

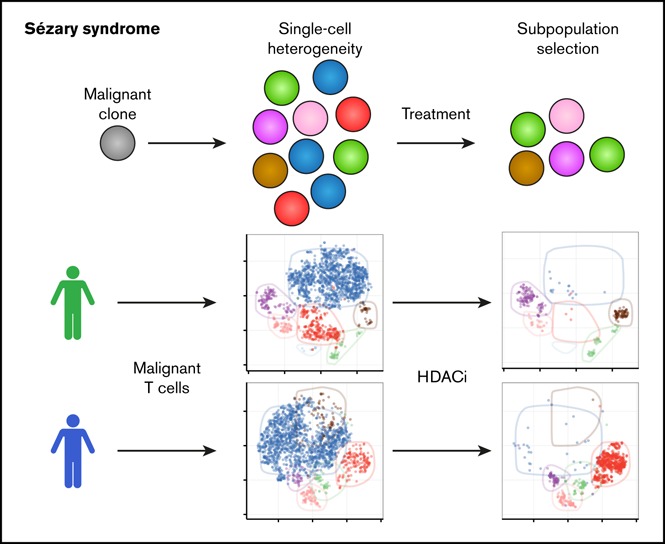
Key Points
Individual patients with Sézary syndrome contain several distinct malignant subpopulations and show marked single-cell heterogeneity.
Malignant subpopulations exhibit differences in their sensitivity to treatment warranting precision therapy.
Abstract
Sézary syndrome (SS) is an aggressive leukemic variant of cutaneous T-cell lymphoma (CTCL) with a median life expectancy of less than 4 years. Although initial treatment responses are often good, the vast majority of patients with SS fail to respond to ongoing therapy. We hypothesize that malignant T cells are highly heterogeneous and harbor subpopulations of SS cells that are both sensitive and resistant to treatment. Here, we investigate the presence of single-cell heterogeneity and resistance to histone deacetylase inhibitors (HDACi) within primary malignant T cells from patients with SS. Using single-cell RNA sequencing and flow cytometry, we find that malignant T cells from all investigated patients with SS display a high degree of single-cell heterogeneity at both the mRNA and protein levels. We show that this heterogeneity divides the malignant cells into distinct subpopulations that can be isolated by their expression of different surface antigens. Finally, we show that treatment with HDACi (suberanilohydroxamic acid and romidepsin) selectively eliminates some subpopulations while leaving other subpopulations largely unaffected. In conclusion, we show that patients with SS display a high degree of single-cell heterogeneity within the malignant T-cell population, and that distinct subpopulations of malignant T cells carry HDACi resistance. Our data point to the importance of understanding the heterogeneous nature of malignant SS cells in each individual patient to design combinational and new therapies to counter drug resistance and treatment failure.
Visual Abstract
Introduction
Cutaneous T-cell lymphoma (CTCL) is a group of non-Hodgkin lymphomas characterized by chronically inflamed skin lesions containing malignant T cells. Sézary syndrome (SS) is an aggressive leukemic variant of CTCL with a median life expectancy of less than 4 years.1,2 Current management of SS comprises a long list of experimental and established treatments including extracorporeal photopheresis and histone deacetylase inhibitors (HDACi).3-5 With the exception of allogenic hematopoietic stem cell transplantation, current treatment options only alleviate symptoms and tumor burden of the disease without the prospect of full remission or cure.5,6 Although initial response rates for most treatments are good, patients with SS often develop resistance to ongoing treatments.3,4 Despite vigorous research and progress in our understanding of the genomic landscape of CTCL, no single common driver mutation has yet been identified.7-9 The lack of recurrent driver mutations is also reflected in the great molecular differences seen between individual patients. However, malignant SS cells from the majority of patients are highly genetically unstable and present with multiple genetic and chromosomal aberrations converging on particular cancer-associated molecular pathways.7-11 Malignant SS cells often exhibit abnormal expression of T-cell surface markers and can be isolated on the basis of their clonal T-cell receptor (TCR) or their characteristic low expression of CD7 and/or CD26.12,13
On the basis of their presence in the blood and lymph nodes and their expression of distinct surface markers such as CD197 (CCR7), CD27, and CD62L (l-selectin), SS cells are suspected to derive from transformed central memory T (TCM) cells.14 However, studies have found that although the majority of malignant cells from most patients with SS do exhibit a TCM surface phenotype, some malignant cells express surface markers inconsistent with the TCM phenotype, such as high levels of CD45RA.15,16 This indicates that the population of malignant SS cells within each patient exhibits some degree of cellular heterogeneity, despite reportedly originating from a single transformed T-cell clone.17
We hypothesize that single-cell heterogeneity within the malignant T-cell population of SS facilitates treatment resistance through selection and may explain the marked recurrence rate in SS. In this study, we establish the presence of cellular heterogeneity within primary malignant T cells from patients with SS at the surface marker and mRNA level. We show that the malignant populations consist of distinct subpopulations that exhibit remarkable differences in their sensitivity toward HDACi treatment.
