

Abstract
The molecular features of hepatocellular carcinoma arising from non‐alcoholic fatty liver disease (NAFLD‐HCC) are not well known. In this study, we investigated the mechanism by which NAFLD‐HCC survives in a fat‐rich environment. We found that caveolin (CAV)‐1 was overexpressed in clinical specimens from NAFLD‐HCC patients. HepG2, HLE, and HuH‐7 HCC cell lines showed decreased proliferation in the presence of the saturated fatty acids palmitic acid and stearic acid, although only HLE cells expressed high levels of CAV‐1. HLE cells treated with oleic acid (OA) showed robust proliferation, whereas CAV‐null HepG2 cells showed reduced proliferation and increased apoptosis. CAV‐1 knockdown in HLE cells attenuated the OA‐induced increase in proliferation and enhanced apoptosis. Liquid chromatography–tandem mass spectrometry analysis revealed that the levels of OA‐containing ceramide, a pro‐apoptotic factor, were higher in HepG2 and CAV‐1‐deficient HLE cells than in HLE cells, suggesting that CAV‐1 inhibits apoptosis by decreasing the level of OA‐containing ceramide. These results indicate that CAV‐1 is important for NAFLD‐HCC survival in fatty acid‐rich environments and is a potential therapeutic target.
Keywords: apoptosis, caveolin‐1, ceramide, hepatocellular carcinoma, non‐alcoholic fatty liver disease
Abbreviations
- CAV
caveolin
- DG
diacylglycerol
- ER
endoplasmic reticulum
- FA
fatty acid
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- LC‐MS/MS
liquid chromatography–tandem mass spectrometry
- MS
mass spectrometry
- NAFLD
non‐alcoholic fatty liver disease
- OA
oleic acid
- PA
palmitic acid
- qRT‐PCR
quantitative RT‐PCR
- SA
stearic acid
- TG
triglyceride
1. INTRODUCTION
Liver cancer is the third leading cause of cancer‐related death worldwide. Hepatocellular carcinoma comprises more than 90% of primary liver cancers and is the most common primary hepatic malignancy.1, 2, 3 Non‐alcoholic fatty liver disease, which is the hepatic manifestation of obesity and metabolic syndrome, is a highly prevalent condition that affects 15–45% of persons in developed nations.4 The spectrum of NAFLD ranges from simple steatosis to steatohepatitis, fibrosis, and cirrhosis.5 Increasing numbers of reports are indicating that NAFLD could be a major cause of HCC, although HCV is the most common cause of HCC.6, 7
Hepatocellular carcinoma can develop even in NAFLD patients without cirrhosis.5, 8, 9 The typical features of HCC arising from NAFLD (NAFLD‐HCC) are moderate or high differentiation, the absence of encapsulation, and better postoperative prognosis when compared with HCV‐associated or alcohol abuse‐associated HCC.10 Although the reasons why NAFLD‐HCCs show such features are largely unknown, a fat‐rich environment might be related to NAFLD‐HCC features. The liver of NAFLD patients typically shows excessive accumulation of TGs within hepatocytes. Although TGs themselves are non‐toxic, their components, FAs, exert toxicity by causing ER stress. Therefore, TG synthesis might provide a buffer against the accumulation of toxic FAs.11 Therefore, NAFLD‐HCC is presumed to have some mechanisms for survival in a cytotoxic FA‐rich environment.12, 13
We hypothesized that lipids or lipid‐related proteins would have some influence on the proliferation of NAFLD‐HCC. In this study, CAV‐1 was identified as a significant molecule of NAFLD‐HCC based on the association between lipids and proteins affecting them. We then elucidated the role of CAV‐1 for the proliferation of NAFLD‐HCC.
2. MATERIALS AND METHODS
2.1. Patient characteristics and clinical specimens
Human HCC tissues and adjacent non‐HCC tissues were separately excised from the resected specimens of 20 patients with HCC. The patients' clinicopathological characteristics are listed in Table 1. In this study, the definition of NAFLD encompassed the entire spectrum of fatty liver disease in individuals without significant alcohol consumption (men, <30 g/day; women, <20 g/day), ranging from fatty liver to steatohepatitis and cirrhosis.14 Hepatitis C infection was considered to be present when patients showed positivity for HCV antibody but not hepatitis B virus antigen. Samples were snap‐frozen with RNAlater (Applied Biosystems, Grand Island, NY, USA) in liquid nitrogen and stored at −80°C. The study was undertaken in accordance with the guidelines for pathological specimen handling and approved by the ethics committee of the Hamamatsu University School of Medicine (Hamamatsu, Japan). All patients provided informed consent for the procedures.
Table 1.
Clinicopathological characteristics of 20 patients with hepatocellular carcinoma (HCC)
| HCC arising from NAFLD (n = 10) | HCC arising from HCV‐infected liver (n = 10) | P‐value | |
|---|---|---|---|
| Sex | |||
| Male | 9 | 7 | |
| Female | 1 | 3 | n.s. |
| Age, years | |||
| Median (range) | 72 (68–90) | 70 (57–78) | n.s. |
| BMI | 25.0 (20.4–27.4) | 20.9 (19.1–27.4) | 0.014 |
| Blood examination | |||
| T. Bil, mg/dL (range) | 0.75 (0.4–1.8) | 0.85 (0.4–1.7) | n.s. |
| AST, IU/mL (range) | 35 (18–72) | 42 (22–72) | n.s. |
| ALT, IU/mL (range) | 36.5 (13–79) | 29.5 (13–82) | n.s. |
| Alb, mg/dL (range) | 4.25 (3.1–4.8) | 4.15 (3.8–4.7) | n.s. |
| PT (%) (range) | 95.5 (73–125) | 90 (78–132) | n.s. |
| Alpha‐fetoprotein, ng/mL | |||
| Median (range) | 4 (1–1074) | 6.5 (4–119 665) | n.s. |
| PIVKA‐II, mAU/mL | |||
| Median (range) | 277 (13–15 905) | 37.5 (20–2130) | n.s. |
| Tumor size, cm | |||
| Median (range) | 3.8 (1.7–14) | 2.9 (1.5–8.5) | n.s. |
| Tumor number | |||
| Solitary | 7 | 5 | |
| Multiple | 3 | 5 | n.s. |
| Stage (UICC) | |||
| I | 6 | 5 | |
| II | 4 | 3 | |
| IIIa | 0 | 2 | n.s. |
Alb, albumin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; HCV, hepatitis C virus; NAFLD, non‐alcoholic fatty liver disease; n.s., not significant; PIVKA‐II, protein induced by vitamin K absence‐II; PT, prothrombin time.
2.2. Cell cultures and treatment
HepG2, HuH‐7, and HLE cells were purchased from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). The cells were maintained in DMEM supplemented with 10% FBS. HLE cells were transfected with siRNAs specific for CAV‐1 or CAV‐2 siRNA (Dharmacon, Lafayette, CO, USA) and Stealth RNAi Negative Control Medium GC Duplex (Invitrogen, Carlsbad, CA, USA) using Lipofectamine RNAiMAX transfection reagent (Invitrogen).
2.3. Cell proliferation assay
HLE, HepG2, and HuH‐7 cells were cultured at a density of 3500, 6125, and 8750 cells/well, respectively, in 96‐well plates in DMEM with 10% FBS overnight in the presence of various concentrations of FAs (0–1000 μmol/L) for 72 h. The cells were washed with PBS, fixed with 4% paraformaldehyde for 30 min, and stained with DAPI for 3 min. The cells were then imaged with an automated microscope (IN Cell Analyzer 2200; GE Healthcare, Little Chalfont, UK). Cell counting was carried out using IN Cell Investigator software (GE Healthcare).
2.4. Microarray analysis
Total RNA was extracted from frozen resected tissue specimens using the RNeasy Mini kit (Qiagen, Valencia, CA, USA). Total RNA quality and quantity were assessed with a NanoDrop ND‐1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Gene expression profiles of HCV‐HCC and NAFLD‐HCC were determined with a Affymetrix GeneChip Human Gene 2.0 ST array (Affymetrix, Santa Clara, CA, USA), and data were analyzed using GeneSpring GX version 12.6 software (Agilent Technologies, Santa Clara, CA, USA). Hierarchical clustering analysis was carried out with a fold change of at least 2 and P < 0.05. We also used gene set enrichment analysis software (Broad Institute of Massachusetts Institute of Technology and Harvard University, http://www.broad.mit.edu/gsea) to identify groups of genes that share a common biological function using the curated c2.cgp.v2.5.symbols.gmt database. Gene sets with false discovery rates of <25% or nominal P < 0.01 were defined as significant. Normalized microarray data were deposited in the Gene Expression Omnibus (accession no. GSE99131).
2.5. Quantitative RT‐PCR
Total RNA was extracted from resected specimens and cultured cells as described above, and reverse transcription was undertaken using the PrimeScript RT Reagent kit (Takara Bio, Otsu, Japan). The cDNA was amplified by qRT‐PCR on a Thermal Cycler Dice Real Time System II (Takara Bio) using the Thunderbird qPCR Mix (Toyobo Life Science, Osaka, Japan). Sequences of primers used for amplification are shown in Table S1.
2.6. Immunohistochemistry
Every 10th section of 4‐mm‐thick sections containing both cancer and adjacent non‐cancer regions, prepared from formalin‐fixed paraffin‐embedded tissue, was used to examine CAV‐1 and CAV‐2 expression. Sections were deparaffinized with sequential xylene and ethanol treatment followed by rehydration; antigen retrieval was carried out by heating the samples at 96°C for 40 min in Tris/EDTA buffer (pH 6). Slides were rinsed with PBS and incubated with 3% hydrogen peroxide in absolute methanol for 30 min to quench endogenous peroxidase activity. After an additional washing step with PBS, sections were incubated overnight at 4°C with antibodies diluted in REAL Substrate Buffer (Dako, Glostrup, Denmark), followed by incubation for 30 min in Dako REAL EnVision/HRP for rabbit/mouse. Immune complexes were detected using Dako REAL diaminobenzidine + chromogen. The sections were lightly counterstained with hematoxylin for 5 min and mounted with permanent mounting medium. Antibodies used in this study are listed in Table S2. Immunostaining intensity was evaluated as follows: 0, no staining; 1, weak staining; 2, medium staining; and 3, strong staining.
2.7. Liquid chromatography–MS/MS
Liquid chromatography–MS/MS was undertaken with an Agilent 1100 binary high‐pressure LC system (Agilent Technologies) and a Q Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Resolutions of 70 000 (full width at half maximum) and 17 500 at m/z 200 were used for full‐scan MS and MS/MS events, respectively. Full‐scan MS data were acquired using a mass range of m/z 220–2000. The MS parameters were as follows: spray voltage, 3.5 kV; capillary temperature, 250°C; sheath gas pressure, 50 psi; auxiliary gas pressure, 10 psi; probe heater temperature, 350°C; S‐lenz radiofrequency level, 50%; normalized collision energy, 30; and stepped normalized collision energy, 35% (both electrospray ionization‐positive and ionization‐negative). Separation was carried out using an Acclaim 120 C18 column (150 × 2.1 mm inner diameter, 3‐μm particle size; Thermo Fisher Scientific) with reverse‐phase LC elution maintained at 40°C. The mobile phases were 5 mmol/L ammonium formate in acetonitrile/methanol/water (1/1/2 v/v/v) and 0.1% formic acid (A), and 5 mmol/L ammonium formate in isopropanol/acetonitrile (9/1 v/v) and 0.1% formic acid (B). The gradient program was as follows: initial elution with 20% B followed by a linear gradient to 100% B from 0 to 50 min, additional elution with 100% B for 10 min, and an immediate decrease to 20% B at 60 min. We then returned to the initial conditions for 10 min until the end of the run. The total run time was 70 min. The mobile phase flow rate was 300 μL/min. Column effluent was introduced into the system from 2 to 60 min after injection. A 5‐μL volume of filtered solution was injected into the system. A targeted inclusion list was used to trigger all data‐dependent events. The composition values were calculated by the internal standard ratio. Untreated HepG2 cells cultured in DMEM + 3% BSA without OA or with 100 μmol/L OA, or 500 μmol/L OA; untreated HLE cells cultured in DMEM + 3% BSA without OA or with 100 μmol/L OA, or 500 μmol/L OA; and siRNA‐transfected HLE cells treated with DMEM + 3% BSA without OA or with 500 μmol/L OA were compared by anova. Representative lipid species in each condition and cell line were assigned with Lipid Search software (Mitsui Knowledge Industry, Tokyo, Japan) using additional independent high‐resolution data obtained with a Q Exactive spectrometer (Thermo Fisher Scientific).
2.8. Western blot analysis
Cells were lysed in chilled lysis buffer supplemented with a complete protease and phosphatase inhibitor cocktail (Roche, Basel, Switzerland). Protein extracts (20–40 μg) were subjected to 12% PAGE followed by electroblotting onto an Immobilon‐P PVDF membrane (Millipore, Billerica, MA, USA). Immunoreactive bands were visualized using Enhanced Chemiluminescence Plus Western Blotting Detection Reagent (GE Healthcare) and ChemiDoc Touch (Bio‐Rad, Hercules, CA, USA). Antibodies used in this study are listed in Table S2.
2.9. Statistical analysis
Statistical analyses were carried out with spss Statistics 22 (IBM, Armonk, NY, USA). Values are expressed as the mean ± SD. Student's t‐test or the Mann–Whitney U‐test was used for comparison of the measurable variation between two groups. Analyses of variance and multiple comparison were carried out to compare the measurable variation of three or more groups. P < 0.05 was defined as statistically significant.
2.10. Additional methods
Preparation of an FA–BSA complex, lipid extraction, double knockdown, expression plasmids, and transmission electron microscopy are described in Documents S1–S5.
3. RESULTS
3.1. Distinct gene expression profiles of NAFLD‐HCC and HCV‐HCC
To identify genes that are up‐ and downregulated in NAFLD‐HCC, we undertook mRNA microarray analysis using three sets of clinical NAFLD‐HCC and HCV‐HCC specimens. Patient profiles are shown in Table S3. Hierarchical clustering and principal component analysis (Figs. 1,S1) revealed that the two HCC groups had distinct gene expression profiles. Of 48 227 genes analyzed, 498 showed at least twofold differences in expression between the two groups (P < 0.05), with 260 genes that were upregulated and 238 that were downregulated in NAFLD‐HCC relative to HCV‐HCC. A gene set enrichment analysis showed that, of the upregulated genes, 70 were associated with biological processes, 30 with cellular components, and 32 with molecular function gene sets, with a false discovery rate of <25% and nominal P < 0.01 (Fig. S2). We focused on the gene sets associated with lipid synthesis and metabolism (Figs. S2,S3A) and identified CAV‐1 and CAV‐2 as two genes that were markedly upregulated in NAFLD‐HCC (Fig. S3B).
Figure 1.
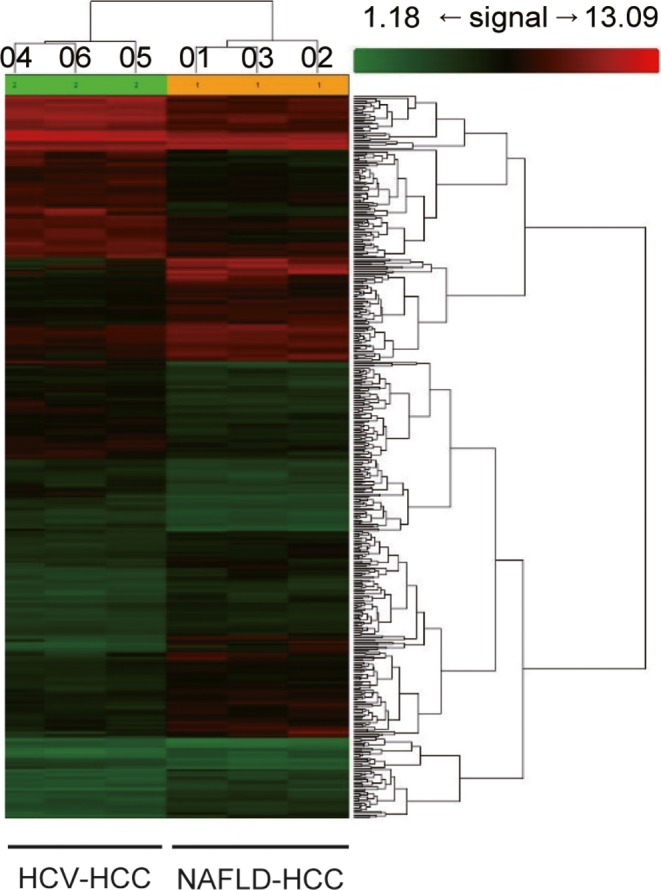
RNA microarray analysis of resected specimens from patients with hepatocellular carcinoma (HCC) arising from non‐alcoholic fatty liver disease (NAFLD) and hepatitis C virus (HCV) infection. Hierarchical clustering revealed differences in gene expression profiles between HCC arising from NAFLD (01–03) and HCV infection (04–06)
3.2. CAV‐1 and CAV‐2 abundantly expressed in NAFLD‐HCC
To confirm the mRNA microarray results, we undertook a qRT‐PCR analysis and found that CAV‐1 and CAV‐2 were significantly overexpressed in NAFLD‐HCC compared to HCV‐HCC (Fig. 2A). In patients with NAFLD, CAV‐1 transcript levels were higher in cancerous than in non‐cancerous lesions (P < 0.05) (Fig. 2B). Conversely, in patients with HCV infection, non‐cancerous tissue showed higher mRNA levels of CAV‐1 and CAV‐2 than cancerous lesions (Fig. 2B). There were no differences in liver function between patients with NAFLD‐HCC and HCV‐HCC (Table 1).
Figure 2.
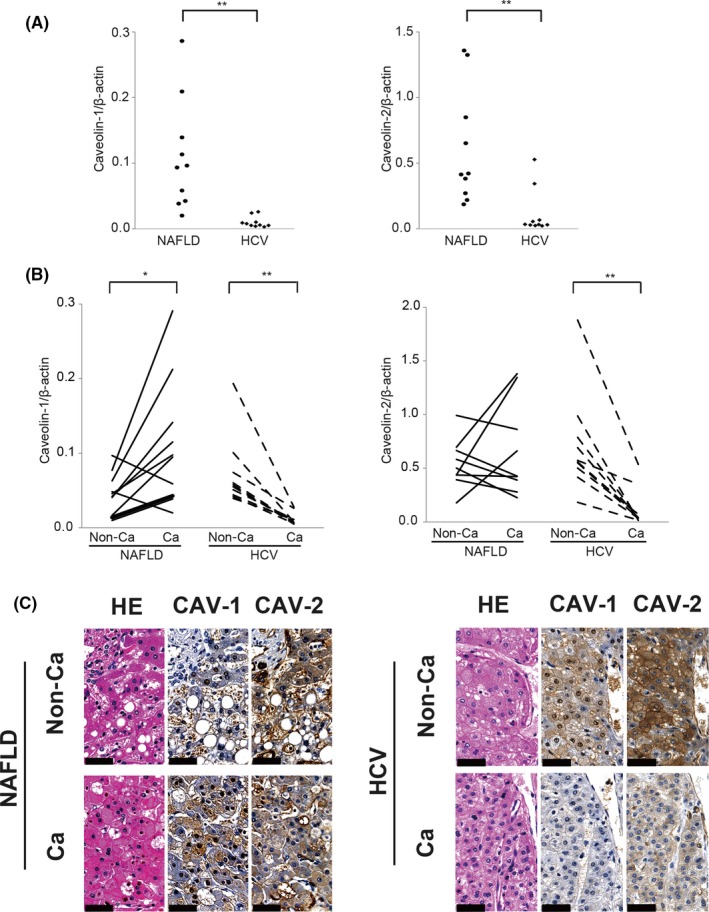
Quantitative RT‐PCR and immunohistochemical analysis of caveolin (CAV) expression in resected hepatocellular carcinoma (HCC) specimens. (A) Quantitative RT‐PCR analysis of CAV‐1 (left panel) and CAV‐2 (right panel) in HCC derived from non‐alcoholic fatty liver disease (NAFLD) and hepatitis C virus (HCV) infection. **P < 0.01, Mann–Whitney U‐test. (B) Comparison of CAV‐1 (left panel) and CAV‐2 (right panel) expression between non‐cancer (Non‐Ca) tissue and cancer tissue (Ca). *P < 0.05, **P < 0.01, Student's t‐test and Mann–Whitney U‐test. (C) In the immunohistochemical analysis, Non‐Ca and Ca lesions derived from NAFLD‐HCC or HCV‐HCC were stained with H&E or antibodies against CAV‐1 or CAV‐2. Caveolin immunoreactivity is visible as a brown color in the membrane or cytosol; nuclei are visible as a blue color by hematoxylin staining. Results are expressed as the mean ± SD. Scale bar = 50 μm
In NAFLD‐HCC samples, non‐cancerous lesions showed typical lipid accumulation in hepatocytes by H&E staining, and faint CAV‐1 expression was observed in hepatocytes by immunohistochemistry (Figs. 2C,S4). In contrast, HCC cells in NAFLD‐HCC showed strong CAV‐1 immunoreactivity. The opposite trends were observed in HCV‐HCC specimens, with HCC cells expressing low levels of CAV‐1 and CAV‐2 and hepatocytes in non‐cancerous lesions showing strong CAV‐1 and CAV‐2 staining (Figs. 2C,S4).
3.3. Caveolin expression affects cell proliferation in the presence of monounsaturated FA
To evaluate the function of CAV in HCC cells, we carried out experiments using three different HCC cell lines, HLE, HepG2, and HuH‐7. The qRT‐PCR and Western blot analyses revealed that HLE cells abundantly expressed CAV‐1 and CAV‐2, whereas HepG2 and HuH‐7 cells showed no CAV‐1 and CAV‐2, faint CAV‐1 and CAV‐2 expression, respectively (Fig. 3A,B). To determine whether CAV expression levels affect HCC progression in an FA‐rich environment, we evaluated proliferation in cells cultured with or without PA, SA, and OA. In the presence of saturated FAs (PA and SA), all three cell lines showed a concentration‐dependent decrease in cell proliferation (Fig. S5). However, CAV‐rich HLE cells, but not CAV‐null HepG2 and HuH‐7 cells, cultured with the monosaturated FA OA, showed increased cell proliferation (Fig. 3C).
Figure 3.
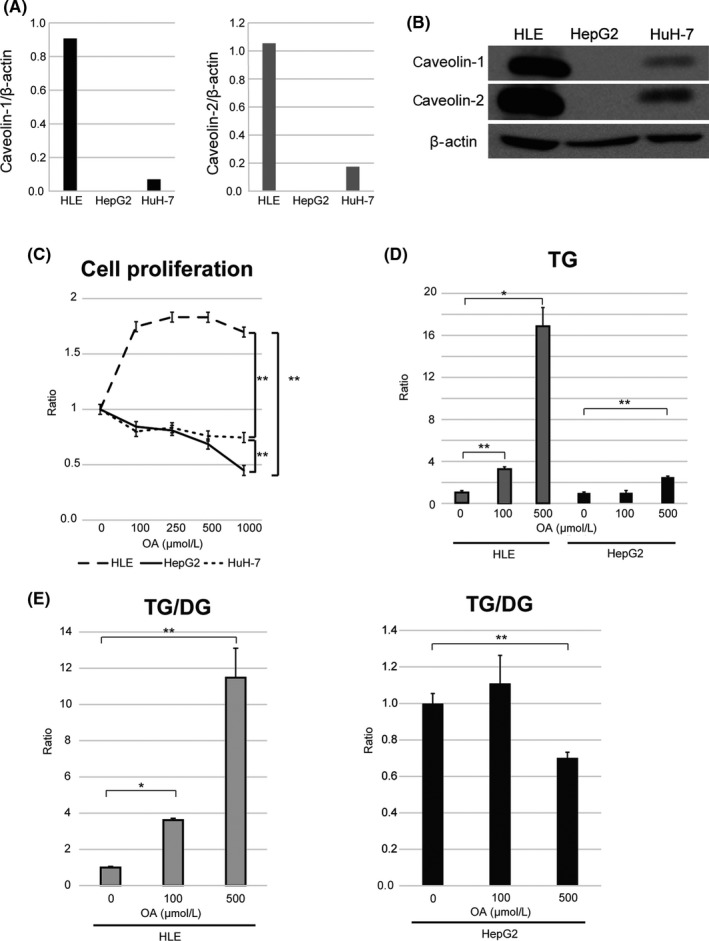
Role of caveolins (CAVs) in hepatocellular carcinoma cell proliferation and triglyceride (TG) accumulation in the presence of monounsaturated fatty acid (oleic acid [OA]). (A,B) Quantitative RT‐PCR (A) and Western blot analyses (B) of CAV‐1 and CAV‐2 expression in three hepatocellular carcinoma cell lines (HLE, HepG2, and HuH‐7). (C) Cell proliferation in medium containing OA. **P < 0.01 (n = 5; two‐way anova). (D,E) Liquid chromatography–tandem mass spectrometry analysis of intracellular TG levels (D) and TG/diacylglycerol (DG) ratio (E) in HepG2 and HLE cells exposed to OA. TG/DG ratio was determined as the ratio in HLE cells cultured in 100 or 500 μmol/L OA to that in cells cultured in 0 μmol/L OA. *P < 0.05, **P < 0.01, one‐way anova. Results are expressed as the mean ± SD
We examined the intracellular accumulation of TG by LC‐MS/MS to determine whether OA is taken up by cells. A 16.8‐fold increase in total TG level was observed in HLE cells treated with 500 μmol/L OA, compared to the 2.5‐fold increase observed in HepG2 cells (Fig. 3D). In particular, OA (C18:1)‐containing TG was highly enriched by OA supplementation (Table S4A).
Triglyceride is generated by adding one FA leg to diacylglycerol (DG), whereas DG is generated by removing one FA leg from TG. We therefore examined the TG/DG ratio and found that it was increased by 11.4‐fold in HLE cells but decreased by 0.7‐fold in HepG2 cells cultured with 500 μmol/L OA (Fig. 3E). These results suggest that CAV‐1 induces OA incorporation of the TG form to prevent FA‐induced cytotoxicity. In the absence of CAV‐1, OA supplementation promotes the conversion of TG to DG, thereby releasing cytotoxic FA.
3.4. CAV silencing abrogates OA‐induced increase in cell proliferation and change in intracellular TG levels
To examine their role in HCC, we silenced CAV‐1 and CAV‐2 genes in HLE cells using siRNAs (Fig. 4A,B). CAV‐1 and CAV‐2 knockdown abolished the OA‐induced increase in HLE cell proliferation (Fig. 4C). Interestingly, intracellular TG level was increased 1.7‐fold in these cells (Fig. 4D, Table S5), whereas the TG/DG ratio was decreased 0.58‐fold (Fig. 4E) by CAV‐1 knockdown. These results suggest that CAV‐1 influences the intracellular TG/DG ratio rather than OA incorporation in HCC cells.
Figure 4.
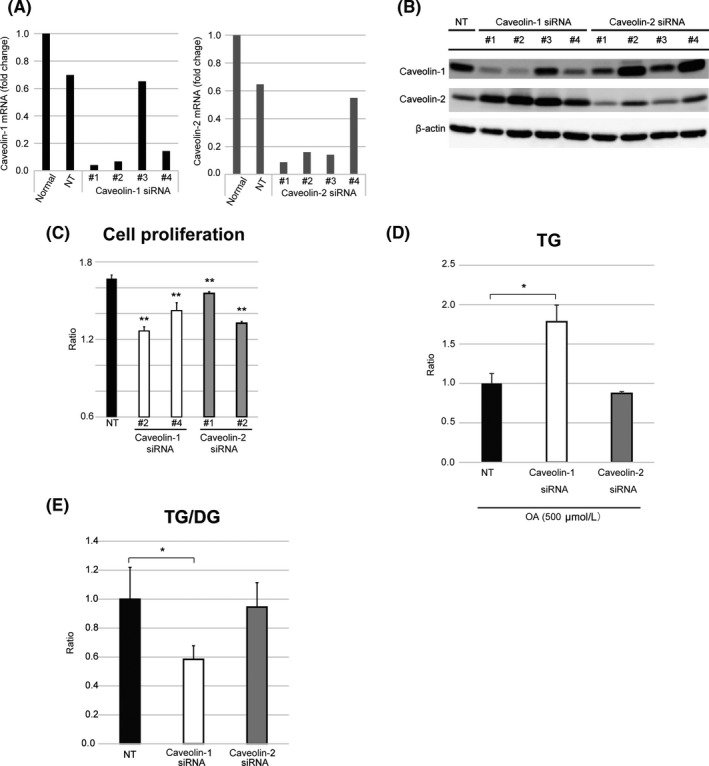
Caveolin (CAV) knockdown affects hepatocellular carcinoma cell proliferation and triglyceride (TG) accumulation. (A,B) Quantitative RT‐PCR (A) and Western blot (B) analyses of cells transfected with siRNAs against CAV‐1 or CAV‐2 or with a non‐targeting (NT) siRNA. (C) Proliferation of HLE cells cultured in the presence of oleic acid (OA) after siRNA transfection (48 h). The ratio was determined as the number of cells cultured in DMEM + 3% BSA + 500 μmol/L OA relative to the number of cells cultured in DMEM + 3% BSA. **P < 0.01, Student's t‐test and Mann–Whitney U‐test. (D) Liquid chromatography–tandem mass spectrometry analysis of intracellular TG level in HLE cells cultured in medium containing 500 μmol/L OA after CAV‐1 or CAV‐2 knockdown. (E) TG/diacylglycerol (DG) ratio. *P < 0.05, Student's t‐test and Mann–Whitney U‐test. Results are expressed as the mean ± SD
As shown in Figure 4(B), the expression of the other caveolin proteins seemed to increase when the targeted caveolin gene was knocked down, suggesting a compensatory mechanism (#1–4 of CAV‐1 siRNA, and #2 and #4 of CAV‐2 siRNA). To eliminate such compensation, we undertook double knockdown of both CAV‐1 and CAV‐2 in HLE cells. Similarly to the single knockdown experiments, proliferation of double knockdown HLE cells was inhibited (Fig. S6).
3.5. Caveolin‐1 suppresses OA‐induced apoptosis
We next examined the expression of apoptosis‐related proteins and found that OA supplementation did not affect the phosphorylation of the anti‐apoptotic protein AKT in CAV‐null HepG2, CAV‐rich HLE, or CAV‐1/‐2‐silenced HLE cells (Fig. 5A‐C). Moreover, the level of the pro‐apoptotic protein cleaved caspase‐3 was increased by exposure to OA (≥500 μmol/L) in HepG2 cells (Fig. 5A) but not in HLE cells (Fig. 5B). Knockdown of CAV‐1 but not of CAV‐2 resulted in increased cleaved caspase‐3 levels in OA‐treated cells (Fig. 5C). In transmission electrical microscopy, only CAV‐1‐silenced HLE cells morphologically showed apoptotic changes (Fig. S7). Moreover, CAV‐1‐overexpressing HepG2 cells showed increased cell proliferation in 500 μmol/L OA‐containing medium (Fig. S8).
Figure 5.
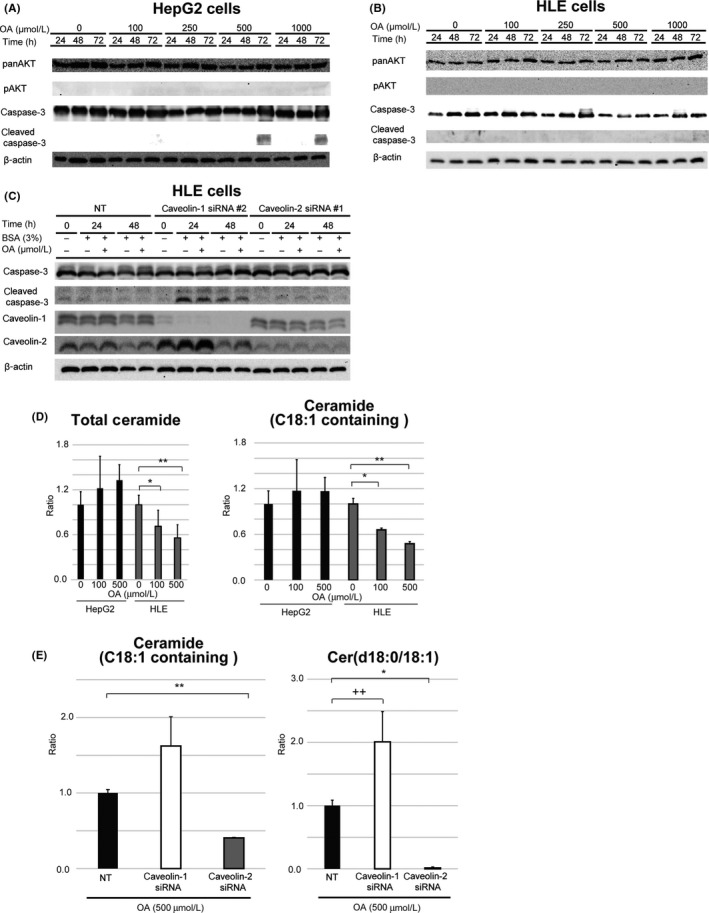
Expression of apoptosis‐related proteins and lipids in hepatocellular carcinoma cells cultured in the presence or absence of oleic acid (OA). (A–C) Western blot analysis of apoptosis‐related proteins in caveolin (CAV)‐null HepG2 (A), CAV‐rich HLE (B), and CAV‐1 or CAV‐2‐deficient HLE (C) cells. (D) Liquid chromatography–tandem mass spectrometry analysis of intracellular ceramide levels in HepG2 and HLE cells. Ratios of ceramide level at indicated concentrations of OA to that at 0 μmol/L OA are shown. Left panel, total ceramide; right panel, OA (C18:1)‐containing ceramide. *P < 0.05, **P < 0.01, one‐way anova. (E) Effect of CAV knockdown on ceramide composition in HLE cells after OA supplementation. Left panel, OA (C18:1)‐containing ceramide ratio; right panel, ceramide composed of OA and C18 sphingosine (d18:0). *P < 0.05, **P < 0.01, ++ P = 0.050, Student's t‐test and Mann–Whitney U‐test. Results are expressed as the mean ± SD
Liquid chromatography–MS/MS analysis showed that the total intracellular level of ceramide, a pro‐apoptotic sphingolipid, was decreased in a concentration‐dependent manner in CAV‐rich HLE but not CAV‐null HepG2 cells exposed to OA (Fig. 5D, left panel). The level of OA (C18:1)‐containing ceramide was also decreased in a concentration‐dependent manner in HLE cells by OA exposure (Fig. 5D, right panel). Additionally, the OA‐containing ceramide level was increased 1.68‐fold by CAV‐1 knockdown (Fig. 5E, left panel), with the greatest increase observed for ceramide composed of OA and C18‐sphingosine (d18:0) (Fig. 5E, right panel; P = 0.05). In contrast, CAV‐2 silencing caused a decrease in OA‐containing ceramide level.
4. DISCUSSION
In this study, we investigated the mechanisms by which NAFLD‐HCC survives in a fat‐rich environment by identifying the specific genes involved by microarray profiling. CAV‐1 and CAV‐2 were identified as novel NAFLD‐HCC‐related genes (Figs. 1,S1,S2). Our qRT‐PCR and immunohistochemical analyses of resected HCC specimens showed that CAV mRNA and protein levels were higher in NAFLD‐HCC than in HCV‐HCC (Fig. 2A). Furthermore, in NAFLD‐HCC specimens, CAV mRNA and protein levels were higher in cancerous lesions compared to adjacent non‐cancerous lesions, whereas the opposite was true in HCV‐HCC specimens (Figs. 2B,C,S4). These results suggest that CAVs are not oncogenic factors.
Caveolins in lipid rafts of the HCC cell membrane are thought to be important for HCV‐RNA replication, as HCV non‐structural proteins are incorporated by phagocytosis through lipid rafts.15 This is supported by our observation that the CAV‐1 mRNA level was higher in non‐cancerous lesions of HCV‐HCC as compared to NAFLD‐HCC (Fig. 2B). In contrast, the CAV‐1 mRNA level was very low in cancerous HCV‐HCC lesions compared to non‐cancerous NAFLD lesions (Fig. 2B). Hepatitis C virus replication is reduced in cancerous HCV‐HCC tissue.16 The low CAV‐1 expression in HCV‐HCC suggests that both HCV replication and CAV‐1 function are dispensable for HCC carcinogenesis. Nonetheless, our results suggest that CAVs play an important role in NAFLD‐HCC progression by promoting survival in an FA‐rich environment.
We used three HCC cell lines to investigate whether the degree of CAV expression affects HCC survival in an FA‐rich environment. In the presence of saturated FAs (PA and SA), none of the three cell lines showed robust proliferation (Fig. S5). Plasma concentrations of total and free PA or SA in human non‐alcoholic steatohepatitis patients were approximately 4200 and 150 μmol/L or 1500 and 60 μmol/L, respectively.17 Excessive PA and SA concentrations are considered to induce apoptosis through ER stress. Our results reflect clinical conditions.18 In contrast, CAV‐rich HLE cells exposed to a monounsaturated FA (OA) showed high proliferation capacity (Fig. 3C). Oleic acid supplementation has been used to mimic NAFLD;19, 20 therefore, our findings indicate that CAVs are important for HCC survival in the FA‐rich environment observed in NAFLD patients.
Caveolin proteins are important for the formation of plasma membrane caveolae, which are involved in endocytosis, transcytosis, viral and bacterial entry, signal transduction, and lipid and glucose metabolism. The CAV protein family in mammals includes three isoforms (CAV‐1, CAV‐2, and CAV‐3). Caveolin‐3 is predominantly expressed in striated muscle, whereas CAV‐1 and CAV‐2 are generally co‐expressed in other cell types. Caveolin‐1 can form a homo‐ or heterodimer with CAV‐2, which does not homodimerize; this suggests that CAV‐1 is indispensable in all tissues in which it is expressed.21 Caveolin‐1 and caveolae are thought to have tumor suppressor or oncogenic properties in various cancers.22, 23 Although some studies have implicated CAV‐1 in HCC, its exact function remains unknown.24, 25, 26, 27, 28, 29, 30 In the present study, we addressed this question by comparing the proliferation of CAV‐rich HLE cells with that of CAV‐null HepG2 cells in the presence or absence of OA. Long‐chain FAs such as OA are utilized by cells for FA beta‐oxidation, TG synthesis, and phospholipid biosynthesis.31 In this study, we evaluated TG accumulation in HCC cells exposed to OA by LC‐MS/MS and found that the levels of TGs, especially those containing OA, were drastically elevated in HLE cells by treatment with OA, whereas in HepG2 cells, total TG and OA‐containing TG levels were enhanced by 500 μmol/L OA supplementation (Fig. 3D, Table S4). A much smaller increase was observed in HepG2 cells. Interestingly, the TG/DG ratio was increased in HLE cells but decreased in HepG2 cells (Fig. 3E), although the TG level was enhanced in both cell lines (Fig. 3D). This effect was abolished by CAV‐1 silencing (Fig. 4E), implying that OA is conjugated to DG as soon as it is internalized by cells through the activity of CAV‐1. Moreover, these results suggest that OA uptake is independent of CAV; candidate factors that mediate this process include FA translocase/cluster of differentiation 36, FA‐binding protein, and FA transport protein.31 We speculate that CAV‐1 helps HCC cells escape the cytotoxic effects of FA by producing TG through the addition of OA chains to DG. This is supported by the finding that CAVs sequester FAs on the cytoplasmic membranous leaflet, which enhances TG formation and protects cells against FA lipotoxicity.32
In HepG2 and CAV‐1 knockdown HLE cells, cleaved caspase‐3 expression was upregulated in the presence of OA at concentrations >500 μmol/L (Fig. 5A,C). These results indicate that apoptosis is induced by high extracellular concentrations of OA. Plasma concentrations of total and free OA in human non‐alcoholic steatohepatitis patients are approximately 2500 and 150 μmol/L, respectively.17 Thus, the conditions in our experiments reflect clinical NAFLD.
Ceramide is a component of the cell membrane that serves as a bioactive intermediate promoting cell death.33, 34 Recent studies have shown that ceramides with long‐chain FAs (C16–C20) have antiproliferative and/or pro‐apoptotic functions.35, 36 In particular, C18 ceramide has been shown to act as a tumor suppressor in preclinical and clinical studies.37, 38, 39, 40 Ceramide is synthesized de novo by coupling of FA to sphingosine as well as through sphingomyelinase‐dependent degradation of sphingomyelin.41 Extracellular OA supplementation can affect the regulation of ceramide, especially if it contains OA. In this study, total and OA‐containing ceramide levels were increased in the presence of OA to some extent in CAV‐null HepG2 cells and CAV‐1‐silenced HLE cells, in which apoptosis was enhanced (Figs. 5A,C,D,G,S7). Unexpectedly, in HLE cells, OA supplementation decreased the levels of intracellular ceramide in a concentration‐dependent manner, especially in ceramide containing OA (Fig. 5D). Moreover, the OA‐induced reduction in the level of OA‐containing ceramide, especially ceramide (d18:0/18:1), was abrogated by CAV‐1 knockdown in HLE cells (Fig. 5E). The reason for this unusual regulation of ceramide is unclear; however, CAV‐1 might be important for the incorporation and sequestration of OA as a form of TG and might suppress increases in ceramide levels by blocking OA translocation to the cell membrane. When CAV‐1 expression is inhibited in an FA‐rich environment, extracellular OA is incorporated into the cell membrane as a form of ceramide, leading to apoptosis.
Recent studies have reported the importance of lipid metabolism‐related genes in HCC.42, 43, 44, 45, 46 Our group has shown that lysophosphatidylcholine acyltransferase 1, which converts lysophosphatidylcholine to phosphatidylcholine through the addition of one FA chain, alters phospholipid composition and modulates hepatoma progression.47 We also found that elongation of long‐chain FA member six, an enzyme that converts PA to SA, is abundantly expressed in NAFLD‐HCC and reduces ER stress and apoptosis induced by PA and SA.18 Thus, HCC cells may acquire mechanisms that promote cell proliferation and invasion by reducing lipotoxicity.
In conclusion, this study shows for the first time a role for CAV‐1 in NAFLD‐HCC progression: CAV‐1 overexpression prevents apoptosis and induces HCC cell proliferation in an OA‐rich environment such as that observed in NAFLD. These findings suggest that CAV‐1, lipids, and lipid‐regulating enzymes are suitable therapeutic targets for the treatment of HCC.
DISCLOSURE STATEMENT
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This study was supported by Grants‐in‐Aid for Scientific Research (Nos. 25462085 and 15K10132) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT). This work was also supported by MEXT/Japan Society for the Promotion of Science KAKENHI (Grant No. JP15H05898B1), Imaging Platform supported by MEXT, and the Japan Agency for Medical Research and Development (Grant No. JP17gm0910004).
Takeda M, Sakaguchi T, Hiraide T, et al. Role of caveolin‐1 in hepatocellular carcinoma arising from non‐alcoholic fatty liver disease. Cancer Sci. 2018;109:2401–2411. 10.1111/cas.13659
Funding information
Ministry of Education, Culture, Sports, Science and Technology of Japan; Japan Society for the Promotion of Science; Japan Agency for Medical Research and Development.
REFERENCES
- 1. El‐Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264‐1273.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adams LA, White SW, Marsh JA, et al. Association between liver‐specific gene polymorphisms and their expression levels with nonalcoholic fatty liver disease. Hepatology. 2013;57(2):590‐600. [DOI] [PubMed] [Google Scholar]
- 3. European Association For The Study Of The Liver ; European Organisation For Research And Treatment Of Cancer . EASL‐EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56(4):908‐943. [DOI] [PubMed] [Google Scholar]
- 4. Beyoglu D, Idle JR. The metabolomic window into hepatobiliary disease. J Hepatol. 2013;59(4):842‐858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis‐new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10(11):627‐636. [DOI] [PubMed] [Google Scholar]
- 6. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10(11):656‐665. [DOI] [PubMed] [Google Scholar]
- 7. Younossi ZM, Otgonsuren M, Henry L, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology. 2015;62(6):1723‐1730. [DOI] [PubMed] [Google Scholar]
- 8. Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol. 2012;56(3):704‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non‐alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56(6):1384‐1391. [DOI] [PubMed] [Google Scholar]
- 10. Margini C, Dufour JF. The story of HCC in NAFLD: from epidemiology, across pathogenesis, to prevention and treatment. Liver Int. 2016;36(3):317‐324. [DOI] [PubMed] [Google Scholar]
- 11. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118(3):829‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metabol. 2006;291(2):E275‐E281. [DOI] [PubMed] [Google Scholar]
- 13. Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl‐CoA desaturase. J Biol Chem. 2009;284(9):5637‐5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non‐alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142(7):1592‐1609. [DOI] [PubMed] [Google Scholar]
- 15. Kim JY, Wang L, Lee J, Ou JJ. Hepatitis C virus induces the localization of lipid rafts to autophagosomes for its RNA replication. J Virol. 2017;91(20):e00541‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harouaka D, Engle RE, Wollenberg K, et al. Diminished viral replication and compartmentalization of hepatitis C virus in hepatocellular carcinoma tissue. Proc Natl Acad Sci USA. 2016;113(5):1375‐1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Almeida IT, Cortez‐Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non‐alcoholic steatohepatitis. Clin Nutr. 2002;21(3):219‐223. [DOI] [PubMed] [Google Scholar]
- 18. Shibasaki Y, Horikawa M, Ikegami K, et al. Stearate‐to‐palmitate ratio modulates endoplasmic reticulum stress and cell apoptosis in non‐B non‐C hepatoma cells. Cancer Sci. 2018;109(4):1110‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Araya J, Rodrigo R, Videla LA, et al. Increase in long‐chain polyunsaturated fatty acid n ‐ 6/n ‐ 3 ratio in relation to hepatic steatosis in patients with non‐alcoholic fatty liver disease. Clin Sci (Lond). 2004;106(6):635‐643. [DOI] [PubMed] [Google Scholar]
- 20. Gomez‐Lechon MJ, Donato MT, Martinez‐Romero A, Jimenez N, Castell JV, O'Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact. 2007;165(2):106‐116. [DOI] [PubMed] [Google Scholar]
- 21. Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14(2):98‐112. [DOI] [PubMed] [Google Scholar]
- 22. Martinez‐Outschoorn UE, Sotgia F, Lisanti MP. Caveolae and signalling in cancer. Nat Rev Cancer. 2015;15(4):225‐237. [DOI] [PubMed] [Google Scholar]
- 23. Fernandez‐Rojo MA, Ramm GA. Caveolin‐1 function in liver physiology and disease. Trends Mol Med. 2016;22(10):889‐904. [DOI] [PubMed] [Google Scholar]
- 24. Cokakli M, Erdal E, Nart D, et al. Differential expression of Caveolin‐1 in hepatocellular carcinoma: correlation with differentiation state, motility and invasion. BMC Cancer. 2009;9:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tse EY, Ko FC, Tung EK, et al. Caveolin‐1 overexpression is associated with hepatocellular carcinoma tumourigenesis and metastasis. J Pathol. 2012;226(4):645‐653. [DOI] [PubMed] [Google Scholar]
- 26. Yang SF, Yang JY, Huang CH, et al. Increased caveolin‐1 expression associated with prolonged overall survival rate in hepatocellular carcinoma. Pathology. 2010;42(5):438‐445. [DOI] [PubMed] [Google Scholar]
- 27. Korhan P, Erdal E, Kandemis E, et al. Reciprocal activating crosstalk between c‐Met and caveolin 1 promotes invasive phenotype in hepatocellular carcinoma. PLoS ONE. 2014;9(8):e105278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gai X, Lu Z, Tu K, Liang Z, Zheng X. Caveolin‐1 is up‐regulated by GLI1 and contributes to GLI1‐driven EMT in hepatocellular carcinoma. PLoS ONE. 2014;9(1):e84551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Senetta R, Stella G, Pozzi E, Sturli N, Massi D, Cassoni P. Caveolin‐1 as a promoter of tumour spreading: when, how, where and why. J Cell Mol Med. 2013;17(3):325‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu WR, Jin L, Tian MX, et al. Caveolin‐1 promotes tumor growth and metastasis via autophagy inhibition in hepatocellular carcinoma. Clin Res Hepatol Gastroenterol. 2016;40(2):169‐178. [DOI] [PubMed] [Google Scholar]
- 31. Saini‐Chohan HK, Mitchell RW, Vaz FM, Zelinski T, Hatch GM. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders: thematic Review Series: Genetics of Human Lipid Diseases. J Lipid Res. 2012;53(1):4‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simard JR, Meshulam T, Pillai BK, et al. Caveolins sequester FA on the cytoplasmic leaflet of the plasma membrane, augment triglyceride formation, and protect cells from lipotoxicity. J Lipid Res. 2010;51(5):914‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bieberich E. Ceramide signaling in cancer and stem cells. Future Lipidol. 2008;3(3):273‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oskouian B, Saba JD. Cancer treatment strategies targeting sphingolipid metabolism. Adv Exp Med Biol. 2010;688:185‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grosch S, Schiffmann S, Geisslinger G. Chain length‐specific properties of ceramides. Prog Lipid Res. 2012;51(1):50‐62. [DOI] [PubMed] [Google Scholar]
- 36. Grammatikos G, Schoell N, Ferreiros N, et al. Serum sphingolipidomic analyses reveal an upregulation of C16‐ceramide and sphingosine‐1‐phosphate in hepatocellular carcinoma. Oncotarget. 2016;7(14):18095‐180105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8(10):831‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saddoughi SA, Garrett‐Mayer E, Chaudhary U, et al. Results of a phase II trial of gemcitabine plus doxorubicin in patients with recurrent head and neck cancers: serum C(1)(8)‐ceramide as a novel biomarker for monitoring response. Clin Cancer Res. 2011;17(18):6097‐6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karahatay S, Thomas K, Koybasi S, et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): attenuation of C(18)‐ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 2007;256(1):101‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koybasi S, Senkal CE, Sundararaj K, et al. Defects in cell growth regulation by C18:0‐ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J Biol Chem. 2004;279(43):44311‐44319. [DOI] [PubMed] [Google Scholar]
- 41. Ponnusamy S, Meyers‐Needham M, Senkal CE, et al. Sphingolipids and cancer: ceramide and sphingosine‐1‐phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010;6(10):1603‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kudo Y, Tanaka Y, Tateishi K, et al. Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3‐kinase pathway. J Hepatol. 2011;55(6):1400‐1408. [DOI] [PubMed] [Google Scholar]
- 43. Hill‐Baskin AE, Markiewski MM, Buchner DA, et al. Diet‐induced hepatocellular carcinoma in genetically predisposed mice. Hum Mol Genet. 2009;18(16):2975‐2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL‐6 and TNF expression. Cell. 2010;140(2):197‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tessitore A, Cicciarelli G, Del Vecchio F, et al. MicroRNA expression analysis in high fat diet‐induced NAFLD‐NASH‐HCC progression: study on C57BL/6J mice. BMC Cancer. 2016;16:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vinciguerra M, Carrozzino F, Peyrou M, et al. Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN. J Hepatol. 2009;50(6):1132‐1141. [DOI] [PubMed] [Google Scholar]
- 47. Morita Y, Sakaguchi T, Ikegami K, et al. Lysophosphatidylcholine acyltransferase 1 altered phospholipid composition and regulated hepatoma progression. J Hepatol. 2013;59(2):292‐299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials