secretion. However, nab‐PTX in the coculture system canceled CAF‐induced migration and invasion promotion and EMT‐related gene changes. Moreover, nab‐PTX increased CXCL10 expression of cancer cells which blocked CAF IL‐6 expression and secretion. Nab‐PTX treatment could increase CXCL10 expression of cancer cells which blocks CAF cancer cell migration and invasion‐promoting effect by inhibiting IL‐6 expression.
Keywords: 130 nm albumin‐bound paclitaxel, cancer‐associated fibroblast, chemokine CXCL10, epithelial‐mesenchymal transition, pancreatic neoplasms
Abbreviations
- CAF
cancer‐associated fibroblast
- CXCL10
C‐X‐C motif chemokine 10
- EMT
epithelial‐mesenchymal transition
- nab‐PTX
nab‐paclitaxel
1. INTRODUCTION
Pancreatic cancer is the fourth leading cause of cancer and only 10%‐20% of patients survive more than 5 years after surgery.1 Pathological fibrosis in pancreatic cancer, which compromises drug delivery, impedes immune cell accessibility and promotes disease aggression and therapy resistance, is one of the key reasons for high mortality.2 Fibroblasts arising from tumor stroma, so‐called CAF, are the main non‐parenchymal cell related to the excessive stromal formation and interact with cancer cells. In pancreatic cancer, cancer cell‐derived cytokines activate pancreatic stellate cells (PSC) forming the CAF phenotype and contribute to pancreatic extensive desmoplasia.3 Also, CAF can secrete many kinds of extracellular matrix components, cytokines, growth factors, and proteases participating in tumor‐stroma cross‐talk. Recent studies showed that CAF could not only promote tumorigenesis, growth, and angiogenesis but also facilitate invasion and metastasis.4, 5, 6 Therefore, a new target for cancer chemotherapy is to interrupt cancer cells and CAF interaction.7
Nab‐paclitaxel is a new paclitaxel nanoparticle formulation in which the paclitaxel is bound to albumin. Nab‐PTX, approved by the Food and Drug Administration (FDA) in 2012 as the first line in neoadjuvant therapy for pancreatic cancer, is widely used in clinical work and significantly improves outcome.8 Taking advantage of albumin‐binding, nab‐PTX efficiently accumulates in peri‐ and intratumoral areas dependent or independent of secreted protein acidic and rich in cysteine (SPARC) mediating.9, 10 Many clinical and animal studies showed that nab‐PTX treatment could disrupt tumor stroma, decrease PSC activity, and inflammatory cytokines.11 However, the role of nab‐PTX in tumor cells and CAF communication is unclear.
The aim of the present study was to investigate the effect of nab‐PTX on CAF‐induced tumor cell migration and invasion.
2. MATERIALS AND METHODS
2.1. Cell culture and treatment
Pancreatic cancer cell lines MIA PaCa‐2 and Panc‐1 were obtained from the Riken Cell Bank (Tsukuba, Japan). CAF were isolated from human pancreatic tissue resected from a pancreatic cancer patient by out‐growing methods as previously reported.12 The patient was newly diagnosed and had not received any relevant treatment prior to surgery. This was authorized in advance by the Institutional Review Board of the University of Tokushima Graduate School (approved ID number: 2790), and the patient provided written informed consent prior to donating the specimens. Pancreatic adenocarcinoma cancer cell lines MIA PaCa‐2 and Panc‐1 were cultured in DMEM (Life Technologies Ltd, Tokyo, Japan) with 10% FBS (Life Technologies Ltd) at 37°C and 5% CO2. CAF were purchased from Cellular Engineering Technologies Inc. (Coralville, IA, USA) Cells were cultured in HLCAF.E.Media‐450 (Cellular Engineering Technologies Inc.) with 10% FBS.
Nab‐paclitaxel (Abraxis; Taiho Pharmaceutical Co., Tokyo, Japan) was dissolved with PBS. Cancer cells were treated with 5 ng/mL nab‐paclitaxel‐containing medium for 24 hours.
2.2. CAF conditioning medium preparation
Cancer‐associated fibroblasts were grown with complete media in 6‐well dishes for 24 hours to reach 70%‐80% confluency. The 80% confluency cancer cells (MIA PaCa‐2 and Panc‐1) were cultured with 1% FBS DMEM for 24 hours and then changed to complete medium of CAF to cancer cells with 1% FBS culture medium for another 24‐hour culture. After that, the FBS‐free medium culture was followed. After 24‐hour culture, the supernatant was collected and filtered through a 0.2‐μm filter before use. For the CXCL10 or interleukin (IL)‐6 of conditioned medium neutralization, CXCL10 (266‐IP; R&D, Minneapolis, MN, USA) or IL‐6 (7270‐IL; R&D) antibody was added to the conditioned medium with a concentration of 0.2 μg/mL and incubated for 1 hour before use.
2.3. Migration and invasion assay
Transwell inserts (Corning, NY, USA) of 8 μm pore size were used for migration assay and precoated with Matrigel matrix (Corning) for invasion assay. MIA PaCa‐2 (5 × 104) or Panc‐1 (1 × 105) were seeded in the upper chamber, and 1‐2 × 104 CAF were seeded in the lower chamber separately. After cell attachment, both chambers were gently washed twice with DMEM, and then the upper chambers were inserted and incubated with serum‐free DMEM. After 24‐hour incubation, the cells which moved through the members of Transwells were fixed in 4% paraformaldehyde and stained with 0.2% crystal violet. The stained cells were counted under a microscope.