Methods
Malignant cells from patients with SS
Peripheral blood mononuclear cells (PBMCs) were collected from the blood of patients diagnosed with SS in accordance with the World Health Organization and European Organization for Research and Treatment of Cancer classification.13 None of the patients received treatment with HDACi or have previously been treated with HDACi. A full list of patient characteristics including past and current treatments is shown in Table 1. PBMCs were isolated by density gradient centrifugation, using LymphoPrep and SepMate-50 tubes (Stem Cell Technologies, catalog #07851 and #85460). Malignant T cells were identified by their expression of a dominant TCRVβ clone (SS6, SS8, SS9, SS10, and SS11) and their characteristic low expression of CD7 and/or CD26.12,13 In accordance with the Declaration of Helsinki, the samples were obtained with informed consent after approval by the Committee on Health Research Ethics (H-16025331).
Table 1.
Patient characteristics
| Patient | Sex | Age, y | Diagnosis | Year SS of diagnosis | ISCL classification | Leukocytes (×1000/µL blood) | CD4:CD8 ratio | TCR clone | Current treatment | Previous treatment |
|---|---|---|---|---|---|---|---|---|---|---|
| SS01 | F | NA | SS | NA | NA | NA | 93.5* | NA | NA | NA |
| SS02 | M | 67 | SS | 2016 | B2 | 2.4 | 9.2 | Yes (BM, Skin) | ECP, IFN-α, Bexarotene, TSEI | Acitretin, Klorokin, Doxycyklin, Prednisolone |
| SS03 | M | 79 | SS | 2016 | B2 | 4.1 | 16.6 | Yes (BM, Blood, Skin) | ECP | Ifliximab, Etanercept, Adalimumab, Ustekinumab, Cyclosporin, PUVA |
| SS04 | M | 85 | SS | 2012 | NA | NA | 32.4* | Yes (BM, Blood, Skin) | ECP, IFN-α | PUVA, IFN-α, Bexarotene |
| SS05 | F | 74 | SS | 2016 | B2 | 85.5 | 90.0 | Yes (BM, Blood, Skin) | ECP† | Prednisolone, Metotrexate, PUVA |
| SS06 | M | NA | SS | 2013 | B2 | NA | 35.2 | Yes (Blood‡) | NA† | NA |
| SS07 | F | 79 | SS | 2016 | B2 | 4 | 30.0 | Yes (BM, Blood, Skin) | ECP, Acitretin | Metotrexate, Allitretionin, Azathioprin, Acitretin |
| SS08 | M | 65 | SS | 2016 | B2 | 1.7 | 16.5 | Yes (BM, Blood, Skin) | ECP, IFN-α, Prednisolone | Ifliximab, Ustekinumab, Secukinumab, Adalimumab, Acitretin, Allitretionin, Prednisolone |
| SS09 | M | 58 | SS | 2018 | B2 | 1.3 | 13.0 | Yes (Blood, LN) | ECP, IFN-α, Prednisolone | Prednisolone, Metotrexate |
| SS10 | M | 80 | SS | 2018 | B2 | 8.1 | 28.0 | Yes (BM, Blood, Skin) | ECP, IFN-α† | PUVA |
| SS11 | M | 76 | SS | 2017 | B2 | 3.3 | 26.0 | Yes (BM, Blood, Skin) | ECP, IFN-α, Bexarotene | PUVA, UVB, Methotrexate, IFN-α, Bexarotene |
BM, bone marrow; ECP, extracorporal photopheresis; IFN, interferon; LN, lymph node; NA, Not available; PUVA, psoralen and ultraviolet A; and TSEI, total skin electron irradiation.
Measured after initiation of treatment.
Cells from these patients were collected before initiation of SS treatment.
Detected by flow cytometry
Flow cytometry
Primary conjugated monoclonal antibodies against CD2, CD3, CD4, CD7, CD15s, CD20, CD25, CD26, CD39, CD43, CD45RA, CD45RO, CD49b, CD62L, CD70, CD93, CD127, CD164, CD197, CD200, CD226, CD279, CCR10, cutaneous lymphocyte-associated antigen, TCRVβ1, TCRVβ2, TCRVβ8, and TCRVβ18 were purchased from BD Biosciences, BioLegend, Miltenyi Biotec, Beckman Coulter, or R&D Systems. Dead cells were excluded using propidium iodide or 7-amino-actinomycin D (BioLegend, catalog #420404). Single-cell suspension was ensured by filtering cells through a 100-µm cell strainer. Antibody panels were diluted in BD Brilliant Stain Buffer (BD Biosciences, catalog #563794), and cells were stained for 30 minutes at room temperature protected from light. Flow cytometric analysis was conducted using a 3- or 5-laser BD LSR-Fortessa at the Core Facility for Flow Cytometry at the University of Copenhagen.
Surface marker screening
Malignant SS cells were screened for the expression of 240 surface markers, using BD Lyoplate Human Surface Marker Screening Panel (BD Biosciences, catalog #560747). Up to 7 samples were tagged using separable combinations of CFSE, CellTrace Violet, CellTracker Violet BMQC, and Fixable Viability Dye eFluor780 (Thermo Fisher, catalog #C34557, #C10094, and #65-0865-14) to allow multiplexed analysis. After careful washing, tagging was verified to allow clear separation of the samples by flow cytometry, and samples were pooled. Staining with primary and secondary Lyoplate antibodies was conducted in accordance with manufacturer instructions. To enable identification of malignant cells, each well was stained with primary conjugated monoclonal antibodies against CD3, CD4, CD7, and CD26 after staining with the secondary Lyoplate antibodies. Dead cells were removed using 7-amino-actinomycin D, and after demultiplexing, malignant cells were gated based on low expression of CD7 and/or CD26.12,13 The 240 screened surface markers were filtered as “expressed” by having at least 10% of cells from at least 1 patient expressing more than the median fluorescence intensity (MFI) of the isotype plus 10 standard deviations (SDs) of the isotype: MFI90% quantile > (MFIIsotype + 10 × SDIsotype).
