

Abstract
Although the deregulation of epidermal growth factor receptor (EGFR) is one of the most common molecular mechanisms of glioblastoma (GBM) pathogenesis, the efficacy of anti‐EGFR therapy is limited. Additionally, response to anti‐EGFR therapy is not solely dependent on EGFR expression and is more promising in patients with reduced activity of EGFR downstream signaling pathways. Thus, there is considerable interest in identifying the compensatory regulatory factors of the EGFR signaling pathway to improve the efficacy of anti‐EGFR therapies for GBM. In this study, we confirmed the low efficacy of EGFR inhibitors in GBM patients by meta‐analysis. We then identified a negative correlation between connexin 43 (Cx43) expression and Akt/ERK activation, which was caused by the direct interactions between Akt/ERK and Cx43. By comparing the interactions between Akt/ERK and Cx43 using a series of truncated and mutated Cx43 variants, we revealed that the residues T286/A305/Q308/Y313 and S272/S273 at the carboxy terminus of Cx43 are critical for its binding with Akt and ERK, respectively. In addition, Kaplan–Meier survival analysis using data from The Cancer Genome Atlas datasets indicated that the expression of Cx43 significantly improved the prognosis of GBM patients who express EGFR. Together, our results suggested that Cx43 acts as an inhibitory regulator of the activation of growth factor receptor downstream signaling pathways, indicating the potential of Cx43 as a marker for predicting the efficacy of EGFR inhibitor treatments for GBM. Targeting the interaction between the carboxy terminus of Cx43 and Akt/ERK could be an effective therapeutic strategy against GBM.
Keywords: Akt, connexin 43, epidermal growth factor receptor, ERK, glioblastoma
1. INTRODUCTION
Glioblastoma (GBM) is the most common central nervous system tumor in adults. Routine therapy of GBM includes surgical resection and radiotherapy followed by temozolomide administration.1, 2 However, the overall survival (OS) of patients is <15 months, with a 2‐year survival rate rarely exceeding 30%.3, 4, 5, 6 Recently, elucidation of GBM molecular pathogenesis indicated that GBM malignancy partially resulted from the deregulation of the epidermal growth factor receptor (EGFR) pathway.7, 8, 9 Amplification of EGFR can be found in approximately 40–50% of primary GBM patients, and 60% of primary GBM patients have EGFR overexpression.7, 10 Furthermore, EGFR overexpression has been implicated in the development and aggressiveness of GBM and found to be associated with unfavorable outcomes.11, 12 This molecular characteristic of GBM makes it a compelling candidate for new therapeutic strategies.
In vitro, the genetic inhibition of EGFR expression can significantly attenuate the proliferation and invasion of GBM cells.13, 14 Exogenous EGFR inhibitors can inhibit aberrant EGFR tyrosine kinase activity and selectively attenuate EGFR‐mediated tumor invasion.15, 16 However, the prognostic value of anti‐EGFR treatment in GBM patients remains controversial because anti‐EGFR therapies fail to decrease the activation of the downstream signaling molecules in EGFR pathways.9, 17, 18, 19, 20 Moreover, the response to EGFR inhibitors is more marked in GBM patients with low levels of Akt phosphorylation.21, 22 Thus, treatment of patients with erlotinib combined with PI3K/Akt pathway inhibitors could be highly beneficial.23, 24, 25 Therefore, efforts to improve the efficacy of anti‐EGFR treatments in GBM patients should focus on the activation of downstream pathways.
Connexin 43 (Cx43, encoded by the gap junction α 1 gene) is the most highly expressed isoform of the gap junction protein family in central nervous system tumors.26, 27 Signals are transferred from adjacent tumor cells through gap junction intercellular communication mediated by the channel structure of Cx43.28, 29 In addition, Cx43 can interact with a large number of signaling and scaffolding proteins through its carboxy terminus (CT) to regulate the adhesion, migration, and proliferation of tumor cells.30, 31 Both Akt and ERK have also been reported to bind and phosphorylate Cx43 CT, resulting in the closure of the hemichannels formed by Cx43.32, 33 Of greater interest, the overexpression of Cx43 has been shown to inhibit the EGF‐induced proliferation and invasion of different cancer cells.34, 35 However, it is not clear whether Cx43 is involved in the regulation of the phosphorylation of Akt and ERK, the main downstream signaling effectors of EGFR.
In this study, we found that the expression level of Cx43 was negatively correlated with the activation of Akt/ERK in GBM patients with EGFR overexpression. Connexin 43 CT was found to directly interact with Akt and ERK1/2 to inhibit their hyperphosphorylation and to attenuate the activation of the epidermal growth factor (EGF)/EGFR signaling pathway. Truncated fragments containing specific residues successfully mimicked the interactions between Cx43 CT and Akt/ERK. Finally, Kaplan–Meier survival analysis showed a significant improved effect of Cx43 expression on the prognosis of GBM patients expressing EGFR.
2. MATERIALS AND METHODS
2.1. Cell lines, primary tumors cells, and glioma patient samples
The human GBM cell line U87 was purchased from ATCC (Rockefeller, Manassas, VA, USA). Paraffin‐embedded samples from glioma patients were obtained during surgery at the Daping Hospital, Army Medical University (Third Military Medical University, Chongqing, China) (23 cases from 2009 and 2012). Two samples (GBM1 and GBM2) were successfully used for primary culture as previously described.36 Written informed consent was obtained from all patients. The Institutional Research Medical Ethics Committee of the Army Medical University granted approval for this study.
2.2. Meta‐analysis
We carried out this meta‐analysis following the guidelines of the Meta‐analysis of Observational Studies in Epidemiology group. PubMed was systematically searched to identify relevant studies using the following keywords and their combination: “glioma,” “EGFR,” and “clinical trial.” The combined hazard ratios (HRs) with their 95% confidence intervals (CIs) and P‐values were determined using Stata data analysis and statistical software version 12.0 (Lakeway, TX, USA).
2.3. Differentially expressed genes and gene set enrichment analysis
We undertook a gene set enrichment analysis (GSEA) using expression data derived from The Cancer Genome Atlas (TCGA) database. The detailed protocol for GSEA is available on the Broad Institute Gene Set Enrichment Analysis website ( http://www.broad.mit.edu/gsea), and the gene sets representing the activation of Akt and ERK are available on the Molecular Signatures Database ( http://software.broadinstitute.org/gsea/msigdb). Briefly, two biological states were separated according to receiver operating characteristic (ROC) curves. In our study, the activated states of Akt and ERK were determined by the significant expression of their target gene sets. By comparing the gene expression in response to activated and non‐activated Akt and ERK, we identified differentially expressed genes.
2.4. Immunohistochemistry
Tumor samples from 23 glioma patients were immunohistochemically studied for the expression of Cx43, EGFR, phosphorylated (p‐)Akt, and p‐ERK. Briefly, tumor samples were prepared as previously described.37After an overnight incubation using antibodies against EGFR (1:100 dilution; Cell Signaling Technology, Boston, MA, USA), p‐Akt (1:100 dilution; Cell Signaling Technology), p‐ERK (1:100 dilution; Cell Signaling Technology), and Cx43 (1:100; Sigma, USA), the slides were incubated with secondary antibodies (Cell Signaling Technology). Immunostaining was carried out with the Envision System using diaminobenzidine (Dako, Denmark). The slides were counterstained with Mayer's hematoxylin, dehydrated, and mounted. Finally, the results were analyzed by Image‐Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MA, USA).