2.4. Enzyme‐linked immunosorbent assay
Level of IL‐6 and CXCL10 was determined using IL‐6 and CXCL10 Quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's protocol. Absorbance at 450 nm was measured using a plate reader (SpectraMax i3; Molecular Devices, Tokyo, Japan) at a correction wavelength of 540 nm.
2.5. Polymerase chain reaction analysis
Total RNA of each sample was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. cDNA was synthesized using a reverse transcription kit (Applied Biosystems, Foster City, CA, USA). The following TaqMan assays (assay identification number) were used: CDH1 (Hs01023895_m1), CDH12 (Hs00362037_m1), vimentin (Hs00958111_m1), IL‐6 (Hs00174131_m1), and CXCL10 (Hs00171042_m1). GAPDH (4352339E) was selected as internal control. StepOnePlus Real‐Time PCR System (Applied Biosystems) was used to carry out qRT‐PCR.
2.6. Cytokine array
Supernatants of cancer cells (Panc‐1) with or without nab‐PTX treatment were collected, and the particulates were removed by filtration through a 0.2‐μm filter. Cytokines in supernatants were detected with a Proteome Profiler Human Cytokine Array Kit (ARY005B; R&D Systems). After blocking, membranes were incubated with samples and antibody cocktail overnight at 4°C. After incubation, the membranes were washed and incubated with streptavidin‐HRP at room temperature for 30 minutes. Chemiluminescent detection reagents were incubated with the membrane for 1 minute, and the signal intensities on the membranes were detected with chemiluminescence (GE Healthcare, Little Chalfort, UK).
2.7. Western blotting
A RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA) containing both the protease inhibitor cocktail (Sigma, St Louis, MO, USA) and the PhosSTOP phosphatase inhibitor cocktail (Roche, Tokyo, Japan) was used to lyse cells. Protein concentrations were then measured with a BCA kit (Thermo Fisher Scientific) and equal amounts of extracted proteins were separated on 10% SDS‐PAGE gels and transferred onto PVDF membranes (Bio‐Rad, Hercules, CA, USA). The membranes were incubated with the indicated primary antibody including E‐cadherin (ab15148; Abcam, Cambridge, MA, USA), N‐cadherin (ab76011; Abcam), Vimentin (ab92547; Abcam), and β‐actin (#4967; Cell Signaling Technology, Danvers, MA, USA), followed by the appropriate HRP‐conjugated secondary antibody. Proteins were detected with chemiluminescence (GE Healthcare).
2.8. Statistical analysis
Data are presented as mean ± SD. Statistical analyses were carried out by ANOVA or Student's t test by SPSS 19.0 (IBM, NY, USA). Student's t test was used to compare the difference between 2 groups. One‐way analysis of variance (one‐way ANOVA) followed by Bonferroni test was used to compare the differences between more than 2 groups. P‐value <.05 indicated a statistically significant difference.
3. RESULTS
3.1. CAF promote pancreatic cancer cell migration and invasion
To investigate the effect of CAF on cancer cell migration and invasion, the cancer cells were cocultured with CAF for 24 hours. Compared with monoculture, cancer cells cocultured with CAF significantly increased the cell number transferred through the polyester or Matrigel matrix‐coated polyester (Figure 1A,B). Also, the conditioned medium derived from CAF which were stimulated with cancer cell‐cultured medium (CAF‐CM) significantly increased cancer cell migration and invasion ability.
Figure 1.
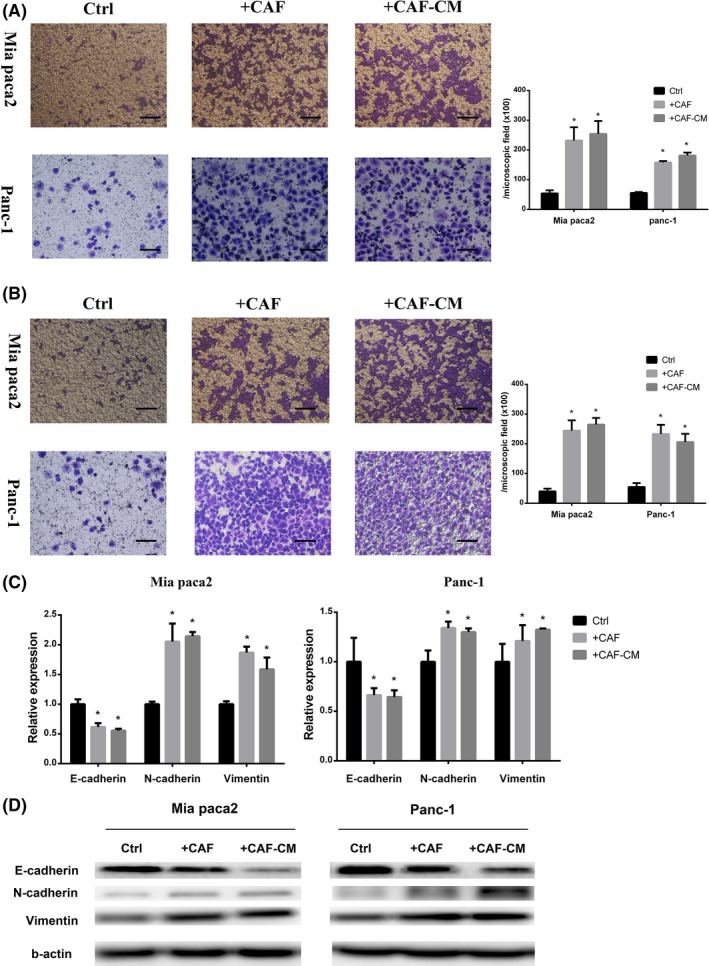
Cancer‐associated fibroblasts (CAF) promoted pancreatic cancer cell migration and invasion. Pancreatic cancer cell lines (MIA PaCa‐2 and Panc‐1) cocultured with CAF (+CAF) or cultured in CAF‐conditioned medium (+CAF‐CM). A,B, Migration assay (A) and invasion assay (B) of cancer cells were monitored at 24 h (*significantly different from Ctrl group, P < .05, n = 4). Scale bar, 100 μm. C,D, PCR analysis and western blot for detection of epithelial‐mesenchymal transition‐related gene (E‐cadherin, N‐cadherin, and vimentin) expression of cancer cells (*significantly different from Ctrl group, P < .05, n = 4)
Epithelial‐mesenchymal transition is essential for cancer cell migration and invasion.13 We detected EMT‐related gene changes of cancer cells after coculture with CAF or stimulation with CAF‐CM. Both coculture with CAF and stimulation with CAF‐CM decreased E‐cadherin but increased N‐cadherin and vimentin expression of both MIA PaCa‐2 and Panc‐1 (Figure 1C,D).