Cell sorting
Cell sorting was conducted on a 3-laser BD FACS Aria-II, using a 100-µm nozzle at the Core Facility for Flow Cytometry at the University of Copenhagen. After staining, unsorted and sorted samples were kept at 4°C throughout. Malignant cells from each patient were single-cell index sorted directly into 3 BD Precise 96-well plates (BD Genomics, catalog #634411), following the manufacturer’s instructions. Remaining malignant cells were sorted into tubes. Purity of sorted cells was consistently above 95%. Immediately after sorting, cells in tubes were pelleted, resuspended in TRI Reagent (Sigma-Aldrich, catalog #T9424), and together with BD Precise plates, stored at −80°C until library construction.
Targeted single-cell RNA sequencing
Targeted single-cell RNA sequencing libraries were constructed using the BD Precise assay, following the manufacturer’s instructions. In short, after lysis and barcoded reverse transcription, all 96 wells were pooled. Subsequently, targeted amplification was performed using the T-reg Precise Panel, consisting of probes targeting 110 T-cell-relevant genes (BD Genomics, catalog #PRC-PNL-IMM). This was followed by final library preparation by the addition of sequencing adapters and plate indices. Pooled libraries were sequenced using the Illumina MiSeq or HiSeq2500 platform at the Center for Genomic Medicine, Rigshospitalet. Demultiplexing, read mapping, and extraction of unique molecular identifier counts were performed using the BD: Precise Targeted Analysis Pipeline at the Seven Bridges Platform. Three wells randomly distributed on each 96-well plate were kept empty. Unlike wells containing single cells, these empty wells showed consistent low background read counts, with practically no reads being mapped to the target transcripts. To discard low-quality samples, only wells with mapping efficiency above 50% were included in the subsequent analysis. Further analysis and data visualization were performed using R.
Bulk RNA sequencing
RNA from sorted malignant cells was isolated using TRI Reagent/BCP (Sigma-Aldrich, catalog #B9677) purification and isopropanol precipitation, as previously described.18 Total RNA sequencing libraries were constructed using the Illumina TruSeq kit and sequenced on the Illumina HiSeq2500 platform at the Center for Genomic Medicine, Rigshospitalet. Raw RNA-Seq reads were pseudoaligned to the human transcriptome (ENSEMBL, GRCh38), using the Kallisto software. Expression values were calculated using the tximport and DEseq2 R packages, adhering to the guidelines suggested in the DEseq2 manual.19
Histone deacetylase inhibitor treatment
PBMCs isolated from patients with SS were kept in RPMI-1640 (Sigma-Aldrich, catalog #R204) containing 10% human serum (Copenhagen Hospital Bloodbank) and 1% Penicillin/Streptomycin (Sigma, catalog #P7539). Cells were treated with 4 concentrations of 2 different HDACi, starting from 4 nM depsipeptide (romidepsin) or 4 µM suberanilohydroxamic acid (SAHA/vorinostat) in fourfold dilution series. Because of dilution error, cells from SS5 were treated with slightly different HDACi concentrations than the other patients (4.0, 0.8, 0.16, and 0.032 instead of 4.0, 1.0, 0.25, and 0.06, respectively). Cells were treated for 8 days, after which they were counted and analyzed by flow cytometry. As some malignant T cells upregulate CD26 and CD7 in response to stimulation, CD26 was included in the clustering of TCRVβ+ cells. For patients for whom antibodies toward TCRVβ clone were not available, the malignant cells were gated as non-CD26+CD7+. Malignant populations were downsampled to 1000 events per treatment before dimension reduction to faithfully represent the populations present in the treated samples (with lower event counts).
Flow cytometry data analysis and visualization
Flow cytometry data were gated and manually inspected, using FlowJo (TreeStar), and subsequently loaded into R using the flowCore and flowWorkspace packages.20 t-distributed stochastic neighbor embedding (t-SNE) dimension reduction (perplexity set to 30) and automated PhenoGraph clustering were performed, using the Cytofkit package.21 All code used in the analyses included in this study is available from the authors on request.