2.5. Glioblastoma cell sphere culture and EGF treatment
Tumorspheres from U87 cells and primary specimens (GBM1, GBM2) were cultured as previously described.36 The cells were starved for 24 h prior to treatment with recombinant human EGF (200 ng/mL; Sigma). Treatment with EGF for 30 min was carried out according to methods described in previous studies.38
2.6. Plasmid constructs and transfection
The full‐length cDNA of human Cx43 was a kind gift from the Han Laboratory ( http://hanlab.xmu.edu.cn/cdna/). The CT and truncated Cx43 fragments were amplified using PCR with the following primer sets: (a) forward, TTGAATATCATTGAACTCTT and reverse, CCAGGAGGAGACATAGGC; (b) forward, GGGTACAAGCTGGTTACTG and reverse, AGGTCGCTGGTCCACAAT; (c) forward, GACCAGCGACCTTCAAGCAGAGCCA and reverse, CTAGATCTCCAGGTCATCAGGC CGA; or CT forward, TTGAATATCATTGAACTCTT and reverse, CTAGATCTCCAGGTCATCAGGCCGA. All fragments were cloned into the pGEX‐4T‐1 expression vectors. Akt‐HA and ERK‐HA vectors (ID 9008 and ID 39225, respectively) were obtained from Addgene (Cambridge, MA, USA). Connexin 43 overexpression adenovirus and empty adenovirus vectors were constructed as previously described.37 Connexin 43 mutants (S272A, S273A, Y286D, A305S, Q308K, and Y313D) with Myc Flag tags were generated using the two‐step PCR method for mutagenesis by Pandabio, Inc. (ChongQing, China).
2.7. Western blot analysis and immunoprecipitation
For immunoprecipitation (IP), 300 μg cell lysate was incubated with 1 μg corresponding antibody (or 1 μg IgG in the negative control [Santa Cruz Biotechnology, Dallas, TX, USA]) bound to protein G‐Sepharose beads (Santa Cruz Biotechnology). Proteins were subjected to western blot analysis as previously described.37 Primary antibodies against Cx43 (Invitrogen, Waltham, MA, USA), PhosphoPlus p44/42 MAPK (T202/T204), and PhosphoPlus Akt (S473) were used with the dilution of 1:1000. After incubation with HRP‐conjugated secondary antibodies (1:3000; Cell Signaling Technology), the intensity of chemiluminescence (SuperSignal West Pico; Pierce, Waltham, MA, USA) was quantified using ImageQuant 5.0 (UpdateStar, Berlin, Germany).
2.8. Immunofluorescence staining
The cells were fixed with 4% paraformaldehyde and incubated with antibodies against Cx43, Akt, and ERK1/2 overnight (dilution of 1:100). Additionally, frozen sections were directly incubated with antibodies against Cx43 (Invitrogen) and EGFR (Cell Signaling Technology) in the same concentration. Appropriate secondary antibodies (Cy3 goat anti‐rabbit [red] and FITC goat anti‐mouse [green]; Molecular Probes, Waltham, MA, USA) were used with the dilution of 1:500. The cell nuclei were counterstained with DAPI (Sigma).
2.9. Glutathione S‐transferase pull‐down assay
Glutathione S‐transferase, or GST‐bound Cx43 fragments, fusion proteins were generated using Escherichia coli BL21 (DE3) cells. The purification of GST‐fused proteins for GST pull‐down assays was carried out according to the instructions of the Pierce GST Protein Interaction Pull‐Down Kit (Thermo Fisher Scientific, Waltham, MA, USA). The GST‐fused proteins bound to glutathione magnetic beads were then incubated with lysates from HEK293 cells transfected with Akt‐HA and ERK‐HA vectors. Finally, protein–protein interactions were detected by western blotting with anti‐HA mAbs (Cell Signaling Technology).
2.10. Homology modeling and protein–protein docking
Homology modeling was carried out in Molecular Operating Environment (MOE) version 2015.1001. Multiple template crystal structures were identified through blast and downloaded from the RCSB Protein Data Bank (PDB) (codes 1R5S and 2LL2). The intermediate model that has the best GB/VI (generalized born/volume integral) scoring function was chosen as the final model and was submitted for further energy minimization using the AMBER12:EHT force field. The protein–protein docking module in MOE‐Dock was used for docking simulations of the two proteins (Akt1 and ERK1/2) and for predicting their binding affinities with a homology model of Cx43. The structures of Akt1 and ERK1/2 were downloaded from the RCSB PDB and prepared using MOE. The corresponding PDB codes were 4EJN (resolution, 2.19 Å) and 3SA0 (resolution, 1.59 Å), respectively.
2.11. Survival analysis
For survival analysis, the expression data for 540 GBM patients were obtained from TCGA database (glioblastoma multiforme TCGA, provisional, http://www.cbioportal.org/.39, 40 During the screening process, each patient was classified into a high or low expression group for any particular gene based on the cut‐off value obtained from the corresponding receiver operating characteristic curve. Using data on OS and survival time, the Kaplan–Meier method was used to assess the association between the particular protein and the survival outcomes of GBM patients.
2.12. Statistical analysis
Statistical analysis were under taken using spss version 16.0 software (SPSS, Chicago, IL, USA) and GraphPad Prism version 6.0 (GraphPad, San Diego, CA, USA). Student's t‐test was used for comparisons between two groups. Survival analysis was carried out using the Kaplan–Meier method and the log–rank test. Correlations between the expression of Cx43 and p‐Akt/p‐ERK were assessed with Pearson's correlation analyses. P‐values <0.05 were considered statistically significant.
3. RESULTS
3.1. Efficacy of anti‐EGFR treatment is limited for GBM patients
As the efficacy of anti‐EGFR treatment for GBM is controversial,9, 17, 18, 19, 20 four studies involving a total of 367 GBM patients were included in this primary meta‐analysis to evaluate the prognostic role of anti‐EGFR treatment in high‐grade glioma. For studies evaluating OS, there was notable heterogeneity regarding anti‐EGFR treatment. Hence, a random‐effects model was used to calculate a pooled HR and its 95% CI. As shown in Figure 1 , meta‐analysis of pooled HR and its 95% CI indicated a lack of significant difference (HR = 0.98; 95% CI, 0.76–1.27), implying that anti‐EGFR treatment is not predictive of favorable outcomes.
Figure 1.