3.2. CAF‐derived IL‐6 increased cancer cell migration and invasion by promoting EMT
A previous study reported that IL‐6 is a protumor cytokine, which can promote cancer cell migration.14 To determine the role of IL‐6 during CAF tumor cell interaction, we measured IL‐6 expression and secretion by CAF. Results showed that coculture with cancer cells or stimulation with cancer cell‐cultured medium significantly increased IL‐6 secretion and expression (Figure 2A,B). Next, we used an IL‐6 antibody to neutralize IL‐6 in the coculture system or CAF‐CM. After IL‐6 neutralization, migration and invasion ability of cancer cells showed a significant difference compared with the coculture or CAF‐CM group (Figure 2C,D). Moreover, the epithelial and mesenchymal markers of cancer cells also appeared statistically different after IL‐6 neutralization (Figure 2E‐G).
Figure 2.
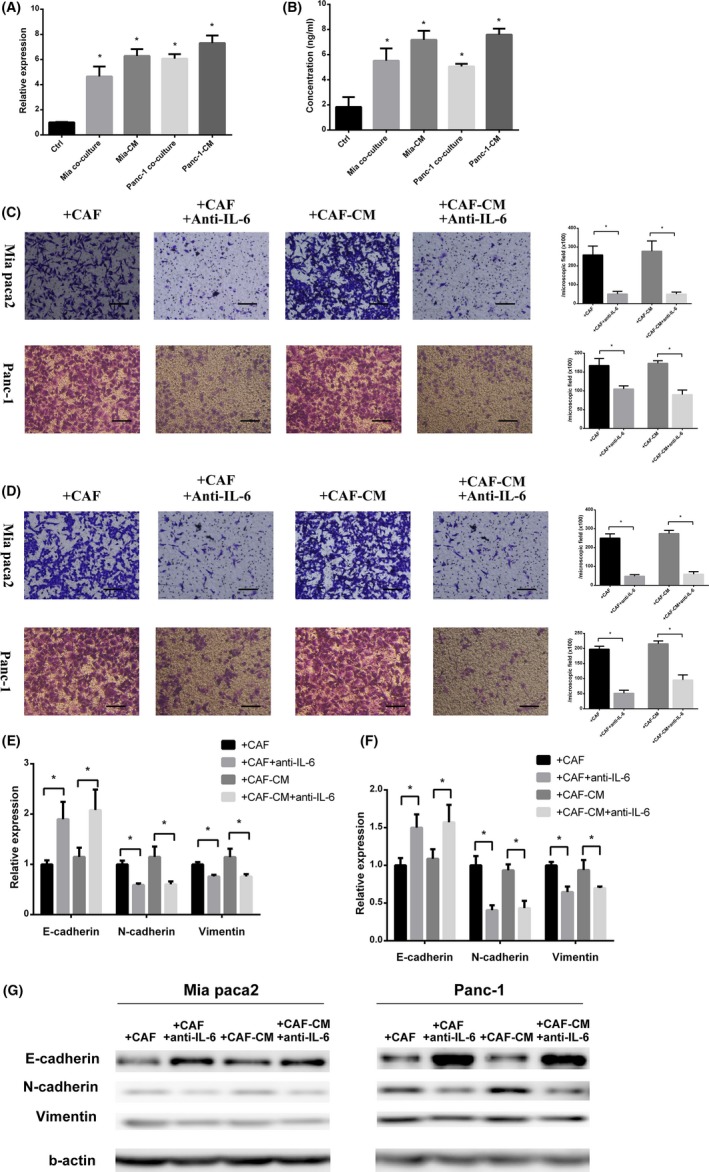
Cancer‐associated fibroblast (CAF) derived interleukin‐6 (IL‐6) promoted pancreatic cancer cell migration and invasion. A,B, PCR analysis (A) and ELISA (B) for IL‐6 expression and secretion of CAF showed that IL‐6 expression of CAF significantly increased after coculture with cancer cells (MIA PaCa‐2 coculture and Panc‐1 coculture) or stimulation with cancer cell‐cultured medium (Mia‐CM and Panc‐1‐CM) for 24 h (*significantly different from Ctrl group, P < .05, n = 4). C,D, Migration assay (C) and invasion assay (D) for cancer cells cocultured with CAF in normal or IL‐6 neutralized coculture system and stimulation by CAF‐CM with or without IL‐6 neutralization (*significantly different between 2 groups, P < .05, n = 4). Scale bar, 100 μm. E,F, PCR analysis for epithelial‐mesenchymal transition‐related gene expression of MIA PaCa‐2 and Panc‐1 (*significantly different between 2 groups, P < .05, n = 4). G, Western blot for E‐cadherin, N‐cadherin, and vimentin expression of MIA PaCa‐2 and Panc‐1
3.3. Nab‐PTX canceled CAF‐induced cancer cell migration and invasion
In order to investigate the effect of nab‐PTX on the interaction between cancer cells and CAF, during cancer cell and CAF coculture, 5 ng/mL nab‐PTX (this dosage had no effect on the cell viability of MIA PaCa‐2, Panc‐1, and CAF, data not shown) treatment was carried out during cancer cell and CAF coculture. After nab‐PTX treatment, cancer cell migration and invasion ability obviously decreased compared with that cocultured with CAF only (Figure 3A,B). Concomitantly, EMT marker changes caused by CAF were reversed after nab‐PTX treatment (Figure 3C,D).