Results
Malignant T cells display heterogeneous surface marker expression
Despite its expected origin from a single transformed clone, we hypothesize that the malignant T-cell population from patients with SS exhibits a high degree of single-cell heterogeneity within each individual patient. Loss of CD7 and CD26 are hallmark characteristics of malignant T cells from patients with SS.12,13 In our SS cohort (see Table 1 for patient characteristics), we found that neither CD7 nor CD26 alone was sufficient to define the malignant T-cell population in all patients (Figure 1A-C). However, we found that the non-CD26+CD7+ population is dramatically enriched for cells expressing the dominant TCRVβ chain (Figure 1B). For the patients for whom no dominant TCRVβ chain could be detected, the non-CD26+CD7+ population consisted almost completely of cells unmarked by the TCRVβ screening kit (Figure 1B). As the TCRVβ screening kit covers about 70% of the normal T-cell receptor repertoire, large proportions of nonreactivity to the kit have been proposed as surrogate marker of clonality.12,22 Although the dominant TCRVβ+ population is highly enriched for the malignant T-cell clone, it also contains a minor population of nonmalignant cells using the same TCRVβ chain (Figure 1C). Thus, we gated malignant cells as the (TCRVβ+) non-CD26+CD7+ population.
Figure 1.

Identification of the malignant population. (A) Gating strategy of nonmalignant CD3+CD4+CD7+CD26+ cells from 10 patients with SS (SS2-SS11). (B) TCRVβ distribution within the CD4+ non-CD26+CD7+ (malignant), CD4+CD26+CD7+ (nonmalignant), and CD8+ T cell populations. Because of incomplete antibody coverage by the TCRVβ screening kit, not all T cells can be assigned to a TCRVβ and are included in the “unmarked” fraction. (C) Assessment of the fraction of CD26+CD7+ cells within the CD4+ population expressing the dominant TCRVβ clone detected in 5 of the 10 patients with SS.
To identify surface markers that are heterogeneously expressed by the malignant T-cell population, we screened malignant cells from 8 patients with SS for the expression of 240 surface antigens, using a flow cytometry-based assay. Of these 240 markers, 86 were expressed by malignant cells (Figure 2A). Using interquantile range (90% quantile − 10% quantile fluorescence; IQR90) as a measure of heterogeneity, we could visualize the heterogeneity in surface marker expression across different patients and identify the markers that are highly heterogeneously expressed (Figure 2B). Although some markers are heterogeneously expressed in all investigated patients, we found that many markers were specific to individual patients or groups of patients (Figure 2A-B). This is consistent with the great differences in gene expression, single-nucleotide mutations, and chromosomal aberrations often observed between patients with SS.7-9,11,23
Figure 2.

Surface marker screening identifies heterogeneity within malignant population. (A-B) Heat map showing (A) MFI or (B) interquantile range (numeric difference between 90% and 10% quantiles: IQR90) of 86 surface markers expressed above isotype levels by at least 10% of the malignant population assayed by flow cytometry. (C) Histograms showing the single-cell distribution of the top 20 most heterogeneously expressed surface markers selected by highest IQR90 within at least 3 patients. Dashed lines display the relevant isotype expression. Histograms are colored by IQR90.
Looking at the cellular distribution of the top 20 most heterogeneously expressed surface markers, we observed that although some markers were expressed in a continuum, others showed clear bimodal separation indicative of distinct subpopulations (Figure 2C).
This shows that malignant cell populations of all investigated patients exhibit high degrees of heterogeneity in their surface marker expression, and that this heterogeneity can be assayed using flow cytometry.
Malignant population contains multiple distinct subpopulations
The heterogeneous surface marker expression strongly argues for the presence of distinct subpopulations within the malignant cell population in each individual patient. We analyzed coexpression of some of the most heterogeneously expressed markers by flow cytometry to formally determine whether distinct subpopulations can be isolated. Using t-SNE, we visualized single-cell expression of several markers at the same time. This allowed identification of populations exhibiting distinct combinations of surface markers within the malignant cell population (Figure 3A). We analyzed the combined expression using an automated clustering algorithm (PhenoGraph), resulting in several clusters that could be merged to convincingly represent subpopulations with distinct surface marker expression (“Reduced”; Figure 3B). Although all included markers were differentially expressed within at least 1 patient, not all patients showed similar heterogeneity of all markers (Figure 3A,C). Only cutaneous lymphocyte-associated antigen exhibited consistent bimodal expression in the malignant populations in all patients (Figure 3A,C).
Figure 3.

Coexpression of surface markers divide malignant cells into subpopulations. (A-B) Coexpression of a panel of surface markers within the malignant population of 7 patients with SS visualized by t-SNE plots colored by (A) fluorescence intensity of the indicated markers or (B) automated clustering using the PhenoGraph algorithm showing all (left) or a reduced number of clusters (right). (C) Single-cell heat maps distributed by reduced PhenoGraph cluster (left bar) and colored by fluorescence intensity of the indicated markers. Violin plots (top) display the overall expression range of the indicated markers within the total malignant population. CLA, cutaneous lymphocyte-associated antigen.
Malignant T cells display heterogeneous mRNA expression
The data presented here establish that cells from a given malignant population can be divided into different subpopulations on the basis of their surface marker expression. We conducted targeted single-cell RNA sequencing of malignant cells to determine whether the heterogeneous surface phenotypes found within the malignant population could be correlated with distinct mRNA transcription profiles. We index sorted 1.566 single malignant cells from 6 patients and analyzed their expression of a range of T-cell-relevant genes. To verify the single-cell expression, the use of index sorting allowed us to correlate surface protein and mRNA expression in each individual cell. We found reasonable correlation between the mRNA and protein expression at the single-cell level for the genes encoding proteins included in our flow cytometry panel (supplemental Figure 1A). Furthermore, we compared the average single-cell mRNA expression with bulk RNA-Seq expression from the malignant population of the same patients and found good correlation (R2 = 0.51-0.62; supplemental Figure 1B).
As was the case for the surface marker expression, the malignant cells from different patients exhibited different expression patterns. However, all patients showed heterogeneous expression of several mRNA transcripts (Figure 4A). To determine whether the single-cell mRNA expression profiles were consistent with the existence of distinct malignant subpopulations, we visualized the expression of select genes using t-SNE. Similar to their surface marker expression, single-cell mRNA expression divided the malignant population into clusters of cells exhibiting differential coexpression (Figure 4B).
Figure 4.

Single-cell mRNA sequencing confirm heterogeneity at the transcriptional level. (A) Heat maps showing the single-cell mRNA expression of malignant cells from 6 patients with SS. (B) Single-cell mRNA coexpression of CCR7, CXCR4, IL7R, LGALS1, PSMB4, PTPRC, S100A10, S100A4, and SELL within the malignant population of 6 patients with SS visualized by t-SNE plots. Color indicate the log2 transformed number of unique molecular identifier (UMI) counts for each gene from the individual cells.
Thus, despite the interpatient differences in expression of mRNA and surface proteins, the malignant population in all investigated patients could be divided into subpopulations, with distinct expression profiles at the mRNA and protein levels.
HDAC inhibitor treatment differentially affects malignant subpopulations
Having established the presence of distinct subpopulations within the malignant cells in all investigated patients with SS, we hypothesized that the high occurrence of treatment resistance found in these patients could be a result of differences in treatment sensitivity of these malignant subpopulations. Such differences in treatment sensitivity would allow treatment to eliminate some subpopulations, and thus reduce the overall malignant cell counts, but leave other subpopulations unaffected. To investigate this, we treated PBMCs from 6 patients with SS with increasing concentrations of 2 HDACi, SAHA (vorinostat), and romidepsin,3,4 and followed changes in malignant subpopulations.
As shown earlier, although we found malignant subpopulations in all investigated patients with SS, their abundance and expression pattern varied greatly from patient to patient. Thus, to capture changes in malignant subpopulations with higher precision, we expanded and customized our flow cytometry panel to each patient on the basis of the heterogeneously expressed markers identified in the surface marker screening experiment (Figure 2).
We found that cells from all patients responded to treatment, as demonstrated by the reduced malignant cell counts, in a dose-dependent manner. However, the magnitude of response to HDACi treatment varied greatly between the investigated patients. We present results from each individual patient in Figure 5 and supplemental Figures 2-6. On the basis of the customized panels, malignant cells from all patients could be divided into at least 5 subpopulations with distinct surface marker expression (Figure 4A; supplemental Figures 2A, 3A, 4A, 5A, and 6A). As exemplified by patient SS8, 0.25 nM romidepsin treatment selectively and drastically reduced in some (ie, blue and red) subpopulations while leaving the remaining subpopulations largely unaffected (ie, brown, purple, and green; Figure 5B-D). This effect was also seen using 1 µM SAHA treatment (Figure 5E-G). Indeed, although the highest concentrations of both HDACi almost completely eliminated the malignant population, the remaining few cells were all from the subpopulations that were also unaffected by lower concentrations (Figure 5C,F). Importantly, the surviving subpopulations showed largely unchanged cell numbers and exhibited constant single-cell surface marker expression after the treatment (Figure 5D,G-H). This indicates that the observed changes in the malignant population are a result of selection of subpopulations, rather than an overall change in surface marker expression. Importantly, although characterized by coexpression of other surface markers, similar differences in the treatment sensitivity of the subpopulation were found in SS cells from all investigated patients (supplemental Figures 2-6).
Figure 5.

HDAC inhibitor treatment affect some malignant subpopulations, but not all. (A) Coexpression of surface marker expression within malignant cells from a patient with SS (SS8) visualized by t-SNE plots colored by fluorescence intensity of the indicated markers or by automated clustering using the PhenoGraph algorithm showing all (left) or a reduced number of clusters (right). (B-G) Changes in the malignant subpopulations after treatment with increasing concentrations of 2 HDAC inhibitors. (B-D) Romidepsin or (E-G) SAHA colored by reduced PhenoGraph clusters. (B,E) Visualized by changes in t-SNE plots of clustered cells. (C-D,F-G) Visualized by stacked bar plots of (C,F) cluster frequency or (D,G) total cell counts. (H) Single-cell heat maps of malignant T cells treated with increasing concentrations of romidepsin. Rows are distributed by reduced PhenoGraph clusters (left) and colored by fluorescence intensity of the indicated markers. Violin plots (top) display the overall expression range of the indicated markers within the total malignant population. (I) Normalized quantification of the population diversity within the malignant population of 6 patients with SS after treatment with increasing concentrations of romidepsin or SAHA, based on distribution among PhenoGraph clusters, using different diversity indices (Shannon, Simpson, and inverse Simpson indices and Fisher’s α diversity). Diversity indices were normalized to the diversity of the untreated sample from each patient. Bars depict mean percentage ± standard error of mean of 6 patients with SS (n = 6). Note that cells from SS5 were treated with slightly different concentrations (see “Methods”).