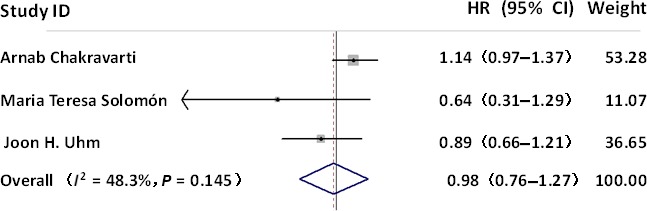
Forest plots of studies evaluating hazard ratios (HR) of anti‐epidermal growth factor receptor (EGFR) therapies. CI, confidence interval
3.2. Expression of GJA1 is associated with suppression of Akt/ERK pathways
Previously, it was observed that the phosphorylation status of Akt/ERK was independent of EGFR expression and could be considered a predictor of the efficacy of anti‐EGFR treatment in glioma patients.21, 41 To investigate this further, we identified GBM patients with EGFR amplification from TCGA database (Glioblastoma Multiforme TCGA, Provisional) (n = 223). We then obtained lists of differentially expressed genes, among which GJA1 encodes Cx43, the most abundant member of the gap junction protein family in the central nervous system, and reported to reduce EGF‐induced proliferation in GBM and several cancer cells.34, 35, 42 Thus, GSEA was carried out to determine the associations between GJA1 expression and the activation of EGFR downstream pathways. As shown in Figure 2A,B, the target genes associated with the activation of the Akt and ERK did not show significant enrichment (P = 0.63 and 0.26, respectively) in EGFR amplification patients, nor in non‐amplification patients. This result confirmed our assumption that the anti‐EGFR treatment was inefficient because the EGFR inhibitors can not inhibit the activation of Akt and ERK stimulated by other pathways. The GBM patients with EGFR amplification were divided into two groups according to the levels of GJA1. As shown in Figure 2C,D, the target genes associated with the activation of Akt and ERK showed a negative significant enrichment in patients with low levels of GJA1 (normalized enrichment score [NES] = −1.52, P = 0.04; NES = −1.43, P = 0.03). These results indicated that the expression of GJA1 might inhibit the activation of Akt and ERK in the patients with EGFR amplification.
Figure 2.
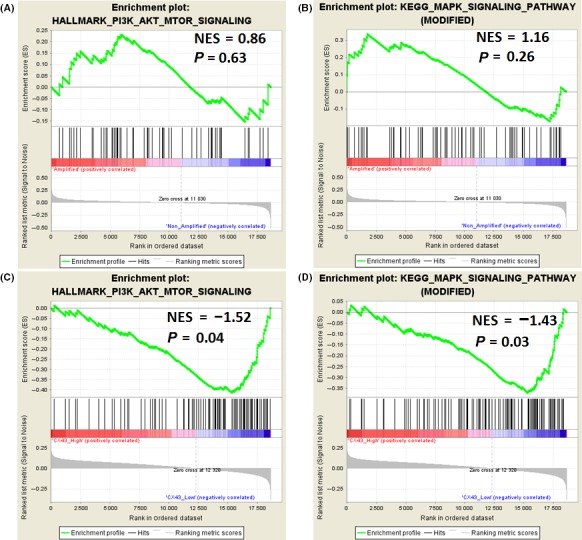
Gene set enrichment analysis plots depicting enrichment of the phosphorylated (p‐)Akt and p‐ERK target gene signature in epidermal growth factor receptor (EGFR)‐amplified glioblastomas in The Cancer Genome Atlas cohort (n = 540). Normalized enrichment score (NES) represents the enrichment of the gene set. Blue, low level of expression; red, high level of expression. A, B, EGFR amplification was not associated with activation of Akt or ERK. C, D, GJA1 expression levels were inversely associated with the activation of Akt and ERK in patients with EGFR amplification
3.3. Connexin 43 expression is inversely correlated with phosphorylation of EGFR downstream pathway effectors in GBM patients
Tumor specimens were obtained from 23 GBM patients with positive EGFR expression (Figure 3A) to analyze the correlation of Cx43 with p‐Akt and p‐ERK. The levels of p‐Akt and p‐ERK were much lower in specimens with high Cx43 expression than in specimens with low Cx43 expression (p‐Akt, correlation coefficient = 0.3987, P = 3.63 × 10−5; p‐ERK, correlation coefficient = 0.4275, P = 1.53 × 10−5) (Figure 3B–E). Moreover, the nuclear accumulation of p‐Akt and p‐ERK was also reduced in patients with high Cx43 expression compared to those with low Cx43 expression (Figure 3B,D, red arrowheads). This result indicates that Cx43 upregulation is inversely correlated with Akt/ERK phosphorylation in human GBM.
Figure 3.
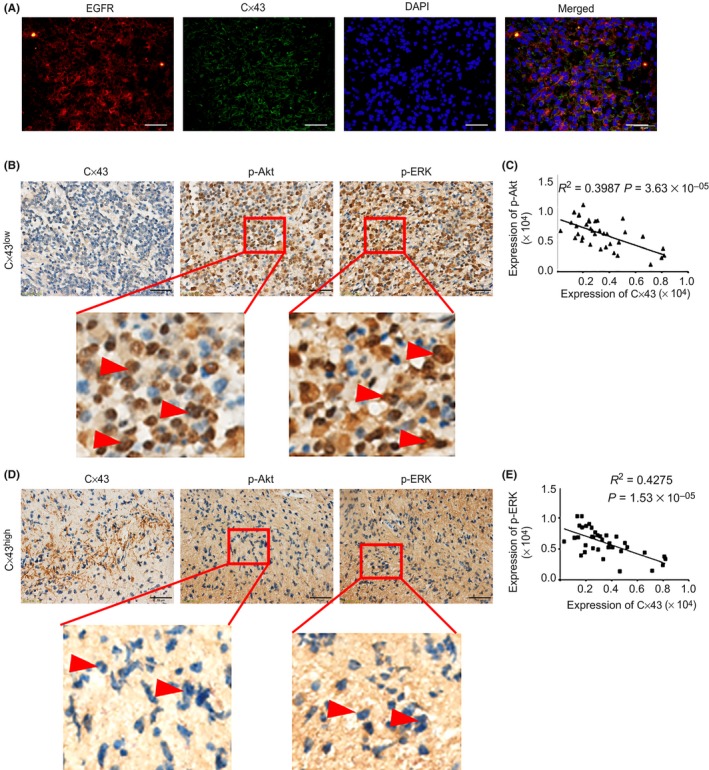
Connexin 43 (Cx43) expression inversely correlates with Akt and ERK phosphorylation in glioblastoma (GBM) patients with epidermal growth factor receptor (EGFR) overexpression. A, Double immunofluorescence staining of EGFR and Cx43 in GBM patients. Scale bar = 50 μm. B–E, Immunohistochemical analysis of Cx43, phosphorylated (p‐)Akt, and p‐ERK in EGFR+ GBM patients. Scale bar = 50 μm. Nuclear accumulation of p‐Akt and p‐ERK is indicated by red arrowheads. Correlations between the expression of Cx43 and p‐Akt/p‐ERK were calculated with Pearson's correlation analyses
3.4. Overexpression of Cx43 inhibits EGF‐induced phosphorylation of Akt and ERK in GBM cell spheres
To clarify whether changes in Cx43 levels influence the activation of Akt and ERK induced by EGF, we examined U87 GBM tumorspheres with nearly absent Cx43 expression that were cultured in stem cell medium. After treatment with PBS, adenovirus vector‐mediated overexpression of Cx43 did not affect the phosphorylation levels of Akt or ERK in any of the tested GBM cells. In contrast, compared to that in GBM tumorspheres transfected with control vector, EGF‐induced phosphorylation of Akt and ERK was inhibited by the overexpression of Cx43 in GBM tumorspheres (Figure 4). Therefore, Cx43 overexpression exerted an inhibitory effect on Akt and ERK phosphorylation in GBM tumorspheres in response to EGFR activation by EGF.