Figure 3.
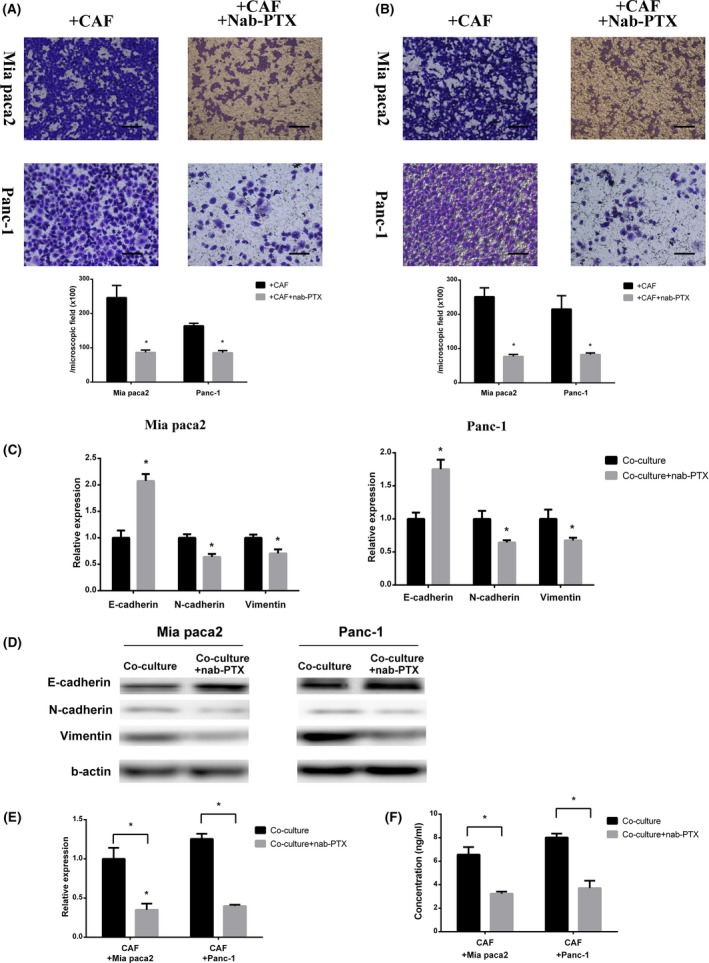
Nab‐paclitaxel (Nab‐PTX) canceled the cancer‐associated fibroblast (CAF)‐induced cancer cell migration and invasion. A,B, Migration assay (A) and invasion assay (B) for cancer cells cocultured with CAF with or without 5 ng/mL nab‐PTX (*significantly different between 2 groups, P < .05, n = 4). Scale bar, 100 μm. C,D, PCR analysis and western blot of E‐cadherin, N‐cadherin, and vimentin expression of cancer cells cocultured with CAF with or without 5 ng/mL nab‐PTX (*significantly different between 2 groups, P < .05, n = 4). E,F, PCR analysis (E) and ELISA (F) for IL‐6 expression and secretion of CAF cocultured with cancer cells with or without 5 ng/mL nab‐PTX (*significantly different between 2groups, P < .05, n = 4)
We showed that coculture with cancer cells or stimulation by cancer cell‐cultured medium upregulated CAF IL‐6 expression, which might correlate with cancer migration and invasion promotion. Therefore, we detected IL‐6 expression after nab‐PTX treatment. After coculture with cancer cells and nab‐PTX treatment combination, CAF remarkably decreased IL‐6 expression and IL‐6 protein level in cultured medium (Figure 3E,F).
To determine whether nab‐PTX predominantly acted on CAF to downregulate IL‐6 expression or on cancer cells to inhibit their ability to stimulate CAF IL‐6 expression, we treated CAF with cancer cell‐cultured medium and nab‐PTX simultaneously (nab‐PTX1) or nab‐PTX pretreated cancer cell‐cultured medium (nab‐PTX2) and then detected their IL‐6 expression and secretion (Figure 4A). Interestingly, compared with stimulation with cancer cell‐cultured medium (CM) only, IL‐6 expression and secretion was significantly reduced in CAF treated with nab‐PTX pretreated cancer cell‐cultured medium (Figure 4B,C). This showed that nab‐PTX acted on cancer cells to decrease their ability to stimulate CAF IL‐6 expression. Then, the conditioned medium derived from CAF of nab‐PTX1 (CAF‐CM + nab‐PTX1) and of nab‐PTX2 (CAF‐CM + nab‐PTX2) was used to stimulate cancer cells during a migration and invasion assay. Along with IL‐6 reduction, the conditioned medium of CAF‐CM + nab‐PTX2 did not increase cancer cell migration and invasion ability (Figure 4D,E). Also, change of EMT markers confirmed that CAF‐conditioned medium prepared with nab‐PTX pretreated cancer cell‐cultured medium had significant effects (Figure 4F,G).
Figure 4.
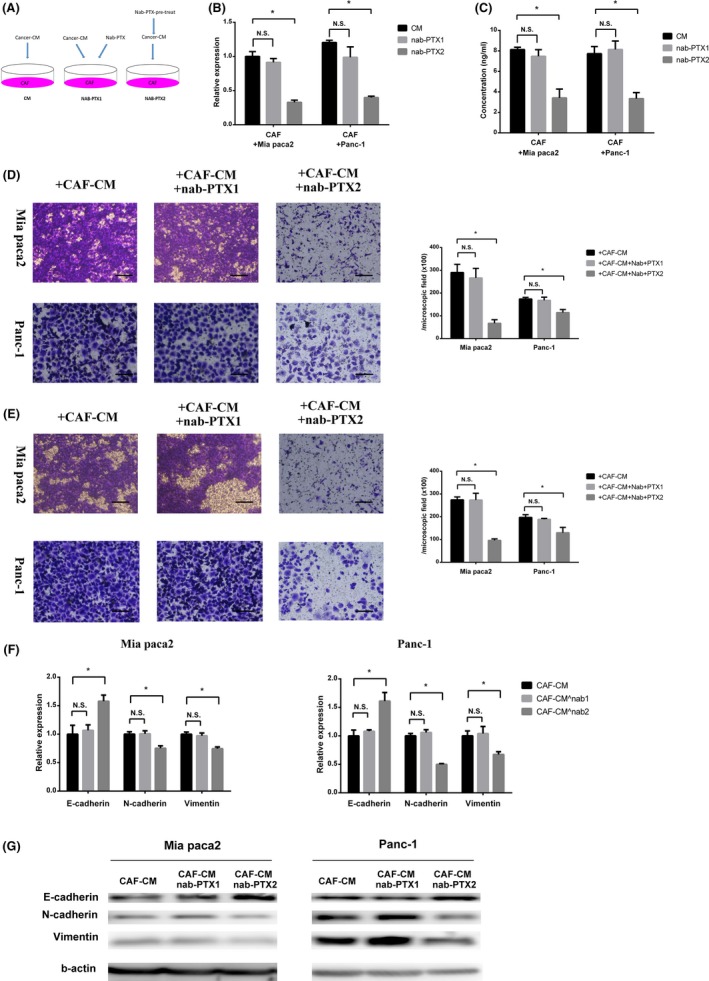
Nab‐paclitaxel (Nab‐PTX) functioned on cancer cells to inhibit cancer‐associated fibroblast (CAF) migration and invasion‐promoting effect. A, CAF were stimulated with cancer cell‐cultured medium (CM) and nab‐PTX simultaneously (nab‐PTX1) or nab‐PTX pretreated cancer cell‐cultured medium (nab‐PTX2). B,C, PCR analysis (B) and ELISA (C) for interleukin‐6 (IL‐6) expression and secretion of CAF stimulated with cancer cell‐cultured medium and nab‐PTX simultaneously (nab‐PTX1) or nab‐PTX pretreated cancer cell‐cultured medium (nab‐PTX2) (*significantly different between 2 groups, P < .05, N.S., not significantly different between 2 groups, P > .05; n = 4). D,E, Migration assay (D) and invasion assay (E) for cancer cells stimulated with conditioned medium derived from CAF of nab‐PTX1 (CAF‐CM + nab‐PTX1) or that of nab‐PTX2 (CAF‐CM + nab‐PTX2) (*significantly different between 2 groups, P < .05, N.S., not significantly different between 2 groups, P > .05; n = 4). Scale bar, 100 μm. F,G, PCR analysis and western blot of E‐cadherin, N‐cadherin, and vimentin expression of cancer cells stimulated with conditioned medium derived from CAF of nab‐PTX1 (CAF‐CM + nab‐PTX1) or that of nab‐PTX2 (CAF‐CM + nab‐PTX2) (*significantly different between 2 groups, P < .05, N.S., not significantly different between 2 groups, P > .05; n = 4)
3.4. Nab‐PTX decreased IL‐6 secretion of CAF by increasing cancer cell CXCL10 secretion
To detect cytokine secretion profile changes in cancer cells after nab‐PTX treatment, we carried out cytokine arrays of cancer cell (Panc‐1) culture supernatants (Figure S1A,B). CCL2, CCL5, CCL12, IL‐13, MIF, and PAI showed a decrease after nab‐PTX treatment and CXCL10 showed a dramatic increase. It has been reported that CXCL10 has an antifibrosis effect.15 Hence, we considered that cancer cell CXCL10 expression promoted by nab‐PTX might decrease CAF IL‐6 expression and cancer cell migration‐promoting ability. We measured CXCL10 expression and secretion of cancer cells after nab‐PTX treatment 3 times separately and confirmed that nab‐PTX treatment upregulated CXCL10 expression and secretion (Figure 5A,B). CXCL10 antibody was used to neutralize secreted CXCL10 by cancer cells after nab‐PTX treatment. After CXCL10 neutralization, nab‐PTX pretreated cancer cell supernatant did not impede IL‐6 secretion of CAF (Figure 5C,D). Subsequently, it did not cancel migration and invasion promotion and EMT marker expression regulation of CAF (Figure 5E‐H). Also, the cancer cell supernatant with or without exogenous CXCL10 was used to stimulate CAF, and IL‐6 expression and secretion of CAF were observed. This showed that exogenous CXCL10 in the cancer cell supernatant could inhibit the expression and secretion of IL‐6 of CAF (Figure S2A,B).