We further hypothesized that the selective elimination of some, but not all, subpopulations would also be reflected in a reduction in the overall diversity of the malignant population. Consistent with this, we found that the diversity within the total malignant population measured by 4 diversity indices calculated from the full set of PhenoGraph clusters was reduced in all investigated patients (Figure 5I).
Together, these findings strongly support our hypothesis that some malignant subpopulations are more resistant to treatments than others, which thus strengthens the model in which malignant heterogeneity is a driver of treatment resistance in SS.
Discussion
Facilitated by the emergence of powerful single-cell methods, the importance of malignant heterogeneity in various cancers is becoming increasingly clear.24,25 Here, we employed single-cell RNA sequencing and high-level multicolor flow cytometry analysis to establish the heterogeneity of malignant cells in SS. We screened malignant SS cells for the expression of 240 surface markers by flow cytometry and confirmed that the surface marker landscape of malignant SS cells largely resembles that of memory T cells. It has been proposed that SS cells originate from central memory T (TCM) cells because of their high expression of CD45RO, CD62L, and CD197.14 This was a novel paradigm, which sparked a number of important studies elucidating the nature and the origin of the malignant clone in CTCL. In accordance, we found that a large proportion of the malignant cells from most patients have this TCM phenotype. However, this hypothesis was disputed by studies showing that the malignant population of many patients with SS also includes a population of cells expressing CD45RA, which usually marks naive T (TN) or stem-cell memory T (TSCM) cells, indicating plasticity in expression of these markers.15,16 It is appealing to assign malignant SS cells to a known T-cell phenotype to infer their cellular origin. However, our data show that the malignant populations exhibit highly heterogeneous expression of many classical T-cell markers and contain multiple populations with coexpression patterns not matching the conventional T-cell classifications (ie, CD45RA+CD45RO+ and CD45RA+CD279+ cells). Furthermore, in 7 of the 9 patients with SS included in the analysis, we found a notable population of CD197+CD62Lneg not resembling classical memory cells. Indeed, despite surface expression matching TN, TCM, or TSCM, Roelens and colleagues reported that at the transcriptional level, SS cells cluster together, rather than with TN, TCM, or TSCM cells from healthy donors.16 Together with our data, this warrants caution when inferring functional similarity between malignant and healthy cells, based on common surface marker expression.
Several studies have identified features of malignant T cells from patients with SS that differentiate them from the majority of nonmalignant T cells. These include high scatter characteristics with abnormal Sézary cell morphology, as well as increased expression of CD60, CD158k (KIR3DL2), CD164, and CD307c (FCRL3) and loss of expression of CD2, CD7, CD26, and CD49d.15,26-31 In our SS cohort, we obtained the most consistently defined malignant population using CD7 and CD26 together with the dominant TCRVβ clone (when possible). Although the vast majority of nonmalignant T cells are CD26+CD7+, a minority of these cells may lose expression of CD7 and CD26.27 As a consequence, in the absence of more selective markers of malignancy, gating malignant cells as non-CD26+CD7+ cells potentially allows a minor fraction of nonmalignant T cells to be included in the malignant population. We have not directly assessed this possible contamination, but the magnitude can be estimated by the presence of polyclonal T cells within the non-CD26+CD7+ population for patients with a malignant population unmarked by the TCRVβ screening kit (2.5%-12.9%; Figure 1B). For the TCRVβ+ gated patients, the contamination can be assumed to be smaller than the size of the TCRVβ+CD26+CD7+ population (0.0%-7.7%; Figure 1C), as this is the dominant phenotype of nonmalignant T cells. Thus, nonmalignant contamination may contribute to the identified heterogeneity. However, as this estimated contamination is much smaller than the identified malignant subpopulations, we find it highly unlikely to be a major contributor.
We analyzed the single-cell mRNA expression of malignant SS cells with probes targeting a panel of 110 T-cell-related genes. Surprisingly, the vast majority of the expressed genes contained in this panel were heterogeneously expressed between different malignant cells. Only a cluster of 5 genes (S100A4, S100A10, IL7R, CCR7, and CXCR4) was highly expressed by most malignant cells. Among these genes, IL7R, CCR7, and CXCR4 encode well-established molecules with defined function in growth and migration of malignant T cells.14,32-35 Yet, even these “classical” biomarkers of CTCL displayed heterogeneous expression within some patients, suggesting that cellular heterogeneity among malignant T cells may also pose difficulties using the novel therapeutic strategies targeting these proteins. Although some of this heterogeneity is likely reflecting the limitations in the sensitivity of the used single-cell RNA sequencing assay, it is intriguing that the 2 cancer-related S100A genes (S100A4 and S100A10),36 which have not been previously investigated in CTCL, show ubiquitous high expression within the malignant population.
It has recently been shown that HDACi treatment is associated with global increases in DNA accessibility and that HDACi accentuate the existing accessibility rather than evoking new accessible DNA elements.37 Qu et al also demonstrate that although HDACi treatment in vivo reduced malignant T-cell counts, the DNA accessibility changes in the nonmalignant compartment are best aligned with the observed normalization of the chromatin signature.37 In the present study, we did not investigate DNA accessibility, but found consistent reductions in malignant T-cell numbers after HDACi treatment in vitro, whereas the nonmalignant T-cell numbers were less affected. This could indicate a dual effect of HDACi treatment: both reducing malignant cell numbers and normalizing the nonmalignant immune system. Future studies should aim at verifying the DNA accessibility results from Qu et al at the single-cell level, as such studies can distinguish changes within the entire malignant population from changes within or as a result of elimination of distinct malignant subpopulations.
Development of resistance to treatment is a major issue in the management of advanced-stage CTCL.5,38,39 We show that the treatment with HDACi can efficiently reduce cell numbers by eliminating the major malignant subpopulations. Yet certain subpopulations displayed considerable resistance to the effect of HDACi in vitro, and we hypothesize that similar subpopulations may escape treatment in patients in vivo, founding the cellular basis for a subsequent outgrowth of malignant SS cells. As some subpopulations are unaffected, HDACi treatment effectively selects for the least-sensitive malignant cells, potentially leading to full-scale treatment resistance and relapse of uncontrollable disease, as commonly seen in these patients.
Such a mechanism for treatment resistance is analogous to how bacteria develop resistance toward most antibiotics. As a consequence, albeit not without pitfalls (such as drug-drug interactions), our results support the simultaneous use of multiple treatments targeting different malignant subpopulations that could lead to full eradication of the malignant T cells overall. To determine the right treatment combinations, a targeted approach would greatly benefit from detailed characterization of the subpopulations of malignant cells in a personalized manner. In this study, we show that such characterization is indeed feasible, using fast and high-throughput methods such as flow cytometry, which is already implemented at most treatment facilities.
Acknowledgments
The authors thank the patients who donated the samples and made this study possible. They also thank the extracorporal photopheresis team at Copenhagen University Hospital, Bispebjerg, for their participation in providing the patient material, as well as the Core Facility for Flow Cytometry at University of Copenhagen and the Center for Genomic Medicine at Rigshospitalet for their technical assistance.
This work was supported by research funding from The Danish Cancer Society, the Fight Cancer Program (Knæk Cancer), Novo Nordisk Research Foundation, Novo Nordic Foundation Tandem Program, The Lundbeck Foundation, The Danish Council for Independent Research (Danmarks Frie Forskningsfond), 2 project grants for N.Ø., a Sapere Aude Talent Grant (DFF-4092-00122) for T.K., LINAK A/S, and Dansk Kræftforsknings Fond.
Supplementary Material
The full-text version of this article contains a data supplement.
Authorship
Contribution: T.B.B and N.Ø. conceived the study and interpreted the data; T.B.B. performed the research and analyzed the data; T.B.B., A.W.-O., S.F., E.B., C.N., M.G., T.H., I.V.L., J.L.P., C.M.B., C.G., T.K., A.W., and N.Ø. designed the research and wrote the manuscript; H.F. and R.G. were responsible for patient material and, together with L.M.L. and L.I., provided important clinical insights; T.L. assisted in bioinformatical data analysis; J.P.C. provided technical expertise; and all authors read, commented on, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Niels Ødum, Blegdamsvej 3, Building: 07-12-62, DK-2200 Copenhagen N, Denmark; e-mail: ndum@sund.ku.dk.
References
- 1.Krejsgaard T, Lindahl LM, Mongan NP, et al. Malignant inflammation in cutaneous T-cell lymphoma-a hostile takeover. Semin Immunopathol. 2017;39(3):269-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28(31):4730-4739. [DOI] [PubMed] [Google Scholar]
- 3.Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109(1):31-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410-5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilcox RA. Cutaneous T-cell lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91(1):151-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janiga J, Kentley J, Nabhan C, Abdulla F. Current systemic therapeutic options for advanced mycosis fungoides and Sézary syndrome. Leuk Lymphoma. 2018;59(3):562-577. [DOI] [PubMed] [Google Scholar]
- 7.da Silva Almeida AC, Abate F, Khiabanian H, et al. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat Genet. 2015;47(12):1465-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi J, Goh G, Walradt T, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47(9):1011-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ungewickell A, Bhaduri A, Rios E, et al. Genomic analysis of mycosis fungoides and Sézary syndrome identifies recurrent alterations in TNFR2. Nat Genet. 2015;47(9):1056-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fanok MH, Sun A, Fogli LK, et al. Role of dysregulated cytokine signaling and bacterial triggers in the pathogenesis of cutaneous T-cell lymphoma. J Invest Dermatol. 2018;138(5):1116-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weed J, Gibson J, Lewis J, et al. FISH panel for leukemic CTCL. J Invest Dermatol. 2017;137(3):751-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibson JF, Huang J, Liu KJ, et al. Cutaneous T-cell lymphoma (CTCL): Current practices in blood assessment and the utility of T-cell receptor (TCR)-Vβ chain restriction. J Am Acad Dermatol. 2016;74(5):870-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105(10):3768-3785. [DOI] [PubMed] [Google Scholar]
- 14.Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010;116(5):767-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moins-Teisserenc H, Daubord M, Clave E, et al. CD158k is a reliable marker for diagnosis of Sézary syndrome and reveals an unprecedented heterogeneity of circulating malignant cells. J Invest Dermatol. 2015;135(1):247-257. [DOI] [PubMed] [Google Scholar]
- 16.Roelens M, Delord M, Ram-Wolff C, et al. Circulating and skin-derived Sézary cells: clonal but with phenotypic plasticity. Blood. 2017;130(12):1468-1471. [DOI] [PubMed] [Google Scholar]
- 17.Kirsch IR, Watanabe R, O’Malley JT, et al. TCR sequencing facilitates diagnosis and identifies mature T cells as the cell of origin in CTCL. Sci Transl Med. 2015;7(308):308ra158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buus TB, Ødum N, Geisler C, Lauritsen JPH. Three distinct developmental pathways for adaptive and two IFN-γ-producing γδ T subsets in adult thymus. Nat Commun. 2017;8(1):1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hahne F, LeMeur N, Brinkman RR, et al. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics. 2009;10(1):106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, Chen J. Cytofkit: a bioconductor package for an integrated mass cytometry data analysis pipeline. PLOS Comput Biol. 2016;12(9):e1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morice WG, Katzmann JA, Pittelkow MR, el-Azhary RA, Gibson LE, Hanson CA. A comparison of morphologic features, flow cytometry, TCR-Vbeta analysis, and TCR-PCR in qualitative and quantitative assessment of peripheral blood involvement by Sézary syndrome. Am J Clin Pathol. 2006;125(3):364-374. [PubMed] [Google Scholar]
- 23.Litvinov IV, Tetzlaff MT, Thibault P, et al. Gene expression analysis in cutaneous T-cell lymphomas (CTCL) highlights disease heterogeneity and potential diagnostic and prognostic indicators. OncoImmunology. 2017;6(5):e1306618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323-334. [DOI] [PubMed] [Google Scholar]
- 25.Qian M, Wang DC, Chen H, Cheng Y. Detection of single cell heterogeneity in cancer. Semin Cell Dev Biol. 2017;64:143-149. [DOI] [PubMed] [Google Scholar]
- 26.Scala E, Narducci MG, Amerio P, et al. T cell receptor-Vbeta analysis identifies a dominant CD60+ CD26- CD49d- T cell clone in the peripheral blood of Sézary syndrome patients. J Invest Dermatol. 2002;119(1):193-196. [DOI] [PubMed] [Google Scholar]
- 27.Scala E, Abeni D, Pomponi D, et al. The role of 9-O-acetylated ganglioside D3 (CD60) and alpha4beta1 (CD49d) expression in predicting the survival of patients with Sezary syndrome. Haematologica. 2010;95(11):1905-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernengo MG, Novelli M, Quaglino P, et al. The relevance of the CD4+ CD26- subset in the identification of circulating Sézary cells. Br J Dermatol. 2001;144(1):125-135. [DOI] [PubMed] [Google Scholar]
- 29.Harmon CB, Witzig TE, Katzmann JA, Pittelkow MR. Detection of circulating T cells with CD4+CD7- immunophenotype in patients with benign and malignant lymphoproliferative dermatoses. J Am Acad Dermatol. 1996;35(3 Pt 1):404-410. [DOI] [PubMed] [Google Scholar]
- 30.Benoit BM, Jariwala N, O’Connor G, et al. CD164 identifies CD4+ T cells highly expressing genes associated with malignancy in Sézary syndrome: the Sézary signature genes, FCRL3, Tox, and miR-214. Arch Dermatol Res. 2017;309(1):11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark RA, Shackelton JB, Watanabe R, et al. High-scatter T cells: a reliable biomarker for malignant T cells in cutaneous T-cell lymphoma. Blood. 2011;117(6):1966-1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narducci MG, Scala E, Bresin A, et al. Skin homing of Sézary cells involves SDF-1-CXCR4 signaling and down-regulation of CD26/dipeptidylpeptidase IV. Blood. 2006;107(3):1108-1115. [DOI] [PubMed] [Google Scholar]
- 33.Kremer KN, Dinkel BA, Sterner RM, Osborne DG, Jevremovic D, Hedin KE. TCR-CXCR4 signaling stabilizes cytokine mRNA transcripts via a PREX1-Rac1 pathway: implications for CTCL. Blood. 2017;130(8):982-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foss FM, Koc Y, Stetler-Stevenson MA, et al. Costimulation of cutaneous T-cell lymphoma cells by interleukin-7 and interleukin-2: potential autocrine or paracrine effectors in the Sézary syndrome. J Clin Oncol. 1994;12(2):326-335. [DOI] [PubMed] [Google Scholar]
- 35.Hahtola S, Tuomela S, Elo L, et al. Th1 response and cytotoxicity genes are down-regulated in cutaneous T-cell lymphoma. Clin Cancer Res. 2006;12(16):4812-4821. [DOI] [PubMed] [Google Scholar]
- 36.Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer. 2015;15(2):96-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qu K, Zaba LC, Satpathy AT, et al. Chromatin accessibility landscape of cutaneous t cell lymphoma and dynamic response to HDAC inhibitors. Cancer Cell. 2017;32(1):27-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakraborty AR, Robey RW, Luchenko VL, et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with an MEK inhibitor. Blood. 2013;121(20):4115-4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fantin VR, Loboda A, Paweletz CP, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68(10):3785-3794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.