Figure 4.
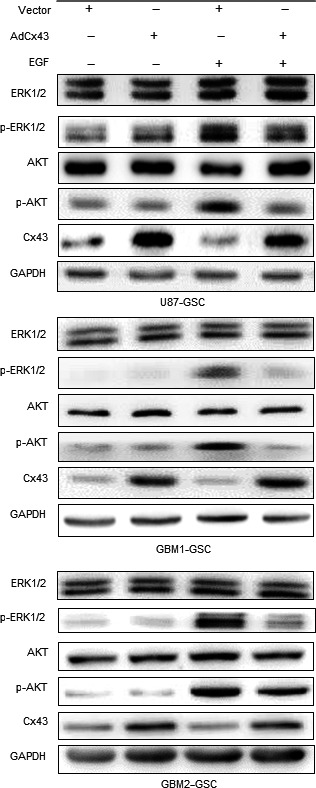
Connexin 43 (Cx43) inhibits epidermal growth factor (EGF)‐induced Akt and ERK phosphorylation in glioblastoma (GBM) spheres. Immunoblot analysis of Cx43 and total or phosphorylated (p‐)Akt and ERK expression in GBM spheres transfected with control or Cx43 overexpression adenovirus. Different groups of cells were treated with EGF (200 ng/mL) or PBS for 15 min
3.5. Connexin 43 CT interacts with Akt and ERK
It has been observed that Cx43 can promote the phosphorylation of Akt and ERK between adjacent cells through its channel structure‐dependent function.43, 44, 45, 46 However, here, we found that the phosphorylation of Akt and ERK was inhibited by Cx43 overexpression. Connexin 43 CT is known to interact with some kinases, such as SRC, to inhibit their phosphorylation.47, 48 We also found that the high expression of Cx43 was inversely related to the nuclear accumulation of p‐Akt and p‐ERK (Figure 3B,D, red arrowheads). Thus, we speculated that the inhibitory effects of Cx43 on the phosphorylation of Akt or ERK might be related to protein–protein interactions between Cx43 CT and Akt/ERK. Therefore, the association between the expression of Cx43 and Akt/ERK was further investigated by IP experiments. Indeed, Cx43 specifically precipitated Akt and ERK in U87 cell lysates, indicating that Cx43 can interact with Akt and ERK (Figure 5A). These results indicated interactions between Cx43 with Akt and ERK. Moreover, immunofluorescence was used to confirm the subcellular colocalization of these three proteins in U87 cells (Figure 5B). Positive yellow signals representing the colocalization of Cx43 (green) and Akt or ERK (red) could be detected near the membrane of GBM cells (Figure 5B, white arrowheads). These results indicated interactions between Cx43 with Akt and ERK in GBM cells.
Figure 5.
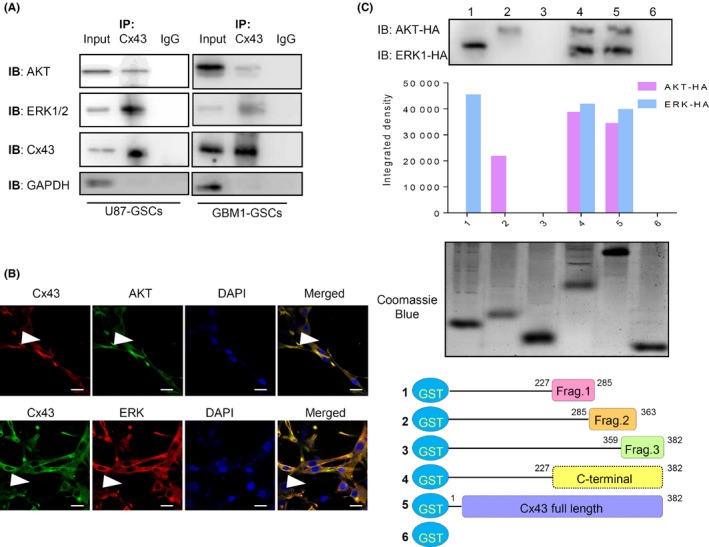
Connexin 43 (Cx43) directly interacts with Akt and ERK. A, Lysate of U87 and GBM1 cell transfected with Cx43 overexpression adenovirus was immunoprecipitated (IP) with anti‐Cx43 antibodies and then analyzed by western blot assays. B, Double immunofluorescence staining of Cx43, total Akt, and ERK expression in U87 cells transfected with Cx43 overexpression adenovirus. The colocalization of Cx43 with Akt or ERK at the plasma membrane is indicated by the white arrowhead. Scale bar = 10 μm. C, Different Cx43 fragments (Frag.) were constructed into a GST‐fused expression vector. Full‐length and deletion fragments of GST–Cx43 fusion proteins were purified for the GST pull‐down assay. Presence of Akt and ERK in the GST pull‐down products was examined through western blot assays with quantification. IB, immunoblot
3.6. Connexin 43 interacts with Akt and ERK through specific regions of CT
Connexin 43 CT has been reported to be phosphorylated on S369–S373 by Akt and on S255–S262/S279–S282 by ERK.31, 32, 33, 49 We wondered whether Akt and ERK can interact with the same region of Cx43 CT. To this end, Cx43 CT was divided into three parts (fragment 1, including 227–285 amino acids [a.a.] of Cx43 CT; fragment 2, including 285–363 a.a of Cx43 CT; and fragment 3, including 359–382 a.a. of Cx43 CT). Sequences encoding these fragments, full‐length Cx43, and Cx43 CT were then cloned in to GST tag‐encoding vectors to obtain fusion proteins (Figure 5C). Consistent with abovementioned results from co‐IP experiments, purified GST‐fused full‐length Cx43 as well as Cx43 CT precipitated Akt and ERK from cell lysates, indicating direct interactions between Cx43 CT and Akt/ERK (Figure 5C, lines 4 and 5). However, only the GST‐fused fragment 1 and fragment 2 precipitated ERK and Akt, respectively (Figure 5C, lines 1 and 2), indicating that fragment 1 can bind to ERK and that fragment 2 can bind to Akt. This result indicated that the specific regions of Cx43 CT (227–285 and 285–363 a.a.) were involved in protein–protein binding between Cx43 and Akt/ERK.
3.7. Connexin 43 inhibits phosphorylation of Akt and ERK through site‐specific interaction
To investigate the exact interaction sites on Cx43 CT for Akt/ERK binding, protein–protein docking experiments were carried out. First, homology modeling was undertaken based on available crystal structures to obtain the 3‐D structure of Cx43. The structure of the C‐terminal domain (residues K234–I382) including a 26‐kDa peptide of the human Cx43 was modeled. The calculated Phi‐Psi backbone angles and side‐chain rotamers were evaluated to discard structures with high‐energy, disallowed bonds, and the best model was selected for the final energy minimization. The final structure of Cx43 CT and the Ramachandran plot are shown in Figure 6A,B.
Figure 6.
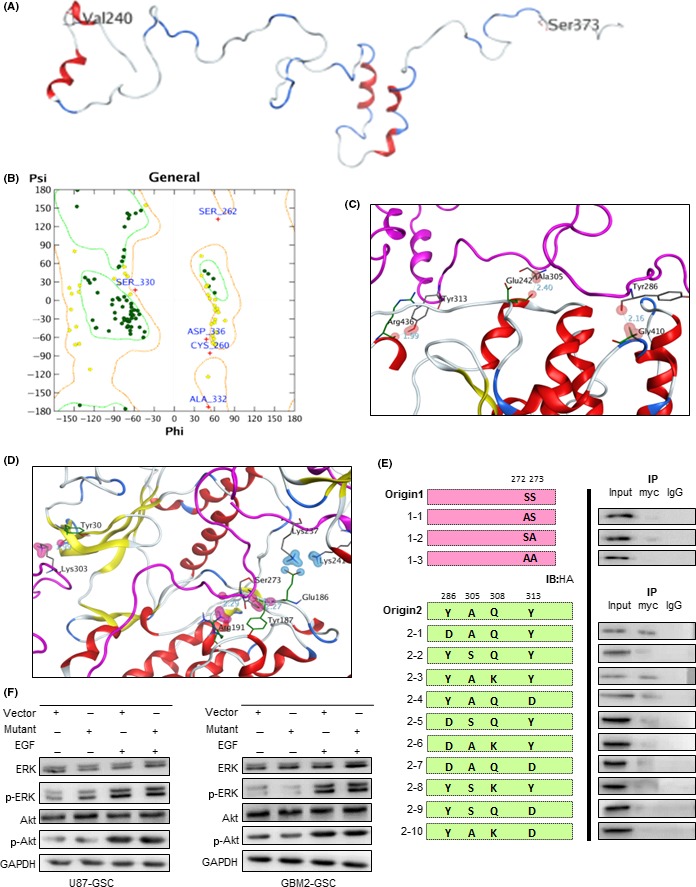
Connexin 43 (Cx43) inhibits the phosphorylation of Akt and ERK through site‐specific interaction. A, Structure of Cx43 carboxy terminus (CT) (K234–I382). B, Ramachandran plot of the final homology model. Ramachandran plot was generated with the Protein Geometry module in Molecular Operating Environment version 2015.1001. C, Structural model of the Cx43CT–Akt complex. D, Structural model of the Cx43CT–ERK complex. E, In vivo mutant binding assay was examined by co‐immunoprecipitation (IP). U87 cells were cotransfected with myc‐Cx43 mutant and Akt1‐HA or ERK1‐HA plasmids. F, Total or phosphorylated Akt and ERK in U87 (left) and GBM2 (right) tumorspheres transfected with mutant fragments was examined through western blot assays. Glioblastoma tumorspheres were transfected with control or Cx43‐mutant plasmid. Before collection cell spheres were treated with EGF (200 ng/mL) or PBS for 15 min
To reveal the specific site for the interactions of Cx43 with Akt and ERK1/2 proteins, two protein–protein docking experiments were carried out by using MOE‐Dock. The docking poses with the highest scores were selected for analysis. During the interaction between Cx43 and Akt protein (Figure 6C), Y286, A305, and Y313 residues of Cx43 could form hydrogen bonds with G410, E242, and R436 residues of Akt, respectively. The distances of these hydrogen bonds were 2.16 Å, 2.40 Å, and 1.99 Å, respectively. In addition, Q308 of Cx43 participated in a weak interaction with R241 of the Akt protein. S272 and S273 residues of Cx43 could form hydrogen bonds with the side chain of Y187 and R191 of ERK1/2, respectively (Figure 6D). The distances of these hydrogen bonds are 2.27 Å and 2.29 Å, respectively.
Based on these results, site‐directed mutagenesis was carried out to generate S272A, S273A, Y286D, A305S, Q308K, and Y313D within Cx43 deletion fragments (Figure 6E), and the mutated sequences were cotransfected with HA‐Akt or HA‐ERK plasmids before co‐IP assays. In detail, both S272 and S273 were crucial for the binding of Cx43 to ERK, and A305 was indispensable for the binding of Cx43 to Akt. However, these interactions were almost abolished by mutations of any two predictive sites. Furthermore, we constructed fragments containing all the mutant residues as mentioned above and then transfected them into U87 and GBM2 tumorspheres. As shown in Figure 6F, the inhibitory function exerted by the overexpression of Cx43 CT was diminished. These results indicate that S272, S273, Y286, A305, Q308, and Y313 of Cx43 CT are the key sites for the interaction between Cx43 and Akt/ERK and mediated the inhibitory effects of Cx43 on EGF‐induced phosphorylation of Akt and ERK.
3.8. Connexin 43 expression improves the OS of EGFR‐expressing GBM patients
Using Kaplan–Meier analysis and data from TCGA database (n = 540), we assessed the value of the combination of EGFR and Cx43 as prognostic biomarkers of GBM (Figure 7). Overall, patients with low EGFR levels had significantly longer OS than patients with high EGFR levels (P = 0.0009). However, in groups characterized by low Cx43 expression, patients with low EGFR levels had significantly better OS than patients with high EGFR levels (P = 0.0102). In contrast, in groups characterized by high Cx43 expression, there was no difference in OS between patients with low and high EGFR expression (P = 0.2379). When EGFR was non‐amplified, the high amplification of Cx43 predicted a worse prognosis (Cx43 high vs. Cx43 low: 13.6 months vs. 15.1 months). However, when the EGFR pathway was activated by the amplification of EGFR, the high amplification of Cx43 brought a better prognosis (Cx43 high vs. Cx43 low, 13.9 months vs. 12.6 months), which indicated the inhibitory function exerted by Cx43 on the activation of the EGFR pathway. These results showed that Cx43 expression significantly improves the prognosis of GBM patients who express EGFR.
Figure 7.
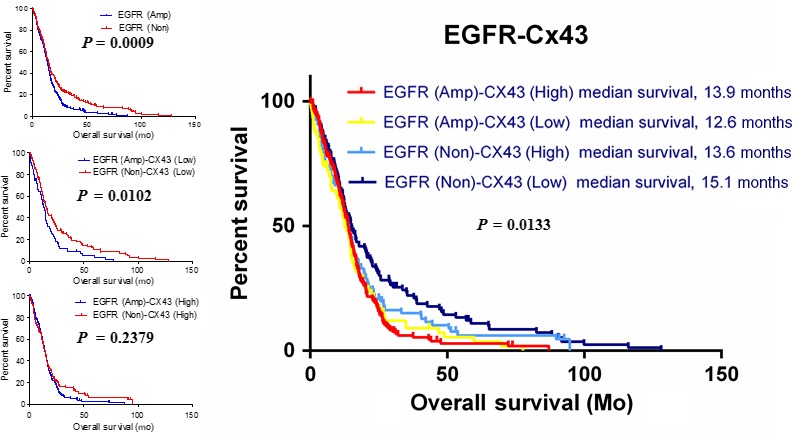
Expression of connexin 43 (Cx43) improves the overall survival of patients with epidermal growth factor receptor (EGFR)‐expressing glioblastoma. Kaplan–Meier survival curves of overall survival duration were produced according to EGFR amplification and Cx43 expression in glioblastoma patients in The Cancer Genome Atlas cohort (n = 540). Statistical significance between two groups was evaluated using the log–rank test
4. DISCUSSION
The persistent activation of Akt independent of EGFR has been a difficult problem during therapy using anti‐EGFR agents and reduces the response to EGFR inhibitors in GBM patients.21, 50, 51 In this study, we showed that Cx43 CT directly interacts with Akt and ERK to inhibit their hyperphosphorylation caused by the activation of EGFR. This inhibitory function of Cx43 CT represents a new molecular feature that could improve the prognosis of GBM patients with EGFR overexpression. These findings might also help us to improve the efficacy of anti‐EGFR treatment in GBM patients.
Our findings are consistent with those of other studies, indicating that GBM patients not selected for any biological characteristic show modest response to EGFR inhibitors.50, 51, 52, 53 The association of EGFR amplification with GBM tumors led to early optimism that EGFR inhibition would be beneficial. This initial optimism was mitigated, however, by the realization that only a subset of patients with EGFR‐amplified GBM actually respond to EGFR blockade.52, 53 Many researchers have observed that the efficacy of EGFR inhibitors in GBM is independent of EGFR amplification status.21, 51 The failure of EGFR inhibitors in a majority of GBM patients with EGFR amplification could stem from inefficient blockade of the receptor or from the inability to reverse downstream signaling abnormalities associated with EGFR amplification.52, 53 Glioblastoma patients expressing low levels of phosphorylation of Akt respond well to erlotinib. Combination of rapamycin, PI‐103, triciribine, and other Akt/mTOR inhibitors with EGFR inhibitors can significantly improve the therapeutic efficacy.23, 24, 25 Therefore, it is promising to determine the compensatory factors that influence EGFR downstream pathway activation, for the development of more effective anti‐EGFR treatment for GBM.
Previous studies suggested that higher Cx43 expression levels in both in vivo tissues and in vitro cultured cells were related to greater levels of ERK1/2 phosphorylation.43, 44, 45, 46 In contrast with the results obtained from normal salivary epithelial and osteoblast cells, Qiu et al. found that Cx43 overexpression reduced EGF‐induced proliferation in ovarian cancer cells.35 Our results also indicated that the expression of Cx43 was inversely correlated with the EGF‐induced phosphorylation of Akt and ERK. Furthermore, cotreatment with the gap junction inhibitor carbenoxolone did not alter the suppressive effects of Cx43 overexpression on EGF‐induced proliferation in cancer cells, suggesting the presence of a gap junction‐independent mechanism for Cx43 in regulation of EGF/EGFR signaling pathway activation.35 In this study, we found that the inhibitory effects exerted by Cx43 involved protein–protein interactions between Cx43 CT with Akt and ERK.
Previously, Cx43 was reported to be phosphorylated by Akt at S369–S373 and by ERK1/2 at S255–S262/S279–S282.31, 32, 33, 49 As expected, fragment 1 (227–285 a.a. of Cx43 CT) could interact with ERK1/2. This corresponds to the region where Cx43 CT is phosphorylated by ERK1/2. However, Akt interacted with fragment 2 (285–363 a.a. of Cx43 CT) but not with fragment 3 (359–382 a.a. of Cx43 CT), the latter of which contains residues that are phosphorylated by Akt. Usually, the binding region for Akt, termed the pleckstrin homology (PH) domain, is located upstream of its phosphorylation site.54 By interacting with the PH domain of Akt, phosphatidylinositol‐(3,4,5)‐P3 (PIP3), Tcl1, Grb10, JIP1, and RasGAP facilitate growth factor‐induced Akt activation.54, 55, 56, 57, 58 Therefore, we speculated that the phosphorylation‐mediated activation of Akt by PIP3 can be inhibited by competitive interaction between the Cx43 CT and the PH domain of Akt. Although the gap junctional function of Cx43 enables adjacent cells to share second messengers, ions, and metabolites and to promote the activation of Akt and ERK,43, 44, 45, 46 the overactivation of Akt and ERK can lead to the phosphorylation of Cx43 to shut down the hemichannels31, 32, 33, 49 and thus, in turn, reduce the activation of Akt and ERK. Moreover, the direct interactions between Cx43 CT and Akt/ERK inhibit the phosphorylation of Akt and ERK in a competitive manner. Thus, Cx43 serves as a negative regulator of Akt and ERK activation.
Connexin 43 can directly interact with various oncoproteins through its CT.59, 60, 61 Therefore, the therapeutic value of Cx43 CT in the treatment of cancer is promising. For example, Tabernero et al. linked the region containing 266–283 a.a. of Cx43 CT with transactivator of transcription to construct a cell‐penetrating peptide called PEP‐2.47, 48, 61 This cell‐penetrating peptide‐based recruited PTEN and Csk to inhibit c‐Src and focal adhesion kinase and dramatically reduced the growth, migration, and survival of glioma stem cells. Because our results indicated that Cx43 can directly interact with Akt and ERK, cell‐penetrating peptides based on 285–363 a.a. (Cx43285–363) and 227–285 a.a. (Cx43227‐285) of Cx43 fragments could be constructed to inhibit the phosphorylation of Akt and ERK, respectively, through protein–protein interactions. The competitive inhibitory effects exerted by these exogenous fragments might result in better outcomes when combined with EGFR inhibitors.
Actually, the therapeutic potential of Cx43 has attracted focus in recent years due to the Cx43‐based tumor suppression of GBM cancer stem cells by reverting tumor cells to a normal phenotype.62, 63 In addition to PEP‐2, mentioned above, several mimic peptides have been produced to exert the tumor‐suppressive function. Juxtamembrane 2 and α‐connexin carboxyl‐terminal 1, for example, were constructed to mimic the interaction between Cx43 with microtubules and ZO‐1, respectively, which were proved to suppress tumor cells though inhibiting the epithelial–mesenchymal transition process.64 However, the therapeutic potential could be limited because the phenotype of tumor cells was not determined by Cx43 alone. Even Cx46, another subtype in the connexin family, played an essential role in CSC maintenance and GBM growth.65 Most current pharmacological agents targeting Cx43 are non‐specific and lack effect, making it difficult to ascribe the antitumor function to Cx43 alone. Thus, there is an undeniable need for more selective co‐inhibitors or co‐activators with Cx43 to exert a more defined action.
In summary, we report a novel finding that Cx43 can bind with Akt and ERK through its CT domain to inhibit EGF‐induced phosphorylation of Akt and ERK. Thus, Cx43 could serve as a promising predictor for the efficacy of anti‐EGFR treatment in GBM patients. More importantly, the identification of specific interaction sites between Cx43 and Akt/ERK also implies an important therapeutic potential for synthesizing mimic peptides, although the current study did not define the specific sites on Akt or ERK1/2 that mediate their interactions with Cx43.
DISCLOSURE STATEMENT
The authors have no conflict of interest.
ACKNOWLEDGMENTS
This study was supported by grants from the National Key Research and Development Program of China (2016YFA0202104 to SCY), the National Natural Science Foundation of China (81572880 to SCY), the Outstanding Youth Science Foundation of Chongqing (CSTC2013JCYJJQ10003 to SCY), the Postgraduate Education Foundation of Chongqing (YJG153062 to SCY), and the Key Clinical Research Program of Southwest Hospital (SWH2016ZDCX1005 to SCY), Technological Innovation Projects in Major Areas of Southwest Hospital (SWH2017ZDCX1003 to SCY).
Kuang J‐Y, Guo Y‐F, Chen Y, et al. Connexin 43 C‐terminus directly inhibits the hyperphosphorylation of Akt/ERK through protein–protein interactions in glioblastoma. Cancer Sci. 2018;109:2611–2622. 10.1111/cas.13707
Funding information
National Key Research and Development Program of China (2016YFA0202104); Outstanding Youth Science Foundation of Chongqing (CSTC2013JCYJJQ10003); Technological Innovation Projects in Major Areas of Southwest Hospital (SWH2017ZDCX1003); National Natural Science Foundation of China (81572880); Key Clinical Research Program of Southwest Hospital (SWH2016ZDCX1005); Postgraduate Education Foundation of Chongqing (YJG153062).
Contributor Information
Shi‐Cang Yu, Email: yushicang@163.com.
Xiu‐Wu Bian, Email: bianxiuwu@263.net.
REFERENCES
- 1. Nachbichler SB, Schupp G, Ballhausen H, Niyazi M, Belka C. Temozolomide during radiotherapy of glioblastoma multiforme : daily administration improves survival. Strahlenther Onkol. 2017;193:890‐896. [DOI] [PubMed] [Google Scholar]
- 2. Liu YL, Liu PF, Shao W, et al. Effect of temozolomide on survival in elderly patients with glioblastoma and impaired performance status: a propensity score‐matching analysis. Onco Targets Ther. 2017;10:4029‐4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nizamutdinov D, Dandashi JA, Stock EM, et al. Survival outcomes prognostication in glioblastoma diagnosed patients. World Neurosurg. 2017;109:e67‐e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shabihkhani M, Telesca D, Movassaghi M, et al. Incidence, survival, pathology, and genetics of adult Latino Americans with glioblastoma. J Neurooncol. 2017;132:351‐358. [DOI] [PubMed] [Google Scholar]
- 5. Pretanvil JA, Salinas IQ, Piccioni DE. Glioblastoma in the elderly: treatment patterns and survival. CNS Oncol. 2017;6:19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kelly C, Majewska P, Ioannidis S, Raza MH, Williams M. Estimating progression‐free survival in patients with glioblastoma using routinely collected data. J Neurooncol. 2017;135:621‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683‐2710. [DOI] [PubMed] [Google Scholar]
- 8. Juratli TA, Qin N, Cahill DP, Filbin MG. Molecular pathogenesis and therapeutic implications in pediatric high‐grade gliomas. Pharmacol Ther. 2017;182:70‐79. [DOI] [PubMed] [Google Scholar]
- 9. Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110:13‐24. [DOI] [PubMed] [Google Scholar]
- 10. Pelloski CE, Ballman KV, Furth AF, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25:2288‐2294. [DOI] [PubMed] [Google Scholar]
- 11. Chen C, Huang R, MacLean A, et al. Recurrent high‐grade glioma treated with bevacizumab: prognostic value of MGMT methylation, EGFR status and pretreatment MRI in determining response and survival. J Neurooncol. 2013;115:267‐276. [DOI] [PubMed] [Google Scholar]
- 12. Lo HW. EGFR‐targeted therapy in malignant glioma: novel aspects and mechanisms of drug resistance. Curr Mol Pharmacol. 2010;3:37‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Penar PL, Khoshyomn S, Bhushan A, Tritton TR. Inhibition of epidermal growth factor receptor‐associated tyrosine kinase blocks glioblastoma invasion of the brain. Neurosurgery. 1997;40:141‐151. [DOI] [PubMed] [Google Scholar]
- 14. Mishima K, Johns TG, Luwor RB, et al. Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Can Res. 2001;61:5349‐5354. [PubMed] [Google Scholar]
- 15. Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non‐small cell lung cancer: a randomized trial. JAMA. 2003;290:2149‐2158. [DOI] [PubMed] [Google Scholar]
- 16. Zhu AX, Rosmorduc O, Evans TR, et al. SEARCH: a phase III, randomized, double‐blind, placebo‐controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2015;33:559‐566. [DOI] [PubMed] [Google Scholar]
- 17. Sathornsumetee S, Rich JN, Reardon DA. Diagnosis and treatment of high‐grade astrocytoma. Neurol Clin. 2007;25:1111‐1139. [DOI] [PubMed] [Google Scholar]
- 18. Sathornsumetee S, Vredenburgh KA, Lattimore KP, Rich JN. Malignant glioma drug discovery ‐ targeting protein kinases. Expert Opin Drug Discov. 2007;2:1‐17. [DOI] [PubMed] [Google Scholar]
- 19. Chi AS, Wen PY. Inhibiting kinases in malignant gliomas. Expert Opin Ther Targets. 2007;11:473‐496. [DOI] [PubMed] [Google Scholar]
- 20. Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med. 2011;13:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haas‐Kogan DA, Prados MD, Tihan T, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880‐887. [DOI] [PubMed] [Google Scholar]
- 22. Yang SX, Costantino JP, Kim C, et al. Akt phosphorylation at Ser473 predicts benefit of paclitaxel chemotherapy in node‐positive breast cancer. J Clin Oncol. 2010;28:2974‐2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartholomeusz C, Yamasaki F, Saso H, Kurisu K, Hortobagyi GN, Ueno NT. Gemcitabine overcomes erlotinib resistance in EGFR‐overexpressing cancer cells through downregulation of Akt. J Cancer. 2011;2:435‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hagerstrand D, Lindh MB, Pena C, et al. PI3K/PTEN/Akt pathway status affects the sensitivity of high‐grade glioma cell cultures to the insulin‐like growth factor‐1 receptor inhibitor NVP‐AEW541. Neuro Oncol. 2010;12:967‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fan QW, Cheng CK, Nicolaides TP, et al. A dual phosphoinositide‐3‐kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN‐mutant glioma. Can Res. 2007;67:7960‐7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crespin S, Fromont G, Wager M, et al. Expression of a gap junction protein, connexin43, in a large panel of human gliomas: new insights. Cancer Med. 2016;5:1742‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ye XY, Jiang QH, Hong T, et al. Altered expression of connexin43 and phosphorylation connexin43 in glioma tumors. Int J Clin Exp Pathol. 2015;8:4296‐4306. [PMC free article] [PubMed] [Google Scholar]
- 28. Hong X, Sin WC, Harris AL, Naus CC. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget. 2015;6:15566‐15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Osswald M, Jung E, Sahm F, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528:93‐98. [DOI] [PubMed] [Google Scholar]
- 30. Leithe E, Mesnil M, Aasen T. The connexin 43 C‐terminus: a tail of many tales. Biochem Biophys Acta. 2017;1860:48‐64. [DOI] [PubMed] [Google Scholar]
- 31. Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588:1423‐1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Riquelme MA, Burra S, Kar R, Lampe PD, Jiang JX. Mitogen‐activated protein kinase (MAPK) activated by prostaglandin E2 phosphorylates connexin 43 and closes osteocytic hemichannels in response to continuous flow shear stress. J Biol Chem. 2015;290:28321‐28328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnstone SR, Kroncke BM, Straub AC, et al. MAPK phosphorylation of connexin 43 promotes binding of cyclin E and smooth muscle cell proliferation. Circ Res. 2012;111:201‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ionta M, Ferreira RA, Pfister SC, Machado‐Santelli GM. Exogenous Cx43 expression decrease cell proliferation rate in rat hepatocarcinoma cells independently of functional gap junction. Cancer Cell Int. 2009;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qiu X, Cheng JC, Klausen C, Chang HM, Fan Q, Leung PC. EGF‐induced connexin43 negatively regulates cell proliferation in human ovarian cancer. J Cell Physiol. 2016;231:111‐119. [DOI] [PubMed] [Google Scholar]
- 36. Wang Z, Wang B, Shi Y, et al. Oncogenic miR‐20a and miR‐106a enhance the invasiveness of human glioma stem cells by directly targeting TIMP‐2. Oncogene. 2015;34:1407‐1419. [DOI] [PubMed] [Google Scholar]
- 37. Yu SC, Xiao HL, Jiang XF, et al. Connexin 43 reverses malignant phenotypes of glioma stem cells by modulating E‐cadherin. Stem Cells. 2012;30:108‐120. [DOI] [PubMed] [Google Scholar]
- 38. Zheng Y, Li X, Qian X, et al. Secreted and O‐GlcNAcylated MIF binds to the human EGF receptor and inhibits its activation. Nat Cell Biol. 2015;17:1348‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012‐2024. [DOI] [PubMed] [Google Scholar]
- 42. McDonough WS, Johansson A, Joffee H, Giese A, Berens ME. Gap junction intercellular communication in gliomas is inversely related to cell motility. Int J Dev Neurosci. 1999;17:601‐611. [DOI] [PubMed] [Google Scholar]
- 43. Yamada A, Futagi M, Fukumoto E, et al. Connexin 43 is necessary for salivary gland branching morphogenesis and FGF10‐induced ERK1/2 phosphorylation. J Biol Chem. 2016;291:904‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stains JP, Civitelli R. Gap junctions regulate extracellular signal‐regulated kinase signaling to affect gene transcription. Mol Biol Cell. 2005;16:64‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen D, Liu Y, Yang H, Zhang X, Fermandes JC, Chen Y. Connexin 43 promotes ossification of the posterior longitudinal ligament through activation of the ERK1/2 and p38 MAPK pathways. Cell Tissue Res. 2016;363:765‐773. [DOI] [PubMed] [Google Scholar]
- 46. Yang H, Shi L, Shi G, Guo Y, Chen D, Shi J. Connexin 43 affects osteogenic differentiation of the posterior longitudinal ligament cells via regulation of ERK activity by stabilizing Runx2 in ossification. Cell Physiol Biochem. 2016;38:237‐247. [DOI] [PubMed] [Google Scholar]
- 47. Gangoso E, Thirant C, Chneiweiss H, Medina JM, Tabernero A. A cell‐penetrating peptide based on the interaction between c‐Src and connexin43 reverses glioma stem cell phenotype. Cell Death Dis. 2014;5:e1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gonzalez‐Sanchez A, Jaraiz‐Rodriguez M, Dominguez‐Prieto M, Herrero‐Gonzalez S, Medina JM, Tabernero A. Connexin43 recruits PTEN and Csk to inhibit c‐Src activity in glioma cells and astrocytes. Oncotarget. 2016;7:49819‐49833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shishido SN, Nguyen TA. Induction of apoptosis by PQ1, a gap junction enhancer that upregulates connexin 43 and activates the MAPK signaling pathway in mammary carcinoma cells. Int J Mol Sci. 2016;17:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Raizer JJ, Abrey LE, Lassman AB, et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12:95‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gallego O, Cuatrecasas M, Benavides M, et al. Efficacy of erlotinib in patients with relapsed gliobastoma multiforme who expressed EGFRVIII and PTEN determined by immunohistochemistry. J Neurooncol. 2014;116:413‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Uhm JH, Ballman KV, Wu W, et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys. 2011;80:347‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roth P, Weller M. Challenges to targeting epidermal growth factor receptor in glioblastoma: escape mechanisms and combinatorial treatment strategies. Neuro Oncol. 2014;16(Suppl 8): viii14‐viii19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto‐oncogene product by phosphatidylinositol‐3,4‐bisphosphate. Science. 1997;275:665‐668. [DOI] [PubMed] [Google Scholar]
- 55. Laine J, Kunstle G, Obata T, Sha M, Noguchi M. The protooncogene TCL1 is an Akt kinase coactivator. Mol Cell. 2000;6:395‐407. [DOI] [PubMed] [Google Scholar]
- 56. Jahn T, Seipel P, Urschel S, Peschel C, Duyster J. Role for the adaptor protein Grb10 in the activation of Akt. Mol Cell Biol. 2002;22:979‐991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim AH, Sasaki T, Chao MV. JNK‐interacting protein 1 promotes Akt1 activation. J Biol Chem. 2003;278:29830‐29836. [DOI] [PubMed] [Google Scholar]
- 58. Yue Y, Lypowy J, Hedhli N, Abdellatif M. Ras GTPase‐activating protein binds to Akt and is required for its activation. J Biol Chem. 2004;279:12883‐12889. [DOI] [PubMed] [Google Scholar]
- 59. Ambrosi C, Ren C, Spagnol G, et al. Connexin43 forms supramolecular complexes through non‐overlapping binding sites for Drebrin, Tubulin, and ZO‐1. PLoS ONE. 2016;11:e0157073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fujimoto K, Nagafuchi A, Tsukita S, Kuraoka A, Ohokuma A, Shibata Y. Dynamics of connexins, E‐cadherin and alpha‐catenin on cell membranes during gap junction formation. J Cell Sci. 1997;110(Pt 3):311‐322. [DOI] [PubMed] [Google Scholar]
- 61. Jaraiz‐Rodriguez M, Tabernero MD, Gonzalez‐Tablas M, et al. A short region of connexin43 reduces human glioma stem cell Migration, invasion, and survival through Src, PTEN, and FAK. Stem Cell Reports. 2017;9:451‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Esseltine JL, Laird DW. Next‐generation connexin and pannexin cell biology. Trends Cell Biol. 2016;26:944‐955. [DOI] [PubMed] [Google Scholar]
- 63. Aasen T, Mesnil M, Naus CC, Lampe PD, Laird DW. Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer. 2017;17:74. [DOI] [PubMed] [Google Scholar]
- 64. Naus CC, Giaume C. Bridging the gap to therapeutic strategies based on connexin/pannexin biology. J Transl Med. 2016;14:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hitomi M, Deleyrolle LP, Mulkearns‐Hubert EE, et al. Differential connexin function enhances self‐renewal in glioblastoma. Cell Rep. 2015;11:1031‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]