Figure 5.
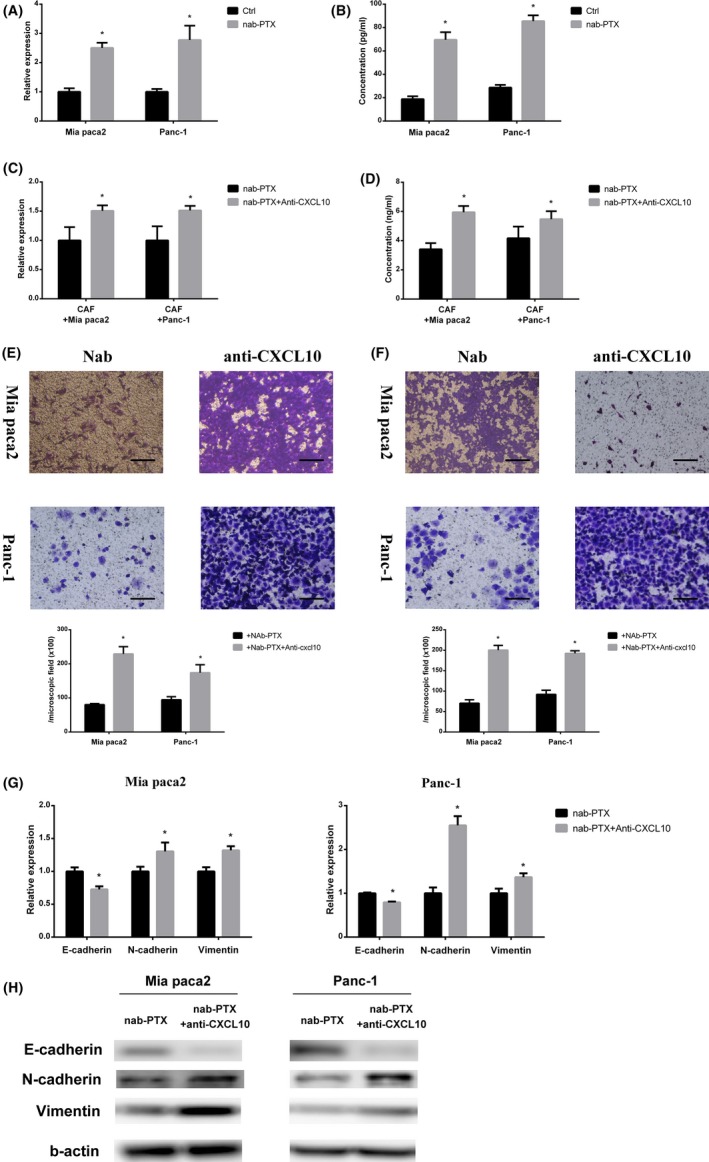
Nab‐paclitaxel (Nab‐PTX) decreased interleulin‐6 (IL‐6) secretion of cancer‐associated fibroblasts (CAF) by increased cancer cell C‐X‐C motif chemokine 10 (CXCL10) secretion. A,B, PCR analysis (A) and ELISA (B) for CXCL10 expression and secretion of cancer cells stimulated with or without nab‐PTX (*significantly different from Ctrl group, P < .05, n = 4). C,D, PCR analysis (C) and ELISA (D) for IL‐6 expression and secretion of CAF stimulated by nab‐PTX pretreated cancer cell medium in which CXCL10 was neutralized (nab‐PTX + anti‐CXCL10) or not (nab‐PTX) (*significantly different between 2 groups, P < .05, n = 4). E,F, Migration assay (E) and invasion assay (F) of cancer cells stimulated with conditioned medium derived from CAF treated with nab‐PTX pretreated cancer cell medium in which CXCL10 was neutralized (anti‐CXCL10) or not (ctrl) (*significantly different between 2 groups, P < .05). Scale bar, 100 μm. G,H, PCR analysis and western blot of E‐cadherin, N‐cadherin, and vimentin expression of cancer cells stimulated with conditioned medium derived from CAF treated with nab‐PTX pretreated cancer cell medium in which CXCL10 was neutralized (anti‐CXCL10) or not (ctrl) (*significantly different between 2 groups, P < .05)
4. DISCUSSION
Nab‐paclitaxel used in clinical practice directly impedes cancer cell mitosis, promotes apoptosis, and synergizes other anti‐cancer agents.16, 17 In the present study, we demonstrated that nab‐PTX could increase cancer cell CXCL10 expression which canceled CAF migration and invasion‐promoting function through decreasing its IL‐6 expression.
The effect of pancreatic cancer cells on stroma reprogramming has been elucidated in many studies. Cancer cell‐derived cytokines including transforming growth factor beta (TGF‐β) and fibroblast growth factor (FGF) families or microRNAs (mi‐RNAs) could promote normal fibroblasts to transform into CAF.18, 19, 20 Also, cancer cells could maintain the CAF phenotype and promote functions such as proliferation, migration, invasion, or angiogenesis.21 In our study, after stimulation with cancer cell‐cultured medium, the migration and invasion‐promoting function of CAF clearly increased.
Cancer‐associated fibroblasts, the predominant non‐parenchymal cells in the tumor stroma, play a key role in tumor‐stromal cross‐talk. Many studies have shown that isolated CAF can promote proliferation, migration and invasion, angiogenesis, and chemoresistance in many types of cancer including pancreatic adenocarcinoma.22, 23, 24 IL‐6, as one of the major proinflammatory cytokines secreted by CAF, involves the modulation of growth of pancreatic cancer. It has been confirmed that IL‐6 is important for the initiation and progression of pancreatic cancer.25 After exogenous IL‐6 stimulation, cancer cells increased vascular endothelial growth factor, neuropilin‐2, and MMP expression, which correlated with cell proliferation and angiogenesis.26 Therefore, high serum or tumor stroma IL‐6 concentrations indicate advanced tumor stage and poor prognosis.27 In the present study, cancer cell migration and invasion ability were related to IL‐6 secretion by CAF. IL‐6‐secreted CAF had migration and invasion‐promoting ability but this ability obviously decreased after IL‐6 antibody neutralization. Also, CAF IL‐6 expression difference was found between cancer cells and nab‐PTX pretreated cancer cells stimulated according to the difference of migration and invasion‐promoting ability.
CXCL10 is upregulated after immune and non‐immune‐mediated tissue injury involving tissue repair and remodeling.28 It has been suggested that CXCL10 is important in regulating the fibrogenesis response. A previous study indicated that CXCL10 knockout mice showed an excessive fibrogenic response to stimulation and exogenous CXCL10 inhibited fibroblast recruitment and subsequent fibrosis.29 Although it is commonly believed that antifibrosis of CXCL10 correlates to inhibit fibroblast migration, the effect of CXCL10 on α‐smooth muscle actin and IL‐6 expression to prevent myofibroblast differentiation is also reported.30 In our study, after nab‐PTX treatment, cancer cells increase CXCL10 expression, and this CXCL10 might decrease CAF IL‐6 expression and suppress CAF tumor‐promoting ability when the nab‐PTX pretreated cancer cell‐cultured medium was used to stimulate CAF.
Even though the dose of nab‐PTX used in the present study was relatively low compared with the clinical setting and the dose of nab‐PTX in our study did not decrease cell proliferation, there were already some changes in transcription of cancer cells and inhibition of cancer cells and CAF interaction caused by our dose of nab‐PTX. It has been shown that paclitaxel could stabilize microtubules and reduce transcription regulator, such as S100A4, nuclear import to block metastatic‐related gene expression in cholangiocarcinoma cells.31 Also, low‐dose paclitaxel could increase nuclear factor kappa B expression and nuclear transition which, as a transcription factor, can combine with the CXCL10 promoter and promote CXCL10 expression.32, 33 In our study, we confirmed that low‐dose nab‐PTX could change the expression of the EMT‐related gene along with decreasing cancer cell migration and invasion. Moreover, CXCL10 expression was increased. Unfortunately, low‐dose nab‐PTX had no direct effect on CAF during cancer cell medium stimulation.
There are some limitations in the present study. The dosage used in our study was not as high as that which is clinically adopted, but even the relatively low dose of CAF had an antitumor effect. The CAF we used in this study is derived from only 1 patient which might limit the external validity of our results. Also, in vivo experiments were not included in this study.
In conclusion, our study showed that nab‐PTX could stimulate CXCL10 expression of pancreatic cancer cells which inhibited IL‐6 expression of CAF and decreased cancer cell migration and invasion‐promoting ability.
CONFLICTS OF INTEREST
This study was funded by Taiho Pharmaceutical Co., Ltd (Tokyo, Japan).
Supporting information
Feng R, Morine Y, Ikemoto T, et al. Nab‐paclitaxel interrupts cancer‐stromal interaction through C‐X‐C motif chemokine 10‐mediated interleukin‐6 downregulation in vitro. Cancer Sci. 2018;109:2509–2519. 10.1111/cas.13694
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583‐592. [DOI] [PubMed] [Google Scholar]
- 3. Tien YW, Wu YM, Lin WC, Lee HS, Lee PH. Pancreatic carcinoma cells stimulate proliferation and matrix synthesis of hepatic stellate cells. J Hepatol. 2009;51:307‐314. [DOI] [PubMed] [Google Scholar]
- 4. Masamune A, Shimosegawa T. Pancreatic stellate cells: a dynamic player of the intercellular communication in pancreatic cancer. Clin Res Hepatol Gastroenterol. 2015;39(suppl 1):S98‐S103. [DOI] [PubMed] [Google Scholar]
- 5. Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer‐associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36:1770‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hwang RF, Moore T, Arumugam T, et al. Cancer‐associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paraiso KH, Smalley KS. Fibroblast‐mediated drug resistance in cancer. Biochem Pharmacol. 2013;85:1033‐1041. [DOI] [PubMed] [Google Scholar]
- 8. Ramanathan RK, Goldstein D, Korn RL, et al. Positron emission tomography response evaluation from a randomized phase III trial of weekly nab‐paclitaxel plus gemcitabine versus gemcitabine alone for patients with metastatic adenocarcinoma of the pancreas. Ann Oncol. 2016;27:648‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim H, Samuel S, Lopez‐Casas P, et al. SPARC‐independent delivery of Nab‐paclitaxel without depleting tumor stroma in patient‐derived pancreatic cancer xenografts. Mol Cancer Ther. 2016;15:680‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neesse A, Frese KK, Chan DS, et al. SPARC independent drug delivery and antitumour effects of nab‐paclitaxel in genetically engineered mice. Gut. 2014;63:974‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alvarez R, Musteanu M, Garcia‐Garcia E, et al. Stromal disrupting effects of nab‐paclitaxel in pancreatic cancer. Br J Cancer. 2013;109:926‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walter K, Omura N, Hong SM, Griffith M, Goggins M. Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol Ther. 2008;7:882‐888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265‐273. [DOI] [PubMed] [Google Scholar]
- 14. Hamada S, Masamune A, Yoshida N, Takikawa T, Shimosegawa T. IL‐6/STAT3 plays a regulatory role in the interaction between pancreatic stellate cells and cancer cells. Dig Dis Sci. 2016;61:1561‐1571. [DOI] [PubMed] [Google Scholar]
- 15. Saxena A, Bujak M, Frunza O, et al. CXCR3‐independent actions of the CXC chemokine CXCL10 in the infarcted myocardium and in isolated cardiac fibroblasts are mediated through proteoglycans. Cardiovasc Res. 2014;103:217‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li JA, Xu XF, Han X, et al. Nab‐paclitaxel plus S‐1 shows increased antitumor activity in patient‐derived pancreatic cancer xenograft mouse models. Pancreas. 2016;45:425‐433. [DOI] [PubMed] [Google Scholar]
- 17. Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor‐free, albumin‐bound paclitaxel, ABI‐007, compared with cremophor‐based paclitaxel. Clin Cancer Res. 2006;12:1317‐1324. [DOI] [PubMed] [Google Scholar]
- 18. Coleman DT, Gray AL, Stephens CA, Scott ML, Cardelli JA. Repurposed drug screen identifies cardiac glycosides as inhibitors of TGF‐beta‐induced cancer‐associated fibroblast differentiation. Oncotarget. 2016;7:32200‐32209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen H, Yu X, Yang F, et al. Reprogramming of normal fibroblasts into cancer‐associated fibroblasts by miRNAs‐mediated CCL2/VEGFA signaling. PLoS Genet. 2016;12:e1006244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakai S, Iwata C, Tanaka HY, et al. Increased fibrosis and impaired intratumoral accumulation of macromolecules in a murine model of pancreatic cancer co‐administered with FGF‐2. J Control Release. 2016;230:109‐115. [DOI] [PubMed] [Google Scholar]
- 21. Lin ZY, Chuang WL. Hepatocellular carcinoma cells cause different responses in expressions of cancer‐promoting genes in different cancer‐associated fibroblasts. Kaohsiung J Med Sci. 2013;29:312‐318. [DOI] [PubMed] [Google Scholar]
- 22. Qiao A, Gu F, Guo X, Zhang X, Fu L. Breast cancer‐associated fibroblasts: their roles in tumor initiation, progression and clinical applications. Front Med. 2016;10:33‐40. [DOI] [PubMed] [Google Scholar]
- 23. Pan B, Liao Q, Niu Z, Zhou L, Zhao Y. Cancer‐associated fibroblasts in pancreatic adenocarcinoma. Future Oncol. 2015;11:2603‐2610. [DOI] [PubMed] [Google Scholar]
- 24. Kubo N, Araki K, Kuwano H, Shirabe K. Cancer‐associated fibroblasts in hepatocellular carcinoma. World J Gastroenterol. 2016;22:6841‐6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL‐6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456‐469. [DOI] [PubMed] [Google Scholar]
- 26. Feurino LW, Zhang Y, Bharadwaj U, et al. IL‐6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP‐1 expression in pancreatic cancer cells. Cancer Biol Ther. 2007;6:1096‐1100. [DOI] [PubMed] [Google Scholar]
- 27. Miura T, Mitsunaga S, Ikeda M, et al. Characterization of patients with advanced pancreatic cancer and high serum interleukin‐6 levels. Pancreas. 2015;44:756‐763. [DOI] [PubMed] [Google Scholar]
- 28. Lee EY, Lee ZH, Song YW. CXCL10 and autoimmune diseases. Autoimmun Rev. 2009;8:379‐383. [DOI] [PubMed] [Google Scholar]
- 29. Tager AM, Kradin RL, LaCamera P, et al. Inhibition of pulmonary fibrosis by the chemokine IP‐10/CXCL10. Am J Respir Cell Mol Biol. 2004;31:395‐404. [DOI] [PubMed] [Google Scholar]
- 30. Liu M, Zeng X, Wang J, et al. Immunomodulation by mesenchymal stem cells in treating human autoimmune disease‐associated lung fibrosis. Stem Cell Res Ther. 2016;7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cadamuro M, Spagnuolo G, Sambado L, et al. Low dose paclitaxel reduces S100A4 nuclear import to inhibit invasion and hematogenous metastasis of cholangiocarcinoma. Cancer Res. 2016;76:4775‐4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bellarosa D, Binaschi M, Maggi CA, Goso C. Sabarubicin‐ (MEN 10755) and paclitaxel show different kinetics in nuclear factor‐kappaB (NF‐kB) activation: effect of parthenolide on their cytotoxicity. Anticancer Res. 2005;25:2119‐2128. [PubMed] [Google Scholar]
- 33. Li Y, Zhang H, Kosturakis AK, et al. MAPK signaling downstream to TLR4 contributes to paclitaxel‐induced peripheral neuropathy. Brain Behav Immun. 2015;49:255‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials